


Abstract
RNA secondary structure is important for designing therapeutics, understanding protein–RNA binding and predicting tertiary structure of RNA. Several databases and downloadable programs exist that specialize in the three-dimensional (3D) structure of RNA, but none focus specifically on secondary structural motifs such as internal, bulge and hairpin loops. The RNA Characterization of Secondary Structure Motifs (RNA CoSSMos) database is a freely accessible and searchable online database and website of 3D characteristics of secondary structure motifs. To create the RNA CoSSMos database, 2156 Protein Data Bank (PDB) files were searched for internal, bulge and hairpin loops, and each loop's structural information, including sugar pucker, glycosidic linkage, hydrogen bonding patterns and stacking interactions, was included in the database. False positives were defined, identified and reclassified or omitted from the database to ensure the most accurate results possible. Users can search via general PDB information, experimental parameters, sequence and specific motif and by specific structural parameters in the subquery page after the initial search. Returned results for each search can be viewed individually or a complete set can be downloaded into a spreadsheet to allow for easy comparison. The RNA CoSSMos database is automatically updated weekly and is available at http://cossmos.slu.edu.
INTRODUCTION
Deoxyribonucleic acid (DNA) and ribonucleic acid (RNA) are fundamental components of all cellular life, and much of their functionality is due to their 3D structure. While DNA is double stranded, RNA is normally single stranded and folds back upon itself, creating sites of mismatched nucleotides within the center and at the end of a duplex. These loops, including internal, bulge and hairpin loops, contribute to the functionality as well as the overall tertiary folding of the nucleic acid. Knowledge of these specific structural motifs can aid in the design of therapeutics (1,2), the understanding of RNA–protein interactions (3,4) and the prediction of tertiary structure folding of RNA (5,6). For example, myotonic dystrophy types 1 and 2 (DM1 and DM2) are diseases occurring from the expression of a tandem repeat in the genome resulting in a 1 × 1 U·U or a 2 × 2 5′CU3′/3′ UC5′ symmetric internal loop, respectively. Specifically- designed small molecules could target these loops and act as effective therapeutics against these two diseases (1). Additionally, C·A 1 × 1 internal loops are a recognition site for adenosine deaminase acting on RNA type 2 (ADAR2) (7) and can be targeted with small molecules to help prevent misediting by the enzyme (2). In some RNA macromolecules, bulge loops and hairpin loops play an important role in protein–RNA binding (3,4), such as the 5′CCCG3′ hairpin in pre-mRNA that is selectively bound by nucleolin (8).
Related databases and programs
Currently, there are several databases that focus on RNA 3D structure. RNAFRAbase 2.0 (9,10) is a powerful online tool capable of analyzing large oligomers and displaying the torsional angles and coordinates. RNAFRAbase also allows users to input structural information and displays selected secondary structures. RNAJunction (11) contains structural information about three way junctions and kissing hairpins found in the Protein Data Bank (PDB) (12), while SCOR (13) categorizes several hundred PDB files by both structural and functional classifications. FR3D (14) and the online version, WebFR3D (15), give users the ability to specify sequence and structural characteristics of desired RNA 3D motifs in order to run a real-time scan of PDB files. Additionally, several programs use sequence alignment against a known 3D structure in order to predict an unknown tertiary structure (16–23). Others scan through the 3D structure of a PDB file to identify local motifs within the specific file (24–28).
The RNA Characterization of Secondary Structure Motifs (RNA CoSSMos) database can be used by researchers as a complementary resource to those previously mentioned. The RNA CoSSMos database focuses on the 3D characteristics of secondary structure motifs in RNA; these include symmetric and asymmetric internal loops, bulge loops and hairpin loops. The structural information is pre-compiled to allow for faster searches, and the graphical user interface is designed to be intuitive, limiting pre-requisite knowledge of specialized syntax. False positives, as defined later, have been omitted from the database or reclassified to create the most accurate database possible. RNA CoSSMos is automatically updated with new PDB files and their structural characterizations weekly, keeping the database current.
