

Abstract
Aging represents a triple threat for myocardial infarction (MI). Not only does the incidence of MI increase with age, but the heart becomes more susceptible to MI induced damage and protective interventions such as ischemic preconditioning (IPC) become less effective. Therefore, any rational therapeutic strategy must be built around the ability to combat the detrimental effects of ischemia in aged individuals. To accomplish this, we need to develop a better understanding of how ischemic damage, protection, and aging are linked. In this regard, mitochondria have emerged as a common theme. First, mitochondria contribute to cell damage during ischemia-reperfusion (IR), and are central to cell death. Second, the protective signaling pathways activated by IPC converge on mitochondria, and the opening of mitochondrial ion channels alone is sufficient to elicit protection. Finally, mitochondria clearly influence the aging process and specific defects in mitochondrial activity are associated with age-related functional decline. This review will summarize the effects of aging on myocardial IR injury and discuss relevant and emerging strategies to protect against MI with an emphasis on mitochondrial function.
Keywords: Myocardial infarction, Mitochondria, Ischemic Preconditioning, Sirtuins
1. Introduction
Stoppage of blood flow through the coronary arteries results in an acute lack of oxygen and deprives cardiomyocytes of substrates. When coupled with the restoration of blood flow, this results in ischemia-reperfusion (IR) injury. The subsequent development of myocardial infarction (MI) is a major cause of death, and the annual incidence of first-time MI in the US is 610,000. There are multiple risk factors for MI, but by far the greatest risk is age (Lloyd-Jones et al., 2010). Despite an increased focus on “heart health” in recent years, and a number of interventions that may reduce the risk of MI, there are few if any therapeutic avenues that reduce damage to the heart following an MI. Furthermore, co-morbidities such as obesity, type II diabetes, and aging, ensure that MI is likely to remain a major clinical problem globally.
In 1986 it was discovered that “preconditioning” the heart with short periods of ischemia could alleviate damage incurred by a subsequent prolonged ischemic insult (Murry et al., 1986). It is also known that the prognosis following an MI is better for patients with unstable angina than those suffering an unpredicted MI (Ottani et al., 1995). These fundamental observations suggest that endogenous mechanisms exist in the heart to protect against future ischemic events. Many studies have attempted to define the signaling pathways induced by ischemic preconditioning (IPC) with the goal of developing pharmacologic mimics to provide protection (e.g. reduced infarct size, recovery of contractile function). The central problem has been that IPC itself is not effective in the aged heart (Figure 1) (Boengler et al., 2009a; Jahangir et al., 2007; Bartling et al., 2003).
Fig. 1.
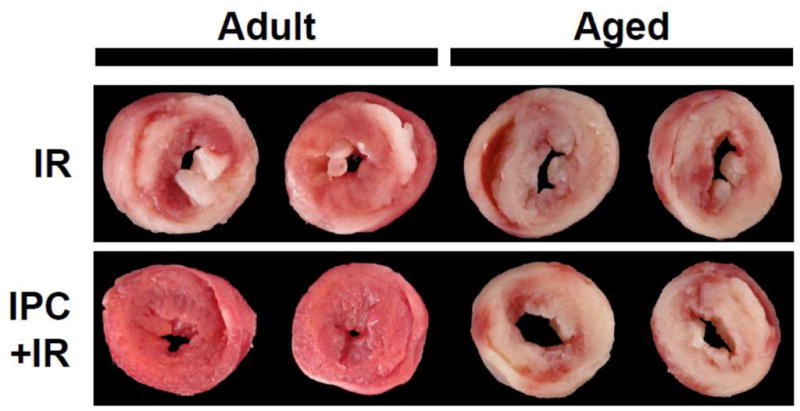
Loss of ischemic preconditioning (IPC) protection in the aged heart. Adult (2 mo.) and Aged (18 mo.) C57BL/6 mouse perfused hearts were subjected to 25 min. ischemia and 60 min. reperfusion (IR) or preconditioned with three cycles of 5 min. ischemia and 5 min. reperfusion followed by index IR. Following protocols, hearts were sliced transversely, stained with triphenyltetrazolium chloride to delineate live (red) and infarcted (white) tissue. Typical slices demonstrate that while both adult and aged hearts were subject to damage from IR injury, IPC protected the adult but not the aged heart from damage.
Aging is associated with an accumulation of pathologic changes leading to a progressive decline in cellular, organ and whole organism function. In addition, aging results in a lower resistance to stress (Shih et al., 2007). Mitochondria have been recognized to play a prominent role in aging, and a decline in mitochondrial function is thought to underlie some of the decline in tissue function with age. In addition, the mitochondrial signaling pathways leading to cell injury and death may be disrupted during aging.
Counteracting the role of mitochondria in pathology however, is the fact that they are known to be critically involved in the protection afforded by IPC. Thus developing a clear understanding of how aging influences the mitochondrial balance between ischemic damage and protection is essential to devise effective therapeutic strategies for cardioprotection in older adults.
2. Cardiac Mitochondria: Basic Function, and Role in IR Injury
Cardiac contractile function relies on mitochondrial oxidative phosphorylation (Ox-Phos) for the bulk of its ATP. While mitochondria are the metabolic hub of all eukaryotic cells, mitochondria from different organs are adapted to specific functions (Johnson et al., 2007). Specifically, cardiac mitochondria are known to preferentially oxidize fatty acids (Stanley et al., 2005). Moreover, cardiac mitochondria exist in two subpopulations: subsarcolemmal (SSM) and interfibrillar mitochondria (IFM), which differ not only in their location but also function (Palmer et al., 1977). For example, IFM have an increased maximal respiration rate and ability to handle Ca2+ (Hofer et al., 2009; Lesnefsky et al., 2001b).
In addition to their central metabolic role, mitochondria are also key players in IR injury. IR injury is characterized by two events: ischemia and reperfusion. At the cellular level, ischemia is characterized by a lack of O2 and substrates, and ischemic cells accumulate metabolites such as lactate. Via alterations in pH, Na+, and ATP levels, ischemia and reperfusion then trigger a cascade of events including massive reactive oxygen species (ROS) generation, loss of nucleotide homeostasis, and disruption of Ca2+ homeostasis, culminating in the formation of the mitochondrial permeability transition (PT) pore and the activation of cell death signaling pathways (reviewed in detail (Lemasters et al., 2009; Murphy et al., 2008; Brookes et al., 2004)). Thus, mitochondria have emerged as key arbiters of cellular survival in IR injury.
3. Cardiac Mitochondria & IR Injury: the Effects of Aging
As shown in Figure 1, the aging heart is more sensitive to IR injury (Lesnefsky et al., 2006; Lesnefsky et al., 2001b). In addition, a significant decline in cardiac mitochondrial function is seen in aging, and this appears to differ between IFM and SSM (Judge et al., 2005; Fannin et al., 1999). This section will focus on two mitochondrial commonalties between aging and IR injury: (i) mitochondrial Ca2+ handling and (ii) mitochondrial ROS generation/oxidative damage.
The Ca2+ insult associated with pathology of cardiac IR injury results in mitochondrial Ca2+ overload and ultimately cell death (Lemasters et al., 2009; Murphy et al., 2008; Brookes et al., 2004). In this regard, it is notable that mitochondria from the aged heart display a decreased capacity to accumulate and retain Ca2+, resulting in a decreased tolerance to Ca2+ insult (Jahangir et al., 2001). This inability to handle a pathological Ca2+ insult may predispose the aged myocardium to IR injury.
In a similar manner, large scale mitochondrial ROS production is associated with IR injury (Fridovich, 1983), and increased mitochondrial ROS and oxidative damage, particularly in IFM, are also found in the aged heart (Judge et al., 2005; Lesnefsky et al., 2001b; Fannin et al., 1999).
