

Abstract
Bacterial pathogens evolve during the infection of their human hosts1-8, but separating adaptive and neutral mutations remains challenging9-11. Here, we identify bacterial genes under adaptive evolution by tracking recurrent patterns of mutations in the same pathogenic strain during the infection of multiple patients. We conducted a retrospective study of a Burkholderia dolosa outbreak among people with cystic fibrosis, sequencing the genomes of 112 isolates collected from 14 individuals over 16 years. We find that 17 bacterial genes acquired non-synonymous mutations in multiple individuals, which indicates parallel adaptive evolution. Mutations in these genes illuminate the genetic basis of important pathogenic phenotypes, including antibiotic resistance and bacterial membrane composition, and implicate oxygen-dependent gene regulation as paramount in lung infections. Several genes have not been previously implicated in pathogenesis, suggesting new therapeutic targets. The identification of parallel molecular evolution suggests key selection forces acting on pathogens within humans and can help predict and prepare for their future evolutionary course.
During acute and chronic infections, bacterial pathogens can accumulate mutations that allow them to better adapt to their human host1,2, evade the immune response12,13, and become more resistant to antibiotic therapy3,4. The spectrum of beneficial mutations that arise during the course of a bacterial infection is likely to reflect genetic pathways critical to bacterial pathogenesis in vivo, and therefore may inspire new therapeutic directions. Recent advances in high-throughput sequencing make it possible to follow the genome evolution of bacterial pathogens7-9,14-17, but it is still difficult to tease apart the adaptive driver mutations from the neutral passenger mutations that have been fixed by chance9-11. In the laboratory, this is addressed by following several populations grown in parallel cultures under identical conditions; the adaptive role of mutations is indicated by their recurrence in replicate experiments17-19. In natural and clinical environments, such studies are more difficult and have not yet been systematically performed on a genome-wide scale. As a result, global patterns of adaptive evolution that underlie bacterial pathogenesis in humans are not well characterized.
Here, we systematically identify recurrent patterns of evolution implicated in pathogenesis by comparing the genetic adaptation of a single bacterial strain in multiple human subjects during the spread of an epidemic. The airways of people with cystic fibrosis (CF, a lethal genetic disorder) are particularly prone to long-term bacterial infections. Most individuals with CF become colonized by a dominant bacterial strain that persists for many years20, allowing significant time for genetic adaption2. In the 1990s, a small epidemic of Burkholderia dolosa – a rare CF pathogen21,22 that can be transmitted from person to person23 – broke out among individuals with CF in Boston24,25. A total of 39 individuals were infected with B. dolosa (Fig. 1a); and all were followed in a Boston hospital, where bacteria isolated during normal care were routinely frozen.
Figure 1. Whole-genome sequencing of 112 Burkholderia dolosa isolates recovered from 14 epidemic patients indicates steady accumulation of mutations over years.
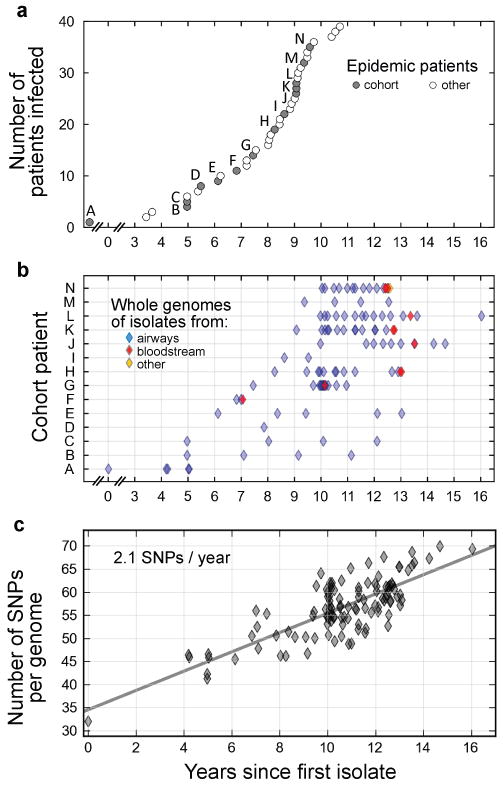
a, An epidemic of B. dolosa spread to 39 people with cystic fibrosis over decades (circles). Time of first attested infection for each patient is indicated in years since the collection of an isolate from patient zero (labeled ‘A’). We studied retrospectively a cohort of 14 patients from this epidemic (gray circles, and labels). b, The genomes of 112 bacterial isolates were sequenced (diamonds; each horizontal line corresponds to a patient). Isolates were recovered over time from the patients' airways (blue), bloodstream (red) or other body compartments (for instance tissue obtained during surgery; orange). c, The number of SNPs between each isolate and the outgroup is plotted as a function of time (years since first isolate). Linear fit is plotted (slope = 2.1 mutations fixed per year).
