

Abstract
Efficient T cell activation depends on the engagement of both TCR and CD28, although the molecular mechanisms that control this signal integration are not fully understood. Using fluorescence resonance energy transfer, we show that T cell activation can drive a reorientation of the cytosolic tails of the CD28 dimer. However, this is not mediated through CD28 ligand binding. Rather, TCR signaling itself mediates this conformation change in CD28. We also show that TCR signaling can induce CD28-ligand interactions. Although the CD28 dimer appears to bind ligand monovalently in solution, we show that both ligand-binding sites are required to efficiently recruit CD28 to the immunological synapse. These results suggest, that analogous to the cross talk from TCR that regulates integrin activation, TCR-initiated inside-out signaling may induce a conformational change to the extracellular domains of CD28, enabling ligand binding and initiating CD28 signaling.
Introduction
Efficient T cell activation requires the spatial and temporal coordination of several different cell surface proteins. Initial signaling through the TCR results in the rapid upregulation of both the affinity and avidity of LFA-1 for its ligand, ICAM-1 (1, 2). This increased adhesion results in arrest of migration and the formation of an immunological synapse (3, 4). T cell activation occurs within the context of this synapse and involves signal integration between the TCR and costimulatory receptors, most notably, CD28 (5–7). Although the biological significance of CD28 costimulation is well documented (8, 9), the cellular and molecular mechanisms that regulate CD28 ligand binding, CD28 triggering, and initiation of CD28 signaling are poorly understood.
Most of the functions of CD28 are associated with modulation of TCR function, primarily by enhancing the magnitude of TCR signaling (10). However, it is becoming clear that there is significant cross-talk and coregulation between these two receptors. TCR signaling in the absence of CD28 does not lead to effective T cell activation and rather favors the induction of tolerance. At the same time, ligands for CD28 are expressed on dendritic cells and macrophages in lymphoid tissue (11, 12), yet CD28 ligand binding and signaling is not induced. Although it is possible to activate T cells through engagement of CD28 alone (13), under normal conditions coengagement of TCR is required to initiate CD28 signaling. Finally, sustained TCR signaling is required to retain CD28 localization within the immunological synapse (14, 15). In this report we show that TCR signaling can induce a reorientation of the cytosolic tail domains of CD28, which is independent of CD28 ligand binding. Additionally, TCR signaling can induce CD28-ligand interactions, suggesting that TCR mediated inside-out signaling may coordinate ligand binding and signal transduction of both adhesion and costimulatory receptors that are essential for T cell activation.
Materials and Methods
Cells
CD4-positive lymph node T cells were purified by negative selection from wild type (WT) or CD28-deficient (CD28KO), DO11.10 TCR transgenic mice as previously described (16) and activated in vitro with OVA peptide 323–339 presented by irradiated BALB/c splenocytes and 10 U/ml of rhIL-2. 6132 Pro cell transfectants co-expressing I-Ad, ICAM-1, and CD80 (6) were used as APC in all experiments, except for where transfectants expressing IAd and ICAM-1 were used as B7-negative APC. Cells were maintained in DMEM (Invitrogen Life Tech) supplemented with 10% FCS, 2 mM glutamine, 0.1 mM non-essential amino acids, and 50 μM 2-ME. All mouse experiments were approved by the University Committee on Animal Resources at the University of Rochester.
T cell transfection
Monomeric cyan fluorescent protein (mCFP) and monomeric yellow fluorescent protein (mYFP) N1 plasmids were described previously (17). Murine CD28 was mutated at Y104A, within the ligand binding site, by overlapping PCR and the mutation was confirmed by DNA sequencing. Mouse and human WT CD28 and mouse Y104A CD28 were cloned upstream of the fluorescent proteins with a 4 amino acid linker (RSTG), to produce the CD28-mCFP or CD28-mYFP fusion proteins. For bi-molecular fluorescence complementation, the monomeric variant of Venus or N-terminal (1–154) or C-terminal (155–238) fragments of Venus was cloned downstream of mouse WT or Y104A CD28. Previously activated T cells were rested for 7–14 days and electroporated using Amaxa Nucleofection (Lonza Cologne AG). Briefly, 4μg of plasmid DNA were transfected into 5 ×106 cells, the cells were rested for 5 hours or overnight to allow CD28 fusion protein expression and then analyzed by flow cytometry and fluorescence microscopy.
