

Abstract
Purpose
Camptothecin (CPT) has potent broad spectrum antitumor activity by inhibiting type I DNA topoisomerase (DNA topo I). It has not been used clinically because it is water-insoluble and highly toxic. As a result, irinotecan (CPT-11), a water-soluble analogue of CPT, has been developed and used as salvage chemotherapy in patients with relapsed/refractory lymphoma, but with only modest activity. Recently, we have developed a cyclodextrin-based polymer conjugates of 20-(S)-CPT (IT-101). In this study, we evaluated the preclinical antilymphoma efficacy of IT-101 as compared to CPT-11.
Experimental Design
We determined an in vitro cytotoxicity of IT-101, CPT-11 and their metabolites against multiple human lymphoma cell lines. In human lymphoma xenografts, the pharmacokinetics, inhibitions of tumor DNA topo I catalytic activity, and antilymphoma activities of these compounds were evaluated.
Results
IT-101 and CPT had very high in vitro cytotoxicity against all lymphoma cell lines tested. As compared to CPT-11 and SN-38, IT-101 and CPT had longer release kinetics and significantly inhibit higher tumor DNA topo I catalytic activities. Furthermore, IT-101 showed significantly prolonged the survival of animals bearing s.c. and disseminated human xenografts when compared to CPT-11 at its maximum tolerated dose in mouse.
Conclusions
The promising present results provide the basis for a phase I clinical trial in patients with relapsed/refractory lymphoma.
Introduction
Although great advances have been made in the treatment of malignant lymphoma, more than half of the patients with aggressive non-Hodgkin lymphoma (NHL) and a vast majority of patients with indolent lymphoma have resistant diseases or relapse after the initial treatment and eventually require salvage chemotherapy. In general, patients with Burkitt lymphoma, anaplastic large T-cell lymphoma (ALTC), and advanced-stage Hodgkin lymphoma (HL) who receives first-line combination chemotherapies can achieve 5-year overall survival rate in 65–90%, 37–93%, and 66–82% of patients, respectively (1–5). However, only a small number of these patients can achieve long-term disease-free survival (DFS) after high-dose therapy and hematopoietic stem cell rescue. The limitation of this approach is that not all patients respond to widely used salvage therapies including EPOCH (6), ESHAP (7), and MINE-ESHAP (8). Therefore, a novel agent for the salvage setting in these patients is needed. The development of salvage regimens are based on the combination of non-cross resistant agents from the first-line chemotherapy regimens. The DNA topoisomerase I (Topo I) inhibitors have been explored as candidates for salvage therapy in patients with relapsed/refractory NHL due to an increase of DNA Topo I activity in lymphoma cells. 20(S)-Camptothecin (CPT) is a plant alkaloid present in fruit, bark, and wood of the Camptotheca acuminata. CPT has a board spectrum of antitumor activity which mediates through interaction with the nuclear enzyme Topo I and prevents it from resealing the DNA break, resulting in a double strain DNA break and cell death (9–12). Moreover, it is a poor substrate for P-glycoprotein, a class of drug efflux pumps that is upregulated in many multi-drug resistant (MDR) cancer cells. However, the clinical use of CPT has been precluded by its significant treatment-related toxicity (TRT) and low antitumor efficacy (13,14). Irinotecan (CPT-11), an analogue of CPT, has been used alone or in combination with other cytotoxic agents as salvage regimens for patients with relapsed/refractory NHL (15–18). In spite of the high response demonstrated in the phase II study of CPT-11 against a board range of solid tumors, it usually has not been employed in the treatment of malignant lymphoma. This is mainly because of its common TRT including grade 3/4 leukopenia and grade 3/4 diarrhea caused by the recommended dosing schedule of this agent (16–19). Although prolonged intravenous (i.v.) infusion of CPT-11 has been reported to enhance antitumor activity (20, 21), a disadvantage of this delivery method observed in xenograft models and early clinical trials was again a high incidence of TRT including diarrhea, nausea/vomiting, neutropenia, anemia, and pulmonary toxicity (22–25). IT-101, a nanoparticulate conjugate of 20(S)-camptothecin and a β-cyclodextrin-based polymer, has recently been developed to improve biodistribution to tumor tissue, minimize TRT, and increase antitumor activity (26, 27). In a preclinical study, IT-101 has been able to demonstrate antitumor activity against a broad range of solid tumors (28). Therefore, we have performed this study to determine the preclinical efficacy of this novel compound in human lymphoma xenografts.
Materials and Methods
DNA construct
The bifunctional ffLuc:Zeocin fusion gene which coexpresses the firefly luciferase (ffLuc) and zeocin (Zeo) resistance genes was cloned into pcDNA3.1+ (Invitrogen, Carlsbad, CA) to generating plasmid ffLuc:Zeocin-pcDNA3.1+ as previously described (29).
Cell culture and genetic modification
Daudi cell (human Burkitt lymphoma line) was obtained from American Type Culture Collection (Manassas, VA). Karpas 299 cell (human anaplastic large T cell lymphoma line) was obtained from Deutsche Sammlung von Mikroorganism und Zellkulturen GmbH (Graunschweig, Germany). L540 cell (human Hodgkin lymphoma line) was kindly provided by Dr. Andreas Engert (University Clinic of Cologne, Cologne, Germany). These cell lines were maintained in sterile culture media (CM) as previously described (29). 8 × 106 Daudi and Karpas cells were genetically modified with 10 μg of ffLuc:Zeocin-pcDNA3.1+ linearized DNA plasmid as previously described (29). Begnining on the third day after electrotransfer, zeocin (InvivoGen, San Diego, CA) was added to the culture at a concentration of 0.1–0.4 mg/mL to maintain stable transfection.
In vitro cytotoxicity of IT-101 against human lymphoma cell lines
The toxicities of CPT, IT-101, SN-38 (active metabolite of CPT-11), and CPT-11 were determined in Daudi, Karpas 299, and L540 cells after 72 hours of incubation in RPMI 1640 supplemented with 10% heat-inactivated FBS (Daudi cell and Karpas cell) or 20% FBS (L540 cell) using the MTS assay.