The molecular mechanisms underlying defects in Ca2+ and ROS homeostasis find their roots at the level of the respiratory chain. Both aging and IR injury result in damage to the Ox-Phos machinery (Kwong et al., 2000; Fannin et al., 1999; Lenaz et al., 1997; Guerrieri et al., 1993), raising the possibility that an already age-diminished Ox-Phos may be susceptible to further decline with IR.
There are similarities between the molecular damage to Ox-Phos that occurs in aging and IR injury. For example, IR causes a loss of cardiolipin (Lesnefsky et al., 2001c). Similar effects on cardiolipin have been reported in the aging heart (Petrosillo et al., 2001; Pepe et al., 1999). In addition to its metabolic role, cardiolipin loss is also associated with increased ROS generation and cytochrome c release (Kagan et al., 2004; Pepe et al., 1999).
Alterations in complex III activity have been reported in both IR injury (Lesnefsky et al., 2001a) and aging (Lesnefsky et al., 2001b), although the molecular mechanisms may be distinct; complex III’s cytochrome c binding site is the target of functional decline in aging, while ischemic damage is primarily at the iron-sulfur center within the complex (Lesnefsky et al., 2001a). Notably, both of these mechanisms manifest in increased ROS production. While this review focuses on changes in ROS production at the level of the Ox-Phos machinery, other mitochondrial ROS modulating components such as monoamine oxidase and p66Shc are also associated with aging. The inhibition of monoamine oxidase and p66Shc result in protection from IR injury (Carpi et al., 2009), their expression levels increase with age (Pandolfi et al., 2005; Saura et al., 1994), and genetic deletion of p66Shc increases life span (Migliaccio et al., 1999).
In addition to changes in ROS generation by the respiratory chain, a state of oxidative stress can also be precipitated by a decrease in antioxidant defenses. In this regard, IR injury is known to decrease the activity of enzymes involved in ROS removal, such as manganese superoxide dismutase and glutathione peroxidase (Shlafer et al., 1987). Antioxidant defenses are similarly compromised with aging (Ferrara et al., 2008; van der Loo et al., 2005; Moghaddas et al., 2003).
The increased mitochondrial ROS generation associated with aging and IR injury can have a profound impact on the formation mitochondrial permeability transition (PT) pore. The induction of the mitochondrial PT pore is associated with irreversible damage and the initiation of cell death (Lemasters et al., 2009). The increased ROS and altered calcium handling conditions set forth in the aged myocardium may be interrelated and ultimately sensitize the heart to the induction of mitochondrial PT pore opening.
It is not clear whether differences in mitochondrial ROS generation are an underlying factor in the differential responses to IR injury that occur between animals of varying aging rates. It is known that mitochondria from short-lived animals generate more ROS (Lambert et al., 2007). Interestingly, these animals also develop myocardial infarction more quickly (Downey et al., 2009; Manintveld et al., 2007; Gersh et al., 2005; Barja et al., 2000). While this connection between the rate of aging and infarct development is appealing, other hemodynamic parameters (e.g. collateral flow) should be taken into consideration when comparing different species. In addition, there have been a number of recent observations that ROS can be beneficial as well as detrimental and that ROS signaling may be important for adaptive responses to ischemia (see below). Hence, the role of ROS depends upon the context in which it is presented, including that of aging.
Collectively, a dysregulation of mitochondrial Ox-Phos, Ca2+ handling and ROS generation occurs in both IR injury and aging, and it is logical to suggest that the molecular mechanisms underlying these phenomena may be shared.
4. Ischemic Preconditioning, Aging and Mitochondria
One strategy to protect the heart from IR injury is IPC (Murry et al., 1986), which can be used clinically in transplant and coronary surgeries (Ji et al., 2007). Similar to IPC, protection can also be achieved via ischemic postconditioning during reperfusion (Zhao et al., 2003). This is a process whereby short periods of ischemia are applied following the index ischemic insult. Like IPC, the effectiveness of postconditioning also decreases with age (Boengler et al., 2008), though whether these similarities persist at the mechanistic level is unclear.
IPC triggers a diverse array of signaling cascades (reviewed in (Heusch et al., 2008; Downey et al., 2007)) (Figure 2), many of which converge at the mitochondrion (Garlid et al., 2009; Murphy et al., 2007b). In addition to factors such as gender (Murphy et al., 2007a), diabetes (Kersten et al., 2000), and many prescription drugs (Wojtovich et al., 2010; Shim et al., 2008), a major confounder for the effectiveness of IPC is age (Boengler et al., 2009a; Jahangir et al., 2007; Bartling et al., 2003). Furthermore, even the effectiveness of drugs which activate similar protective signaling pathways as IPC (e.g. volatile anesthetics) is diminished with age (Schulman et al., 2001). As relates to IPC, the mechanism underlying its loss of effectiveness in aged individuals is thought to include: (i) disruption of the machinery that maintains cardiac redox status, (ii) inadequate energy supply due to impaired Ox-Phos function, (iii) defective IPC signaling, or (iv) a change in the trigger/threshold for IPC activation.
Fig. 2.

Ischemic preconditioning (IPC): signaling pathways and targets. IPC elicits protection through a complex signaling cascade that converges on the mitochondrion. This schematic, while not comprehensive, focuses on functional and regulatory interactions discussed in this review. Aging results in the loss of IPC which may result from modifications to this signal transduction pathway, many components of which have been shown to be altered with age. For a comprehensive overview of IPC kinase signaling please refer to (Downey et al., 2007). Abbreviations: IPC, ischemic preconditioning; GPCRs, G protein coupled receptors; GFRs, growth factor receptors; SIRT1, Sirtuin 1; eNOS, endothelial nitric oxide synthase; MAPKs, Mitogen-activated protein kinases; mTOR, mammalian target of rapamycin; PI3K, phosphatidylinositol 3-kinase; Akt, Protein Kinase B; PDK-1, pyruvate dehydrogenase kinase; PKA, protein kinase A; PTEN, Phosphatase and tensin homolog; PLC/D, Phospholipase C/D; p90RSK, p90 ribosomal S6 kinase; PKCδ/ε, protein kinase C δ/ε; p70S6K, p70S6 kinase; AMPK, AMP-activated protein kinase; ROS, reactive oxygen species; GSK-3β, glycogen synthase kinase-3β; ETC, electron transport chain; KATP, ATP-sensitive potassium channel; PT Pore, permeability transition pore.
A mild generation of ROS is required for IPC induced protection, and notably antioxidants render IPC ineffective (Dost et al., 2008). The aging heart, with a large increase in ROS production (Juhaszova et al., 2005; Judge et al., 2005; Lesnefsky et al., 2001b; Muscari et al., 1990) coupled with decrease in mitochondrial antioxidant defenses (e.g. manganese superoxide dismutase) (Ferrara et al., 2008) may render IPC ineffective by disrupting redox signaling. Several components of the IPC signal cascade (e.g. mitochondrial KATP channel) are redox sensitive (Queliconi et al., 2011), although the question remains whether antioxidants can restore redox balance and IPC to the aged heart.
The recovery from IR requires an adequate energy supply. The underlying loss of IPC efficacy in aging may be due to impaired Ox-Phos since, as previously discussed, components of the Ox-Phos machinery such as complex I activity are diminished with aging via irreversible oxidative modifications (Navarro et al., 2007; Choksi et al., 2007; Fannin et al., 1999).
-
The protection resulting from IPC depends on a diverse array of signaling cascades (Garlid et al., 2009) and the loss of one or more of these with aging may compromise preconditioning. For example, the mitochondrial KATP channel (Wojtovich et al., 2009; Facundo et al., 2006) and sirtuin activity (SIRT1) (Nadtochiy et al., 2011; Nadtochiy et al., 2010) are critical components of IPC, and both exhibit an age-dependent decline in activity (Braidy et al., 2011; Krylova et al., 2006).
