

Abstract
Checkpoints are cellular surveillance and signaling pathways that coordinate the response to DNA damage and replicative stress. Consequently, failure of cellular checkpoints increases susceptibility to DNA damage and can lead to profound genome instability. This study examines the role of a human RECQ helicase, WRN, in checkpoint activation in response to DNA damage. Mutations in WRN lead to genomic instability and the premature aging condition Werner syndrome. Here, the role of WRN in a DNA-damage-induced checkpoint was analyzed in U-2 OS (WRN wild type) and isogenic cells stably expressing WRN-targeted shRNA (WRN knockdown). The results of our studies suggest that WRN has a crucial role in inducing an S-phase checkpoint in cells exposed to the topoisomerase I inhibitor campthothecin (CPT), but not in cells exposed to hydroxyurea. Intriguingly, WRN decreases the rate of replication fork elongation, increases the accumulation of ssDNA and stimulates phosphorylation of CHK1, which releases CHK1 from chromatin in CPT-treated cells. Importantly, knockdown of WRN expression abolished or delayed all these processes in response to CPT. Together, our results strongly suggest an essential regulatory role for WRN in controlling the ATR–CHK1-mediated S-phase checkpoint in CPT-treated cells.
Key words: WRN, Topoisomerase I, Checkpoint, S-phase, CPT
Introduction
The BLM, WRN and RECQL4 helicase genes are mutated in Bloom's syndrome (BS), Werner's syndrome (WS) and Rothmund–Thomson syndrome (RTS), respectively. These syndromes are autosomal recessive disorders associated with predisposition to the development of cancer. Cells from BS and WS patients have a hyper-recombination phenotype and display significant genomic instability (Bohr, 2008). Mutations in the SGS1 gene, the RECQ helicase in Saccharomyces cerevisiae, cause a similar phenotype (Gangloff et al., 1994; Watt et al., 1995; Watt et al., 1996; Frei and Gasser, 2000) that can be partially suppressed by expression of human BLM or WRN (Watt et al., 1996; Yamagata et al., 1998; Heo et al., 1999). Recently, Bernstein and co-workers suggested that the role of SGS1 in DNA replication can be uncoupled from its role in homologous recombination (Bernstein et al., 2009). Moreover, redundant roles of RECQ helicases in the resolution of recombination intermediates is unlikely to explain the link of recombination with the clinical symptoms of WS, BS and RTS.
Defects in RECQ helicases cause hypersensitivity to replication-blocking agents (Cobb et al., 2003; Bernstein et al., 2009). The S-phase checkpoint blocks ongoing replication and late replication firing, and promotes the repair of damaged replication forks (Branzei and Foiani, 2005; Willis and Rhind, 2009a). Two key proteins, ATM (ataxia telangiectasia mutated) and ATR (ataxia and Rad related) kinases, play distinct but overlapping roles in DNA-damage induced checkpoints. ATM–CHK2 kinases are primary players in the response to ionizing radiation, and the ATR–CHK1-mediated S-phase checkpoint is pivotal for the response to UV, methyl methane sulfonate (MMS), hydroxyurea (HU) and campthothecin (CPT) (Kastan and Bartek, 2004; Willis and Rhind, 2009a). SGS1-deficient yeast (S. cerevisiae) cells have a minor but reproducible defect in the intra-S-phase checkpoint response, allowing spindle elongation and fork progression in the presence of HU and MMC, respectively (Frei and Gasser, 2000). The fission yeast (Schizosaccharomyces pombe) RECQ homolog RQH1, plays a similar role, inducing the S-phase checkpoint in response to MMS (Willis and Rhind, 2009b) and HU (Stewart et al., 1997). In Caenorhabditis elegans, the loss-of-function mutation in HIM-6 (BLM ortholog) results in a partially defective cell cycle arrest in response to HU (Wicky et al., 2004). Interestingly, RNAi-mediated depletion of WRN-1 or CHK1 in C. elegans leads to accelerated progression through S phase in the developmental stage. A similar phenotype is also observed in double RNAi knockdown of WRN-1 and CHK1, suggesting that WRN and CHK1 are involved in the same S-phase checkpoint pathway (Lee et al., 2004).
The evidence that human RECQ helicases play a role in checkpoint activation is limited. After release from HU arrest, BS cells recover normally but with a slight delay compared with wild-type cells, which indicates that BLM is not required for the recovery from S-phase arrest (Davies et al., 2004). A recent study also suggests a role for RECQL4 in S-phase checkpoints (Sangrithi et al., 2005). Previous studies documented that WS cells, which are hyper-sensitive to CPT, HU and PUVA (psoralen plus UVA), arrest and resume DNA replication normally in response to CPT (Poot et al., 1992; Pichierri et al., 2001). It has been reported that WRN-depleted human fibroblasts show a marked delay in completing the cell cycle after treatment with MMS or HU (Sidorova et al., 2008). The authors of this study suggested that WRN might be a downstream target of ATR–CHK1 and/or ATM–CHK2 checkpoint signaling, rather than an upstream sensor (Sidorova et al., 2008). However, it was recently revealed that in C. elegans, WRN-1 acts upstream of ATM and ATR to facilitate RPA foci formation and CHK1 phosphorylation upon HU stress (Lee et al., 2010). It has also been suggested that the upstream functions of WRN-1 are likely to be conserved, but might be cryptic in human systems as a result of functional redundancy. The precise role for human WRN in replication-stress-induced checkpoint is not yet known (Lee et al., 2010).
We have shown previously that WRN-deficient cells have a defective intra-S-phase checkpoint response to PUVA, HU or CPT (Cheng et al., 2008). This study provided detailed evidence that WRN is recruited to DNA double-strand breaks (DSBs) at replication forks, activating an ATM-dependent checkpoint in response to PUVA, but not in response to γ-irradiation (Cheng et al., 2008). This suggests a differential role of WRN in checkpoint activation in response to DSBs. Alternatively, the paused DNA polymerase and other damage could also be targets for WRN to mediate the S-phase checkpoint.
CPT is a topoisomerase 1 (TOPI) inhibitor that generates transient replication-blocking TOPI–DNA covalent complexes, which undergo rapid reversal after removal of the drug (Covey et al., 1989; Shao et al., 1999). CPT treatment rapidly induces a strong S-phase checkpoint through the ATR–CHK1 pathway (Wan et al., 1999; Cliby et al., 2002; Wang, 2002; Seiler et al., 2007), which can be reversed by repair of TOPI-mediated DNA lesions. Consistent with this, depletion of ATR, CHK1 or WRN, but not ATM, causes hypersensitivity to CPT (Flatten et al., 2005). CPT-treated WS cells accumulate DSBs and unresolved recombination structures during S-phase (Pichierri et al., 2001). Nevertheless, the exact role of WRN, if any, in the ATR–CHK1 pathway in CPT-treated cells is not known. The goal of this study was to examine the role of WRN in CPT-induced checkpoint activation, and test the possibility of its involvement in the ATR–CHK1 pathway. The results indicate that CPT-treated WRN-depleted human fibroblasts complete S phase more quickly and activate the ATR–CHK1-dependent response more slowly than is observed in wild-type cells. Evidence presented in this study suggests that WRN delays replication fork elongation in CPT-treated cells.
Results
Depletion of WRN leads to defective S-phase checkpoint in the presence of CPT but not HU
To maintain low-level expression of WRN, experiments were carried out with U-2 OS cells stably expressing scrambled shRNA (WRN-WT) and WRN-targeted shRNA (WRN-KD), respectively. WRN-KD cells showed ~80% depletion of WRN protein under all experimental conditions used in this study (Fig. 1A). The WRN-KD cells exhibited elevated levels of both spontaneous DSBs and inducedDSBs in response to CPT (1 μM) and HU (1 mM) compared with WRN-WT cells (Fig. 1B,C). A similar increase in spontaneous DSBs and hypersensitivity towards CPT has been reported for WRN-deficient patient cell lines (Pichierri et al., 2001; Franchitto and Pichierri, 2004; Lowe et al., 2004).
Fig. 1.

Depletion of WRN in U-2 OS cells leads to increased accumulation of DSBs in response to DNA-damaging agents. (A) Western blot analysis shows ~80% reduction in the level of WRN protein expression in WRN-KD cells compared with WRN-WT cells. Lamin B was used as a protein loading control. Analysis of DNA DSBs in WRN-WT and WRN-KD cells in response to (B) CPT and (C) HU, respectively. Cells were either left untreated or treated with CPT (1 μM) and HU (1 mM) and DNA DSBs were evaluated by neutral comet assay after 3 hours and 6 hours of respective drug treatment. Lower panels in B and C show the quantification of DSBs in terms of Olive tail moment in comet assay. Points represent mean ± s.d. from at least three experiments (200 nuclei in each experiment).