Human lymphoma xenograft models
Mouse care and experimental procedures were carried out in accordance with the Research Animal Care Committee (RACC) of Beckman Research Institute at City of Hope. We first established three localized s.c. models in six- to eight-week-old female severe combined immunodeficiency (SCID/NCr, BALB/C background) mice (National Cancer Institute, Federick, MD). These animals were injected with 0.2 mL of 1:1 mixture of tumor cell suspension in 1% human serum albumin in Hank’s balanced salt solution (HBSS; Mediatech) and Matrigel (BD Biosciences, San Jose, CA) into their right flanks. The cell dose of Daudi cell, Karpas 299 cell, and L540 cell was 5 × 106, 3 × 106, and 5 × 106 cells, respectively. To establish two disseminated models, six- to eight-week-old female nonobese diabetic severe combined immunodeficiency (NOD.scid/NCr) mice (National Cancer Institute) were injected with 0.2 mL of 1:1 lymphoma cells stably expressing ffLuc activity in a suspension of 1x Dulbecco’s phosphate-buffered saline solution (DPBS; Mediatech) and 1% heat-inactivated FBS via lateral tail vein. The cell dose of Daudi cell and Karpas 299 cell was 7.5 × 106 and 5 × 106 cells, respectively.
Plasma and tumor concentrations of IT-101, CPT, CPT-11, and SN-38
Localized s.c. human xenograft-bearing animals with tumor volumes (TVs) reaching approximately 500–800 mm3 were randomly divided into two treatment groups of 20 animals each; group 1: CPT-11 (100 mg/kg, intraperitoneal [i.p.], single dose) and group 2: IT-101 (10 mg/kg, i.v., single dose). Then tumor specimens and plasma from five mice in each treatment group were corrected at four time points (before dosing, 2, 24, and 48 hours after dosing) to measure plasma and tumor concentrations of the compounds and their active metabolites. Measurements of plasma and tumor concentrations of IT-101 and CPT were carried out for SCID mice treated with IT-101, whereas those of CPT-11 and SN-38 were carried out for mice treated with CPT-11. The method of these measurements has been previously described (26).
Tumor type I DNA topoisomerase (DNA Topo I) catalytic activity inhibited by IT-101 and CPT-11
At each time point along with the measurements of plasma and tumor concentrations of IT-101, CPT-11, and their metabolites, the inhibition of tumor DNA Topo I catalytic activity after administration of either IT-101 or CPT-11 were evaluated. The catalytic activity of DNA Topo I was determined by measuring the relaxation of supercoiled (form I) plasmid substrate DNA using the Topo I assay kit (TopoGEN, Port Orange, FL) essentially according to the method of Liu and Miller (30). First, preparation of crude extracts from tumor tissues was performed as previously described (31). Second, nuclear extraction was performed using celLytic nuclear extraction kit (Sigma-Aldrich, St. Loius, MO). Then tumor DNA Topo I catalytic activity was determined following the instructions accompanying the Topo I assay kit. Briefly, the reaction mixtures used consists of supercoiled (form I) plasmid substrate DNA (0.5 μg), tumor nuclear extract (0.5 μg total protein) and the assay buffer. Supercoiled (form I) plasmid DNA (0.5 μg) and relaxed DNA (0.5 μg) provided by the Topo I assay kit were used as the control markers. The reaction mixtures were incubated at 37 oC for 30 minutes, and terminated by adding 5 μL stop buffer/gel loading buffer. Samples were loaded onto a 1% agarose gel submersed in 1x TAE buffer (50x TAE buffer: 242 g Tris base, 57.1 mL glacial acetic acid, and 100 ml 0.5 M EDTA) and electrophoresed overnight at room temperature. The gel was stained with 0.2 μg/mL ethidium bromide for 20 minutes at room temperature, destained in water for 20 minutes and imaged under UV light. The background of supercoiled DNA band was subtracted, and the density of the supercoiled DNA band was quantified using FluorChem AlphaEaseFC2 software (Alpha Innotech, San Leandro, CA). The percentage of DNA Topo I catalytic activity inhibited by either IT-101 or CPT-11 was equal to the density of supercoiled DNA band from treated tumor divided by the density of supercoiled DNA band from untreated control and timed by 100.
Tumor burden monitoring
In the s.c. model, detection of tumor growth by serial physical measurements was initiated two to seven days after tumor implant and repeated at least twice a week until the average TV was approximately 100–200 mm3 at which time therapy was initiated. This was repeated at least once a week until the end of the study. The TV was calculated as previously described (28). In disseminated model, in vivo biophotonic imaging (see below) was initiated approximately seven days after tumor injection.
Biophotonic imaging
The in vivo ffLuc-derived bioluminescent imaging (BLI) signal was evaluated using an IVIS 100 imaging system (Xenogen, Alameda, CA) at 18 minutes after a single intraperitoneal (i.p.) injection of dissolved D-Luciferin (Xenogen) at a dose of 50 mg/kg (0.1 mL of a 10 mg/mL solution per 20-g mouse). Photons were quantified using the Living Image version 2.5 software (Xenogen). Background bioluminescence signal was defined as < 106 p/s/cm2/sr based on the average ffLuc-derived BLI of normal control mice.
Determination of treatment efficacy
The treatment result for each animal may be pathological complete tumor response (pCTR), complete tumor response (CTR), or partial tumor response (PTR). In a CTR, the TV is < 13.5 mm3 for two consecutive measurements in localized s.c. model, whereas the BLI is < 106 p/s/cm2/sr for two consecutive measurements in the disseminated model. A pCTR is defined as CTR combined with evidence of nonviable tumor on histopathological study. In a PTR, the TV is < 50% of its pretreatment volume for two consecutive measurements and the TV ≥ 13.5 mm3 for one or more of these two measurements, whereas the BLI is < 50% of its pretreatment signal and the BLI signal >106 p/s/cm2/sr for one or both of these two measurements. In accordance with the institutional RACC, the predetermined tumor end point is defined as follows: (1) the TV > 2,000 mm3 and/or ulcerated tumor in localized s.c. model; and (2) the BLI signal > 1010 p/s/cm2/sr and/or weight loss > 20% with evidence of progressive disease and/or hind limb paralysis and or death from tumor progression in disseminated model. Percent animal survival is defined by the percentage of animals that did not die of the tumor or TRT or reach the predetermined tumor end point during the course of the study. At the end of the study, all survived animals were euthanized with CO2. In order to detect some residual tumors by histopathology study, a whole thickness of skin around the tumor injection site with or without gross tumor was harvested from animals with s.c. xenografts; whereas a brain, a heart, a spleen, a liver, ovaries, kidneys, lungs, femurs, and spines were harvested from animals with disseminated xenografts.