Fluorescence resonance energy transfer (FRET)
For T cell:APC conjugates, APC were pre-loaded with or without 2 μg/ml OVA peptide, centrifuged (20 second at 2000 × g) with T cells, incubated for 5 min at 37°C, resuspended in complete media, and plated in Delta T dishes. For anti-CD3 cross-linking, T cells were incubated with 1μg/ml of biotinylated anti-CD3 (2C11; BD Biosciences) for 5 minutes at 37°C, plated in Delta T dishes, and incubated for 10 minutes in 1μg/ml of strepavidin-Cy5 (Jackson ImmunoResearch Laboratories). TCR signaling was inhibited by addition of 5 μM PP2 (Axxora LLC, San Diego, CA). Live cell imaging was done at 37° C on a Nikon Eclipse E2000-E microscope with a 60× oil immersion objective as described (17). CFP and YFP images were captured before and after YFP photobleaching. Image analysis was done within an optical segment of the plasma membrane. After subtraction of background, FRET efficiency was calculated as: FRET Efficiency = [(CFPpost-CFPpre)/CFPpost]*100.
CD28 localization
T cell:APC conjugates were formed as described above or in combination with 4.5μ latex beads coated with 1 μg/ml anti-CD3 (2C11) or anti-MHC class I (34-2-12; BD Biosciences) and plated on poly-L-lysine coated coverslips. Samples were fixed in 3% paraformaldehyde and were imaged at room temperature on a Zeiss Axiovert microscope with a 63× oil immersion objective. Conjugates were scored for recruitment of CD28 to the B7-positive cell and shown as percent of conjugates. For bi-molecular fluorescence complementation, the efficiency of CD28 localization to the immunological synapse was determined on no-neighbor deconvolved images and represented as a ratio of Venus fluorescence within the immunological synapse over Venus fluorescence outside of the immunological synapse.
Results and discussion
To address the molecular events associated with CD28 activation, we have established a FRET-based assay to detect changes in the orientation of the cytosolic tail domains of CD28. Both CD28-CFP and CD28-YFP fusion proteins were expressed at the cell surface (Fig. 1A) and restored CD28 function in CD28KO T cells (Supplemental Fig. 1). Resting T cells coexpressing both CD28-CFP and CD28-YFP showed a high FRET efficiency (Fig. 1B). The level of energy transfer was independent of the overall levels and ratio of CFP and YFP fluorescence, indicating that FRET was likely mediated by intramolecular interactions within the CD28 homodimer, rather than by CD28 clustering (Supplemental Fig. 2, A–D). Mouse and human CD28 do not efficiently form interspecies heterodimers (18). High FRET efficiency was detected in both mouse/mouse and human /human homodimers, but significantly reduced when mouse CD28-CFP and human CD28-YFP were coexpressed (Supplemental Fig. 2E). These results and the relatively high levels of FRET detected indicated that the C-termini of the CD28 cytosolic tails are in close proximity within the CD28 homodimer (Supplemental Fig. 2F).
FIGURE 1.

CD28 FRET efficiency is reduced when CD28 is recruited to the immunological synapse during T cell activation. T cells expressing CD28-YFP and CD28-CFP were analyzed as isolated resting T cells or in conjugates with antigen-pulsed APC. A, Representative images showing efficient recruitment of both CD28-CFP and CD28-YFP to the immunological synapse. B, FRET efficiency of individual T cells in the absence of activation (alone) or within the pool of CD28 that was recruited to the immunological synapse (IS). Lines represent median FRET values; population distributions were compared by nonparametric Mann-Whitney (p<0.0001).
Both CD28-CFP and CD28-YFP were efficiently recruited to the immunological synapse in T cell:APC conjugates (Fig. 1A). Strikingly, CD28 FRET efficiency was significantly reduced within the immunological synapse (Fig. 1B). These results indicate that some reorientation of the cytosolic tail domains within the CD28 homodimer takes places during T cell activation.
Surprisingly the reduction of CD28 FRET was not dependent on CD28 ligand binding (Fig. 2). CD28-CFP and CD28-YFP were mutated within the CD28 ligand-binding site (Y104A) (19, 20). When CD28 cannot bind ligand it is neither recruited to nor excluded from the immunological synapse, so CD28 remains evenly distributed across the plasma membrane (Fig. 2A). Therefore, we measured CD28 FRET efficiency of CD28 that was present at the interface with the APC and within the pool of CD28 that was localized outside of the contact region. FRET efficiency was significantly reduced even in the absence of ligand binding, but only within the pool of CD28 that was localized within the immunological synapse (Fig. 2B). These results were confirmed with WT T cells and B7-deficient APC, where the change in CD28 FRET efficiency was dependent on Ag stimulation, but not CD28-B7 interactions (Fig. 2C). Taken together, these results suggest that the reduced FRET between the cytosolic tail domains in CD28 reflects a conformational change within the CD28 homodimer that is induced prior to CD28 ligand binding and signaling.