We conducted a retrospective study of 112 B. dolosa isolates from 14 individuals with Cystic Fibrosis from this epidemic outbreak – including the first infected subject in the Boston area (patient zero) – over the course of 16 years (Fig. 1b and Supplementary Table 1). During this period, five of these individuals received a lung transplant, and eight died. Most of the 112 isolates were recovered from the subjects' airways; a few were recovered from the blood of subjects with bacteremia. This collection covers the epidemic with high temporal resolution and enables us to study the parallel evolution of the same strain in multiple individuals (Supplementary Fig. 1).
We sequenced the whole-genome of these 112 B. dolosa isolates on an Illumina GAIIx sequencer (75bp single-end reads, average read depth 37×; Supplementary Fig. 2) and aligned the reads onto a B. dolosa reference genome26. We focused our analysis on SNPs; although structural variants and mobile elements may also be important, they are beyond the scope of this study. Our analysis identified 492 polymorphic loci (Supplementary Table 2). These mutations accumulated at a steady rate of ∼2 SNP/year (r=0.79) (Fig. 1c), with no discernible difference between subjects (Supplementary Fig. 3). This rate of mutation accumulation in the presence of selection within the human body is consistent with bacterial mutation fixation rates reported in long-term human infections2,27. The steady accumulation of mutations generated enough genetic diversity to resolve evolutionary relationships between isolates, which were investigated through the creation of a maximum-likelihood phylogenetic tree (Fig. 2a).
Figure 2. Bacterial phylogeny reveals a likely network of transmission between patients, and between organs.
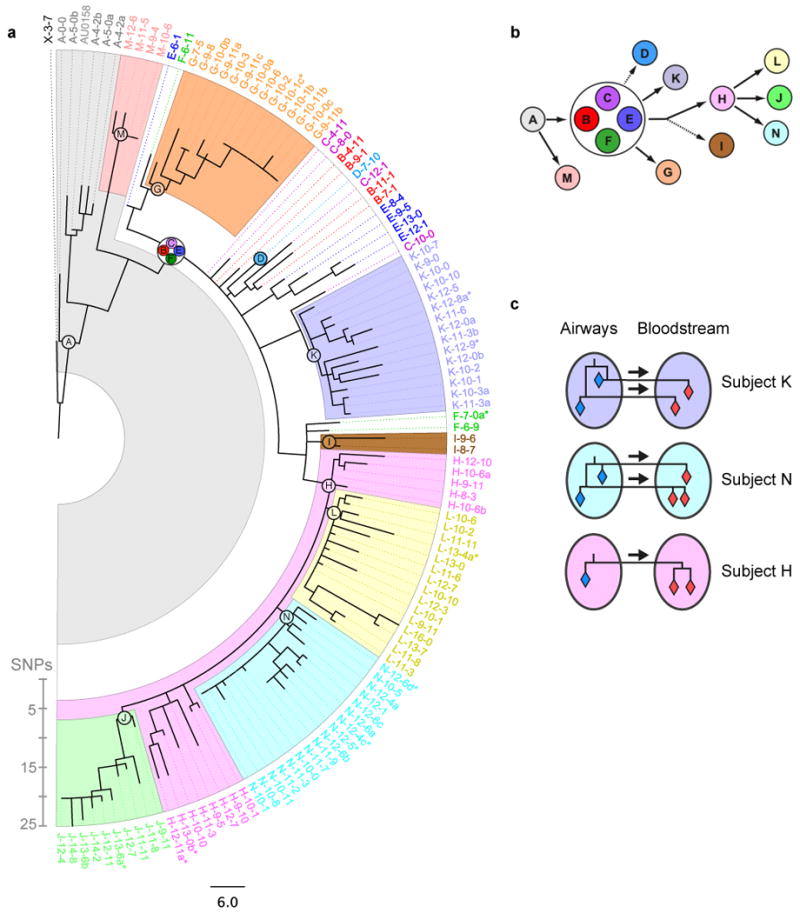
a, The maximum-likelihood phylogenetic tree is displayed (SNP scale shown). The 112 isolates are indicated by thin dashed lines colored according to patient, and labeled according to patient and time (e.g., ‘C-14-5’ was recovered from patient C, fourteen years and five months after the first isolate). Blood isolates are indicated by a dollar sign, and isolates with the same patient and date are distinguished by letters. The last common ancestor (LCA) of isolates from the same patient is represented as a circle of the appropriate color and label. Colored backgrounds indicate patient-specific genetic fingerprints. Patients B, C, E, and F share the same LCA (white background). b, Phylogeny between the inferred LCAs suggests a likely network of infection between patients (arrows). Dashed arrows indicate less certainty (fewer than 3 isolates). c, Phylogeny between blood and lung isolates recovered from the same patient evidences the transmission of multiple clones to the bloodstream during bacteremia (multiple arrows, patients K and N).