In addition to upstream signaling cascades that converge on mitochondria, intrinsic features of mitochondria themselves may also modulate their ability to receive/respond to cardioprotective signaling cascades. For example, a prerequisite for IPC is the ability of the respiratory chain to undergo reversible modulation of its activity. Indeed, several reversible inhibitors of Ox-Phos are cardioprotective (Burwell et al., 2009). It has been shown that complex I mutant mice cannot be preconditioned, suggesting that pre-existing deficiencies in Ox-Phos may compromise the plasticity of electron transport chain activity during IPC. As such, age induced modifications to the Ox-Phos machinery (Choksi et al., 2007) may have a similar effect.
Similarly, the cellular processes that remove damaged mitochondria could also play a role in IPC. Autophagy acts as quality control by targeting and removing dysfunctional macromolecules and organelles (Gottlieb et al., 2010). This turnover is critical to maintaining a healthy pool of functional mitochondria. It is well documented that autophagy is compromised with aging, and the resulting accumulation of damaged mitochondria may lead to impaired Ox-Phos function in older individuals (reviewed in (Green et al., 2011)). Moreover, autophagy is required for IPC (Huang et al., 2011) and the inhibition of autophagy results in exacerbated IR injury (Weber et al., 2010). A simple hypothesis is that the accumulation of damaged mitochondria resulting from a reduction in autophagy during aging may impact the effectiveness of IPC. Coupled with an increased sensitivity to IR injury caused by damaged mitochondria, reductions in autophagy may represent a critical risk factor for MI. Since multiple IPC signaling pathways are also affected by age, however, it is not clear that stimulating autophagy alone would be sufficient to restore IPC in aged individuals. Nevertheless, it is likely that ridding the myocytes of age-associated dysfunctional mitochondria would increase ischemic tolerance and alter the threshold for inducing damage.
It is interesting that a particular sub-population of mitochondria has been implicated in IPC signaling. Specifically, it has been shown that the gap junctional protein connexin 43 (Cx43) is required for IPC, and cardiac Cx43 is localized exclusively in SSMs, not in IFMs (Rottlaender et al., 2010; Boengler et al., 2009b). This suggests that SSM rather than IFM are more influential in the IPC signal transduction cascade. While the overall expression of Cx43 is indeed decreased in aged myocardium (Boengler et al., 2007), this observation poses a dilemma, since aging appears to primarily affect the metabolic function of IFM, not SSM (Lesnefsky et al., 2006; Lesnefsky et al., 2001b). Clearly, there may be subtle functions of SSM (e.g. their possession of Cx43), which render them more important in aging induced loss of IPC, versus the more generalized metabolic changes in aging which are assigned to IFM, and may not be important in loss of IPC. Alternatively, signaling between mitochondrial populations may play a role in IPC and this signaling could become defective with age.
It is known that the degree of protection afforded by a fixed IPC stimulus varies depending on the severity of index IR injury which follows (Hanley et al., 2005). Thus one reason underlying the loss of IPC efficacy in aging may be that a different amount of IPC stimulus is required to elicit protection in aging vs. young hearts. Interestingly, one study has found that a greater than normal IPC stimulus was able to elicit protection in elderly patients undergoing angioplasty (Lee et al., 2002).
In summary, there are several mechanisms which may underlie the loss of IPC in aging. Further elucidation of these mechanisms may aid in the development of therapeutics to restore IPC in the aged heart.
5. Current research
Despite numerous IPC mimicking drugs discovered in the laboratory, none has yet made the transition to clinical use, with the result that there are currently no FDA approved drugs for the indication of reducing myocardial infarct size. One reason for this may be that most MI patients are neither young nor healthy, whereas pharmacologic agents are often developed in the laboratory using young animals (Hausenloy et al., 2010; Downey et al., 2009). Examples of normally-efficacious agents which are ineffective at providing cardioprotection in aged individuals include volatile anesthetics, mitochondrial K+ channel openers and adenosine A1 agonists (Sniecinski et al., 2004; Schulman et al., 2001). Thus, an important avenue for current research is to develop broadly applicable therapies, particularly agents that work in the aging heart. In this regard, two areas of research that are currently promising are studies of caloric restriction and the use of model organisms with short life spans.
5.1 Caloric restriction (CR) and sirtuins
Recent research has shown that caloric restriction (CR) may activate signaling pathways which restore the ability to precondition aged hearts (Abete et al., 2010). A major advance in the CR field was the discovery of the silent information regulator (Sirtuin) family of proteins. There are 7 mammalian Sirtuins (SIRTs), some of which are up-regulated and activated by CR and may be responsible for the effects of CR (Morris et al., 2011).
SIRT3, which is localized in mitochondria, may decrease oxidative stress via deacetylation and activation of MnSOD (Qiu et al., 2010). In addition, SIRT3 dependent deacetylation of isocitrate dehydrogenase can elevate NADPH levels, which may keep antioxidant enzymes in a reduced state (Schlicker et al., 2008). SIRT3 can also deacetylate cyclophilin D and prevent its interaction with adenine nucleotide translocase (Hafner et al., 2010), which may inhibit mitochondrial PT pore opening.
SIRT1 is a cytosolic/nuclear protein, which has been implicated in cardiac stress responses in aging (Hsu et al., 2008). We found that pharmacologic inhibition or partial genetic ablation of SIRT1 blocked IPC in adult mouse hearts (Nadtochiy et al., 2011; Nadtochiy et al., 2010), suggesting an endogenous role for SIRT1 in protection. Thus, it is possible that age-dependent decline in SIRT1 activity (Braidy et al., 2011) may contribute to loss of IPC. Furthermore, over-expression of SIRT1 elicits resistance to oxidative stress and protects heart from IR injury (Nadtochiy et al., 2011; Hsu et al., 2008). This suggests that boosting SIRT1 activity in aging may be able to restore IPC.
The molecular mechanisms of SIRT1-mediated cardioprotection have not been fully elucidated, but SIRT1 is known to regulate a variety of potential protective events, such as eNOS activation and autophagy (Nadtochiy et al., 2011). In addition, SIRT1 is at the crossroad of the CR-mediated regulation of mammalian target of rapamycin (mTOR) and AMP-activated protein kinase (AMPK) pathways (Ghosh et al., 2010; Shinmura et al., 2007), all of which are cardioprotective. The complexity and intertwinement of signaling cascades involved in reducing IR injury is illustrated by the finding that the cardioprotective effect of rapamycin was reversed by the mitochondrial KATP channel inhibitors (Khan et al., 2006), suggesting that both SIRT1 and CR may indirectly be involved in mitochondrial KATP activation. In addition, mTOR mediated protection may also include activation of autophagy (Gurusamy et al., 2010). Intriguingly, exercise has been shown to increase SIRT1 activity in aged rats (Ferrara et al., 2008) which raises the possibility that SIRT1 may contribute to exercise induced cardioprotection (Lawler et al., 2009).
5.2 Model Systems of Aging
As mentioned above, an important and emerging approach is to study IPC in aging animals. In this regard, invertebrate animal models are of increasing interest, owing to their short generation times and ease of use. Mechanistically, the process of triggering protection by IPC may exhibit certain species-dependent components, but the fundamental phenomenon of IPC is evolutionarily conserved (Dasgupta et al., 2007), and readily reproduced in organisms such as the nematode C. elegans.
C. elegans has been used extensively to study the relationship between aging, ROS and mitochondrial dysfunction, and more recently to identify genetic determinants of hypoxic sensitivity and hypoxic preconditioning (HPC). Early work showed that a mutation in mitochondrial complex II (mev-1) increased ROS production and decreased lifespan (Ishii et al., 1998). In addition, mutations that increased lifespan, such as in the canonical insulin signaling pathway, also increased resistance to ROS. These data were consistent with the free radical theory of aging, which states that increased mitochondrial metabolism and oxygen consumption will result in increased ROS production and a subsequently reduced lifespan.