FIGURE 2.

The reduction in CD28 FRET detected during T cell activation is mediated through TCR signaling, not by CD28 ligand binding. A, Representative images of T cells expressing Y104A CD28-YFP and Y104A CD28-CFP. B, FRET efficiency was measured in isolated T cells (alone) and for CD28 localized within the contact region (IS) and distal to the contact region (Back) in T cell:APC conjugates. C, T cells expressing WT CD28-YFP and WT CD28-CFP in conjugates with APC that do not express B7. FRET efficiency was measured in isolated T cells (alone) and for CD28 localized within the contact region (IS) and distal to the contact region (Back) in T cell:APC conjugates in the presence and absence of Ag. D, T cells expressing WT CD28-YFP and WT CD28-CFP were stimulated by anti-CD3 cross-linking (CD3CL) or with anti-CD3 and the Src family kinase inhibitor, PP2. FRET values, population medians, and p values (nonparametric Mann-Whitney) are shown.
These data suggest that the reorientation of cytosolic tail domains within the CD28 homodimer is not mediated by CD28 ligand binding, but rather by some proximal signaling event associated with T cell activation. To address the role of TCR signaling, we activated T cells expressing CD28-CFP and CD28-YFP by anti-CD3 cross-linking. T cells stimulated with anti-CD3 showed a substantial decrease in FRET around the entire T cell plasma membrane (Fig. 2D). The difference in the distribution of FRET change between antigen (Fig. 2, B and C) and anti-CD3 (Fig. 2D) stimulation could reflect differences in the magnitude or kinetics of TCR signaling or simply the delivery of polarized (APC) or non-polarized TCR signals (anti-CD3 cross-linking). Inhibition of TCR signaling by addition of the Src-family kinase inhibitor, PP2, blocked the anti-CD3-induced reduction in CD28 FRET (Fig. 2D). These results indicate that the reorientation of the CD28 cytosolic tail domains detected during T cell activation can be mediated by TCR signaling.
The ability of TCR signaling to drive a change in the orientation of the cytosolic tail domains of CD28 is reminiscent of inside-out signaling that regulates integrin activation and ligand binding (1, 2). We and others have previously shown that inhibition of TCR signaling after a functional immunological synapse is formed results in a rapid loss of CD28 polarization from the synapse, even though the T cell:APC conjugate remains intact and CD28 ligands on the APC are still available for CD28 binding (14, 15). To determine whether TCR signaling could induce the initiation of CD28-ligand binding, we formed conjugates with B7-positive cells in the absence of Ag (Fig. 3). Under these conditions in the absence of any TCR signaling, CD28 is not recruited to the B7-positive cell, in spite of the availability of CD28 ligand (Fig. 3A). Strikingly, T cell activation with anti-CD3 coated beads induced the rapid redistribution of CD28 toward the B7-positve cell (Fig. 3, A and B). Control anti-MHC class I-coated beads (Fig. 3B) and addition of anti-CD3-coated beads in the absence of an interacting cell had no effect on CD28 localization (not shown). Thus, TCR signaling at a localized site (anti-CD3 coated beads) can induce CD28-ligand interactions at a distal site (B7-positive cell), suggesting that TCR signaling can enhance CD28 ligand binding.
FIGURE 3.

TCR engagement can induce CD28 ligand binding. CD28KO T cells were transduced with CD28-YFP and used to form conjugates with B7-positive APC in the presence of Ag or in the absence of Ag alone or with anti-MHC class I or anti-CD3 coated beads. Representative images (A) and the frequency of conjugates (n=30–40) with CD28 polarized toward the APC (B) are shown. P values are from two population proportion Z-student tests.