At the epidemic level, the phylogeny suggests a network of transmission between subjects. Isolates from the same subject tend to form genetically related clusters in the phylogeny (Fig. 2a, Supplementary Fig. 3). These clusters define subject-specific genetic fingerprints, from which transmission history can be inferred. We constructed the last common ancestor (LCA) for each subject's set of isolates, which bears the subject-specific fingerprints. The phylogenetic relationships between these inferred strains indicated the likely network of transmissions among the 14 subjects (Fig. 2b). Because these data account for only 14 of the 39 subjects in this epidemic, we cannot determine whether transmission occurred directly from one subject to another, or indirectly through a subject not in our study, via a healthcare worker, or through a medical device. Nevertheless, this analysis shows that this specific epidemic was transmitted through several people during its spread and demonstrates the strength of the approach in identifying the infection network of an epidemic.
At the level of individual subjects, the phylogenetic analysis evidenced the transfer of multiple B. dolosa clones to the subject's bloodstream during bacteremia. We examined isolates from the three subjects for whom we obtained more than one blood isolate (subjects H, K, and N). In two of these individuals, we found pairs of blood isolates that evolved from distinct lung isolates (Fig. 2c), which is inconsistent with the transmission of a single clone from lungs to blood (Supplementary Fig. 4a). This evidence for multiclonal transmission is consistent either with a punctuated transmission of multiple clones from the genetically diverse lung28-31 (Supplementary Fig. 4b), or with multiple transmissions occurring over time. These different possibilities would lead to recommendations for distinct therapeutic actions: whereas a lung transplant might be effective in preventing the continuous leak of bacteria through lesions of the lung, it would not block the proliferation of bacteria already within the bloodstream. This analysis thus brings into focus unresolved questions about the mechanistic basis of bacteremia.
Finally, we investigated evolution at the gene level. We looked for genetic correlates of known pathogenic phenotypes. We first assayed resistance to ciprofloxacin, a fluoroquinolone frequently prescribed to CF subjects (Fig. 3a). Resistance among the 112 isolates varied over two orders of magnitudes (Supplementary Fig. 5a). We scored each gene for correlation between the presence of mutations and drug resistance (Fig. 3a, inset). This genome-wide association study implicated a single gene in the phenotype, BDAG_02180, homolog to Escherichia coli gyrA. All the genotypes associated with resistance had nonsynonymous mutations in T83 or D87, known for their role in fluoroquinolone resistance4,32,33. Mutations in these residues occurred in six subjects. In each case, phylogenetic analysis indicated that mutations were independently acquired within the subject, after initial infection (Supplementary Fig. 5b). In some cases, we even found in the same subject mutations in both residues, each carried by a different isolate. These findings support the presence of a strong selective pressure from fluoroquinolones but suggest that there are only few genetic paths to resistance in vivo.
Figure 3. Pathogenic phenotypes are associated with point mutations in key genes.