DATABASE DESIGN AND CONTENT
Data extraction and motif identification
The extraction of the 3D RNA structures from the PDB was similar to that previously described by Davis et al. (29). Briefly stated, all PDB structures containing RNA, including RNA–protein structures and RNA–DNA hybrids, were downloaded. At the time of publication, 2156 PDB structures were included. Input descriptors were written based upon the 1 × 1 symmetric internal mismatch input descriptor designed by Davis et al. (29). An input descriptor is necessary to designate which base pairings are valid, e.g. canonical base pairs for nearest neighbors and non-canonical base pairs for the mismatch, and which base pairings are invalid, e.g. non-canonical base pairs for nearest neighbors and canonical base pairs for the mismatch. MC-Search (24,25) was used to identify motifs based upon these input descriptors and then to ‘clip’ the motif so all nucleotides that were not directly part of the loop or the closing base pair(s) were removed. MC-Search works by searching through the three-dimensional structure for specific nucleotide interactions, defined by Saenger (30), Westhof (31,32) or Major (25). After MC-Search ‘clipped’ the structure, MC-Annotate 1.6.2 (24,25) was then used to determine structural data including sugar puckers, base pairings and stacking interactions between the closing base pairs and the loop nucleotides. Standard code was used to process MC-Annotate output and compile it into the database.
Removal of false positives found by MC-Search
A unique feature of the RNA CoSSMos database is the removal of false positives found by MC-Search before the data is exported. For the purposes of the RNA CoSSMos database, there are two criteria for a false positive; if either is met, then the data are excluded from that subcategory of the database and reclassified. The first criterion requires that the nearest neighbors of the loop must be an A–U, C–G, G–C, G–U, U–A or U–G base pair. The second criterion requires that the mismatched nucleotides themselves cannot potentially form an A–U, C–G, G–C, G–U, U–A, or U–G base pair. These criteria and definitions of false positives appear to be unique to the RNA CoSSMos database; therefore, for researchers who agree with these criteria, the reclassification of false positives offers a distinct advantage over other available databases and software.
In symmetric internal loops, for instance, MC-Search may classify 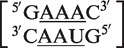 as a 3 × 3 symmetric internal loop, with the mismatch nucleotides underlined. According to the previously stated definition, this would be considered a false positive on the basis that the last mismatch of the loop, the 3′ adenine of the upper strand and the 5′ uracil of the lower strand, has the ability to form an A–U pair. In the RNA CoSSMos database, this has been reclassified as the 2 × 2 symmetric internal loop
as a 3 × 3 symmetric internal loop, with the mismatch nucleotides underlined. According to the previously stated definition, this would be considered a false positive on the basis that the last mismatch of the loop, the 3′ adenine of the upper strand and the 5′ uracil of the lower strand, has the ability to form an A–U pair. In the RNA CoSSMos database, this has been reclassified as the 2 × 2 symmetric internal loop 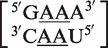 . As shown here, symmetric internal loops that are determined to be false positives are reclassified as either smaller symmetric loops or as having no mismatches.
. As shown here, symmetric internal loops that are determined to be false positives are reclassified as either smaller symmetric loops or as having no mismatches.
False positives are also possible for asymmetric internal loops. With asymmetric internal loops, either smaller asymmetric internal loops or bulges are possible results from the screenings. For example, the sequence 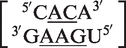 was classified as a 2 × 3 internal loop by MC-Search. Using the same methodology as with the symmetric internal loops described above, this would be classified in the RNA CoSSMos database as the 1 × 2 internal loop
was classified as a 2 × 3 internal loop by MC-Search. Using the same methodology as with the symmetric internal loops described above, this would be classified in the RNA CoSSMos database as the 1 × 2 internal loop 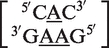 . Another internal loop classified as a 2 × 3 internal loop by MC-Search,
. Another internal loop classified as a 2 × 3 internal loop by MC-Search, 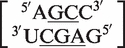 , is reclassified in the RNA CoSSMos database as the single nucleotide bulge loop
, is reclassified in the RNA CoSSMos database as the single nucleotide bulge loop 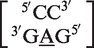 .
.
While the majority of the asymmetric false positives are able to be categorized into a specific smaller loop or bulge, some remained that resulted in an indeterminate mismatch. In order to maintain equivalent data analysis between both the symmetric and asymmetric internal loops, the undetermined loops were excluded from the RNA CoSSMos database. For instance, the 2 × 3 internal loop classified by MC-Search
 is a false positive; however, this loop was excluded from the RNA CoSSMos database. Due to the possibility of the adenosine in the top strand forming a base pair with either of the uracils on the bottom strand, which would result in two distinct single nucleotide bulges, loops such as this would need to be considered on an individual basis and therefore were omitted from the RNA CoSSMos database. Hairpin loops of 5–7 nt were evaluated for false positives in a similar manner. Bulge loops were assessed to ensure the closing base pairs were an A–U, C–G, G–C, G–U, U–A or U–G base pair. Within the RNA CoSSMos database after reclassifications, there are 11,860 symmetric internal loops, 7,781 asymmetric internal loops, 30,231 bulge loops, and 22,257 hairpin loops (Table 1).