One potential mechanism whereby TCR signaling could impact CD28 ligand interaction is to alter the valency of CD28 ligand binding. CD28 is expressed as a disulfide linked homodimer, however biochemical and structural analysis of recombinant, soluble CD28 indicate that it can only interact with ligand monovalently (21, 22). Although both binding sites in the CD28 dimer are accessible to ligand, their orientation is thought to preclude simultaneous ligand binding, because of steric interference at the distal end of the ligand. However, when CD28 is expressed as a transmembrane protein, TCR inside-out signaling may transduce a conformational change to the ligand binding domains, enabling bivalent binding. To address the role of bivalent CD28 binding during recruitment of CD28 to the immunological synapse, we utilized bimolecular fluorescence complementation (23) to selectively label CD28 heterodimers that express only a single, functional ligand-binding site. WT CD28 was fused to intact Venus, the N-terminal fragment of Venus (CD28-VN), or to the C-terminal fragment of Venus (CD28-VC). Coexpression of WT CD28-VN and WT CD28-VC results in acquisition of Venus fluorescence and CD28-VN/CD28-VC dimers were efficiently recruited to the immunological synapse (Supplemental Fig. 3). These results indicate that reassembly of the fragments of Venus on the C-terminus of CD28 does not interfere with CD28 expression, ligand binding, or synapse localization.
When CD28 Y104A-VN and Y104A-VC were coexpressed, the heterodimer reconstituted Venus fluorescence and was expressed at the plasma membrane, but this dimer does not have a ligand-binding site and was not recruited to the immunological synapse (Fig. 4). To determine whether a single ligand-binding site was sufficient to drive CD28 localization to the immunological synapse, T cells were co-transfected with WT CD28-VN and Y104A CD28-VC. In this case, Venus fluorescence was only reconstituted in CD28 dimers that contained a single, functional ligand-binding site. Consistent with previous results (20), coexpression of WT CD28 did facilitate recruitment of Y104A CD28 to the immunological synapse. The selective labeling of CD28 dimers containing two, one or no ligand-binding sites using bimolecular fluorescence complementation allowed us to quantify the relative recruitment efficiency of these dimers. This analysis revealed a clear defect in the recruitment of dimers with only a single ligand-binding site (Fig. 4). Thus, the presence of two functional ligand-binding sites is critical for efficient recruitment of CD28 dimers to the immunological synapse.
FIGURE 4.
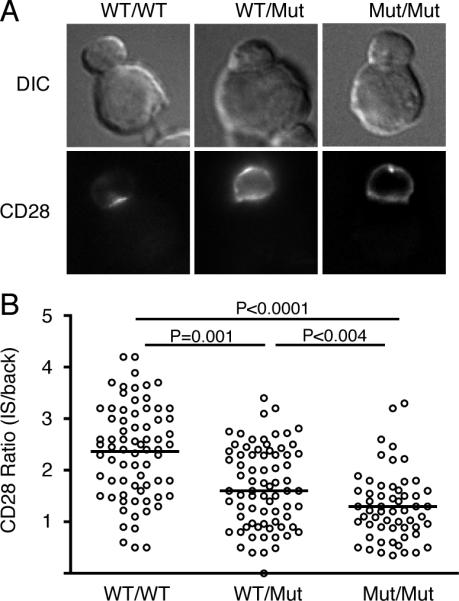
CD28 requires both ligand-binding sites within a dimer to be efficiently recruited to the immunological synapse. CD28KO T cells were transfected with WT-VN and WT-VC (WT/WT), with WT-VN and Y104A-VC (WT/Mut), or with Y104A-VN and Y104A-VC (Mut/Mut) and T cell:APC conjugates were analyzed for localization of CD28 dimers by fluorescence of reassembled VN/VC fragments. A, Representative images. B, The efficiency of CD28 recruitment to the immunological synapse (ratio of CD28 IS/back), population medians and p values (Mann-Whitney) are shown.
In this report we show that TCR signaling can drive reorientation of the cytosolic tail domains of CD28 and induce CD28 ligand binding suggesting that TCR may regulate bidirectional CD28 signaling. The ability of cell surface receptor signaling to dynamically regulate ligand binding of a different receptor is well established in the regulation of integrin activation (1, 2). For integrins, TCR-dependent activation of RAP-1 leads to recruitment of cytosolic proteins to the integrin cytosolic tails. This drives separation of the integrin cytosolic tails, which can be detected as a loss in FRET efficiency (24), and a corresponding change in the conformation of the extracellular domains that increases the affinity for ligand and allows for the induction of integrin signaling. Whether TCR-induced changes in the orientation of the CD28 cytosolic tail domains is also mediated through downstream signaling or some localized change within the plasma membrane is not known. Our results suggest that inside-out signaling from TCR may regulate both the primary adhesion molecule, LFA-1, and the primary costimulatory molecule, CD28, involved in T cell activation and provides a molecular paradigm to coordinate the sequential receptor activation events that control complex cellular processes.