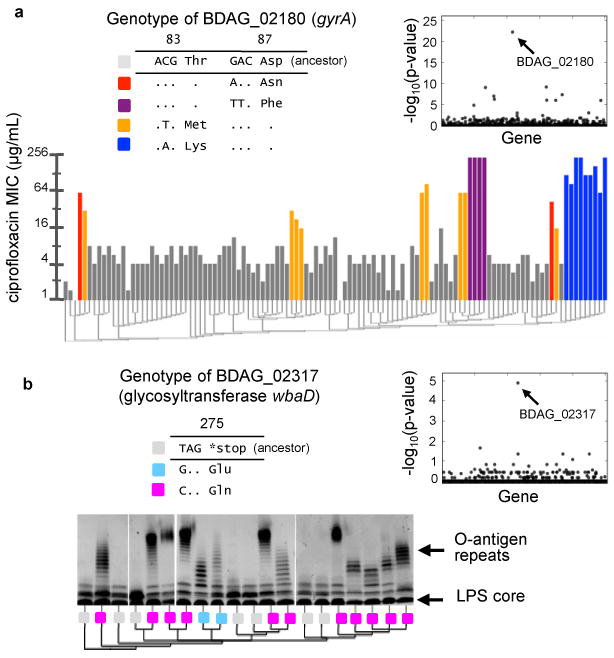
a, Minimal inhibitory concentration (MIC) of ciprofloxacin for each isolate (vertical bars) is correlated with genotypic changes in BDAG_02180, a homolog to E. coli gyrA, which result in coding changes at residues 83 and 87. Phylogeny is indicated below as a dendrogram and genotypes at BDAG_02180 are shown in the legend. Inset: P values for correlation between the presence of mutations in each gene and resistance levels (Kendall's tau). b, Silver-stained gels showing the presence of O-antigen repeats (banded pattern) in the LPS of twenty isolates. Genotypes of BDAG_02317, a hoolog of glycosyltransferase wbaD, are shown in the legend. The presentation of O-antigen repeats corresponds to a recurrent gain-of-function mutation. Inset: P values for correlation between the presence of mutations in each gene and O-antigen presentation (Fischer's exact test).
We then focused on a second pathogenic phenotype, the presentation of O-antigen repeats in the lipopolysaccharide (LPS) of the bacterium's outer membrane, known for its importance to virulence in related species34-36. We found that some of our isolates present the O-antigen while others do not (Fig. 3b). A single nucleotide in the glycosyltransferase gene BDAG_02317 correlated exactly with the presentation of O-antigen repeats (Fig. 3b, inset). The ancestral genotype at this locus, a stop codon, corresponds to the absence of O-antigen repeats; two different mutations at the same amino acid position – each restoring a full-length protein – are associated with presence of the repeats. We confirmed this association experimentally; we found that complementation with the full-length glycosyltransferase gene could restore O-antigen presentation (Supplementary Fig. 6a, Supplementary Note). Harnessing the phylogenetic information, we determined that the last common ancestors of strains from each subject presented the truncated genotype. Thus, the gain-of-function mutations occurred in nine subjects independently (Supplementary Fig. 6b), highlighting the strength of the selective pressure for O-antigen presentation during the infection. These results identify a previously uncharacterized genetic mechanism for O-antigen switching and hint at a tradeoff during person-to-person transmission.
We recognize that the human body challenges bacteria with many selective pressures beyond those discussed above. We therefore developed a systematic approach for identifying genes under positive selection without prior knowledge of the phenotypes being selected. At the genome level, we found no evidence for selection in coding regions (dN/dS∼1) and no significant intragenic bias (Supplementary Note). However, we reasoned that genes under selection would be mutated independently in different subjects17-19. We leveraged the phylogeny to calculate the number of mutations each gene received, distinguishing genes mutated multiple times from those mutated once but appearing in several subjects through the expansion of the lineage that carried them. We counted 561 independent mutational events in 304 genes (Supplementary Table 3). Assuming neutral evolution, we would expect that these mutations would distribute randomly among the 5,014 B. dolosa genes, and that genes would rarely acquire more than a single mutation. Instead, we observed that many more genes than expected contained multiple mutations (Fig. 4a, inset). Seventeen genes were found to have three or more different mutations (neutral expectation: ∼1, Online Methods), and four genes had over ten different mutations (expected: 0 genes).