is a false positive; however, this loop was excluded from the RNA CoSSMos database. Due to the possibility of the adenosine in the top strand forming a base pair with either of the uracils on the bottom strand, which would result in two distinct single nucleotide bulges, loops such as this would need to be considered on an individual basis and therefore were omitted from the RNA CoSSMos database. Hairpin loops of 5–7 nt were evaluated for false positives in a similar manner. Bulge loops were assessed to ensure the closing base pairs were an A–U, C–G, G–C, G–U, U–A or U–G base pair. Within the RNA CoSSMos database after reclassifications, there are 11,860 symmetric internal loops, 7,781 asymmetric internal loops, 30,231 bulge loops, and 22,257 hairpin loops (Table 1).
Table 1.
Number of motifs found in the RNA CoSSMos database
| Loop Type and Size | Number of Loops in RNA CoSSMos |
|---|---|
| Internal Symmetric Loops | |
| 1×1 | 7 545 |
| 2×2 | 1 993 |
| 3×3 | 1 568 |
| 4×4 | 558 |
| 5×5 | 196 |
| Internal Asymmetric Loops | |
| 1×2 | 3 329 |
| 1×3 | 817 |
| 1×4 | 553 |
| 1×5 | 55 |
| 2×3 | 1 647 |
| 2×4 | 417 |
| 2×5 | 133 |
| 3×4 | 372 |
| 3×5 | 69 |
| 4×5 | 389 |
| Hairpin Loops | |
| 3 | 3 358 |
| 4 | 10 225 |
| 5 | 4 498 |
| 6 | 2 746 |
| 7 | 1 430 |
| Bulge Loops | |
| 1 | 21 070 |
| 2 | 5 941 |
| 3 | 2 025 |
| 4 | 489 |
| 5 | 706 |
Database search capabilities and results output
There are several ways of searching the RNA CoSSMos database. Fields can be used in any combination and include general PDB information, experimental parameters, motif and sequence (Figure 1A). General PDB information parameters include PDB identification number, the authors of the published structure and keywords found within the PDB file. It is also possible to search by the type of experiment that was used to determine the 3D structure; X-ray diffraction and cryo-electron microscopy can be limited by the resolution of the experiment, while NMR experiments can be limited by the number of structures within the ensemble. Additionally, after the initial search has been run, it is possible to specify preferred structural characteristics in the subquery.
Figure 1.

The CoSSMos (A) search page and (B) results page.
Along with these optional experimental parameter filters, users have the ability to search for specific motifs in differing sizes. These motifs include 1 × 1, 2 × 2, 3 × 3, 4 × 4 and 5 × 5 symmetric internal loops, 1 × 2, 1 × 3, 1 × 4, 1 × 5, 2 × 3, 2 × 4, 2 × 5, 3 × 4, 3 × 5 and 4 × 5 asymmetric internal loops, hairpin loops of 3, 4, 5, 6 or 7 nt and bulge loops of 1, 2, 3, 4 or 5 nt. Within the motif-specific search, it is possible to search for one submotif, several submotifs or all submotifs. For example, selecting only triloops, both triloops and tetraloops, or all hairpins found within the database is allowed by RNA CoSSMos. To search more precisely, the desired sequence can be selected by distinguishing closing base pairs and mismatched nucleotides using the seven standard base abbreviations: A, C, G, U, R (any purine), Y (any pyrimidine) and N (any nucleotide). Once the results from the initial search have been returned, the user can choose to view those results (Figure 1B) or modify the parameters (Figure 2B). Search modifications can be made by either narrowing the standard search parameters or by using subqueries, which allow the user to search through the previous dataset and specify certain structural features, i.e. sugar puckers, glycosidic linkages, interacting edges and stacking conformations (Figure 2B). The subquery page can be located through a tab at the top of the results page.
Figure 2.

The CoSSMos (A) detailed results page, (B) subquery search page, and (C) the NMR overview page.