CPT rapidly induces an intra-S-phase checkpoint (Seiler et al., 2007). However, after acute pulsed exposure to CPT (1 μM, 3 hours), WRN-depleted cells progress through S phase, whereas wild-type cells do not (Cheng et al., 2008). To better understand the role of WRN in the CPT-induced replication checkpoint, we examined cell cycle progression in wild-type and WRN-depleted cells exposed to CPT for 24 hours. Cells were treated with CPT (10 nM to 1 μM) and nocodazole (0.25 μg/ml) for 24 hours and analyzed by FACS. The nocodazole treatment arrests all the cells in G2–M phase, which allows for monitoring S-phase progress within the time of one cell cycle. Nocodazole arrested the same fraction of WRN-WT and WRN-KD cells in G2–M (Fig. 2A), indicating that depletion of WRN does not alter cell cycle progression in the absence of DNA damage. In WRN-WT cells, 10 and 75 nM CPT induced late S-phase arrest, and 250 nM and 1 μM CPT induced predominately early S-phase arrest (Fig. 2A–C). By contrast, WRN-depleted cells treated with 10, 75 or 250 nM CPT progressed normally to G2, escaping the CPT-induced S-phase checkpoint (Fig. 2A–C). The majority of WRN-depleted cells exposed to 1 μM CPT arrested in S phase, although a small fraction of the cells escaped arrest and progressed to G2 (Fig. 2A–C).
Fig. 2.

Depletion of WRN causes defective S-phase checkpoint in the presence of CPT. (A) WRN-WT and WRN-KD cells were treated with increasing concentration of CPT (0–1000 nM) for 24 hours in the presence of nocodazole (0.25 μg/ml) as indicated. After 24 hours cells were harvested to determine cell cycle distributions by flow cytometry as shown in cell count vs DNA content profiles of the cells. (B,C) Quantification of S-phase population (B) and G2–M-phase population (C) in the same experiments as described in Fig. 1A (n=4). Points represent mean ± s.d.
A similar analysis was performed on WRN-KD and WRN-WT cells exposed to hydroxyurea (HU), which induces replication stalling due to nucleotide depletion. The results show that WRN-WT cells treated with 0.1 mM HU progressed through S phase and arrested in late S or S–G2 (Fig. 3A–C). Intriguingly, at the same HU concentration, WRN-depleted cells arrested in early S or G1–S, with a slightly larger fraction of arrested cells at higher HU concentration (Fig. 3A–C). Thus, at low concentration of HU (0.1 mM) the G2–M fraction was higher in WRN-WT than in WRN-KD cells (Fig. 3C). The results demonstrate that WRN-KD cells show an intact S-phase arrest or checkpoint after exposure to HU but not after exposure to CPT. Next, we were interested in probing the exact role of WRN in elicitation of the intra-S-phase checkpoint in response to CPT.
Fig. 3.

WRN is dispensable for HU-induced checkpoint. (A) WRN-WT and WRN-KD cells were treated with increasing concentrations of HU (0.1–1 mM) for 24 hours in the presence of nocodazole (0.25 μg/ml) as indicated. After 24 hours, cells were harvested to determine cell cycle distributions by flow cytometry as shown in cell count vs DNA content profiles of the cells. (B,C) Quantification of S-phase population (B) and G2–M-phase population (C) in the same experiments as described in Fig. 3A (n=3). Points represent mean ± s.d.
Early recruitment of WRN to stalled replication forks in response to CPT but not HU
Because WS cells accumulate collapsed replication forks in response to CPT (Pichierri et al., 2001), it is tempting to speculate that WRN might be an upstream sensor, which facilitates checkpoint activation at the replication fork sites to prevent its collapse. Alternatively, WRN might be a sensor for DSBs to elicit an S-phase checkpoint. To test these possibilities, we asked whether WRN colocalizes with 53BP1, which accumulates rapidly at DSBs in mammalian cells (Schultz et al., 2000). As shown in Fig. 4A, WRN preferentially located in the nucleolus in untreated cells, but is mobilized and distributed, preferentially in foci, throughout the nucleus in approximately 47% of CPT-treated cells. About 31% of WRN foci colocalize with 53BP1 foci (Fig. 4A,B), thus only a fraction of WRN associates with DSBs after CPT treatment. Colocalization studies were also performed for WRN and γH2AX, another DSB marker. Similarly, a small fraction of WRN foci colocalize with CPT-induced γH2AX foci (supplementary material Fig. S1), indicating that WRN does not selectively interact with DSBs in CPT-treated cells.
Fig. 4.

DSB-dependent and -independent localization of WRN in response to CPT. (A) WRN and 53BP1 colocalize to nuclear foci. Indirect immunofluorescence of WRN (green) and 53BP1 (red) in untreated and CPT (1 μM, 3 hours) treated cells are shown. In the merged image, yellow coloration is due to colocalization of WRN and 53BP1. (B) Quantification of WRN foci formation at 53BP1 sites (DSBs) and other sites (non-DSB) in the same experiment as described in A. Bars represent mean ± s.d. (C) Detection of WRN foci formation at replication sites by immunostaining. Indirect immunofluorescence of WRN (green) and RPA32 (red) in untreated and CPT treated cells (1 μM, 3 hours) are shown. In the merged image, yellow coloration is due to colocalization of WRN and RPA32. The DAPI staining (blue) in left panel shows the position of the nucleus. A minimum of 150 cells were assessed for each sample in three independent experiments.
CPT stabilizes TOP1–DNA complexes, which causes replication stalling and collapse, and resulting replication-associated DSBs. Therefore, we further tested whether WRN distribution also associates with stalled replication forks by studying co-immunofluorescence of WRN and the ssDNA binding protein RPA32 in the presence of CPT. Interestingly, most of the WRN foci (~73%) colocalize with RPA32 foci in CPT-treated cells (Fig. 4C), consistent with a previous report and the idea that WRN associates with RPA32 in stalled or collapsed replication forks upon CPT stress (Futami et al., 2008). Moreover, kinetic studies shown in supplementary material Fig. S2 indicate that WRN starts to form foci as soon as 30 minutes after exposure to CPT, but it does not form foci after exposure to HU (1 mM, 30 minutes). Nevertheless, a 30 minute exposure to 1 μM CPT or 1 mM HU caused ~55% or ~82% inhibition of [3H]thymidine incorporation during DNA synthesis, respectively (data not shown). Thus, HU-induced stalled replication forks are not immediate targets of WRN. These data confirm a differential role for WRN in the response to HU- and CPT-induced DNA damage. This might reflect the rapid recruitment of WRN to CPT-induced stalled replication forks (Fig. 4C) to mediate the checkpoint response in order to avoid fork collapse.
WRN regulates ATR–CHK1-induced S-phase checkpoint in response to CPT
The interaction between WRN and the ATR–CHK1 axis was explored by measuring ATR-dependent phosphorylation of CHK1 in CPT-treated WRN-WT and WRN-KD, WRN defective WS-KO-375, and ATR-defective Seckel cells. As shown in Fig. 5A, CPT (1 μM, 3 hours) induced CHK1-ser345 phosphorylation strongly in WRN-WT cells whereas only minimal or no induced CHK1 Ser345 phosphorylation was observed in WRN-KD, WS-KO-375 or Seckel cells. Several reports show that ATR mediates phosphorylation of chromatin bound CHK1 to facilitate its release, as required for checkpoint activation (Smits et al., 2006; Niida et al., 2007). We sought to investigate whether this process might be affected in WRN-depleted cells.
Fig. 5.

WRN regulates ATR-mediated signaling in response to CPT. (A) CHK1 phosphorylation on Ser345 was detected in WRN-WT, WRN-KD, WS-KO-375 cells and Seckel cells treated with CPT (1 μM). Cells were harvested at the indicated time point and cell lysates prepared as described for western analyses. (B) CHK1 phosphorylation was determined at various time points as indicated. (C) CHK1 phosphorylation on Ser345 was detected in control and WRN-KD cells treated with HU (1 mM, 3 hours). (D) Phosphorylation and ubiquitylation of ATR substrates in control and WRN-KD cells after CPT treatment (1 μM, 3 hours). Arrow shows phosphorylated form of RPA32; asterisk indicates monoubiquitylated form of FANCD2. Because endogenous levels of γH2AX and monoubiquitylated FANCD2 are high in WRN-depleted cells at untreated conditions, fold increase (FI) in the levels of induced γH2AX and monoubiquitylated FANCD2 in response to CPT are given by normalizing the fold increase (FI) in WRN-WT cells to 1.