However, this seemingly direct correlation between ROS and lifespan has recently been challenged, and there are multiple lines of evidence that in fact low levels of mitochondrial ROS formation can stimulate adaptive processes that increase both longevity and stress-resistance. The ability of sub-lethal mitochondrial stress to trigger protection has been termed mitohormesis (for comprehensive reviews, see (Ristow et al., 2011; Ristow et al., 2010).
Relevant examples that dispute the free radical theory of aging include demonstrating that overexpression of the mitochondrial superoxide dismutase in C. elegans does not alter lifespan (Doonan et al., 2008) and that lifespan extension in C. elegans mitochondrial (mit) mutants with decreased Ox-Phos activity does not correlate with oxidative stress levels (Rea et al., 2007). In fact, mild mitochondrial dysfunction may signal a beneficial response (Ventura et al., 2009; Rea et al., 2007; Dillin et al., 2002). In this regard, localized ROS in mitochondria (Yang et al., 2010) may coordinate functional responses; it is possible that these responses are not limited to the cell in which the signal originates, either, as recent data suggests that mitochondria can contribute to protection through trans-cellular signaling (Durieux et al., 2011). This may be akin to the phenomenon of “remote preconditioning” observed in mammals.
The ability to elicit biphasic responses (ie adaptive or detrimental) by modulating Ox-Phos activity is consistent with the idea that mitochondria are central to both cell death and to protective signaling. These data also establish clear parallels between C. elegans and mammalian systems. Previous work indicating that ROS production is increased and mitochondrial function is decreased in aged mammals, as detailed in Section 3, leads us to speculate that the balance between the adaptive and detrimental effects of ROS is altered with aging, though whether this is due to differing sites of ROS formation, the ability of signaling pathways to recognize ROS, or functional changes that occur at the level of the mitochondria themselves has yet to be determined.
A significant advantage to working with a genetic model organism is the ability to perform unbiased screens to answer the above type of question. However, C. elegans are extremely resistant to conventional hypoxia (~0.5% O2), and instead relatively anoxic conditions (<0.1% O2) have been used to identify genetic determinants of whole-organism ischemic sensitivity (Hayakawa et al., 2011; Mabon et al., 2009; Mendenhall et al., 2006; Scott et al., 2002) as well as IPC (LaRue et al., 2011; Dasgupta et al., 2007). Novel targets identified thus far include tRNA synthetases (Anderson et al., 2009), Apoptosis Signal Regulating Kinase family proteins (Hayakawa et al., 2011), 5′ AMP-activated protein kinase (LaRue et al., 2011), components of the unfolded protein response machinery (Mao et al., 2010), and the Apaf1 ortholog ced-4, which has been shown to be required for IPC independent of its role in apoptosis (Dasgupta et al., 2007). Whether these results will translate to mammalian models is as-of-yet unclear given the fact that hypoxic and anoxic responses differ in worms
It is not yet clear to what extent mitochondria are necessary to support the role of these proteins, but signaling pathways that coordinate adaptive responses in worms often target mitochondria. Our own work has shown that worm mitochondria, like those from mammals, have both KATP and big conductance potassium (BK) channels whose activities appear to be both necessary and sufficient for protection from ischemic damage (Wojtovich et al., 2008). Interestingly, metabolic profiling in worms with Ox-Phos mutations suggests a transition to alternate means of ATP production that do not require O2, and these worms are both long-lived and resistant to ischemia (Butler et al., 2010; Rea et al., 2007). In this respect, it would be reasonable to ask whether Ox-Phos mutants are endogenously preconditioned.
In addition to the identification of novel targets that regulate anoxic/hypoxic sensitivity, novel mechanisms have been discovered for well-established targets. As in mammals, HIF-1 in worms mediates responses to non-lethal or moderate hypoxic exposure (Shen et al., 2005). HIF-1 was recently shown to regulate the expression of a tyrosinase in C. elegans ASJ sensory neurons. This enzyme is secreted and antagonizes p53-dependent germline apoptosis, thus linking hypoxia and programmed cell death through cell non-autonomous signaling (Sendoel et al., 2010). Whether increased tyrosinase expression is sufficient to confer protection is unknown, nor is it known whether this signaling pathway is similarly protective in other cell death paradigms. However, it is notable that multiple reports have shown that HIF-1 modulates lifespan in worms (Zhang et al., 2009; Chen et al., 2009; Mehta et al., 2009).
While validation in mammalian systems is undoubtedly necessary, these examples illustrate how the use of genetics has great potential to yield previously unheralded protective approaches and molecular targets. Although the understanding of invertebrate aging (Partridge, 2011) and mitohormesis (Ristow et al., 2010) is advancing, significant gaps remain to be addressed. For example, does aging impact on HPC efficacy in worms? Does mitochondrial dysfunction preclude HPC? Can HPC be elicited cell non-autonomously? The use of genetic model organisms has and will continue to yield fundamental new insights into these questions.
5. Conclusions
Multiple signaling pathways interface at the mitochondrion to influence longevity and IPC. Elucidating the mechanisms of loss of IPC efficacy in aging remains a major problem in cardiovascular research, which demands the development of novel treatment strategies for myocardial infarction in aged individuals. Mitochondria both contribute to the pathology of IR injury and are critical mediators of cardioprotective signaling. Current research focusing on lifestyle interventions such as caloric restriction and exercise, pharmacologic approaches, and model organism research are promising avenues for learning how to restore IPC in aged patients.
Highlights.
Mitochondria are central to ischemic cell damage and death.
Mitochondria contribute to healthy aging.
Ischemic preconditioning (IPC) acts via mitochondria to reduce subsequent ischemic damage.
The efficacy of IPC is reduced in aged individuals who most need protective intervention.
Understanding mechanisms linking mitochondria to IPC will aid in the development of therapeutics to mimic IPC in the aged heart.