We also show that CD28 depends on two functional ligand-binding sites for efficient recruitment to the immunological synapse. Our results favor a model whereby TCR-induced inside-out signal through the CD28 cytosolic tail domains drives a corresponding conformational change in the extracellular domain of CD28 that facilitates bivalent ligand binding. Interestingly, bivalent ligand binding to CD28 through superagonist mAb or CTLA-4/CD28 fusion proteins results in T cell activation in the absence of TCR engagement (25), raising the possibility that TCR can activate CD28 both to enhance ligand binding and potentiate CD28 signaling. Although historically CD28 has been thought to modulate TCR signaling, our results support the idea that TCR signaling can also strongly regulate CD28 function. Under super-optimal situations, both TCR and CD28 alone can activate T cells, so both receptors can signal independently. But under normal situations, synergistic cross-talk between TCR and CD28 provides a mechanism for coincidence detection to regulate T cell activation and control the initiation of T cell immune responses.
Supplementary Material
Acknowledgements
We thank Young-Min Hyun for advice and Deb Fowell and Kayla Garrett for comments on the manuscript.
This work was supported by NIH grant (AI090259) to JM.
Abbreviations used in this article
- CD28KO
CD28-deficient
- FRET
fluorescence resonance energy transfer
- WT
wild type
References
- 1.Kinashi T. Intracellular signalling controlling integrin activation in lymphocytes. Nat. Rev. Immunol. 2005;5:546–559. doi: 10.1038/nri1646. [DOI] [PubMed] [Google Scholar]
- 2.Shattil SJ, Kim C, Ginsberg MH. The final steps of integrin activation: the end game. Nat. Rev. Mol. Cell Biol. 2010;11:288–300. doi: 10.1038/nrm2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fooksman DR, Vardhana S, Vasiliver-Shamis G, Liese J, Blair DA, Waite J, Sacristán C, Victora GD, Zanin-Zhorov A, Dustin ML. Functional anatomy of T cell activation and synapse formation. Annu. Rev. Immunol. 2010;28:79–105. doi: 10.1146/annurev-immunol-030409-101308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yokosuka T, Saito T. Dynamic regulation of T-cell costimulation through TCR-CD28 microclusters. Immunol. Rev. 2009;229:27–40. doi: 10.1111/j.1600-065X.2009.00779.x. [DOI] [PubMed] [Google Scholar]
- 5.Huang J, Lo P, Zal T, Gascoigne N, Smith B, Levin S, Grey H. CD28 plays a critical role in the segregation of PKCθ within the immunological synapse. Proc. Natl. Acad. Sci. (USA) 2002;99:9369–9373. doi: 10.1073/pnas.142298399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sanchez-Lockhart M, Marin E, Graf B, Abe R, Harada Y, Sedwick CE, Miller J. CD28-mediated transcriptional and posttranscriptional regulation of IL-2 expression are controlled through different signaling pathways. J. Immunol. 2004;173:7120–7124. doi: 10.4049/jimmunol.173.12.7120. [DOI] [PubMed] [Google Scholar]
- 7.Yokosuka T, Kobayashi W, Sakata-Sogawa K, Takamatsu M, Hashimoto-Tane A, Dustin ML, Tokunaga M, Saito T. Spatiotemporal regulation of T cell costimulation by TCR-CD28 microclusters and protein kinase C theta translocation. Immunity. 2008;29:589–601. doi: 10.1016/j.immuni.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Riley JL, June CH. The CD28 family: a T-cell rheostat for therapeutic control of T-cell activation. Blood. 2005;105:13–21. doi: 10.1182/blood-2004-04-1596. [DOI] [PubMed] [Google Scholar]
- 9.Rudd CE, Taylor A, Schneider H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol. Rev. 2009;229:12–26. doi: 10.1111/j.1600-065X.2009.00770.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Acuto O, Michel F. CD28-mediated co-stimulation; A quantitative support for TCR signaling. Nat. Rev. Immunol. 2003;3:939–951. doi: 10.1038/nri1248. [DOI] [PubMed] [Google Scholar]