Treatment schedule and tumor burden monitoring
For reference purposes, CPT-11 was used as the internal positive control in the current study as previously described (28). Mice with established human lymphoma xenografts were allocated into five different treatment groups of nine animals each, as follows: group 1 (untreated control); group 2 (CPT-11 at 100 mg/kg, i.p., qwk × 3); group 3 (IT-101 at 5 mg/kg, i.v., qwk × 3); group 4 (IT-101 at 10 mg/kg, i.v., qwk × 3); and group 5 (single dose of IT-101 at 15 mg/kg, i.v.).
Tumor burdens were monitored at least once weekly toward the predetermined endpoints or for 126 days postadministration in the localized s.c. models and for 90 days postadministration in the disseminated models.
Tolerability
Mice were weighed twice weekly during treatment and then weekly until the end of the study and were observed for some expected TRT, including progressive weight loss, anorexia, and diarrhea. The unacceptable TRT for the maximum tolerated dose (MTD) was defined as previously described (28).
Statistical and graphical analyses
For DNA Topo I catalytic activity, samples were compared by paired t test. The survival curves were constructed using the Kaplan-Meier method. The log-rank test was used to compare the percent animal survival between treatment groups. P < 0.05 was considered statistically significant.
Results
In vitro cytotoxicity of Topo I inhibitors against three lymphoma cell lines
Based on the Developmental Therapeutics Program of NCI/NIH and our unpublished data, the IC50’s of CPT, IT-101, SN-38, and CPT-11 for colon, breast, and prostate cancer cell lines were 0.02–0.2 μM, 0.22–0.38 μM, 0.001–0.003 μM, and 13–45 μM, respectively. Using MTS assay, the IC50’s ranged from 0.01–0.06 μM and 0.06–0.4 μM after 72 hours of incubation with CPT and IT-101, respectively, indicating that both CPT and IT-101 had very high in vitro cytotoxicity against all lymphoma cell lines tested (Table 1).
Table 1.
IC50’s of CPT, IT-101, SN-38, and CPT-11 in various lymphoma cell lines after a 72-hour incubation period
| Lymphoma cell line | CPT (μM) | IT-101 (μM) | SN-38 (μM) | CPT-11 (μM) |
|---|---|---|---|---|
| Daudi | 0.06 | 0.17 | 0.003 | 62 |
| Karpas 299 | 0.02 | 0.4 | 0.008 | 11 |
| L540 | 0.01 | 0.06 | 0.008 | 2 |
Longer release kinetics of IT-101 and CPT as compared with CPT-11 and SN-38
In plasma, the maximum concentration of polymer-bound CPT, free CPT, CPT-11, and SN-38 was at 2 hours posttreatment in all s.c. tumor xenograft animals tested but the concentration of CPT-11 and SN-38 declined rapidly at 24 hours posttreatment (Fig. 1A). In the s.c. tumor xenografts themselves, the maximum concentration of polymer-bound CPT and free CPT was at 48, 24, and 48 hours posttreatment in Daudi tumors, Karpas 299 tumors, and L540 tumors, respectively; whereas the maximum concentration of SN-38 was at 2, 2, and 24 hours posttreatment in Daudi tumors, Karpas 299 tumors, and L540 tumors, respectively (Fig. 1B). These results indicated that IT-101 has longer release kinetics in tumor than that of CPT-11.
Fig. 1.
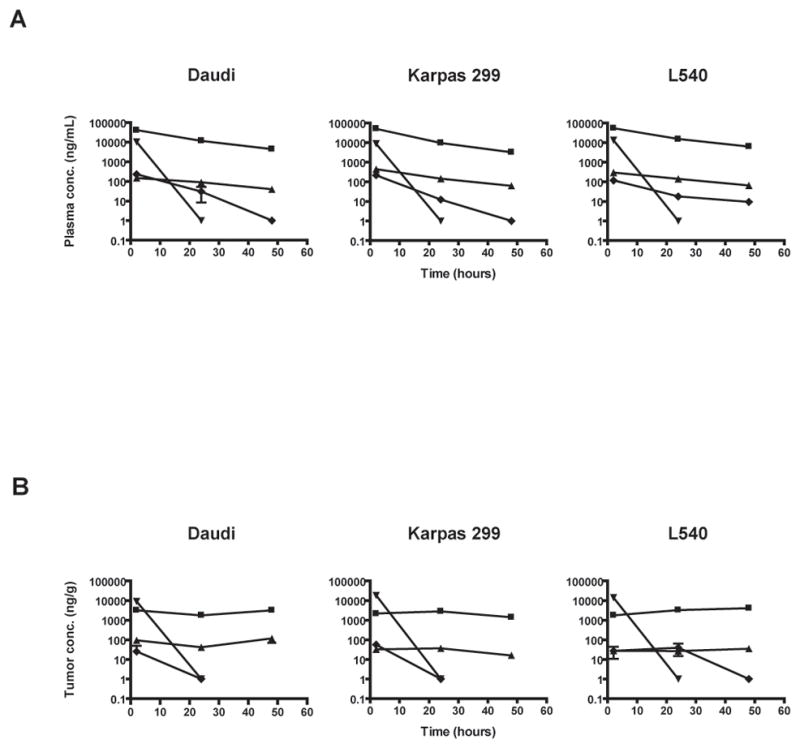
(A) Plasma concentration of polymer-bound (■), free CPT (▲), CPT-11 (▼), and SN-38 (◆) as a function of time after a single bolus i.v. injection of IT-101 (10 mg/kg) and a single i.p. injection of CP-11 (100 mg/kg). (B) Tumor concentration of polymer-bound (■), free CPT (▲), CPT-11 (▼), and SN-38 (◆) as a function of time after a single bolus i.v. injection of IT-101 (10 mg/kg) and a single i.p. injection of CPT-11 (100 mg/kg). Points and error bars indicate mean and standard error of mean, respectively.
Tumor type I DNA Topoisomerase catalytic activity inhibited by IT-101 and CPT-11
Based on the IT-101 kinetic data above, we predicted that IT-101 would be able to inhibit tumor DNA Topo I catalytic activity for longer time than CPT-11. The sensitivity of tumor cells to either IT-101 or CPT-11 was determined by inhibition of tumor nuclear Topo I catalytic activity using a DNA relaxation assay. At 2, 24, and 48 hours posttreatment, both IT-101 (10 mg/kg, i.v., × 1) and CPT-11 (100 mg/kg, i.p., × 1) could significantly inhibit nuclear DNA Topo I catalytic activity in all type of s.c. lymphoma xenografts tested. However, at 48 hours, IT-101 could inhibit nuclear DNA Topo I catalytic activity significantly better than CPT-11 in s.c. Daudi tumors and s.c. Karpas 299 tumors (P = 0.007; 95% CI, 11.2–30.99 and P = 0.037; 95% CI, 2.98–48.08, respectively); though there was only a nonsignificant trend in s.c. L540 tumors (P = 0.07). These observations are associated with longer release kinetics of IT-101 in tumor and are consistent with the maximum concentration of polymer-bound CPT and free CPT at 48 hours posttreatment as compared to SN-38 at 2 hours posttreatment (Fig. 2).