Acknowledgments
Funding was provided by US National Institutes of Health grants RO1-HL071158 (to PSB) and RO1-GM-087483 (to PSB and KN) and an American Heart Association, Founder Affiliate Postdoctoral Fellowship award 11POST7290028 (to APW).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abete P, Cacciatore F, Testa G, la-Morte D, Galizia G, De SD, Calabrese C, Cioppa A, Ferrara N, Rengo F. Ischemic preconditioning in the aging heart: from bench to bedside. Ageing Res Rev. 2010;9 (2):153–162. doi: 10.1016/j.arr.2009.07.001. [DOI] [PubMed] [Google Scholar]
- Anderson LL, Mao X, Scott BA, Crowder CM. Survival from hypoxia in C. elegans by inactivation of aminoacyl-tRNA synthetases. Science. 2009;323 (5914):630–633. doi: 10.1126/science.1166175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barja G, Herrero A. Oxidative damage to mitochondrial DNA is inversely related to maximum life span in the heart and brain of mammals. FASEB J. 2000;14 (2):312–318. doi: 10.1096/fasebj.14.2.312. [DOI] [PubMed] [Google Scholar]
- Bartling B, Friedrich I, Silber RE, Simm A. Ischemic preconditioning is not cardioprotective in senescent human myocardium. Ann Thorac Surg. 2003;76 (1):105–111. doi: 10.1016/s0003-4975(03)00186-3. [DOI] [PubMed] [Google Scholar]
- Boengler K, Buechert A, Heinen Y, Roeskes C, Hilfiker-Kleiner D, Heusch G, Schulz R. Cardioprotection by ischemic postconditioning is lost in aged and STAT3-deficient mice. Circ Res. 2008;102 (1):131–135. doi: 10.1161/CIRCRESAHA.107.164699. [DOI] [PubMed] [Google Scholar]
- Boengler K, Konietzka I, Buechert A, Heinen Y, Garcia-Dorado D, Heusch G, Schulz R. Loss of ischemic preconditioning’s cardioprotection in aged mouse hearts is associated with reduced gap junctional and mitochondrial levels of connexin 43. Am J Physiol Heart Circ Physiol. 2007;292 (4):H1764–H1769. doi: 10.1152/ajpheart.01071.2006. [DOI] [PubMed] [Google Scholar]
- Boengler K, Schulz R, Heusch G. Loss of cardioprotection with ageing. Cardiovasc Res. 2009a;83 (2):247–261. doi: 10.1093/cvr/cvp033. [DOI] [PubMed] [Google Scholar]
- Boengler K, Stahlhofen S, van de SA, Gres P, Ruiz-Meana M, Garcia-Dorado D, Heusch G, Schulz R. Presence of connexin 43 in subsarcolemmal, but not in interfibrillar cardiomyocyte mitochondria. Basic Res Cardiol. 2009b;104 (2):141–147. doi: 10.1007/s00395-009-0007-5. [DOI] [PubMed] [Google Scholar]
- Braidy N, Guillemin GJ, Mansour H, Chan-Ling T, Poljak A, Grant R. Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS One. 2011;6 (4):e19194. doi: 10.1371/journal.pone.0019194. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol. 2004;287 (4):C817–C833. doi: 10.1152/ajpcell.00139.2004. [DOI] [PubMed] [Google Scholar]
- Burwell LS, Nadtochiy SM, Brookes PS. Cardioprotection by metabolic shutdown and gradual wake-up. J Mol Cell Cardiol. 2009;46 (6):804–810. doi: 10.1016/j.yjmcc.2009.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler JA, Ventura N, Johnson TE, Rea SL. Long-lived mitochondrial (Mit) mutants of Caenorhabditis elegans utilize a novel metabolism. FASEB J. 2010;24 (12):4977–4988. doi: 10.1096/fj.10-162941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpi A, Menabo R, Kaludercic N, Pelicci P, Di LF, Giorgio M. The cardioprotective effects elicited by p66(Shc) ablation demonstrate the crucial role of mitochondrial ROS formation in ischemia/reperfusion injury. Biochim Biophys Acta. 2009;1787 (7):774–780. doi: 10.1016/j.bbabio.2009.04.001. [DOI] [PubMed] [Google Scholar]
- Chen D, Thomas EL, Kapahi P. HIF-1 modulates dietary restriction-mediated lifespan extension via IRE-1 in Caenorhabditis elegans. PLoS Genet. 2009;5 (5):e1000486. doi: 10.1371/journal.pgen.1000486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choksi KB, Nuss JE, Boylston WH, Rabek JP, Papaconstantinou J. Age-related increases in oxidatively damaged proteins of mouse kidney mitochondrial electron transport chain complexes. Free Radic Biol Med. 2007;43 (10):1423–1438. doi: 10.1016/j.freeradbiomed.2007.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta N, Patel AM, Scott BA, Crowder CM. Hypoxic preconditioning requires the apoptosis protein CED-4 in C. elegans. Curr Biol. 2007;17 (22):1954–1959. doi: 10.1016/j.cub.2007.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillin A, Hsu AL, rantes-Oliveira N, Lehrer-Graiwer J, Hsin H, Fraser AG, Kamath RS, Ahringer J, Kenyon C. Rates of behavior and aging specified by mitochondrial function during development. Science. 2002;298 (5602):2398–2401. doi: 10.1126/science.1077780. [DOI] [PubMed] [Google Scholar]
- Doonan R, McElwee JJ, Matthijssens F, Walker GA, Houthoofd K, Back P, Matscheski A, Vanfleteren JR, Gems D. Against the oxidative damage theory of aging: superoxide dismutases protect against oxidative stress but have little or no effect on life span in Caenorhabditis elegans. Genes Dev. 2008;22 (23):3236–3241. doi: 10.1101/gad.504808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dost T, Cohen MV, Downey JM. Redox signaling triggers protection during the reperfusion rather than the ischemic phase of preconditioning. Basic Res Cardiol. 2008;103 (4):378–384. doi: 10.1007/s00395-008-0718-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downey JM, Cohen MV. Why do we still not have cardioprotective drugs? Circ J. 2009;73 (7):1171–1177. doi: 10.1253/circj.cj-09-0338. [DOI] [PubMed] [Google Scholar]
- Downey JM, Davis AM, Cohen MV. Signaling pathways in ischemic preconditioning. Heart Fail Rev. 2007;12 (3–4):181–188. doi: 10.1007/s10741-007-9025-2. [DOI] [PubMed] [Google Scholar]
- Durieux J, Wolff S, Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011;144 (1):79–91. doi: 10.1016/j.cell.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Facundo HT, Fornazari M, Kowaltowski AJ. Tissue protection mediated by mitochondrial K+ channels. Biochim Biophys Acta. 2006;1762 (2):202–212. doi: 10.1016/j.bbadis.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Fannin SW, Lesnefsky EJ, Slabe TJ, Hassan MO, Hoppel CL. Aging selectively decreases oxidative capacity in rat heart interfibrillar mitochondria. Arch Biochem Biophys. 1999;372 (2):399–407. doi: 10.1006/abbi.1999.1508. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Rinaldi B, Corbi G, Conti V, Stiuso P, Boccuti S, Rengo G, Rossi F, Filippelli A. Exercise training promotes SIRT1 activity in aged rats. Rejuvenation Res. 2008;11 (1):139–150. doi: 10.1089/rej.2007.0576. [DOI] [PubMed] [Google Scholar]
- Fridovich I. Superoxide radical: an endogenous toxicant. Annu Rev Pharmacol Toxicol. 1983;23:239–257. doi: 10.1146/annurev.pa.23.040183.001323. [DOI] [PubMed] [Google Scholar]
- Garlid KD, Costa AD, Quinlan CL, Pierre SV, Dos SP. Cardioprotective signaling to mitochondria. J Mol Cell Cardiol. 2009;46 (6):858–866. doi: 10.1016/j.yjmcc.2008.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gersh BJ, Stone GW, White HD, Holmes DR., Jr Pharmacological facilitation of primary percutaneous coronary intervention for acute myocardial infarction: is the slope of the curve the shape of the future? JAMA. 2005;293 (8):979–986. doi: 10.1001/jama.293.8.979. [DOI] [PubMed] [Google Scholar]