Figure 4.
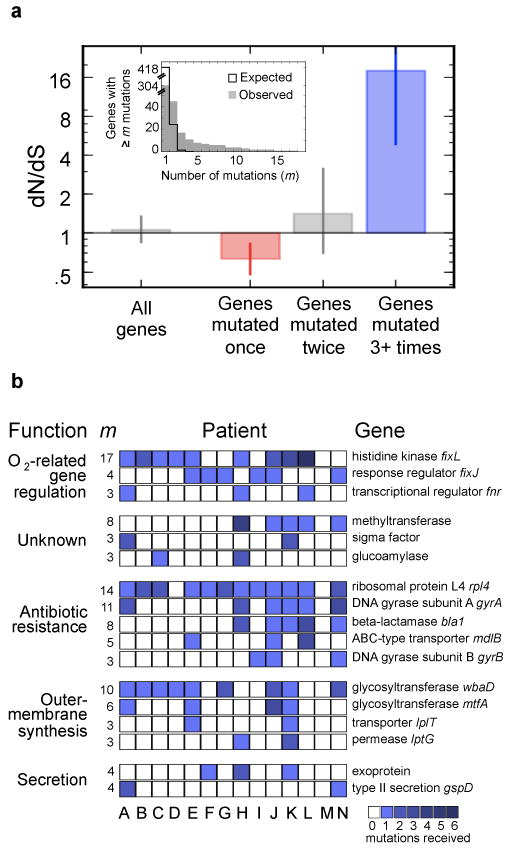
Parallel evolution identifies a set of genes under strong selection during pathogenesis. a, Inset, The number of genes that acquired at least m mutations across the epidemic is plotted as a function of m (gray bars). This distribution contrasts sharply with the distribution expected for neutral evolution (black line). Under neutral evolution, the expectation is that only one gene would receive three or more mutations (m>3); instead, we observed 17 such genes. Main, The canonical signal for selection (dN/dS) is calculated for these 17 genes (109 mutations in these 17 genes), the 28 genes with m=2, and the 247 genes with m=1 (Supplementary Fig. 3 presents the contribution of these mutations to the molecular clock). Values of dN/dS greater than 1 indicate positive selection (blue), values smaller than 1 indicate purifying selection (red). Error bars indicate 95% CIs. Calculated over all genes without regard to m, this analysis would not show a signal for selection. b, Each one of the 17 genes (rows) under positive selection contained an acquired mutated in several patients, signified by squares (color intensity indicate the number of mutations observed within this patient). The total number of mutations observed within that gene, m, is indicated left. Genes are grouped by biological function, and labeled with the annotations of close homologs, and, when available, close homolog names.
To determine whether genes that acquired multiple mutations were under positive selection, or were merely sites of mutational bias, we calculated the canonical measure for selection, dN/dS (Fig. 4a). The large subset of 247 genes which contained only one mutation exhibited a weak but significant signal for purifying (i.e., negative) selection (dN/dS=0.63, p<10-3, Online Methods). The set of genes with two mutations did not show evidence of selection (dN/dS=1.4, CI:0.7-3.1); this set may include a combination of genes that are under some selection and genes that fixed two mutations by chance (22 expected under neutral drift; 28 observed). By contrast, the 17 genes that acquired three or more mutations received 18 times as many non-synonymous mutations as expected by neutral drift, and are under strong positive selection (dN/dS=18, CI:4.9-152.7). This suggests that these seventeen genes are not neutral mutational hotspots; they are undergoing adaptive evolution under the pressure of natural selection.