Consistent with the above result, dissociation of CHK1 from chromatin and its accumulation in non-chromatin soluble fraction was increased after exposure to CPT for 3 hours in WRN-WT cells but not in WRN-KD cells (supplementary material Fig. S3). These results suggest that WRN acts upstream of CHK1 in CPT-treated cells. A time course experiment showed that CHK1 phosphorylation reaches a maximum after exposure for 3 hours and is maintained at this level for up to 12 hours in CPT-treated WRN-WT cells. In CPT-treated WRN-KD cells, CHK1 phosphorylation was delayed and reached its maximum after 6–12 hours (Fig. 5B). By contrast, the level of CHK1 Ser345 phosphorylation after exposure to HU was comparable in WRN-WT and WRN-KD cells (Fig. 5C). The slow kinetics of CHK1 phosphorylation in WRN-KD cells after CPT treatment is not likely to be accounted for by fewer S-phase cells or CHK1 downregulation in the WRN-KD population compared with WRN-WT cells because only 2–5% fewer S-phase cells and comparable CHK1 expression were monitored in the WRN-KD cell population (Figs 2, 3, 5) compared with WRN-WT cells. Moreover CHK1 phosphorylation was not affected in WRN-KD cells, which displayed a similar number of S-phase cells before and after HU stress. Furthermore, unlike WRN-WT most WRN-KD cells completed S-phase (Fig. 2) after CPT treatment, which is consistent with a defective S-phase checkpoint. Our data are therefore consistent with distinct roles of WRN during HU- and CPT-induced cell cycle arrest (Figs 2, 3).
The results shown in Fig. 5A,B predict that early events downstream of ATR might be compromised in CPT-treated WRN-KD cells. To test this possibility, phosphorylation of the ATR targets RPA32 and H2AX, and monoubiquitylation of ATR-phosphorylated FANCD2 (Andreassen et al., 2004; Wu et al., 2005), were examined in CPT-treated WT and WRN-KD cells. Indeed, all these downstream events were compromised in WRN-KD cells after exposure to CPT (Fig. 5D). It should be noted that γH2AX levels in the untreated samples varied significantly between the cell lines. WS-KO-375, Seckel and WRN-KD cells exhibited higher levels of H2AX phosphorylation in untreated cells compared with that in untreated WRN-WT cells. This is in accordance with previous reports showing that WRN depletion activates the proteins involved in the DNA-damage-response pathway (Szekely et al., 2004; Cheng et al., 2008). Therefore, even though the level of H2AX phosphorylation is lower in WRN-WT cells after CPT treatment compared with levels in WRN-KD and WS-KO-375 cells, the level of induced H2AX phosphorylation after CPT treatment is higher in WRN-WT cells.
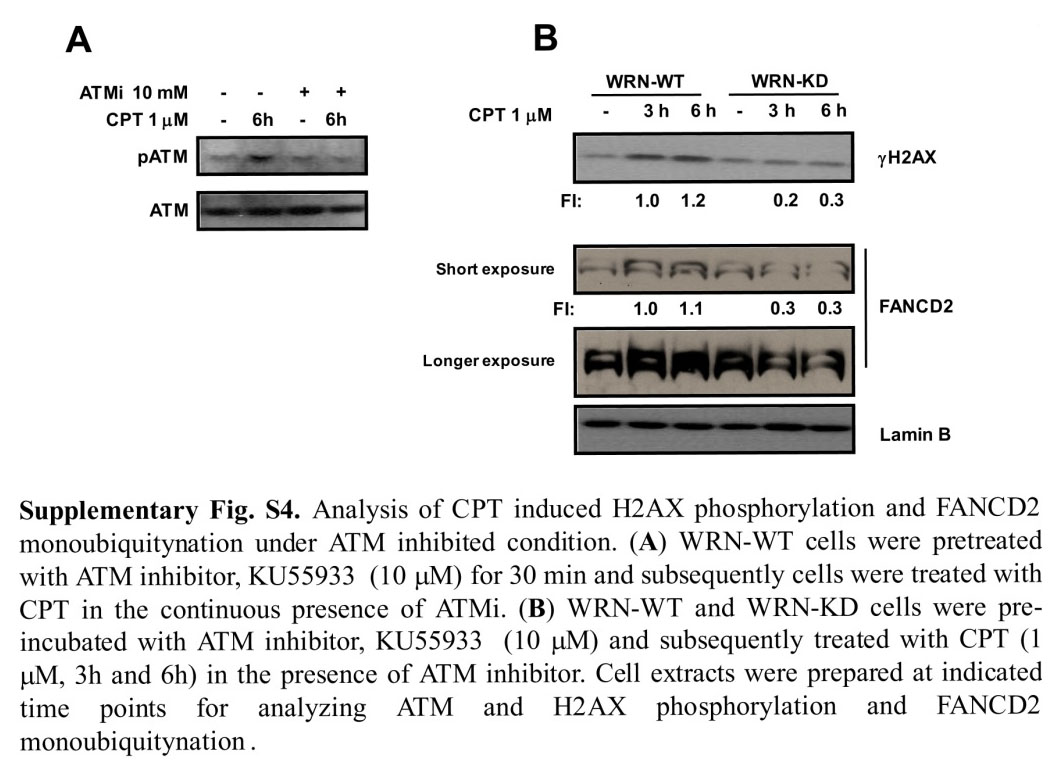
Apart from ATR, ATM also plays a key role in eliciting DNA-damage-response pathways after CPT stress. To determine the role of ATM in WRN-mediated regulation of ATR pathway, we investigated the CPT-induced phosphorylation of H2AX and monoubiquitylation of FANCD2 in WRN-WT and WRN-KD cells in the presence of an ATM-specific inhibitor (KU 55933). In control experiments, KU 55933 (10 μM) inhibited ATM autophosphorylation efficiently in response to CPT (supplementary material Fig. S4A). CPT treatment induced H2AX phosphorylation and FANCD2 monoubiquitylation in WRN-WT cells whether or not ATM inhibitor was present. Furthermore, ATM inhibition did not affect the response to CPT treatment of WRN-KD cells (compare Fig. 5 and supplementary material Fig. S4B). This suggests that the role for WRN in activating early events in the ATR pathway is independent of ATM.
One explanation for these data is that CPT generates fewer DSBs and hence ATR-mediated activation of downstream proteins is less pronounced in WRN-depleted cells. However, as evident from Fig. 1B, WRN-KD cells accumulate more DSBs in response to CPT than seen in WRN-WT cells. DSBs were also measured in WRN-WT and WRN-KD cells using immunostaining for 53BP1. Supplementary material Fig. S5 shows that the 53BP1 foci are significantly higher in CPT-treated WRN-KD cells compared with CPT-treated WRN-WT cells. Therefore, the lack of ATR pathway activation in CPT-treated WRN-KD cells is not a consequence of reduced accumulation of CPT-induced DSBs.
WRN regulates CPT-induced intra-S-phase checkpoint, but not the G2 checkpoint
The ATR–CHK1 checkpoint arrests CPT-treated cells in S and G2 phase (Cliby et al., 2002). As evident from Fig. 3, WRN-deficient WRN-KD cells did not elicit an intact S-phase checkpoint after CPT treatment. However, it remains possible that although WRN-depleted cells, with increased amounts of DSBs (Fig. 1B; supplementary material Fig. S5), complete their bulk DNA synthesis (with a defective S-phase checkpoint) (Fig. 2A, Fig. 5A,B), they might display an intact DSB-mediated G2–M checkpoint. To investigate the integrity of the G2 checkpoint, the fraction of CPT-treated WRN-WT and WRN-KD cells in S and G2 was measured by FACS. Because the FACS analysis does not allow us to distinguish G2 cells from mitotic-arrested cells histone H3 Ser10 phosphorylation, a marker for mitosis, was also analyzed. Fig. 6A,B shows that WRN-WT cells accumulated in late S or G2–M phase, whereas WRN-KD cells progressed through S phase and arrested in G2–M. Interestingly, although nocodazole treatment leads to histone H3 Ser10 phosphorylation and mitotic arrest in both WRN-WT and WRN-KD cell lines, CPT treatment failed to cause any such phosphorylation in WRN-WT and WRN-KD cells (Fig. 6C). CPT treatment in the presence of nocodazole also failed to induce histone H3 Ser10 phosphorylation in WRN-WT and WRN-KD cells, indicating that these cells were arrested in G2 phase. Together, these results show that WRN is required for the CHK1-mediated intra-S-phase checkpoint, but not for the G2 checkpoint in the presence of CPT.