- Ghosh HS, McBurney M, Robbins PD. SIRT1 negatively regulates the mammalian target of rapamycin. PLoS One. 2010;5 (2):e9199. doi: 10.1371/journal.pone.0009199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb RA, Mentzer RM. Autophagy during cardiac stress: joys and frustrations of autophagy. Annu Rev Physiol. 2010;72:45–59. doi: 10.1146/annurev-physiol-021909-135757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science. 2011;333 (6046):1109–1112. doi: 10.1126/science.1201940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrieri F, Capozza G, Fratello A, Zanotti F, Papa S. Functional and molecular changes in FoF1 ATP-synthase of cardiac muscle during aging. Cardioscience. 1993;4 (2):93–98. [PubMed] [Google Scholar]
- Gurusamy N, Lekli I, Mukherjee S, Ray D, Ahsan MK, Gherghiceanu M, Popescu LM, Das DK. Cardioprotection by resveratrol: a novel mechanism via autophagy involving the mTORC2 pathway. Cardiovasc Res. 2010;86 (1):103–112. doi: 10.1093/cvr/cvp384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner AV, Dai J, Gomes AP, Xiao CY, Palmeira CM, Rosenzweig A, Sinclair DA. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging (Albany NY) 2010;2 (12):914–923. doi: 10.18632/aging.100252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanley PJ, Daut J. K(ATP) channels and preconditioning: a re-examination of the role of mitochondrial K(ATP) channels and an overview of alternative mechanisms. J Mol Cell Cardiol. 2005;39 (1):17–50. doi: 10.1016/j.yjmcc.2005.04.002. [DOI] [PubMed] [Google Scholar]
- Hausenloy DJ, Baxter G, Bell R, Botker HE, Davidson SM, Downey J, Heusch G, Kitakaze M, Lecour S, Mentzer R, Mocanu MM, Ovize M, Schulz R, Shannon R, Walker M, Walkinshaw G, Yellon DM. Translating novel strategies for cardioprotection: the Hatter Workshop Recommendations. Basic Res Cardiol. 2010;105 (6):677–686. doi: 10.1007/s00395-010-0121-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa T, Kato K, Hayakawa R, Hisamoto N, Matsumoto K, Takeda K, Ichijo H. Regulation of anoxic death in Caenorhabditis elegans by mammalian apoptosis signal-regulating kinase (ASK) family proteins. Genetics. 2011;187 (3):785–792. doi: 10.1534/genetics.110.124883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heusch G, Boengler K, Schulz R. Cardioprotection: nitric oxide, protein kinases, and mitochondria. Circulation. 2008;118 (19):1915–1919. doi: 10.1161/CIRCULATIONAHA.108.805242. [DOI] [PubMed] [Google Scholar]
- Hofer T, Servais S, Seo AY, Marzetti E, Hiona A, Upadhyay SJ, Wohlgemuth SE, Leeuwenburgh C. Bioenergetics and permeability transition pore opening in heart subsarcolemmal and interfibrillar mitochondria: effects of aging and lifelong calorie restriction. Mech Ageing Dev. 2009;130 (5):297–307. doi: 10.1016/j.mad.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu CP, Odewale I, Alcendor RR, Sadoshima J. Sirt1 protects the heart from aging and stress. Biol Chem. 2008;389 (3):221–231. doi: 10.1515/BC.2008.032. [DOI] [PubMed] [Google Scholar]
- Huang C, Andres AM, Ratliff EP, Hernandez G, Lee P, Gottlieb RA. Preconditioning involves selective mitophagy mediated by Parkin and p62/SQSTM1. PLoS One. 2011;6 (6):e20975. doi: 10.1371/journal.pone.0020975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii N, Fujii M, Hartman PS, Tsuda M, Yasuda K, Senoo-Matsuda N, Yanase S, Ayusawa D, Suzuki K. A mutation in succinate dehydrogenase cytochrome b causes oxidative stress and ageing in nematodes. Nature. 1998;394 (6694):694–697. doi: 10.1038/29331. [DOI] [PubMed] [Google Scholar]
- Jahangir A, Ozcan C, Holmuhamedov EL, Terzic A. Increased calcium vulnerability of senescent cardiac mitochondria: protective role for a mitochondrial potassium channel opener. Mech Ageing Dev. 2001;122 (10):1073–1086. doi: 10.1016/s0047-6374(01)00242-1. [DOI] [PubMed] [Google Scholar]
- Jahangir A, Sagar S, Terzic A. Aging and cardioprotection. J Appl Physiol. 2007;103 (6):2120–2128. doi: 10.1152/japplphysiol.00647.2007. [DOI] [PubMed] [Google Scholar]
- Ji B, Liu M, Liu J, Wang G, Feng W, Lu F, Shengshou H. Evaluation by cardiac troponin I: the effect of ischemic preconditioning as an adjunct to intermittent blood cardioplegia on coronary artery bypass grafting. J Card Surg. 2007;22 (5):394–400. doi: 10.1111/j.1540-8191.2007.00433.x. [DOI] [PubMed] [Google Scholar]
- Johnson DT, Harris RA, French S, Blair PV, You J, Bemis KG, Wang M, Balaban RS. Tissue heterogeneity of the mammalian mitochondrial proteome. Am J Physiol Cell Physiol. 2007;292 (2):C689–C697. doi: 10.1152/ajpcell.00108.2006. [DOI] [PubMed] [Google Scholar]
- Judge S, Jang YM, Smith A, Hagen T, Leeuwenburgh C. Age-associated increases in oxidative stress and antioxidant enzyme activities in cardiac interfibrillar mitochondria: implications for the mitochondrial theory of aging. FASEB J. 2005;19 (3):419–421. doi: 10.1096/fj.04-2622fje. [DOI] [PubMed] [Google Scholar]
- Juhaszova M, Rabuel C, Zorov DB, Lakatta EG, Sollott SJ. Protection in the aged heart: preventing the heart-break of old age? Cardiovasc Res. 2005;66 (2):233–244. doi: 10.1016/j.cardiores.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Kagan VE, Borisenko GG, Tyurina YY, Tyurin VA, Jiang J, Potapovich AI, Kini V, Amoscato AA, Fujii Y. Oxidative lipidomics of apoptosis: redox catalytic interactions of cytochrome c with cardiolipin and phosphatidylserine. Free Radic Biol Med. 2004;37 (12):1963–1985. doi: 10.1016/j.freeradbiomed.2004.08.016. [DOI] [PubMed] [Google Scholar]
- Kersten JR, Toller WG, Gross ER, Pagel PS, Warltier DC. Diabetes abolishes ischemic preconditioning: role of glucose, insulin, and osmolality. Am J Physiol Heart Circ Physiol. 2000;278 (4):H1218–H1224. doi: 10.1152/ajpheart.2000.278.4.H1218. [DOI] [PubMed] [Google Scholar]
- Khan S, Salloum F, Das A, Xi L, Vetrovec GW, Kukreja RC. Rapamycin confers preconditioning-like protection against ischemia-reperfusion injury in isolated mouse heart and cardiomyocytes. J Mol Cell Cardiol. 2006;41 (2):256–264. doi: 10.1016/j.yjmcc.2006.04.014. [DOI] [PubMed] [Google Scholar]
- Krylova IB, Kachaeva EV, Rodionova OM, Negoda AE, Evdokimova NR, Balina MI, Sapronov NS, Mironova GD. The cardioprotective effect of uridine and uridine-5′-monophosphate: the role of the mitochondrial ATP-dependent potassium channel. Exp Gerontol. 2006;41 (7):697–703. doi: 10.1016/j.exger.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Kwong LK, Sohal RS. Age-related changes in activities of mitochondrial electron transport complexes in various tissues of the mouse. Arch Biochem Biophys. 2000;373 (1):16–22. doi: 10.1006/abbi.1999.1495. [DOI] [PubMed] [Google Scholar]
- Lambert AJ, Boysen HM, Buckingham JA, Yang T, Podlutsky A, Austad SN, Kunz TH, Buffenstein R, Brand MD. Low rates of hydrogen peroxide production by isolated heart mitochondria associate with long maximum lifespan in vertebrate homeotherms. Aging Cell. 2007;6 (5):607–618. doi: 10.1111/j.1474-9726.2007.00312.x. [DOI] [PubMed] [Google Scholar]