The 17 genes under positive selection (Fig. 4b, Supplementary Table 4), which are mostly conserved across the Burkholderia genus (Supplementary Fig. 7), indicate genetic pathways that may be involved in pathogenesis. The presence of the two genes previously identified in connection with antibiotic resistance and O-antigen presentation (gyrA: 11 mutations; glycosyltransferase wbaD: 10 mutations) provides a further connection of these genes to pathogenic phenotypes under selection. Eleven of the 17 genes belong to functional categories related to pathogenicity: membrane synthesis (4 genes, including 2 in LPS biosynthesis), secretion (2), and antibiotic resistance (5). The presence of a second glycosyltransferase in the O-antigen cluster (6 mutations) stresses the importance of this pathway to the disease. Notably, the remaining 6 genes had not previously been implicated in pathogenesis of lung infections. Three of these – a glucoamylase, a methyltransferase, and a sigma factor – have no well-annotated close homologs and their roles in pathogenicity are thus unclear. Another gene trio (homologs of fnr, fixL, and fixJ), including the gene most mutated gene (BDAG_01161, a homolog of fixL, that had 17 nonsynonymous mutations), can be linked through homology to oxygen-dependent gene regulation37. The large number of mutations in this pathway resonates with reports of lowered oxygen tension in CF mucus38 and of ties between oxygen sensing and virulence modulation in the gastrointestinal tract39. Homologs of these three genes have been implicated in diverse regulatory processes37,39, but their function and the genes they regulate in B. dolosa are currently unknown. The identification of 17 B. dolosa genes that underwent selective pressure during infection in subjects with cystic fibrosis highlights key pathways involved in pathogenesis and may suggest new therapeutic targets for this and other lung infections.
Tracking the genomic evolution of bacterial pathogens during the infection of their human host provides a direct method for observing evolutionary mechanisms in vivo and identifying genes central to pathogenesis. This study, which harnesses the combination of high-throughput sequencing and parallel evolution in the clinical settings, is a step towards a comprehensive understanding of genetic adaptation during pathogenesis. Systematically identifying selective pressures acting on pathogens within their hosts may help point to new therapeutic directions.
Supplementary Material
Acknowledgments
We are grateful to: M. Caimano, M. Cendron, P. Kokorowski, S. Lory, C. Marx, N. Delany, S. Walker, M. Waldor and R. Ward for insightful discussions and comments; O. Iartchouk, A. Brown, M. Light and their team at PCPGM for Illumina sequencing; J. Deane and L. Williams for technical assistance; S. Vargas for assistance with IRB protocols; M. Baym, M. Ernebjerg, A. Palmer, E. Toprak, K. Vetsigian, Z. Yao, and all of the Kishony Lab members for helpful discussions and general support. JBM was supported by the Foundational Questions in Evolutionary Biology Prize Fellowship and the Systems Biology PhD Program (Harvard Medical School). GPP was supported in part by The Mannion Fund for Research of the Center for the Critically Ill Child of Children's Hospital Boston. JJL was supported by the Cystic Fibrosis Foundation. This work was supported in part by US National Institutes of Health grants GM077052 (to Systems Biology Department, Harvard Medical School), R01 GM081617 (to R.K.), by the New England Regional Center of Excellence for Biodefense and Emerging Infectious Diseases (NERCE) grant AI057159 (to R.K.), and a Harvard Catalyst grant (to R.K., A.J.M., and Marc Cendron).
Footnotes
Accession Codes: Consensus sequences for all 113 sequenced isolates are available at the National Center for Biotechnology Information’s Sequence Read Archive. Accession numbers are listed in Supplementary Table S1.
Contributions: J.-B.M., A.J.M. and R.K. conceived the study. J.J.L., A.J.M. and G.P.P. collected the clinical samples. T.D.L. and N.L. performed resistance phenotyping. J.B.G., D.R., M.R.D., D.S., and G.P.P. performed LPS phenotyping and complementation. M.A., G.P.B., A.J.M. and G.P.P. conducted chart review and provided medical information. T.D.L., J.-B.M. and R.K. performed whole-genome sequencing and data analysis. T.D.L., J.-B.M., J.J.L., A.J.M., G.P.P. and R.K. interpreted the results and wrote the manuscript.
Competing financial interests: The authors declare no competing financial interest.