Within the results pages of the RNA CoSSMos database, the user can look at each specific mismatch found in the PDB or at the entire dataset via the download results option, which exports as a pound delimited file into a spreadsheet. The individual detailed results are useful for specific structural searches, while the downloadable results allow the user to more easily compare groups of structures. As explained in Davis et al. (29), MC-Annotate characterizes each mismatch with four different parameters: residue conformation, base pairing, adjacent stacking and non-adjacent stacking. For each nucleotide, the residue conformation is given by describing the sugar pucker as either endo or exo and the glycosidic linkage as either syn or anti. For the base pairing characterizations that are labeled as ‘Interacting Edges’ in the RNA CoSSMos database, MC-Annotate relies on the nomenclature schemes developed by Major (25), Saenger (30) and Westhof (31,32). Abbreviations [i.e. Watson–Crick (W), Hoogsteen (H) and sugar (S)] are used to identify the edge of the base that is involved in the hydrogen bonding. In most cases, a two-letter designation is used to describe the interacting edges where the first letter describes which edge of the base is pairing, and the second specifies where on that edge the bonding is occurring (25). For example, Ws would be used to designate that the Watson–Crick side of the base near the sugar edge is participating in the hydrogen bond. Some bonding patterns are also designated by either a Roman numeral (Saenger notation) (30) or an Arabic numeral (Westhof notation) (31,32). For interacting edges that are not designated with the two-letter abbreviation, a different, non-standard annotation may be used to describe the hydrogen-bonding patterns, such as 5′O2P/3′Bh to describe a bifurcated hydrogen-bonding pattern between the 2′ oxygen of the sugar and the Hoogsteen face. Stacking interactions, both adjacent and non-adjacent, are described by the terms upward, downward, outward or inward as proposed by Major and Thibault (33). All mismatches within the database are characterized by these four designations.
The detailed results page (Figure 2A) for each motif displays all structural characterizations for every applicable nucleotide and a ‘clipped’ PDB structure in a Jmol (34) applet, which allows for any user with a web browser that supports Java to view the structure. In order to ensure the accurate representation of the motif structure, the PDB file for the ‘clipped’ structure was not altered; instead, the nucleotides and amino acids that are not involved in the mismatch are blacked out, reducing the possibility of unintentionally deleting or modifying the structural data. This ‘clipped’ structure can be manipulated as in any Jmol program and then downloaded as a snapshot. In addition to the structural information and the clipped structure in Jmol, the detailed results page includes an option for printer-friendly results, links to the PDB (12) and links to the Nucleic Acid Database (35), where applicable.
Additional features
Beyond the search capabilities and the results output, the RNA CoSSMos database contains an optional username, a frequently asked questions page and an NMR overview page. Although the RNA CoSSMos database is freely accessible to all, registering with a username is encouraged. Upon registering, users will be able to save up to 10 searches on the RNA CoSSMos database, as well as receive emails about important updates to the RNA CoSSMos database. The frequently asked questions page is linked to the home page of the website and contains information on both the RNA CoSSMos database itself and the structural characterizations, including definitions of and graphics depicting the interactions between nucleotides. For all ensemble structures determined by NMR, the RNA CoSSMos database contains a unique NMR overview page, providing a convenient way to analyze the structural characteristics between ensemble structures (Figure 2C). In the NMR overview, the first structure of the ensemble is arbitrarily defined as the prime structure to which the others are compared. The remaining structures’ characteristics are evaluated and color coded based upon the equivalency. If the interaction is equivalent to that in the prime structure, the box is colored green; if not, the box is colored red. Additionally, each structure is assigned a percentage based upon its contribution to the ensemble character. The structure with the highest percentage may be considered as the representative structure of the ensemble.
CONCLUSIONS AND FUTURE DIRECTIONS
The RNA CoSSMos database is a unique online tool that gives researchers the ability to search for 3D characteristics of RNA secondary structure motifs without creating the need for the user to run a comprehensive search. The database design is a simple and intuitive graphical user interface, which eliminates the need for complex syntax. Within the database, the reclassification of false positives eliminates incorrect identifications of the motifs, allowing for the most accurate database possible. Additionally, multiple search parameters, including sequence and motif structure, make RNA CoSSMos versatile for many different uses. The downloadable results, the detailed results pages and the NMR overview pages create many different options for viewing the structural characterizations of the motifs. Future versions of RNA CoSSMos may extend into DNA secondary structure motifs and higher order RNA motifs, such as A-platforms and U-turns. Additionally, the indeterminate mismatches may be included, and users will be able to search for them. Future directions of the database will also be driven by feedback from the users. As the PDB continues to grow, so will the RNA CoSSMos database and its capabilities.