- LaRue BL, Padilla PA. Environmental and genetic preconditioning for long-term anoxia responses requires AMPK in Caenorhabditis elegans. PLoS One. 2011;6 (2):e16790. doi: 10.1371/journal.pone.0016790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawler JM, Kwak HB, Kim JH, Suk MH. Exercise training inducibility of MnSOD protein expression and activity is retained while reducing prooxidant signaling in the heart of senescent rats. Am J Physiol Regul Integr Comp Physiol. 2009;296 (5):R1496–R1502. doi: 10.1152/ajpregu.90314.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TM, Su SF, Chou TF, Lee YT, Tsai CH. Loss of preconditioning by attenuated activation of myocardial ATP-sensitive potassium channels in elderly patients undergoing coronary angioplasty. Circulation. 2002;105 (3):334–340. doi: 10.1161/hc0302.102572. [DOI] [PubMed] [Google Scholar]
- Lemasters JJ, Theruvath TP, Zhong Z, Nieminen AL. Mitochondrial calcium and the permeability transition in cell death. Biochim Biophys Acta. 2009;1787 (11):1395–1401. doi: 10.1016/j.bbabio.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenaz G, Bovina C, Castelluccio C, Fato R, Formiggini G, Genova ML, Marchetti M, Pich MM, Pallotti F, Parenti CG, Biagini G. Mitochondrial complex I defects in aging. Mol Cell Biochem. 1997;174 (1–2):329–333. [PubMed] [Google Scholar]
- Lesnefsky EJ, Gudz TI, Migita CT, Ikeda-Saito M, Hassan MO, Turkaly PJ, Hoppel CL. Ischemic injury to mitochondrial electron transport in the aging heart: damage to the iron-sulfur protein subunit of electron transport complex III. Arch Biochem Biophys. 2001a;385 (1):117–128. doi: 10.1006/abbi.2000.2066. [DOI] [PubMed] [Google Scholar]
- Lesnefsky EJ, He D, Moghaddas S, Hoppel CL. Reversal of mitochondrial defects before ischemia protects the aged heart. FASEB J. 2006;20 (9):1543–1545. doi: 10.1096/fj.05-4535fje. [DOI] [PubMed] [Google Scholar]
- Lesnefsky EJ, Moghaddas S, Tandler B, Kerner J, Hoppel CL. Mitochondrial dysfunction in cardiac disease: ischemia--reperfusion, aging, and heart failure. J Mol Cell Cardiol. 2001b;33 (6):1065–1089. doi: 10.1006/jmcc.2001.1378. [DOI] [PubMed] [Google Scholar]
- Lesnefsky EJ, Slabe TJ, Stoll MS, Minkler PE, Hoppel CL. Myocardial ischemia selectively depletes cardiolipin in rabbit heart subsarcolemmal mitochondria. Am J Physiol Heart Circ Physiol. 2001c;280 (6):H2770–H2778. doi: 10.1152/ajpheart.2001.280.6.H2770. [DOI] [PubMed] [Google Scholar]
- Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De SG, Ferguson TB, Ford E, Furie K, Gillespie C, Go A, Greenlund K, Haase N, Hailpern S, Ho PM, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott MM, Meigs J, Mozaffarian D, Mussolino M, Nichol G, Roger VL, Rosamond W, Sacco R, Sorlie P, Roger VL, Thom T, Wasserthiel-Smoller S, Wong ND, Wylie-Rosett J. Heart disease and stroke statistics--2010 update: a report from the American Heart Association. Circulation. 2010;121 (7):e46–e215. doi: 10.1161/CIRCULATIONAHA.109.192667. [DOI] [PubMed] [Google Scholar]
- Mabon ME, Mao X, Jiao Y, Scott BA, Crowder CM. Systematic identification of gene activities promoting hypoxic death. Genetics. 2009;181 (2):483–496. doi: 10.1534/genetics.108.097188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manintveld OC, Te Lintel HM, van den Bos EJ, Suurenbroek GM, Dekkers DH, Verdouw PD, Lamers JM, Duncker DJ. Cardiac effects of postconditioning depend critically on the duration of index ischemia. Am J Physiol Heart Circ Physiol. 2007;292 (3):H1551–H1560. doi: 10.1152/ajpheart.00151.2006. [DOI] [PubMed] [Google Scholar]
- Mao XR, Crowder CM. Protein misfolding induces hypoxic preconditioning via a subset of the unfolded protein response machinery. Mol Cell Biol. 2010;30 (21):5033–5042. doi: 10.1128/MCB.00922-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta R, Steinkraus KA, Sutphin GL, Ramos FJ, Shamieh LS, Huh A, Davis C, Chandler-Brown D, Kaeberlein M. Proteasomal regulation of the hypoxic response modulates aging in C. elegans. Science. 2009;324 (5931):1196–1198. doi: 10.1126/science.1173507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendenhall AR, LaRue B, Padilla PA. Glyceraldehyde-3-phosphate dehydrogenase mediates anoxia response and survival in Caenorhabditis elegans. Genetics. 2006;174 (3):1173–1187. doi: 10.1534/genetics.106.061390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliaccio E, Giorgio M, Mele S, Pelicci G, Reboldi P, Pandolfi PP, Lanfrancone L, Pelicci PG. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature. 1999;402 (6759):309–313. doi: 10.1038/46311. [DOI] [PubMed] [Google Scholar]
- Moghaddas S, Hoppel CL, Lesnefsky EJ. Aging defect at the QO site of complex III augments oxyradical production in rat heart interfibrillar mitochondria. Arch Biochem Biophys. 2003;414 (1):59–66. doi: 10.1016/s0003-9861(03)00166-8. [DOI] [PubMed] [Google Scholar]
- Morris KC, Lin HW, Thompson JW, Perez-Pinzon MA. Pathways for ischemic cytoprotection: role of sirtuins in caloric restriction, resveratrol, and ischemic preconditioning. J Cereb Blood Flow Metab. 2011;31 (4):1003–1019. doi: 10.1038/jcbfm.2010.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008;88 (2):581–609. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy E, Steenbergen C. Gender-based differences in mechanisms of protection in myocardial ischemia-reperfusion injury. Cardiovasc Res. 2007a;75 (3):478–486. doi: 10.1016/j.cardiores.2007.03.025. [DOI] [PubMed] [Google Scholar]
- Murphy E, Steenbergen C. Preconditioning: the mitochondrial connection. Annu Rev Physiol. 2007b;69:51–67. doi: 10.1146/annurev.physiol.69.031905.163645. [DOI] [PubMed] [Google Scholar]
- Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74 (5):1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- Muscari C, Caldarera CM, Guarnieri C. Age-dependent production of mitochondrial hydrogen peroxide, lipid peroxides and fluorescent pigments in the rat heart. Basic Res Cardiol. 1990;85 (2):172–178. doi: 10.1007/BF01906970. [DOI] [PubMed] [Google Scholar]
- Nadtochiy SM, Redman E, Rahman I, Brookes PS. Lysine deacetylation in ischaemic preconditioning: the role of SIRT1. Cardiovasc Res. 2010 doi: 10.1093/cvr/cvq287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadtochiy S, Yao H, McBurney M, Gu W, Guarente L, Rahman I, Brookes P. SIRT1 mediated acute cardioprotection. AJP Heart. 2011 doi: 10.1152/ajpheart.00294.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro A, Boveris A. The mitochondrial energy transduction system and the aging process. Am J Physiol Cell Physiol. 2007;292 (2):C670–C686. doi: 10.1152/ajpcell.00213.2006. [DOI] [PubMed] [Google Scholar]
- Ottani F, Galvani M, Ferrini D, Sorbello F, Limonetti P, Pantoli D, Rusticali F. Prodromal angina limits infarct size. A role for ischemic preconditioning. Circulation. 1995;91 (2):291–297. doi: 10.1161/01.cir.91.2.291. [DOI] [PubMed] [Google Scholar]