References
- 1.Suerbaum S, Josenhans C. Helicobacter pylori evolution and phenotypic diversification in a changing host. Nat Rev Microbiol. 2007;5:441–452. doi: 10.1038/nrmicro1658. [DOI] [PubMed] [Google Scholar]
- 2.Smith EE, et al. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc Natl Acad Sci U S A. 2006;103:8487–8492. doi: 10.1073/pnas.0602138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Musher DM, et al. Emergence of macrolide resistance during treatment of pneumococcal pneumonia. N Engl J Med. 2002;346:630–631. doi: 10.1056/NEJM200202213460820. [DOI] [PubMed] [Google Scholar]
- 4.Wong A, Kassen R. Parallel evolution and local differentiation in quinolone resistance in Pseudomonas aeruginosa. Microbiology. 2011;157:937–944. doi: 10.1099/mic.0.046870-0. [DOI] [PubMed] [Google Scholar]
- 5.Zdziarski J, et al. Host imprints on bacterial genomes—rapid divergent evolution in individual patients. PLoS Path. 2010;6:e1001078. doi: 10.1371/journal.ppat.1001078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang L, et al. Evolutionary dynamics of a bacteria in a human host enviornment. Proc Natl Acad Sci U S A. 2011;108:7481–7486. doi: 10.1073/pnas.1018249108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kennemann L, et al. Helicobacter pylori genome evolution during human infection. Proc Natl Acad Sci U S A. 2011;108:5033–5038. doi: 10.1073/pnas.1018444108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mwangi MM, et al. Tracking the in vivo evolution of multidrug resistance in Staphylococcus aureus by whole-genome sequencing. Proc Natl Acad Sci U S A. 2007;104:9451–9456. doi: 10.1073/pnas.0609839104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harris SR, et al. Evolution of MRSA during hospital transmission and intercontinental spread. Science. 2010;327:469–474. doi: 10.1126/science.1182395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goodarzi H, Hottes AK, Tavazoie S. Global discovery of adaptive mutations. Nat Methods. 2009;6:581–583. doi: 10.1038/nmeth.1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pleasance ED, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010;463:191–196. doi: 10.1038/nature08658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moxon ER, Rainey PB, Nowak MA, Lenski RE. Adaptive evolution of highly mutable loci in pathogenic bacteria. Curr Biol. 1994;4:24–33. doi: 10.1016/s0960-9822(00)00005-1. [DOI] [PubMed] [Google Scholar]
- 13.van der Woude MW, Baumler AJ. Phase and antigenic variation in bacteria. Clin Microbiol Rev. 2004;17:581–611. doi: 10.1128/CMR.17.3.581-611.2004. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Croucher NJ, et al. Rapid pneumococcal evolution in response to clinical interventions. Science. 2011;331:430–434. doi: 10.1126/science.1198545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holt KE, et al. High-throughput sequencing provides insights into genome variation and evolution in Salmonella Typhi. Nat Genet. 2008;40:987–993. doi: 10.1038/ng.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pallen MJ, Wren BW. Bacterial pathogenomics. Nature. 2007;449:835–842. doi: 10.1038/nature06248. [DOI] [PubMed] [Google Scholar]
- 17.Elena SF, Lenski RE. Evolution experiments with microorganisms: the dynamics and genetic bases of adaptation. Nat Rev Genet. 2003;4:457–469. doi: 10.1038/nrg1088. [DOI] [PubMed] [Google Scholar]
- 18.Woods R, et al. Tests of parallel molecular evolution in long-term experiment with Escherichia coli. Proc Natl Acad Sci U S A. 2006;103:9107–9112. doi: 10.1073/pnas.0602917103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barrick JE, et al. Genome evolution and adpatation in a long-term experiment with Escherichia coli. Nature. 2009;461:1243–1247. doi: 10.1038/nature08480. [DOI] [PubMed] [Google Scholar]
- 20.Lipuma JJ. The changing microbial epidemiology in cystic fibrosis. Clin Microbiol Rev. 2010;23:299–323. doi: 10.1128/CMR.00068-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vermis K, et al. Proposal to accommodate Burkholderia cepacia genomovar VI as Burkholderia dolosa sp. nov. Int J Syst Evol Microbiol. 2004;54:689–691. doi: 10.1099/ijs.0.02888-0. [DOI] [PubMed] [Google Scholar]