Fig. 2.

Administration of IT-101 (10 mg/kg, i.v., single dose) can significantly inhibit tumor DNA Topo I catalytic activity in lymphoma xenografts as compared to CPT-11. DNA relaxation assay was done with nuclear extracts containing 0.5 μg total protein, isolated from s.c. lymphoma xenografts at four time points (before dosing, 2 hours, 24 hours, and 48 hours after dosing), as depicted. The mean percentage of tumor DNA Topo I catalytic activity inhibited by either IT-101 or CPT-11 was calculated from three or more separate experiments. Error bars indicate standard error of mean.
Tolerabiltiy
Recent studies have demonstrated that at the same accumulative dose, IT-101 qwk × 3 can simultaneously maximize antitumor effect and minimize TRT when compared to multiple daily dosing schedules (28). The maximum tolerated dose (MTD) of IT-101 in solid tumor bearing athymic nude mice (no data in SCID and NOD.scid/NCr mice) was more than 16.1 mg/kg qwk × 3 but less than 25 mg/kg qwk × 3. However, based on our internal study of human lymphoma xenografts in athymic nude mice, the MTD of IT-101 was less than 15 mg/kg qwk × 2 so that three weekly doses of IT-101 at 5 mg/kg (CPT equivalents), three weekly doses of IT-101 at 10 mg/kg (CPT equivalents) and a single dose of IT-101 at 15 mg/kg (CPT equivalents) were planned for use in the present study. Except for three treatment-related deaths (TRDs) in IT-101 (15 mg/kg, i.v., × 1) treated SCID mice, all animals tolerated the treatments well. Treatments with three weekly doses of IT-101 at 5 mg/kg and 10 mg/kg were well tolerated in all animals. Thus, the MTD of i.v. IT-101 in tumor bearing SCID mice was between 10 mg/kg qwk × 3 and 15 mg/kg single dose. As a result, we dropped the IT-101 (15 mg/kg, i.v., × 1) arm from the disseminated models using NOD.scid/NCr mice. Therapy with i.p. CPT-11 was generally well tolerated in both strains of animal. Mean body weight loss of SCID mice and NOD.scid/NCr mice was minimal.
In vivo efficacy of IT-101 against localized s.c. human lymphoma xenograft models
In localized s.c. Daudi tumors, therapy was initiated ten days after tumor cell inoculation. Forty-five SCID mice with established localized s.c. tumor were allocated into five different treatment groups of nine animals per group as described earlier. The average TV among all groups was equally distributed. All untreated controls and animals treated with CPT-11 developed progressive tumor growth with an average TV of more than 2,000 mm3 within 38 days and 59 days after tumor injection, respectively. In contrast, IT-101 (5 mg/kg, i.v., qwk × 3) and IT-101 (10 mg/kg, i.v., qwk × 3) could significantly prolong the survival of the animals as compared to those treated with CPT-11 (P < 0.0001 and P < 0.0001, respectively) (Fig. 3A). Five of nine mice (56%) treated with IT-101 5 mg/kg and seven of nine mice (78%) treated with IT-101 (10 mg/kg, i.v., qwk × 3) survived and were pathologically confirmed disease-free after 126 days posttreatment.
Fig. 3.

Administration of IT-101 (5 mg/kg, i.v., qwk × 3) and (10 mg/kg, i.v., qwk × 3) to animals bearing three distinct s.c. human lymphoma xenografts can result in significant better survival as compared to CPT-11. Treatments were initiated at 10, 4, and 16 days after subcutaneous injection of Daudi cells, Karpas 299 cells, and L540 cells, respectively. Treatment arms included untreated control (■), CPT-11 (100 mg/kg, i.p., qwk × 3) (◇), IT-101 (5 mg/kg, i.v., qwk × 3) (○), and IT-101 (10 mg/kg, i.v., qwk × 3) (x). The tumor burden was monitored longitudinally by physical measurements. Using Kaplan-Meier method, the survival curves were plot for each treatment group and the log-rank test was used to compare the percent animal survival between treatment groups. In Daudi tumors and L540 tumors, a significant difference between the groups treated with either IT-101 (5 mg/kg, i.v., qwk × 3) or IT-101 (10 mg/kg, i.v., × 3) was not observed. However, these two treatment groups achieved significantly better survival as compared to the groups treated with CPT-11 (P < 0.0001 and P < 0.0001, respectively). In Karpas 299 tumors, no significant difference between the groups treated with either IT-101 (5 mg/kg, i.v., qwk × 3) or IT-101 (10 mg/kg, i.v., qwk × 3) was observed; but animals treated with IT-101 at (10 mg/kg, i.v., qwk × 3) had significantly better survival as compared to those treated with CPT-11. (P = 0.0072). Points indicate mean survival.
In s.c. Karpas 299 tumors, therapy was initiated four days after tumor cell inoculation. All untreated control mice developed progressive tumor growth with an average TV more than 2,000 mm3 within 25 days after tumor injection. As shown in Fig. 3B, although the significant difference between the groups treated with either IT-101 (5 mg/kg, i.v., qwk × 3) or IT-101 (10 mg/kg, i.v., qwk × 3) was not observed; the animals treated with IT-101 (10 mg/kg, i.v., qwk × 3) had significantly longer survival than those treated with CPT-11 (P = 0.0072). At the end of the study, 44% of IT-101 (10 mg/kg, i.v., qwk × 3) treated animals, 33% of IT-101 (5 mg/kg, i.v., qwk × 3) treated animals, and 33% of CPT-11 treated animals achieved a pathologically confirmed disease-free status.