Fig. 6.

WRN regulates CPT-induced intra-S-phase checkpoint but not G2 checkpoint. (A) WRN-WT and WRN-KD cells were treated with CPT (0.25 μM) and cells were harvested at indicated time points to determine cell cycle distributions by flow cytometry. Arrows indicate arrested cells in S phase. (B) Quantification of S-phase and G2–M-phase populations in A (n=3). Points represent mean ± s.d. (C) Histone H3 Ser10 phosphorylation was quantified in WRN-WT and WRN-KD cells untreated and treated with CPT and nocodazole alone and together as indicated. Cell lysates were prepared at the indicated time point. (D) WRN-WT and WRN-KD cells were untreated (UT) or treated with CPT (0.25 μM) and nocodazole (0.25 μg/ml) for 24 hours in the presence or absence of UCN 01 (0.1 μM) as indicated. Cell cycle distribution was analyzed by flow cytometry as shown in cell count vs DNA content profiles of the cells. Percentage of cells in S and G2 phase population is indicated under respective samples (n=3).
To confirm the role of WRN in the regulation of the CHK1-mediated S-phase checkpoint, WRN-WT and WRN-KD cells were treated with 250 nM CPT and the CHK1 chemical inhibitor, UCN 01 (Syljuasen et al., 2005; Seiler et al., 2007). FACS analysis showed that ~81% of WRN-WT cells were blocked in S-phase by CPT, but most cells overrode this checkpoint to enter G2 in the presence of UCN 01 plus CPT (Fig. 6D). However, no or minimal effect of UCN-01 was observed in WRN-KD cells (Fig. 6D, 45% G2-cells in CPT vs 52% G2-cells in UCN 01 plus CPT-treated WRN-KD cells). Control experiments showed that UCN 01 had a minimal effect on cell cycle progression of WRN-WT and WRN-KD cells in the absence of CPT (Fig. 6D). This result supports the hypothesis that WRN regulates ATR–CHK1 function in a common intra-S-phase checkpoint pathway in CPT-treated cells.
WRN inhibits replication fork elongation in the presence of CPT
A WRN-regulated ATR–CHK1 pathway could inhibit replicon initiation and/or chain elongation in CPT-treated cells and, in turn, prevent collapse of stalled replication forks. To differentiate between these possibilities, pulse-labeled DNA was analyzed on alkaline sucrose gradients (Wang et al., 2004; Heffernan et al., 2002; Willis and Rhind, 2009a) to determine the steady-state distribution of nascent DNA intermediates in CPT-treated WRN-WT and WRN-KD cells. Fig. 7A shows the experimental design for these experiments. The results show that both replicon initiation intermediates (top of the gradient) and chain elongation intermediates (bottom of the gradient) were less abundant in CPT treated (500 nM, 2 hours) compared with untreated WRN-WT cells (Fig. 7B,C). Interestingly, although the replication initiation process was reduced similarly, replication elongation was only minimally reduced in CPT-treated WRN-KD cells (Fig. 7C; fork elongation ~25% in WRN-WT vs ~60% in WRN-KD cells in response to CPT). This result indicates that the WRN-regulated inhibition of fork elongation might account for S-phase delay in response to CPT.
Fig. 7.

Inhibition of chain elongation of DNA replication is impaired in WRN-depleted cells following exposure to CPT. (A) Schematic representation of [3H]thymidine and CPT treatment. (B) Cells were harvested after CPT treatment as in A, and nascent DNA was separated by velocity sedimentation. Net 3H radioactivity corrected for 14C spillover was normalized to cell number (total 14C radioactivity). Sedimentation is from right to left (i.e. fraction 1 is at the bottom of the gradient and fraction 25 at the top). Percentage fork progress in the last panel was calculated by dividing the value of normalized 3H c.p.m. of each CPT-treated fraction with that of untreated sample and multiplied by 100. The double and single arrows indicate the sedimentation rates of 167 kb and 37 kb DNA marker, respectively.
WRN acts as an upstream sensor to CPT-induced DNA lesions
The above results suggest that WRN plays an early role in ATR–CHK1 activation, thereby inhibiting replication fork progression in CPT-treated cells. Several reports show that ATR activation requires single-stranded DNA (ssDNA) (You et al., 2002; Zou et al., 2003). There are two possible mechanisms by which WRN could regulate ATR-mediated CHK1 phosphorylation at stalled replication forks in CPT-treated cells: (1) WRN might facilitate ssDNA formation, leading to sequential activation of ATR and CHK1, and/or (2) WRN might play a role in recruiting ATR to stalled replication forks. To test the first possibility, ssDNA was quantified by ssDNA-specific BrdU staining in CPT-treated WRN-WT and KD cells. Two types of ssDNA foci were observed in WT cells: large foci that colocalized with 53BP1 foci, representing resected DSB ends; and small foci that did not colocalize with 53BP1 (i.e. not associated with DSBs) (Fig. 8A–C; supplementary material Fig. S6). Additional experiments showed that the ssDNA foci colocalized well with RPA32, suggesting that ssDNA is generated at the sites of replication (supplementary material Fig. S7). Large ssDNA foci at DSBs (colocalization with 53BP1) were significantly more abundant in CPT-treated WRN-depleted cells (Fig. 8B), whereas small ssDNA foci were more abundant in CPT-treated WRN-WT cells (Fig. 8C), suggesting that WRN facilitates ssDNA formation at replication forks in CPT-treated cells.
Fig. 8.

WRN is involved in the generation of ssDNA and ATR activation in response to CPT. (A) Detection of incorporated BrdU specifically in ssDNA after CPT treatment. Cells pre-labeled with BrdU were treated with CPT (1 μM, 3 hours) and stained with antibodies against BrdU (green) and 53BP1 (red). The DAPI staining (blue) in the left panel shows the position of the nucleus. DNase-treated cells are included to show that the whole-cell population was uniformly labeled with BrdU (Syljuasen et al., 2005; Lee et al., 2006). (B,C) Quantification of ssDNA foci formation at DSBs and non-DSB sites in the same experiment as described in A at indicated time points. A minimum of 100–200 cells were assessed for each sample in three independent experiments. Points represent mean ± s.d. (D) WRN co-immunoprecipitated with ATR in human U-2 OS cells. Immunoprecipitations were carried out using anti-ATR antibody, or an immunoglobulin G control in presence of ethidium bromide (30 μg/ml), on extracts from cells exposed to no treatment or CPT treatment (1 μM, 2 and 4 hours). The immunoprecipitates were analyzed by western blot for the presence of WRN using anti-WRN antibodies. WS, WS-KO-375 cells.
We also analyzed whether WRN and ATR co-immunoprecipitate in U-2 OS cells exposed to CPT. Fig. 8D shows that WRN and ATR readily co-immunoprecipitate with anti-ATR antibodies in both untreated and CPT-treated U-2 OS cell extracts, but not with immunoglobulin G control antibodies or in WS-KO-375 (WRN-defective) cells. Reciprocal co-immunoprecipitation with anti-WRN antibodies also confirmed the co-immunoprecipitation of ATR and WRN. This interaction was enhanced in CPT-treated cells (supplementary material Fig. S8A,B), similarly to a previous report showing that WRN-ATR interaction increases in response to interstrand DNA crosslinks (Otterlei et al., 2006). Also, co-immunofluorescence experiments demonstrated an increase in colocalization between WRN and ATR foci in response to CPT (supplementary material Fig. S8C). Taken together, these data suggest that WRN and ATR form a complex that is enhanced and/or stabilized in CPT-treated cells.