AVAILABILITY
The RNA CoSSMos database is freely available online at http://cossmos.slu.edu. Users of the RNA CoSSMos database should cite this article and are encouraged to cite the original references for MC-Search and MC-Annotate from Francois Major's laboratory (24,25).
FUNDING
The National Institutes of Health (1R15GM085699-01A1 to B.M.Z.). Funding for open access charge: NIH.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors would like to thank Francois Major for providing us with executable versions of MC-Search and MC-Annotate and for providing assistance with the software in the context of the work described here. Additionally, the authors would like to thank Nina Zulic Hausmann for her initial work on the input descriptors.
REFERENCES
- 1.Lee MM, Childs-Disney JL, Pushechnikov A, French JM, Sobczak K, Thornton CA, Disney MD. Controlling the specificity of modularly assembled small molecules for RNA via ligand module spacing: targeting the RNAs that cause myotonic muscular dystrophy. J. Am. Chem. Soc. 2009;131:17464–17472. doi: 10.1021/ja906877y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tran T, Disney MD. Molecular recognition of 6′-N-5-hexynoate kanamyin A and RNA 1x1 internal loops containing CA mismatches. Biochemistry. 2011;50:962–969. doi: 10.1021/bi101724h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gupta A, Gribskov M. The role of RNA sequence and structure in RNA-protein interactions. J. Mol. Biol. 2011;409:574–587. doi: 10.1016/j.jmb.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 4.Hermann T, Patel DJ. RNA bulges as architectural and recognition motifs. Structure. 2000;8:47–54. doi: 10.1016/s0969-2126(00)00110-6. [DOI] [PubMed] [Google Scholar]
- 5.Leontis NB, Westhof E. Analysis of RNA motifs. Curr. Opin. Struct. Biol. 2003;13:300–308. doi: 10.1016/s0959-440x(03)00076-9. [DOI] [PubMed] [Google Scholar]
- 6.Reiter NJ, Chan CW, Mondragón A. Emerging structural themes in large RNA molecules. Curr. Opin. Struct. Biol. 2011;21:319–326. doi: 10.1016/j.sbi.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wong SK, Sato S, Lazinski DW. Substrate recognition by ADAR1 and ADAR2. RNA. 2001;7:846–858. doi: 10.1017/s135583820101007x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Allain FHT, Bouvet P, Dieckmann T, Feigon J. Molecular basis of sequence-specific recognition of pre-ribosomal RNA by nucleolin. EMBO J. 2000;19:6870–6881. doi: 10.1093/emboj/19.24.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Popenda M, Blazewicz M, Szachniuk M, Adamiak RW. RNA FRABASE version 1.0: an engine with a database to search for the three-dimensional fragments within RNA structures. Nucleic Acids Res. 2008;36:D386–D391. doi: 10.1093/nar/gkm786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Popenda M, Szachniuk M, Blazewicz M, Wasik S, Burke EK, Blazewicz J, Adamiak RW. RNA FRABASE 2.0: an advanced web-accessible database with the capacity to search the three-dimensional fragments within RNA structures. BMC Bioinformatics. 2010;11:231–242. doi: 10.1186/1471-2105-11-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bindewald E, Hayes R, Yingling Y, Kasprzak W, Shapiro BA. RNAJunction: a database of RNA junctions and kissing loops for three-dimensional structural analysis and nanodesign. Nucleic Acids Res. 2008;36:D392–D397. doi: 10.1093/nar/gkm842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klosterman PS, Tamura M, Holbrook SR, Brenner SE. SCOR: a structural classification of RNA database. Nucleic Acids Res. 2002;30:392–394. doi: 10.1093/nar/30.1.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sarver M, Zirbel CL, Stombaugh J, Mokdad A, Leontis NB. FR3D: finding local and composite recurrent structural motifs in RNA 3D structures. J. Math. Biol. 2008;56:215–252. doi: 10.1007/s00285-007-0110-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Petrov AI, Zirbel CL, Leontis NB. WebFR3D—a server for finding, aligning and analyzing recurrent RNA 3D motifs. Nucleic Acids Res. 2011;39:W50–W55. doi: 10.1093/nar/gkr249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dror O, Nussinov R, Wolfson H. ARTS: alignment of RNA tertiary structures. Bioinformatics. 2005;21:ii47–ii53. doi: 10.1093/bioinformatics/bti1108. [DOI] [PubMed] [Google Scholar]