The antilymphoma activity of IT-101 was also evaluated in s.c. L540 tumors. In this model, therapy was initiated 16 days after tumor cell inoculation. Tumors in all untreated control mice grew rapidly with ulceration within 51 days after tumor injection and were then sacrificed. Although a significantly longer survival could be observed in all CPT-11 treated mice as compared with untreated controls (P = 0.018), all had progressive tumor growth with ulceration within 79 days after tumor implant. Consistent with the other s.c. xenografts, IT-101 (5 mg/kg, i.v., qwk × 3) treated animals and IT-101 (10 mg/kg, i.v., qwk × 3) treated animals had significantly longer survival than those treated with CPT-11 (P < 0.001 and P < 0.0001, respectively) as shown in Fig. 3C. Seven of nine animals (78%) that received either IT-101 (5 mg/kg, i.v., qwk × 3) or IT-101 (10 mg/kg, i.v., qwk × 3) were pathologically confirmed tumor free and survived for at least 126 days after dosing.
In vivo efficacy of IT-101 against disseminated human lymphoma xenograft models
Previous studies have demonstrated that Daudi cells and Karpas 299 cells injected i.v. into mice spread in a pattern comparable with the dissemination of human lymphomas and show preferential localization to the lymph nodes (32, 33).
We further evaluated the antilymphoma efficacy of IT-101 in disseminated Daudi tumors in which the therapy was initiated 11 days after the tumor injection. All untreated controls died of disseminated disease proceeding by progressive weight loss or were sacrificed due to being moribund within 67 days after tumor cell inoculation. In the same xenografts, 55.6% of animals receiving IT-101 (10 mg/kg, i.v., qwk × 3) and 33.3% of animal receiving IT-101 (5 mg/kg, i.v., qwk × 3) could achieve pCTR at day 125 posttreatment, whereas none of animals receiving CPT-11 did so (Fig. 4A). In addition, IT-101 (5 mg/kg, i.v., qwk × 3) and IT-101 (10 mg/kg, i.v., qwk × 3) could significantly prolong the survival of animals bearing disseminated Daudi+ ffLuc tumor when compared to CPT-11 (P = 0.002 and P < 0.0001, respectively) as shown in Fig. 4B.
Fig. 4.

Administration of IT-101 (5 mg/kg, i.v., qwk × 3) and IT-101 (10 mg/kg, i.v., qwk × 3) in disseminated human B-cell lymphoma xenograft-bearing NOD/scid mice could result in significant better survival as compared to CPT-11. Treatments were initiated on 11 days after tumor inoculation. The treatment arms included untreated control (■), CPT-11 (100 mg/kg, i.p., qwk × 3) (◇), IT-101 (5 mg/kg, i.v., qwk × 3) (○), and IT-101 (10 mg/kg, i.v., qwk × 3) (x). The tumor burden was monitored longitudinally by quantification of tumor-derived ffLuc-activity. (A) Total photon flux normalized for exposure time and surface area and expressed in units of photons/second/cm2/steradian (p/s/cm2/sr) for individual mice was graphed over time and serial pseudocolor images representing light intensity from Daudi+ ffLuc in selected mice were shown. (B) Kaplan-Meier survival curves were plot for each treatment group. The log-rank test was used to compare the percent animal survival between treatment groups. A significant difference between the groups treated with either IT-101 (5 mg/kg, i.v., qwk × 3) or IT-101 (10 mg/kg, i.v., qwk × 3) was not observed; but these two treatment groups could significantly establish better survival as compared to the animals treated with CPT-11 (P = 0.0002 and P < 0.0001, respectively). Points indicate mean survival.
We also evaluated the efficacy of IT-101 in disseminated Karpas 299+ ffLuc tumor in which the therapy was initiated eight days after tumor cell injection. All untreated controls died of disseminated disease with severe weight loss or were sacrificed due to being moribund or reaching the predetermined endpoint of ffLuc activity > 1010 p/s/cm2/sr within 29 days after tumor cell inoculation (Fig. 5A). Although all of the animals receiving therapy succumbed from disseminated disease by day 83 after tumor cell injection, IT-101 (10 mg/kg, i.v., qwk × 3) treated animals had significantly longer survival than that of IT-101 (5 mg/kg, i.v., qwk × 3) treated animals and CPT-11 treated animals (P = 0.0009 and P = 0.0049, respectively) as shown in Fig. 5B.
Fig. 5.

IT-101 (10 mg/kg, i.v., qwk × 3) had superior antilymphoma activity as compared to IT-101 (5 mg/kg, i.v., qwk × 3) and CPT-11 in disseminated human anaplastic large T-cell xenografts. NOD.scid/NCr mice were intravenously injected with ffLuc+ Karpas 299 cells to establish disseminated human anaplastic large T-cell xenografts and treated with the indicated cytotoxic agent beginning eight days later. Treatment arm included untreated control (■), CPT-11 (100 mg/kg, i.p., qwk × 3) (◇), IT-101 (5 mg/kg, i.v., qwk × 3) (○), and IT-101 (10 mg/kg, i.v., qwk × 3) (x). The tumor burden was monitored longitudinally by quantification of tumor-derived ffLuc-activity. (A) Total photon flux normalized for exposure time and surface area and expressed in units of photons/second/cm2/steradian (p/s/cm2/sr) for individual mice was graphed over time and serial pseudocolor images representing light intensity from Karpas 299+ ffLuc in selected mice were shown. (B) Kaplan-Meier survival curves for each treatment group. The log-rank test was used to compare the percent animal survival between treatment groups. IT-101 (10 mg/kg, i.v., qwk × 3) treated animals had significantly longer survival as compared to those treated with IT-101 (5 mg/kg, i.v., qwk × 3) and those treated with CPT-11 (P = 0.0009, P = 0.0049, respectively). Points indicate mean survival.
Discussion
In order to mitigate the disadvantages of CPT, i.e. high toxicity and low clinical therapeutic efficacy, we recently attached it to a hydrophilic β-cyclodextrin-based polymer known as IT-101 (26). IT-101 maintains CPT in its active lactone form. This novel compound is too large to pass through normal vessel walls and renal clearance is inhibited. Therefore, the compound can achieve a long plasma half-life, permits a large amount of the compound to reach the tumor site through an abnormally leaky tumor vasculature, and accumulates in tumor tissue due to a lack of effective lymphatic drainage (34–36). Based on our previous study and unpublished data, pharmacokinetics and pharmacodynamics of IT-101 at various dose levels similar to the ones explored here (0.9 – 9 mg/kg in rat, equivalent to 1.8 to 18 mg/kg scaled allometrically to mouse) were previously studied in rats (26). Area under the curve (AUC) and maximum plasma concentration (cmax) for both polymer conjugated and unconjugated CPT scaled linearly with dose and were similar when normalized for the dose administered. Mean terminal half-life of conjugated CPT was 18 hours and independent of dose. These results indicate a linear dose-plasma exposure correlation for both conjugated and unconjugated CPT after administration of IT-101. In animal models, not only is the mean plasma elimination half-life (T1/2) for IT-101 (17–19 hours) significantly longer than that of CPT (1.3 hours), but much higher average 24-hour total CPT levels are achieved in tumors after treatment with IT-101 compared to those treated with CPT alone (26). Since CPT is an S-phase-specific drug, an optimal level of DNA Topo I inhibition is needed in which the tumors are exposed to the compound for a prolonged period of time. In addition, the lower level of freely circulating CPT may reduce the TRT (26).