Discussion
By using CPT, HU and PUVA, several reports show that WRN could be involved in the resolution of recombinational intermediates that arise from replication arrest or collapse (Pichierri et al., 2001; Dhillon et al., 2006; Sidorva et al., 2008). We have recently shown that WRN is involved in facilitating an ATM-mediated checkpoint in response to PUVA induced collapsed forks (Cheng et al., 2008). These previously published reports strongly document a role for WRN, downstream to DSBs, in the repair of collapsed forks. Here, we present data directly implicating a further upstream role of WRN in the S-phase-specific checkpoint to prevent CPT-induced fork collapse. We find that both CPT-induced S-phase checkpoint and ATR signaling are severely compromised in WRN-depleted cells (Figs 2, 5). However, the CPT-induced G2 checkpoint is fully functional in both WT and WRN-KD cells (Fig. 6A,B). Interestingly, a defective S-phase checkpoint and ATR–CHK1 signaling in WRN-KD cells seems to be a CPT-specific response because both wild-type and WRN-KD cells show robust CHK1 phosphorylation and S-phase arrest in the presence of HU (Fig. 3A–C and Fig. 5C).
CPT-induced DNA damage appears to communicate with ATR–CHK1 through WRN by at least three mechanisms. First, stabilized TOPI–DNA complexes could be recognized by WRN. Second, CPT-induced DSBs might elicit a checkpoint response. Third, replication-derived complex DNA structures or unresolved positive supercoils, as suggested by Koster and co-workers (Koster et al., 2007), could also be sensed by WRN. WRN is known to stimulate the TOPI-mediated relaxation cycle (Laine et al., 2003) and collaborate with proteins (RAD51, RAD52, MRN complex, ku70, DNA-PKcs) involved in DSB repair by homologus recombination and non-homologus end joining (Sakamoto et al., 2001; Baynton et al., 2003; Cheng et al., 2004; Otsuki et al., 2007), suggesting a putative role of WRN in the first two mechanisms mentioned above. In this study, however, our results showed that a significantly higher fraction of WRN colocalizes with RPA32 (replication forks sites) compared with 53BP1 (DSBs site) (~73 vs 31%) in response to CPT (Fig. 4A,C). Of note, WRN is found at most of the CPT induced DSB sites (53BP1) (Fig. 4A), supporting its role in DSB repair through homologous recombination and non-homologous end joining. However, we also observed a fraction of WRN foci that did not colocalize with DSB sites, and these are likely to represent WRN molecules engaged in other functions, possibly related to CPT-induced stalled replication forks. A recent study documented the role of WRN in preventing fork collapse and DSBs upon replication arrest (Ammazzalorso et al., 2010). Intriguingly, Ammazzalorso and co-workers demonstrated that ATR-mediated phosphorylation of WRN facilitates proper accumulation of WRN in nuclear foci, colocalization with RPA and prevention of fork collapse upon replication stress (Ammazzalorso et al., 2010). Thus, the role of WRN in CPT-induced toxicity is not just confined to DSB related repair and resolution of recombinational intermediates but it also plays a crucial role in preventing fork collapse. The presence of WRN at replication forks (Fig. 4C), and its role in checkpoint activation (Fig. 5) and prevention of DSB formation (Fig. 1B) raise a compelling argument for an intriguing, previously unconsidered, checkpoint-regulating role of WRN at stalled replication forks.
We further addressed how WRN mediates the slow down of DNA synthesis in response to CPT (Fig. 2). Recent work using DNA fiber labeling showed that the replication initiation was normal in WS primary fibroblast cells whereas the majority of fork elongations were asymmetrical (Rodriguez-Lopez et al., 2002). Our analysis, using alkaline sucrose gradient centrifugation, led to the interesting observation that the frequency of origin firing is reduced to similar extents in WRN-KD and WRN-WT cells, whereas fork elongation is less affected in WRN-KD cells compared with WRN-WT cells in the presence of CPT (Fig. 7B). Implicit in this result is the ability of the WRN-induced checkpoint to impede fork progression selectively in the presence of CPT. However, our results do not overrule the idea that inhibition of the origin firing near the forks also inhibits fork restart in WRN-WT cells in response to CPT, which warrants further study.
Importantly, Kumar and colleagues recently showed the persistence of replication intermediates in MMS-treated fission yeast by using 2D-gel analysis of replication intermediates (Kumar et al., 2009). This suggests the inhibition of fork progress in response to MMS. The effects were dependent on Cds1 (a checkpoint kinase in fission yeast) and were abolished in cells overexpressing cdc25 phosphatase, showing evidence for checkpoint-induced regulation of fork elongation in response to DNA damage. Similarly, unchecked fork progression in WRN-depleted cells might lead to frequent encounters of replication forks with accumulated positive supercoils or TOPI–DNA complexes resulting in enhanced fork collapse (supplementary material Fig. S5; Fig. 1A). Here, we propose that a WRN-dependent checkpoint regulates fork progression by a global mechanism in which the activated checkpoint acts in trans to affect all fork stalling in response to CPT. Few individual pauses at DNA–TOPI complexes would not significantly delay replication, but the S-phase-checkpoint-mediated cumulative effect of forks pausing at many sites would lead to an overall reduction in replication rate.
Mechanistic insight into CPT-induced activation of the ATR–CHK1 pathway by WRN
The ATR–CHK1 pathway is the primary pathway regulating the S-phase checkpoint in response to stalled replication forks induced by UVC, HU, aphidicolin etc. (Zhou and Elledge, 2000; Cortez et al., 2001; Melo and Toczyski, 2002). These agents specifically cause independent recruitment of ATR and an RFC-related protein, Rad17, to RPA-coated single-stranded DNA. Subsequently, the Rad9–Rad1–Hus1 (9-1-1) complex is recruited to chromatin by the Rad17–RFC complex, and ATR phosphorylates Rad17 in a Hus1-dependent manner (Zou et al., 2002; Zou et al., 2003). Together, these events regulate phosphorylation of chromatin-bound CHK1, which in turn causes rapid dissociation of phosphorylated CHK1 from the chromatin (Smits et al., 2006; Niida et al., 2007). Although the ATR–CHK1 pathway is required for S-phase arrest in response to CPT treatment, it is not known how this pathway is activated. Several lines of evidence in this study suggest a role of WRN in CPT-induced checkpoint activation. Thus, we have demonstrated a functional interaction between WRN and ATR by co-immunoprecipitation and colocalization experiments, and shown that WRN regulates CHK1 phosphorylation and subsequent release from chromatin (Fig. 5B, Fig. 8; supplementary material Figs S3, S8). We further show evidence here that WRN depletion intriguingly caused no significant effect on ssDNA levels at DSBs generated by resection, but the ssDNA level was drastically reduced at the replication sites (Fig. 7A–C). These results suggest that the WRN-dependent checkpoint signal does not emanate from the (resected) DSBs per se. Moreover our results are consistent with the hypothesis that WRN has an upstream role to facilitate ssDNA formation for ATR-mediated checkpoint activation upon CPT stress. Specifically, in response to UVC, aphidicolin and cisplatin, the MCM helicase is thought to become functionally uncoupled from DNA polymerase to generate a common ssDNA intermediate. This is in turn coated with RPA to elicit checkpoint signaling (Byun et al., 2005). In the case of CPT-induced lesions (accumulated TOPI–DNA covalent complexes and associated positive supercoilings), it is not possible for the MCM helicase to bypass these lesions to uncouple from DNA polymerase. Thus, it is possible that WRN senses CPT-induced lesions in order to stimulate MCM helicase activity or induce transient uncoupling between leading and lagging strand synthesis to generate ssDNA. Alternatively, WRN might directly generate ssDNA through its intrinsic helicase activity. Although WRN alone shows preference for recessed 3′ ends (Huang et al., 2000), it might also unwind fork substrates without a 3′ single-strand tail in the presence of RPA (Ahn et al., 2009). A third possibility is that WRN helicase promotes reverse branch migration of the nascent templates away from a stalled fork. Any or all of these processes could account for the presence of ssDNA at forks. It is of future interest to determine in detail how WRN causes generation of ssDNA in response to stabilized TOPI-DNA complex.
In summary, we have demonstrated that WRN is involved in the induction of an intra S-phase checkpoint in response to CPT. Our finding that WRN is involved in the generation of ssDNA at replication forks, but not at sites of DSB end resection, shows that WRN plays distinct roles in inducing checkpoint signaling that results from TOPI–DNA complexes encountering forks. This raises the possibility that the CPT sensitivity of WS cells reflects loss of an S-phase surveillance mechanism, in addition to the known function of WRN during resolution of intermediates in recombinational DNA repair and the NHEJ pathway (Otsuki et al., 2007; Kawabe et al., 2006; Pichierri et al., 2001). Our findings have important implications for understanding resistance mechanisms provided by WRN to a clinically important group of cancer therapeutic drugs, specifically TOPI and CHK1 inhibitors in combination with WRN silencing (Futami et al., 2008).