- 11.Inaba K, Witmer-Pack M, Inaba M, Hathcock KS, Sakuta H, Azuma M, Yagita H, Okumura K, Linsley PS, Ikehara S, Muramatsu S, Hodes RJ, Steinman RM. The tissue distribution of the B7-2 costimulator in mice: abundant expression on dendritic cells in situ and during maturation in vitro. J. Exp. Med. 1994;180:1849–1860. doi: 10.1084/jem.180.5.1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vyth-Dreese F, Dellemijn T, Majoor D, de Jong D. Localization in situ of the co-stimulatory molecules B7.1, B7.2, CD40 and their ligands in normal human lymphoid tissue. Eur. J. Immunol. 1995;25:3023–3029. doi: 10.1002/eji.1830251106. [DOI] [PubMed] [Google Scholar]
- 13.Luhder F, Huang Y, Dennehy K, Guntermann C, Muller I, Winkler E, Kerkau T, Ikemizu S, Davis S, Hanke T, Hunig T. Topological requirements and signaling properties of T cell-activting, anti-CD28 antibody superagonists. J. Exp. Med. 2003;197:955–966. doi: 10.1084/jem.20021024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanchez-Lockhart M, Graf B, Miller J. Signals and sequences that control CD28 localization to the central region of the immunological synapse. J. Immunol. 2008;181:7639–7648. doi: 10.4049/jimmunol.181.11.7639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tseng SY, Waite JC, Liu M, Vardhana S, Dustin ML. T cell-dendritic cell immunological synapses contain TCR-dependent CD28-CD80 clusters that recruit protein kinase Ctheta. J. Immunol. 2008;181:4852–4863. doi: 10.4049/jimmunol.181.7.4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zuckerman LA, Pullen L, Miller J. Functional consequences of costimulation by ICAM-1 on IL-2 gene expression and T cell activation. J. Immunol. 1998;160:3259–3268. [PubMed] [Google Scholar]
- 17.Lefort CT, Hyun YM, Schultz JB, Law FY, Waugh RE, Knauf PA, Kim M. Outside-in signal transmission by conformational changes in integrin Mac-1. J. Immunol. 2009;183:6460–6468. doi: 10.4049/jimmunol.0900983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Truitt KE, Nagel T, Suen LF, Imboden JB. Structural requirements for CD28-mediated costimulation of IL-2 production in Jurkat T cells. J. Immunol. 1996;156:4539–4541. [PubMed] [Google Scholar]
- 19.Peach RJ, Bajorath J, Brady W, Leytze G, Greene J, Naemura J, Linsley PS. Complementarity determining region 1 (CDR1)- and CDR3-analogous regions in CTLA-4 and CD28 determine the binding to B7-1. J. Exp. Med. 1994;180:2049–2058. doi: 10.1084/jem.180.6.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pentcheva-Hoang T, Egen JG, Wojnoonski K, Allison JP. B7-1 and B7-2 selectively recruit CTLA-4 and CD28 to the immunological synapse. Immunity. 2004;21:401–413. doi: 10.1016/j.immuni.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 21.Collins AV, Brodie DW, Gilbert RJ, Iaboni A, Manso-Sancho R, Walse B, Stuart DI, van der Merwe PA, Davis SJ. The interaction properties of costimulatory molecules revisited. Immunity. 2002;17:201–210. doi: 10.1016/s1074-7613(02)00362-x. [DOI] [PubMed] [Google Scholar]
- 22.Evans EJ, Esnouf RM, Manso-Sancho R, Gilbert RJ, James JR, Yu C, Fennelly JA, Vowles C, Hanke T, Walse B, Hunig T, Sorensen P, Stuart DI, Davis SJ. Crystal structure of a soluble CD28-Fab complex. Nat. Immunol. 2005;6:271–279. doi: 10.1038/ni1170. [DOI] [PubMed] [Google Scholar]
- 23.Kerppola TK. Bimolecular fluorescence complementation (BiFC) analysis as a probe of protein interactions in living cells. Annu. Rev. Biophys. 2008;37:465–487. doi: 10.1146/annurev.biophys.37.032807.125842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim M, Carman CV, Springer TA. Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science. 2003;301:1720–1725. doi: 10.1126/science.1084174. [DOI] [PubMed] [Google Scholar]
- 25.Dennehy KM, Elias F, Zeder-Lutz G, Ding X, Altschuh D, Luhder F, Hunig T. Cutting edge: monovalency of CD28 maintains the antigen dependence of T cell costimulatory responses. J. Immunol. 2006;176:5725–5729. doi: 10.4049/jimmunol.176.10.5725. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.