Our current data clearly demonstrate the advantage of this nanoparticle technology, namely that IT-101 can provide superior antilymphoma activity than that of CPT-11 against xenografts of human lymphoma. In vitro, both IT-101 and CPT-11 caused marked inhibition of growth of three distinct human lymphoma cell lines. Because the in vitro conditions lacked bio-distribution and drug delivery variables, the observed cytotoxicity of either IT-101 or CPT-11 was most likely due to its direct antiproliferative effect against these lymphoma cell lines tested. Since previous study has shown that the sensitivity of tumor cells to DNA Topo I-targeted cytotoxic agents is related to the level of DNA Topo I catalytic activity in the nucleus, we have chosen the three distinct s.c. lymphoma xenografts which exert a high level of DNA Topo I catalytic activity as the representatives in our study (11). Our short term in vivo studies showed that IT-101 was able to significantly inhibit DNA Topo I catalytic activity at 48 hours postadministration in Daudi tumors and Karpas 299 tumors as compared to CPT-11. Such an effect results from the prolonged release kinetics of CPT from IT-101 in tumor tissue, leading to higher degree of cytotoxicity as compared to CPT-11. This was consistent with our observation of higher levels of both IT-101 prodrug and free CPT observed in tumors at 24 and 48 hours postadministration compared to CPT-11 and its active metabolite SN-38. Moreover, the long-term therapeutic efficacy of IT-101 was clearly superior to CPT-11. In localized s.c. xenografts, upon discontinuation of the IT-101 treatment, most of animals attained pCTR at the end of the study, in agreement with the disseminated Daudi tumors treated with IT-101 (10 mg/kg, i.v., qwk × 3). Among treatment groups, IT-101 (10 mg/kg, i.v., qwk × 3) gave the best results in terms of pCTR and survival benefit in both s.c. and disseminated xenografts.
We also have demonstrated that the MTD of IT-101 in SCID/NCr (BALB/C background) and NOD.scid/NCr mice bearing human lymphoma xenograft was less than that of athymic nude mice (28). This result may be explained by the fact that the SCID/NCr and NOD.scid/NCr mice are more immunocompromised as compared to athymic nude mice. Recently, a novel SN-38–incorporating polymeric micelle, NK012, has been developed (37). This compound produced a much higher cytotoxic effect against lung and colon cancer cell lines as compared to CPT-11, mainly due to an enhancement and prolonged distribution of free SN-38 in the tumor tissues. However, SN-38 is cross-resistant with the first-line chemotherapeutic agents commonly used in NHL such as doxorubicin and mitoxantrone (38, 39). Moreover, in a phase II study of CPT-11, a good response (CR + PR) was seen in only 0%, 38%, and 0% of patients with relapsed/refractory Hodgkin’s lymphoma, Burkitt lymphoma, and T-cell lymphoma (40). As a result, CPT-11 is not a good candidate in treating relapsed/refractory NHL. Therefore, the positive outcome of this study clearly demonstrates the potential clinical benefit of IT-101 and can help in the design of treatment schedules in phase I clinical trials. IT-101 monotherapy should be tested in patients with relapsed/refractory NHL. Many preclinical and clinical studies have demonstrated that a sequential administration of a DNA Topo I inhibitor followed by a DNA Topo II inhibitor exerts a synergistic antilymphoma effect (17, 41, 42). A recent study indicated that single agent fluoroquinolone can inhibit both DNA Topo I and II activities in eukaryotic cells; and in combination with either CPT or etoposide, it led to a synergistic inhibition of DNA Topo I or II activity, respectively (43). As a result, it would be of value to further test an antilymphoma effect of IT-101 combined with a DNA Topo II inhibitor or fluoroquinolone. The dose-limiting toxicity of IT-101 is currently being determined in ongoing phase I clinical trial at City of Hope Comprehensive Cancer Center.
In conclusion, we have demonstrated that IT-101 is able to control and inhibit tumor growth both in vitro and in vivo, and prolong survival of animals bearing multiple distinct human lymphoma xenografts. This preclinical therapeutic efficacy of IT-101 supports its clinical evaluation in patients with NHL and HL.
Statement of Translational Relevance.
Topoisomerase inhibitors have a broad spectrum of activity against human cancers including malignant lymphoma. Although camptothecin (CPT), a type 1 DNA topoisomerase inhibitor, has significant antitumor activity, its water insolubility and toxicity has precluded its clinical use and derivatives, such as irinotecan, have been used clinically with some activity. We have tested the efficacy of IT-101. IT-101 is comprised of a cyclodextrin-containing polymer conjugate of CPT that self assembles into a nanoparticle of ca. 30 nm diameter. The drug was designed to reduce the toxicity of CPT and relies on intracellular chemistry to liberate the drug from the protective polymer once inside the cell. When tested in five different animal models of lymphoma, the drug demonstrated prolongation of survival and the achievement of a complete remission in most of the animals compared to irinotecan, without increased toxicity. As a result, it is our plan to initiate a phase I/II clinical trial in a broad group of patients with relapsed/refractory lymphoma.
Acknowledgments
This study was supported by City of Hope Lymphoma SPORE Grant (P50 CA107399). The authors would like to thank Christopher Ruel for help with statistical analysis; City of Hope’s Animal Resource Center under the direction of Dr. Richard Ermel, Patty Wong, and, Aaron Shoop for help with animal experiments; Dr. Peiguo Chu, Dr. Karen Cheng, and Sofia Loera for help with histopathological staining and reviews; and Linling Chen for help with Topo I catalytic activity study.
Footnotes
Specific Contributions: T. N., J. W., D. C., T.S., M.E.D., L.K., Y.Y., S.J.F., & A.R. designed research; T. N. & J. W., J.D., & L.K. performed research; T.S. provided vital reagent; M.E.D. developed vital reagent, T.N., J.W., D.C., L.K., & S.J.F wrote the manuscript.