Materials and Methods
Cell lines and culture conditions
U-2 OS osteosarcoma cells were routinely grown in DMEM supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine, 1 mM sodium pyruvate, 100 U/ml penicillin and 100 μg/ml streptomycin at 37°C in 5% CO2. The generation and maintenance of U-2 OS based WRN-WT (expressing control shRNA) and WRN-KD (expressing WRN shRNA) cells have been described previously (Cheng et al., 2008). WS-KO-375 (WRN mutated) and ATR-defective Seckel cells (GM18367) were grown in RPMI and MEM medium supplemented with 10–15% FBS, glutamine and antibiotics, as above. All cell culture media were purchased from GIBCO. Nocodazole, CPT, HU, protease inhibitors (leupeptin, pepstatin and aprotinin) and phosphatase inhibitors cocktails were purchased from Sigma (St. Louis, MO). The ATM inhibitor, KU55933 was purchased from Calbiochem. (EMD Bioscience, La Jolla, CA).
Cell extracts, chromatin fractionation, immunoblotting and antibodies
Whole-cell lysates and immunoblot analysis were described previously (Cheng et al., 2004). To analyze histone H3 Ser10 phosphorylation, cells were scraped, pooled with nonattached cells, washed, and lysed with buffer containing 50 mM Tris-HCl, 0.5% NP-40, 150 mM NaCl, protease and phosphatase inhibitors. After incubation for 30 minutes on ice, the extracts were centrifuged (14,000 r.p.m., 20 minutes) and supernatant was collected for blotting. Chromatin fractionation of mammalian cells was performed essentially as described previously (Mandez and Stillman, 2000). In brief, PBS washed (2×106) cells were resuspended in 200 μl of solution A [10 mM HEPES (pH 7.9), 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 1 mM DTT, 0.1% Triton X-100, protease and phosphatase inhibitors]. After incubation on ice for 5 minutes, the cytoplasmic (S1) and nuclear fractions (P1) were harvested by centrifugation at 1300 g for 4 minutes. Next, the P1 fraction was washed twice in solution A, lysed in 150 μl solution B (3 mM EDTA, 0.2 mM EGTA, 1 mM DTT, protease and phosphatase inhibitors). After incubating on ice for 10 minutes, the soluble nuclear (S2) and chromatin fractions (P2) were harvested by centrifugation at 1700 g for 4 minutes. Isolated chromatin (P2) was then washed in solution B, spun down at 10,000 g and resuspended in 150 μl Laemmli sample buffer. The S1 and S2 fractions were pooled for analyzing proteins in non-chromatin fraction. Primary antibodies were used in immunoblotting against the following proteins: WRN, Lamin B, CHK1, TOPI (Santa Cruz Biotechnology, Santa Cruz, CA), WRN, FANCD2, phosphor-histone3-ser10 (Abcam, Cambridge, UK), p345CHK1 (Epitomics, Burlingame, CA and Cell Signaling Technology, Beverly, MA), ATR (Oncogene, Darmstadt, Germany), RPA32 (Bethyl, Montgomery, TX), 53BP1 (Novus, Littleton, CO), ORC2 (BD Biosciences, San Jose, CA) and γH2AX (Upstate Biotechnology, Lake Placid, NY).
Immunoprecipitation
All steps in immunoprecipitations were performed at 4°C. After treating with CPT (1 μM), cells were resuspended in 750 μl of TNN lysis buffer [50 mM Tris-HCl, pH 7.6, 120 mM NaCl and 0.5 % (v/v) NP-40 and protease inhibitors]. Cell lysates were kept on ice for 30 minutes, treated with 50 U of RNase-free DNase I (Roche, BM) at 25°C for 30 minutes and centrifuged at 12,800 g for 10 minutes. Approximately 1 mg/ml protein cell lysates were incubated with antibody against ATR or an IgG control antibody overnight in the presence of ethidium bromide (25 μg/ml). Subsequently, Protein-G–agarose beads (Invitrogen) were added and incubated for an additional 3 hours under rotation. Antibody–protein complexes were harvested by centrifugation, and the pellet was washed four times in 500 μl of TNN buffer. Pellets were boiled in SDS-PAGE loading buffer and subjected to electrophoresis and western blotting by standard procedures. Control experiments showed that the positive control IgG did not interact with ATR and WRN in cells untreated treated with CPT (supplementary material Fig. S8B).
Immunofluorescence
Cells washed in PBS were fixed in ice-cold methanol and permeabilized in Triton X-100 (0.5% in PBS, 10 minutes). Binding with anti-WRN (Santacruz and Abcam), anti-53BP1 (Novus) and anti-ATR (Oncogene) antibodies was performed in 5% BSA in PBS [1 hour, room temperature (RT)]. Primary antibody detection was achieved using donkey anti-mouse Alexa Fluor 488 and donkey anti-rabbit Alexa Fluor 555 secondary antibodies (Invitrogen) (1 hour, RT). Slides were mounted in Antifade Gold containing DAPI (Invitrogen). Nuclei were examined at a magnification of 63× using a Carl Zeiss fluorescence microscope. The ssDNA patch assay was performed as reported previously (Syljuasen et al., 2005; Lee et al., 2006). Briefly, BrdU-prelabeled (10 μg/ml, 30 hours) cells were treated with CPT. Then cells washed in PBS were fixed (chilled in methanol, 30 minutes), permeabilized [chilled in methanol:acetone, 7:3 (v/v), 30 minutes] and blocked in 5% BSA. Cells were then incubated overnight with mouse monoclonal anti-BrdU antibody (which recognizes BrdU in ssDNA only; BD Biosciences, 1:500) and 53BP1 (Novus, 1:4000) or RPA32 (Bethyl, 1:5000). Secondary antibodies, mounting and fluorescence microscopy was performed as described above. Larger BrdU foci were quantified manually by carefully monitoring the foci that colocalized with 53BP1. The remainder of the prominent and bright smaller foci were quantified as smaller ssDNA foci. At least 100–200 nuclei were analyzed in each untreated and treated samples in two to four independent experiments.
Flow cytometry analysis
Single cell suspensions were fixed with 80% ethanol in PBS (24 hours, −20°C). Fixed cells were stained in solution containing 20 μg/ml propidium iodide (Sigma) and 50 μg/ml RNAse (Roche). Data were collected with a Becton Dickinson FACS machine (Mountain View, CA) and analyzed using FlowJo software.
Quantification of inhibition of DNA replication
This assay was performed as previously described (Heffernan et al, 2002) with minor modifications. Briefly, WRN-WT and WRN-KD (1×105) cells were grown in the presence of [14C]thymidine (specific activity, 10 nCi/ml; 50 Ci/mol) for 48 hours, and then in non-radioactive medium overnight to chase and label DNA uniformly. Cells were mock treated or exposed to CPT (0.5 μM), incubated at 37°C for 1 hour 45 minutes, and then [3H]thymidine (10–50 μCi/ml; specific activity, 56 Ci/mmol) was added to the same medium for 15 minutes. Cells were harvested in lysis buffer (1 M NaOH and 0.02 M EDTA) and cell lysate was layered on top of a 36 ml 5–20% alkaline sucrose gradient containing 0.4 M NaOH, 2.0 M NaCl, and 0.01 M EDTA. Gradients were placed under laboratory fluorescent lights for l hour for cell lysis, centrifuged in an SW28 rotor (Beckman Instruments) at 25,000 r.p.m. at 20°C for 12 hours. Gradients were fractionated into 24 equal fractions through the bottom of the centrifuge tube. Acid-insoluble 3H/14C radioactivity was determined for each fraction by liquid scintillation spectrometry. After correction for spillover of 14C into the 3H channel, net 3H c.p.m. in each gradient fraction were divided by the total l4C cpm in the gradient to normalize DNA synthesis to cell number.
Neutral comet assay
DSB formation in WRN-WT and WRN-KD cells in response to CPT were measured as reported earlier (Lin et al., 2008).
Supplementary Material
Acknowledgements
We thank W. H. Cheng for critically reading the manuscript and for his valuable suggestions.