- 17.Dror O, Nussinov R, Wolfson HJ. The ARTS web server for aligning RNA tertiary structures. Nucleic Acids Res. 2006;34:W412–W415. doi: 10.1093/nar/gkl312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferrè F, Ponty Y, Lorenz WA, Clote P. DIAL: a web server for the pairwise alignment of two RNA three-dimensional structures using nucleotide, dihedral angle and base-pairing similarities. Nucleic Acids Res. 2007;35:W659–W668. doi: 10.1093/nar/gkm334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang Y-F, Huang Y-L, Lu CL. SARSA: a web tool for structural alignment of RNA using a structural alphabet. Nucleic Acids Res. 2008;36:W19–W24. doi: 10.1093/nar/gkn327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Capriotti E, Marti-Renom MA. RNA structure alignment by a unit-vector approach. Bioinformatics. 2008;24:i112–i118. doi: 10.1093/bioinformatics/btn288. [DOI] [PubMed] [Google Scholar]
- 21.Lai C-E, Tsai M-Y, Liu Y-C, Wang C-W, Chen K-T, Lu CL. FASTR3D: a fast and accurate search tool for similar RNA 3D structures. Nucleic Acids Res. 2009;37:W287–W295. doi: 10.1093/nar/gkp330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rother M, Rother K, Puton T, Bujnicki JM. ModeRNA: a tool for comparative modeling of RNA 3D structure. Nucleic Acids Res. 2011;39:4007–4022. doi: 10.1093/nar/gkq1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kirillova S, Tosatto SCE, Carugo O. Open access FRASS: the web-server for RNA structural comparison software. BMC Bioinformatics. 2010;11:327–334. doi: 10.1186/1471-2105-11-327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gendron P, Lemieux S, Major F. Quantitative analysis of nucleic acid three-dimensional structures. J. Mol. Biol. 2001;308:919–936. doi: 10.1006/jmbi.2001.4626. [DOI] [PubMed] [Google Scholar]
- 25.Lemieux S, Major F. RNA canonical and non-canonical base pairing types: a recognition method and complete repertoire. Nucleic Acids Res. 2002;30:4250–4263. doi: 10.1093/nar/gkf540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duarte CM. RNA structure comparison, motif search and discovery using a reduced representation of RNA conformational space. Nucleic Acids Res. 2003;31:4755–4761. doi: 10.1093/nar/gkg682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhong C, Tang H, Zhang S. RNAMotifScan: automatic identification of RNA structural motifs using secondary structural alignment. Nucleic Acids Res. 2010;38:176e. doi: 10.1093/nar/gkq672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Macke TJ, Ecker DJ, Gutell RR, Gautheret D, Case DA, Sampath R. RNAMotif, an RNA secondary structure definition and search algorithm. Nucleic Acids Res. 2001;29:4724–4735. doi: 10.1093/nar/29.22.4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davis AR, Kirkpatrick CC, Znosko BM. Structural characterization of naturally occurring RNA single mismatches. Nucleic Acids Res. 2010;39:1081–1094. doi: 10.1093/nar/gkq793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saenger W. Principles of Nucleic Acid Structure. New York: Springer; 1984. [Google Scholar]
- 31.Leontis NB, Westhof E. Conserved geometrical base-pairing patterns in RNA. Q Rev. Biophys. 1998;31:399–455. doi: 10.1017/s0033583599003479. [DOI] [PubMed] [Google Scholar]
- 32.Leontis NB, Westhof E. Geometric nomenclature and classification of RNA base pairs. RNA. 2001;7:499–512. doi: 10.1017/s1355838201002515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Major F, Thibault P. In: Bioinformatics: From Genetics to Therapies. Lengauer T, editor. Germany: Wiley-VCH, Weinheim; 2007. pp. 491–539. [Google Scholar]
- 34.Jmol: an open source Java viewer for chemical structures in 3D. http://www.jmol.org/ [Google Scholar]
- 35.Berman HM, Olson WK, Beveridge DL, Westbrook J, Gelbin A, Demeny T, Hsieh SH, Srinivasan AR, Schneider B. The Nucleic Acid Database: a comprehensive relational database of three-dimensional structures of nucleic acids. Biophys. J. 1992;63:751–759. doi: 10.1016/S0006-3495(92)81649-1. [DOI] [PMC free article] [PubMed] [Google Scholar]