References
- 1.Bociek RG. Adult Burkitt’s lymphoma. Clin Lymphoma. 2005;6:11–20. doi: 10.3816/clm.2005.n.021. [DOI] [PubMed] [Google Scholar]
- 2.Tilly H, Gaulard P, Lepage E, et al. Primary anaplastic large-cell lymphoma in adults: clinical presentation, immunophenotype, and outcome. Blood. 1997;90:3727–34. [PubMed] [Google Scholar]
- 3.Gascoyne RD, Aoun P, Wu D, et al. Prognostic significance of anaplastic lymphoma kinase (ALK) protein expression in adults with anaplastic large cell lymphoma. Blood. 1999;93:3913–21. [PubMed] [Google Scholar]
- 4.Duggan DB, Petroni GR, Johnson JL, et al. Randomized comparison of ABVD and MOPP/ABV hybrid for the treatment of advanced Hodgkin’s disease: report of an intergroup trial. J Clin Oncol. 2003;21:607–14. doi: 10.1200/JCO.2003.12.086. [DOI] [PubMed] [Google Scholar]
- 5.Santoro A, Bonadonna G, Bonfante V, Valagussa P. Alternating drug combinations in the treatment of advanced Hodgkin’s disease. N Engl J Med. 1982;306:770–5. doi: 10.1056/NEJM198204013061303. [DOI] [PubMed] [Google Scholar]
- 6.Wilson WH, Bryant G, Bates S, et al. EPOCH chemotherapy: toxicity and efficacy in relapsed and refractory non-Hodgkin’s lymphoma. J Clin Oncol. 1993;11:1573–82. doi: 10.1200/JCO.1993.11.8.1573. [DOI] [PubMed] [Google Scholar]
- 7.Velasquez WS, McLaughlin P, Tucker S, et al. ESHAP--an effective chemotherapy regimen in refractory and relapsing lymphoma: a 4-year follow-up study. J Clin Oncol. 1994;12:1169–76. doi: 10.1200/JCO.1994.12.6.1169. [DOI] [PubMed] [Google Scholar]
- 8.Rodriguez MA, Cabanillas FC, Velasquez W, et al. Results of a salvage treatment program for relapsing lymphoma: MINE consolidated with ESHAP. J Clin Oncol. 1995;13:1734–41. doi: 10.1200/JCO.1995.13.7.1734. [DOI] [PubMed] [Google Scholar]
- 9.Muggia FM, Dimery I, Arbuck SG. Camptothecin and its analogs. An overview of their potential in cancer therapeutics. Ann N Y Acad Sci. 1996;803:213–23. doi: 10.1111/j.1749-6632.1996.tb26391.x. [DOI] [PubMed] [Google Scholar]
- 10.Hertzberg RP, Caranfa MJ, Hecht SM. On the mechanism of topoisomerase I inhibition by camptothecin: evidence for binding to an enzyme-DNA complex. Biochemistry. 1989;28:4629–38. doi: 10.1021/bi00437a018. [DOI] [PubMed] [Google Scholar]
- 11.Rothenberg ML. Topoisomerase I inhibitors: review and update. Ann Oncol. 1997;8:837–55. doi: 10.1023/a:1008270717294. [DOI] [PubMed] [Google Scholar]
- 12.Pommier Y. DNA topoisomerase I and II in cancer chemotherapy: update and perspectives. Cancer Chemother Pharmacol. 1993;32:103–8. doi: 10.1007/BF00685611. [DOI] [PubMed] [Google Scholar]
- 13.Moertel CG, Schutt AJ, Reitemeier RJ, Hahn RG. Phase II study of camptothecin (NSC-100880) in the treatment of advanced gastrointestinal cancer. Cancer Chemother Rep. 1972;56:95–101. [PubMed] [Google Scholar]
- 14.Muggia FM, Creaven PJ, Hansen HH, Cohen MH, Selawry OS. Phase I clinical trial of weekly and daily treatment with camptothecin (NSC-100880): correlation with preclinical studies. Cancer Chemother Rep. 1972;56:515–21. [PubMed] [Google Scholar]
- 15.Bass AJ, Gockerman JP, Hammett E, et al. Phase I evaluation of prolonged-infusion gemcitabine with irinotecan for relapsed or refractory leukemia or lymphoma. J Clin Oncol. 2002;20:2995–3000. doi: 10.1200/JCO.2002.08.166. [DOI] [PubMed] [Google Scholar]
- 16.Sugiyama K, Omachi K, Fujiwara K, et al. Irinotecan hydrochloride for the treatment of recurrent and refractory non-Hodgkin lymphoma: a single institution experience. Cancer. 2002;94:594–600. doi: 10.1002/cncr.10266. [DOI] [PubMed] [Google Scholar]
- 17.Niitsu N, Iijima K, Chizuka A. Combination therapy with irinotecan (CPT-11), mitoxantrone, and dexamethasone in relapsed or refractory non-Hodgkin’s lymphoma: a pilot study. Ann Hematol. 2001;80:411–6. doi: 10.1007/s002770100313. [DOI] [PubMed] [Google Scholar]
- 18.Saotome T, Takagi T, Sakai C, Kumagai K, Tamaru J. Combination chemotherapy with irinotecan and adriamycin for refractory and relapsed non-Hodgkin’s lymphoma. Ann Oncol. 2000;11:115–6. doi: 10.1023/a:1008368905546. [DOI] [PubMed] [Google Scholar]
- 19.Makino T, Nakahara K, Takatsuka Y, et al. Successful treatment of chemotherapy-resistant adult T cell leukemia/lymphoma by irinotecan hydrochloride (CPT-11) Rinsho Ketsueki. 1994;35:42–8. [PubMed] [Google Scholar]
- 20.Thompson J, Zamboni WC, Cheshire PJ, et al. Efficacy of systemic administration of irinotecan against neuroblastoma xenografts. Clin Cancer Res. 1997;3:423–31. [PubMed] [Google Scholar]
- 21.Houghton PJ, Cheshire PJ, Hallman JD, 2nd, et al. Efficacy of topoisomerase I inhibitors, topotecan and irinotecan, administered at low dose levels in protracted schedules to mice bearing xenografts of human tumors. Cancer Chemother Pharmacol. 1995;36:393–403. doi: 10.1007/BF00686188. [DOI] [PubMed] [Google Scholar]
- 22.Conti JA, Kemeny NE, Saltz LB, et al. Irinotecan is an active agent in untreated patients with metastatic colorectal cancer. J Clin Oncol. 1996;14:709–15. doi: 10.1200/JCO.1996.14.3.709. [DOI] [PubMed] [Google Scholar]