Footnotes
Funding
This research was supported by grants from the European Commission [grant number LSHM-CT-2004-512020]; Lundbeck Foundation [grant number 4-55951-95094019]; The Danish Research Council [grant number 271-08-0697]; The Carlsberg Foundation [grant number 2008_01_0533]; The Danish Cancer Society [grant number DP05024]; and The Danish Aging Research Center (Velux Foundation). This research was also partly supported by funds from the intramural program of the National Institute on Aging, National Institutes of Health [grant number Z01 AG000721-01]. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.081372/-/DC1
References
- Ahn B., Lee J. W., Jung H., Beck G., Bohr V. A. (2009). Mechanism of Werner DNA helicase: POT1 and RPA Stimulates WRN to unwind beyond gaps in the translocating Strand. PLoS One 4, e4673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammazzalorso F., Pirzio L. M., Bignami M., Franchitto A., Pichierri P. (2010). ATR and ATM differently regulate WRN to prevent DSBs at stalled replication forks and promote replication fork recovery. EMBO J. 29, 3156-3169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreassen P. R., D'Andrea A. D., Taniguchi T. (2004). ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes Dev. 18, 1958-1963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baynton K., Otterlei M., Bjørås M., Kobbe C., Bohr V. A., Seeberg E. (2003). WRN interacts physically and functionally with the recombination mediator protein RAD52. J. Biol. Chem. 278, 36476-36486 [DOI] [PubMed] [Google Scholar]
- Bernstein K. A., Shor E., Sunjevaric I., Fumasoni M., Burgess R. C., Foiani M., Branzei D., Rothstein R. (2009). Sgs1 function in the repair of DNA replication intermediates is separable from its role in homologous recombinational repair. EMBO J. 28, 915-925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohr V. A. (2008). Rising from the RecQ-age: the role of human RecQ helicases in genome maintenance Trends Biochem. Sci. 33, 609-620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branzei D., Foiani M. (2005). The DNA damage response during DNA replication. Curr. Opin. Cell Biol. 17, 568-575 [DOI] [PubMed] [Google Scholar]
- Byun T. S., Pacek M., Yee M. C., Walter J. C., Cimprich K. A. (2005). Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 19, 1040-1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng W. H., Kobbe C., Opresko P. L., Arthur L. M., Komatsu K., Seidman M., Carney J. P., Bohr V. A. (2004). Linkage between Werner Syndrome Protein and the Mre11 Complex via Nbs1. J. Biol. Chem. 279, 21169-21176 [DOI] [PubMed] [Google Scholar]
- Cheng W. H., Muftic D., Muftuoglu M., Dawut L., Morris C., Helleday T., Shiloh Y., Bohr V. A. (2008). WRN is required for ATM activation and the S-phase checkpoint in response to interstrand cross-link-induced DNA double-strand breaks. Mol. Biol. Cell 19, 3923-3933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliby W. A., Lewis K. A., Lilly K. K., Kaufmann S. H. (2002). S phase and G2 arrests induced by topoisomerase I poisons are dependent on ATR kinase function. J. Biol. Chem. 277, 1599-1606 [DOI] [PubMed] [Google Scholar]
- Cobb J. A., Bjergbaek L., Shimada K., Frei C., Gasser S. M. (2003). DNA polymerase stabilization at stalled replication forks requires Mec1 and the RecQ helicase Sgs1. EMBO J. 22, 4325-4336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez D., Guntuku S., Qin J., Elledge S. J. (2001). ATR and ATRIP: Partners in checkpoint signaling. Science 294, 1713-1716 [DOI] [PubMed] [Google Scholar]
- Covey J. M., Jaxel C., Kohn K. W., Pommier Y. (1989). Protein-linked DNA strand breaks induced in mammalian cells by camptothecin, an inhibitor of topoisomerase-I. Cancer Res. 49, 5016-5022 [PubMed] [Google Scholar]
- Davies S. L., North P. S., Dart A., Lakin N. D., Hickson I. D. (2004). Phosphorylation of the Bloom's syndrome helicase and its role in recovery from S-phase arrest. Mol. Cell Biol. 24, 1279-1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillon K. K., Sidorova J., Saintigny Y., Poot M., Gollahon K., Rabinovitch P. S., Monnat R. J., Jr (2006). Functional role of the Werner syndrome RecQ helicase in human fibroblasts. Aging Cell 6, 53-61 [DOI] [PubMed] [Google Scholar]
- Flatten K., Dai N. T., Vroman B. T., Loegering D., Erlichman C., Karnitz L. M., Kaufmann S. H. (2005). The role of checkpoint kinase 1 in sensitivity to topoisomerase I poisons. J. Biol. Chem. 280, 14349-14355 [DOI] [PubMed] [Google Scholar]
- Franchitto A., Pichierri P. (2004). Werner syndrome protein and the mre11 complex are involved in a common pathway of replication fork recovery. Cell Cycle. 3, 1331-1339 [DOI] [PubMed] [Google Scholar]
- Frei C., Gasser S. M. (2000). The yeast Sgs1p helicase acts upstream of Rad53p in the DNA replication checkpoint and colocalizes with Rad53p in S-phase-specific foci. Genes Dev. 14, 81-96 [PMC free article] [PubMed] [Google Scholar]
- Futami K., Ishikawa Y., Goto M., Furuichi Y., Sugimoto M. (2008). Role of Werner syndrome gene product helicase in carcinogenesis and in resistance to genotoxins by cancer cells. Cancer Sci. 99, 843-848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangloff S., Mcdonald J. P., Bendixen C., Arthur L., Rothstein R. (1994). The yeast type-I topoisomerase Top3 interacts with Sgs1, a DNA helicase homolog – a potential eukaryotic reverse gyrase. Mol. Cell. Biol. 14, 8391-8398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heffernan T. P., Simpson D. A., Frank A. R., Heinloth A. N., Paules R. S., Cordeiro-Stone M., Kaufmann W. K. (2002). An ATR- and Chk1-dependent S checkpoint inhibits replicon initiation following UVC-induced DNA damage. Mol. Cell. Biol. 22, 8552-8561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo S. J., Tatebayashi K., Ohsugi I., Shimamoto A., Furuichi Y., Ikeda H. (1999). Bloom's syndrome gene suppresses premature ageing caused by Sgs1 deficiency in yeast. Genes Cells 4, 619-625 [DOI] [PubMed] [Google Scholar]
- Huang S., Beresten S., Li B., Oshima J., Ellis N. A., Campisi J. (2000). Characterization of the human and mouse WRN 3′→5′ exonuclease. Nucl. Acids. Res. 28, 2396-2405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastan M. B., Bartek J. (2004). Cell-cycle checkpoints and cancer. Nature 432, 316-323 [DOI] [PubMed] [Google Scholar]
- Kawabe Y., Seki M., Yoshimura A., Nishino K., Hayashi T., Takeuchi T., Iguchi S., Kusa Y., Ohtsuki M., Tsuyama T., et al. (2006). Analyses of the interaction of WRNIP1 with Werner syndrome protein (WRN) in vitro and in the cell. DNA Rep. 5, 816-828 [DOI] [PubMed] [Google Scholar]
- Kumar S., Huberman J. A. (2009). Checkpoint-dependent regulation of origin firing and replication fork movement in response to DNA damage in fission yeast. Mol. Cell. Biol. 29, 602-611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koster D. A., Palle K., Bot E. S. M., Bjornsti M. A., Dekker N. H. (2007). Antitumour drugs impede DNA uncoiling by topoisomerase I. Nature. 448, 213-217 [DOI] [PubMed] [Google Scholar]
- Laine J. P., Opresko P. L., Indig F. E., Harrigan J. A., von Kobbe C., Bohr V. A. (2003). Werner protein stimulates topoisomerase I DNA relaxation activity. Cancer Res. 63, 7136-7146 [PubMed] [Google Scholar]
- Lee S. J., Yook J. S., Han S. M., Koo H. S. (2004). A Werner syndrome protein homolog affects C. elegans development, growth rate, life span and sensitivity to DNA damage by acting at a DNA damage checkpoint. Development 131, 2565-2575 [DOI] [PubMed] [Google Scholar]
- Lee Y., Park S. J., Ciccone S. L. M., Kim C. R., Lee S. H. (2006). An in vivo analysis of MMC-induced DNA damage and its repair Carcinogenesis 27, 446-453 [DOI] [PubMed] [Google Scholar]