- 22.Lipuma JJ. Update on the Burkholderia cepacia complex. Curr Opin Pulm Med. 2005;11:528–533. doi: 10.1097/01.mcp.0000181475.85187.ed. [DOI] [PubMed] [Google Scholar]
- 23.LiPuma JJ, Dasen SE, Nielson DW, Stern RC, Stull TL. Person-to-person transmission of Pseudomonas cepacia between patients with cystic fibrosis. Lancet. 1990;336:1094–1096. doi: 10.1016/0140-6736(90)92571-x. [DOI] [PubMed] [Google Scholar]
- 24.Biddick R, Spilker T, Martin A, LiPuma JJ. Evidence of transmission of Burkholderia cepacia, Burkholderia multivorans and Burkholderia dolosa among persons with cystic fibrosis. FEMS Microbiol Lett. 2003;228:57–62. doi: 10.1016/S0378-1097(03)00724-9. [DOI] [PubMed] [Google Scholar]
- 25.Kalish LA, et al. Impact of Burkholderia dolosa on lung function and survival in cystic fibrosis. Am J Respir Crit Care Med. 2006;173:421–425. doi: 10.1164/rccm.200503-344OC. [DOI] [PubMed] [Google Scholar]
- 26.Broad Institute of Harvard and MIT; Burkholderia dolosa Sequencing Project. ( http://www.broadinstitute.org/) [Google Scholar]
- 27.Morelli G, et al. Microevolution of Helicobacter pylori during prolonged infection of single hosts and within families. PLoS Genet. 2010;6:e1001036. doi: 10.1371/journal.pgen.1001036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sibley CD, et al. A polymicrobial perspective of pulmonary infections exposes an enigmatic pathogen in cystic fibrosis patients. Proc Natl Acad Sci U S A. 2008;105:15070–15075. doi: 10.1073/pnas.0804326105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guss AM, et al. Phylogenetic and metabolic diversity of bacteria associated with cystic fibrosis. ISME J. 2011;5:20. doi: 10.1038/ismej.2010.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mowat E, et al. Psuedomonas aeruginosa population diversity and turnover in cystic fibrosis infections. Am J Respir Crit Care Med. 2011;183:1674–1679. doi: 10.1164/rccm.201009-1430OC. [DOI] [PubMed] [Google Scholar]
- 31.Wilder CN, Allada G, Schuster M. Instantaneous within-patient diversity of Psuedomonas aeruginosa quorum-sensing populations from cystic fibrosis lung infections. Infect Immun. 2009;77:5631–5639. doi: 10.1128/IAI.00755-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weigel LM, Steward CD, Tenover FC. gyrA mutations associated with fluoroquinolone resistance in eight species of Enterobacteriaceae. Antimicrob Agents Chemother. 1998;42:2661–2667. doi: 10.1128/aac.42.10.2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reyna F, Huesca M, Gonzalez V, Fuchs LY. Salmonella typhimurium gyrA mutations associated with fluoroquinolone resistance. Antimicrob Agents Chemother. 1995;39:1621–1623. doi: 10.1128/aac.39.7.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Silhavy TJ, Kahne D, Walker S. The bacterial cell envelope. Cold Spring Harb Perspect Biol. 2010;2:a000414. doi: 10.1101/cshperspect.a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vinion-Dubiel AD, Goldberg JB. Lipopolysaccharide of Burkholderia cepacia complex. J Endotoxin Res. 2003;9:201–213. doi: 10.1179/096805103225001404. [DOI] [PubMed] [Google Scholar]
- 36.Ortega X, et al. Reconstitution of O-Specific Lipopolysaccharide Expression in Burkholderia cenocepacia Strain J2315, Which Is Associated with Transmissible Infections in Patients with Cystic Fibrosis. J Bact. 2005;187:1324–1333. doi: 10.1128/JB.187.4.1324-1333.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Crosson S, McGrath PT, Stephens C, McAdams HH, Shapiro L. Conserved modular design of an oxygen sensory/signaling network with species-specific output. Proc Natl Acad Sci U S A. 2005;102:8018–8023. doi: 10.1073/pnas.0503022102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Worlitzsch D, et al. Effects of reduced mucus oxygen concentration in airway Pseudomonas infections of cystic fibrosis patients. J Clin Invest. 2002;109:317–325. doi: 10.1172/JCI13870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Felsenstein J. PHYLIP (Phylogeny Inference Package) version 3.6. Department of Genome Sciences, University of Washington; Seattle: 2005. Distributed by the author. [Google Scholar]
- 40.Marteyn B, et al. Modulation of Shigella virulence in response to available oxygen in vivo. Nature. 2010;465:355–358. doi: 10.1038/nature08970. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.