- 23.Cunningham D, Pyrhonen S, James RD, et al. Randomised trial of irinotecan plus supportive care versus supportive care alone after fluorouracil failure for patients with metastatic colorectal cancer. Lancet. 1998;352:1413–8. doi: 10.1016/S0140-6736(98)02309-5. [DOI] [PubMed] [Google Scholar]
- 24.Rougier P, Van Cutsem E, Bajetta E, et al. Randomised trial of irinotecan versus fluorouracil by continuous infusion after fluorouracil failure in patients with metastatic colorectal cancer. Lancet. 1998;352:1407–12. doi: 10.1016/S0140-6736(98)03085-2. [DOI] [PubMed] [Google Scholar]
- 25.Fukuoka M, Niitani H, Suzuki A, et al. A phase II study of CPT-11, a new derivative of camptothecin, for previously untreated non-small-cell lung cancer. J Clin Oncol. 1992;10:16–20. doi: 10.1200/JCO.1992.10.1.16. [DOI] [PubMed] [Google Scholar]
- 26.Schluep T, Cheng J, Khin KT, Davis ME. Pharmacokinetics and biodistribution of the camptothecin-polymer conjugate IT-101 in rats and tumor-bearing mice. Cancer Chemother Pharmacol. 2006;57:654–62. doi: 10.1007/s00280-005-0091-7. [DOI] [PubMed] [Google Scholar]
- 27.Cheng J, Khin KT, Davis ME. Antitumor activity of beta-cyclodextrin polymer-camptothecin conjugates. Mol Pharm. 2004;1:183–93. doi: 10.1021/mp049966y. [DOI] [PubMed] [Google Scholar]
- 28.Schluep T, Hwang J, Cheng J, et al. Preclinical efficacy of the camptothecin-polymer conjugate IT-101 in multiple cancer models. Clin Cancer Res. 2006;12:1606–14. doi: 10.1158/1078-0432.CCR-05-1566. [DOI] [PubMed] [Google Scholar]
- 29.Brown CE, Wright CL, Naranjo A, et al. Biophotonic cytotoxicity assay for high-throughput screening of cytolytic killing. J Immunol Methods. 2005;297:39–52. doi: 10.1016/j.jim.2004.11.021. [DOI] [PubMed] [Google Scholar]
- 30.Liu LF, Miller KG. Eukaryotic DNA topoisomerases: two forms of type I DNA topoisomerases from HeLa cell nuclei. Proc Natl Acad Sci U S A. 1981;78:3487–91. doi: 10.1073/pnas.78.6.3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Husain I, Mohler JL, Seigler HF, Besterman JM. Elevation of topoisomerase I messenger RNA, protein, and catalytic activity in human tumors: demonstration of tumor-type specificity and implications for cancer chemotherapy. Cancer Res. 1994;54:539–46. [PubMed] [Google Scholar]
- 32.Ghetie MA, Richardson J, Tucker T, Jones D, Uhr JW, Vitetta ES. Disseminated or localized growth of a human B-cell tumor (Daudi) in SCID mice. Int J Cancer. 1990;45:481–5. doi: 10.1002/ijc.2910450318. [DOI] [PubMed] [Google Scholar]
- 33.Tian ZG, Longo DL, Funakoshi S, et al. In vivo antitumor effects of unconjugated CD30 monoclonal antibodies on human anaplastic large-cell lymphoma xenografts. Cancer Res. 1995;55:5335–41. [PubMed] [Google Scholar]
- 34.Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46:6387–92. [PubMed] [Google Scholar]
- 35.Dvorak HF, Nagy JA, Dvorak JT, Dvorak AM. Identification and characterization of the blood vessels of solid tumors that are leaky to circulating macromolecules. Am J Pathol. 1988;133:95–109. [PMC free article] [PubMed] [Google Scholar]
- 36.Trouet A. Perspectives in cancer research. Increased selectivity of drugs by linking to carriers. Eur J Cancer. 1978;14:105–11. doi: 10.1016/0014-2964(78)90167-6. [DOI] [PubMed] [Google Scholar]
- 37.Koizumi F, Kitagawa M, Negishi T, et al. Novel SN-38-incorporating polymeric micelles, NK012, eradicate vascular endothelial growth factor-secreting bulky tumors. Cancer Res. 2006;66:10048–56. doi: 10.1158/0008-5472.CAN-06-1605. [DOI] [PubMed] [Google Scholar]
- 38.Allen JD, Brinkhuis RF, Wijnholds J, Schinkel AH. The mouse Bcrp1/Mxr/Abcp gene: amplification and overexpression in cell lines selected for resistance to topotecan, mitoxantrone, or doxorubicin. Cancer Res. 1999;59:4237–41. [PubMed] [Google Scholar]
- 39.Brangi M, Litman T, Ciotti M, et al. Camptothecin resistance: role of the ATP-binding cassette (ABC), mitoxantrone-resistance half-transporter (MXR), and potential for glucuronidation in MXR-expressing cells. Cancer Res. 1999;59:5938–46. [PubMed] [Google Scholar]
- 40.Takagi T, Saotome T. Chemotherapy with irinotecan (CPT-11), a topoisomerase-I inhibitor, for refractory and relapsed non-Hodgkin’s lymphoma. Leuk Lymphoma. 2001;42:577–86. doi: 10.3109/10428190109099317. [DOI] [PubMed] [Google Scholar]
- 41.Kano Y, Suzuki K, Akutsu M, et al. Effects of CPT-11 in combination with other anti-cancer agents in culture. Int J Cancer. 1992;50:604–10. doi: 10.1002/ijc.2910500420. [DOI] [PubMed] [Google Scholar]
- 42.Kim R, Hirabayashi N, Nishiyama M, Jinushi K, Toge T, Okada K. Experimental studies on biochemical modulation targeting topoisomerase I and II in human tumor xenografts in nude mice. Int J Cancer. 1992;50:760–6. doi: 10.1002/ijc.2910500516. [DOI] [PubMed] [Google Scholar]
- 43.Reuveni D, Halperin D, Shalit I, Priel E, Fabian I. Quinolones as enhancers of camptothecin-induced cytotoxic and anti-topoisomerase I effects. Biochem Pharmacol. 2008;75:1272–81. doi: 10.1016/j.bcp.2007.11.014. [DOI] [PubMed] [Google Scholar]