- Lee S. J., Gartner A., Hyun M., Ahn B., Koo H. S. (2010). The Caenorhabditis elegans Werner syndrome protein functions upstream of ATR and ATM in response to DNA replication inhibition and double-strand DNA breaks. PLoS Genet. 6, e1000801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C., Ban Y., Lyu Y. L., Desai S. D., Liu L. F. (2008). A ubiquitin-proteasome pathway for the repair of topoisomerase I-DNA covalent complexes. J. Biol. Chem. 283, 21074-21083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe J., Sheerin A., Jennert-Burston K., Burton D., Ostler E. L., Bird J., Green M. H., Faragher R. G. (2004) Camptothecin sensitivity in Werner syndrome fibroblasts as assessed by the COMET technique. Ann. N Y Acad. Sci. 1019, 256-259 [DOI] [PubMed] [Google Scholar]
- Melo J., Toczyski D. (2002). A unified view of the DNA damage checkpoint. Curr. Opin. Cell Biol. 14, 237-245 [DOI] [PubMed] [Google Scholar]
- Mendez J., Stillman B. (2000). Chromatin association of human origin recognition complex, Cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol. Biol. Cell 20, 8602-8612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niida H., Katsuno Y., Banerjee B., Hande M. P., Nakanishi M. (2007). specific role of Chk1 phosphorylations in cell survival and checkpoint activation. Mol. Cell. Biol. 27, 2572-2581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsuki M., Seki M., Kawabe Y., Inoue E., Dong Y. P., Abe T., Kato G., Yoshimura A., Tada S., Enomoto T. (2007). WRN counteracts the NHEJ pathway upon camptothecin exposure. Biochem. Biophys. Res. Comm. 355, 477-482 [DOI] [PubMed] [Google Scholar]
- Otterlei M., Bruheim P., Ahn B., Bussen W., Karmakar P., Baynton K., Bohr V. A. (2006). Werner syndrome protein participates in a complex with RAD51, RAD54, RAD54B and ATR in response to ICL-induced replication arrest. J. Cell Sci. 119, 5137-5146 [DOI] [PubMed] [Google Scholar]
- Pichierri P., Franchitto A., Mosesso P., Palitti F. (2001). Werner's syndrome protein is required for correct recovery after replication arrest and DNA damage induced in S-phase of cell cycle. Mol. Cell. Biol. 12, 2412-2421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poot M., Hoehn H., Runger T. M., Martin G. M. (1992). Impaired S-phase transit of Werner syndrome cells expressed in lymphoblastoid cell-lines. Exp. Cell. Res. 202, 267-273 [DOI] [PubMed] [Google Scholar]
- Rodríguez-López A. M., Jackson D. A., Iborra F., Cox L. S. (2002). Asymmetry of DNA replication fork progression in Werner's syndrome. Aging Cell 1, 30-39 [DOI] [PubMed] [Google Scholar]
- Sakamoto S., Nishikawa K., Heo S. J., Goto M., Furuichi Y., Shimamoto A. (2001). Werner helicase relocates into nuclear foci in response to DNA damaging agents and co-localizes with RPA and Rad5. Genes Cell 6, 421-430 [DOI] [PubMed] [Google Scholar]
- Sangrithi M. N., Bernal J. A., Madine M., Philpott A., Lee J., Dunphy W. G., Venkitaraman A. R. (2005). Initiation of DNA replication requires the RECQL4 protein mutated in Rothmund-Thomson syndrome. Cell 121, 887-898 [DOI] [PubMed] [Google Scholar]
- Schultz L. B., Chehab N. H., Malikzay A., Halazonetis T. D. (2000). p53 binding Protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J. Cell Biol. 151, 1381-1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler J. A., Conti C., Syed A., Aladjem M. I., Pommier Y. (2007). The intra-S-phase checkpoint affects both DNA replication initiation and elongation: Single-cell and -DNA fiber analyses. Mol. Cell. Biol. 27, 5806-5818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao R. G., Cao C. X., Zhang H. L., Kohn K. W., Wold M. S., Pommier Y. (1999). Replication-mediated DNA damage by camptothecin induces phosphorylation of RPA by DNA-dependent protein kinase and dissociates RPA: DNA-PK complexes. EMBO J. 18, 1397-1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidorova J. M., Li N. Z., Folch A., Monnat R. J. (2008). The RecQ helicase WRN is required for normal replication fork progression after DNA damage or replication fork arrest. Cell Cycle 7, 796-807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits V. A. J., Reaper P. M., Jackson S. P. (2006). Rapid PIKK-dependent release of Chk1 from chromatin promotes the DNA-damage checkpoint response. Curr. Biol. 16, 150-159 [DOI] [PubMed] [Google Scholar]
- Stewart E., Chapman C. R., Al-Khodairy F., Carr A. M., Enoch T. (1997). Rqh(1+), a fission yeast gene related to the Bloom's and Werner's syndrome genes, is required for reversible S phase arrest. EMBO J. 16, 2682-2692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syljuasen R. G., Sorensen C. S., Hansen L. T., Fugger K., Lundin C., Johansson F., Helleday T., Sehested M., Lukas J., Bartek J. (2005). Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol. Cell. Biol. 25, 3553-3562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szekely A. M., Bleichert F., Nümann A., Van Komen S., Manasanch E., Ben Nasr A., Canaan A., Weissman S. M. (2005). Werner protein protects nonproliferating cells from oxidative DNA damage. Mol. Cell Biol. 25, 10492-10506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan S. H., Capasso H., Walworth N. C. (1999). The topoisomerase I poison camptothecin generates a Chk1-dependent DNA damage checkpoint signal in fission yeast. Yeast 15, 821-828 [DOI] [PubMed] [Google Scholar]
- Wang J. L., Wang X., Wang H., Iliakis G., Wang Y. (2002). CHK1-regulated S-phase checkpoint response reduces camptothecin cytotoxicity. Cell Cycle 1, 267-272 [PubMed] [Google Scholar]
- Wang X., Guan J., Hu B. C., Weiss R. S., Iliakis G., Wang Y. (2004). Involvement of Hus1 in the chain elongation step of DNA replication after exposure to camptothecin or ionizing radiation. Nucl. Acids Res. 32, 767-775 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Watt P. M., Louis E. J., Borts R. H., Hickson I. D. (1995). Sgs1 - A eukaryotic homolog of Escherichia coli Recq that interacts with Topoisomerase-I in Vivo and is required for faithful chromosome segregation. Cell 81, 253-260 [DOI] [PubMed] [Google Scholar]
- Watt P. M., Hickson I. D., Borts R. H., Louis E. J. (1996). SGS1, a homologue of the Bloom's and Werner's syndrome genes, is required for maintenance of genome stability in Saccharomyces cerevisiae. Genetics 144, 935-945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicky C., Alpi A., Passannante M., Rose A., Gartner A., Muller F. (2004). Multiple genetic pathways involving the Caenorhabditis elegans Bloom's syndrome genes Him-6, Rad-51, and Top-3 are needed to maintain genome stability in the germ line. Mol. Cell. Biol. 24, 5016-5027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis N., Rhind N. (2009a). Regulation of DNA replication by the S-phase DNA damage checkpoint. Cell Div. 4, 1-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis N., Rhind N. (2009b). Mus81, Rhp51 (Rad51), and Rqh1 form an epistatic pathway required for the S-phase DNA damage checkpoint. Mol. Biol. Cell. 20, 819-833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X., Yang Z., Liu Y., Zou Y. (2005). Preferential localization of hyperphosphorylated replication protein A to double-strand break repair and checkpoint complexes upon DNA damage. Biochem. J. 391, 473-480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata K., Kato J., Shimamoto A., Goto M., Furuichi Y., Ikeda H. (1998). Bloom's and Werner's syndrome genes suppress hyperrecombination in yeast sgs1 mutant: Implication for genomic instability in human diseases. Proc. Natl. Acad. Sci. USA 95, 8733-8738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Z., Kong L., Newport J. (2002). The role of single-stranded DNA and polymerase α in establishing the ATR, Hus1 DNA replication checkpoint. J. Biol. Chem. 277, 27088-27093 [DOI] [PubMed] [Google Scholar]
- Zhou B. B., Elledge S. J. (2000). The DNA damage response: Putting checkpoints in perspective. Nature 408, 433-439 [DOI] [PubMed] [Google Scholar]
- Zou L., Elledge S. J. (2003). Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300, 1542-1548 [DOI] [PubMed] [Google Scholar]
- Zou L., Cortez D., Elledge S. J. (2002). Regulation of ATR substrate selection by Rad17-dependent loading of Rad9 complexes onto chromatin. Genes Dev. 16, 198-208 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}