- Palmer JW, Tandler B, Hoppel CL. Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J Biol Chem. 1977;252 (23):8731–8739. [PubMed] [Google Scholar]
- Pandolfi S, Bonafe M, Di TL, Tiberi L, Salvioli S, Monti D, Sorbi S, Franceschi C. p66(shc) is highly expressed in fibroblasts from centenarians. Mech Ageing Dev. 2005;126 (8):839–844. doi: 10.1016/j.mad.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Partridge L. Some highlights of research on aging with invertebrates, 2010. Aging Cell. 2011;10 (1):5–9. doi: 10.1111/j.1474-9726.2010.00649.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepe S, Tsuchiya N, Lakatta EG, Hansford RG. PUFA and aging modulate cardiac mitochondrial membrane lipid composition and Ca2+ activation of PDH. Am J Physiol. 1999;276 (1 Pt 2):H149–H158. doi: 10.1152/ajpheart.1999.276.1.H149. [DOI] [PubMed] [Google Scholar]
- Petrosillo G, Ruggiero FM, Pistolese M, Paradies G. Reactive oxygen species generated from the mitochondrial electron transport chain induce cytochrome c dissociation from beef-heart submitochondrial particles via cardiolipin peroxidation. Possible role in the apoptosis. FEBS Lett. 2001;509 (3):435–438. doi: 10.1016/s0014-5793(01)03206-9. [DOI] [PubMed] [Google Scholar]
- Qiu X, Brown K, Hirschey MD, Verdin E, Chen D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010;12 (6):662–667. doi: 10.1016/j.cmet.2010.11.015. [DOI] [PubMed] [Google Scholar]
- Queliconi BB, Wojtovich AP, Nadtochiy SM, Kowaltowski AJ, Brookes PS. Redox regulation of the mitochondrial K(ATP) channel in cardioprotection. Biochim Biophys Acta. 2011;1813 (7):1309–1315. doi: 10.1016/j.bbamcr.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea SL, Ventura N, Johnson TE. Relationship between mitochondrial electron transport chain dysfunction, development, and life extension in Caenorhabditis elegans. PLoS Biol. 2007;5 (10):e259. doi: 10.1371/journal.pbio.0050259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ristow M, Schmeisser S. Extending life span by increasing oxidative stress. Free Radic Biol Med. 2011;51 (2):327–336. doi: 10.1016/j.freeradbiomed.2011.05.010. [DOI] [PubMed] [Google Scholar]
- Ristow M, Zarse K. How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis) Exp Gerontol. 2010;45 (6):410–418. doi: 10.1016/j.exger.2010.03.014. [DOI] [PubMed] [Google Scholar]
- Rottlaender D, Boengler K, Wolny M, Michels G, Endres-Becker J, Motloch LJ, Schwaiger A, Buechert A, Schulz R, Heusch G, Hoppe UC. Connexin 43 acts as a cytoprotective mediator of signal transduction by stimulating mitochondrial KATP channels in mouse cardiomyocytes. J Clin Invest. 2010;120 (5):1441–1453. doi: 10.1172/JCI40927. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Saura J, Richards JG, Mahy N. Differential age-related changes of MAO-A and MAO-B in mouse brain and peripheral organs. Neurobiol Aging. 1994;15 (4):399–408. doi: 10.1016/0197-4580(94)90071-x. [DOI] [PubMed] [Google Scholar]
- Schlicker C, Gertz M, Papatheodorou P, Kachholz B, Becker CF, Steegborn C. Substrates and regulation mechanisms for the human mitochondrial sirtuins Sirt3 and Sirt5. J Mol Biol. 2008;382 (3):790–801. doi: 10.1016/j.jmb.2008.07.048. [DOI] [PubMed] [Google Scholar]
- Schulman D, Latchman DS, Yellon DM. Effect of aging on the ability of preconditioning to protect rat hearts from ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2001;281 (4):H1630–H1636. doi: 10.1152/ajpheart.2001.281.4.H1630. [DOI] [PubMed] [Google Scholar]
- Scott BA, Avidan MS, Crowder CM. Regulation of hypoxic death in C. elegans by the insulin/IGF receptor homolog DAF-2. Science. 2002;296 (5577):2388–2391. doi: 10.1126/science.1072302. [DOI] [PubMed] [Google Scholar]
- Sendoel A, Kohler I, Fellmann C, Lowe SW, Hengartner MO. HIF-1 antagonizes p53-mediated apoptosis through a secreted neuronal tyrosinase. Nature. 2010;465 (7298):577–583. doi: 10.1038/nature09141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen C, Nettleton D, Jiang M, Kim SK, Powell-Coffman JA. Roles of the HIF-1 hypoxia-inducible factor during hypoxia response in Caenorhabditis elegans. J Biol Chem. 2005;280 (21):20580–20588. doi: 10.1074/jbc.M501894200. [DOI] [PubMed] [Google Scholar]
- Shih PH, Yen GC. Differential expressions of antioxidant status in aging rats: the role of transcriptional factor Nrf2 and MAPK signaling pathway. Biogerontology. 2007;8 (2):71–80. doi: 10.1007/s10522-006-9033-y. [DOI] [PubMed] [Google Scholar]
- Shim YH, Kersten JR. Preconditioning, anesthetics, and perioperative medication. Best Pract Res Clin Anaesthesiol. 2008;22 (1):151–165. doi: 10.1016/j.bpa.2007.08.003. [DOI] [PubMed] [Google Scholar]
- Shinmura K, Tamaki K, Saito K, Nakano Y, Tobe T, Bolli R. Cardioprotective Effects of Short-Term Caloric Restriction Are Mediated by Adiponectin via Activation of AMP-Activated Protein Kinase. Circulation. 2007 doi: 10.1161/CIRCULATIONAHA.107.725697. [DOI] [PubMed] [Google Scholar]
- Shlafer M, Myers CL, Adkins S. Mitochondrial hydrogen peroxide generation and activities of glutathione peroxidase and superoxide dismutase following global ischemia. J Mol Cell Cardiol. 1987;19 (12):1195–1206. doi: 10.1016/s0022-2828(87)80530-8. [DOI] [PubMed] [Google Scholar]
- Sniecinski R, Liu H. Reduced efficacy of volatile anesthetic preconditioning with advanced age in isolated rat myocardium. Anesthesiology. 2004;100 (3):589–597. doi: 10.1097/00000542-200403000-00019. [DOI] [PubMed] [Google Scholar]
- Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005;85 (3):1093–1129. doi: 10.1152/physrev.00006.2004. [DOI] [PubMed] [Google Scholar]
- van der Loo B, Bachschmid M, Labugger R, Schildknecht S, Kilo J, Hahn R, Palacios-Callender M, Luscher TF. Expression and activity patterns of nitric oxide synthases and antioxidant enzymes reveal a substantial heterogeneity between cardiac and vascular aging in the rat. Biogerontology. 2005;6 (5):325–334. doi: 10.1007/s10522-005-4807-1. [DOI] [PubMed] [Google Scholar]
- Ventura N, Rea SL, Schiavi A, Torgovnick A, Testi R, Johnson TE. p53/CEP-1 increases or decreases lifespan, depending on level of mitochondrial bioenergetic stress. Aging Cell. 2009;8 (4):380–393. doi: 10.1111/j.1474-9726.2009.00482.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber TA, Reichert AS. Impaired quality control of mitochondria: aging from a new perspective. Exp Gerontol. 2010;45 (7–8):503–511. doi: 10.1016/j.exger.2010.03.018. [DOI] [PubMed] [Google Scholar]
- Wojtovich AP, Brookes PS. The complex II inhibitor atpenin A5 protects against cardiac ischemia-reperfusion injury via activation of mitochondrial KATP channels. Basic Res Cardiol. 2009;104 (2):121–129. doi: 10.1007/s00395-009-0001-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtovich AP, Burwell LS, Sherman TA, Nehrke KW, Brookes PS. The C. elegans mitochondrial K+(ATP) channel: a potential target for preconditioning. Biochem Biophys Res Commun. 2008;376 (3):625–628. doi: 10.1016/j.bbrc.2008.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtovich AP, Williams DM, Karcz MK, Lopes CM, Gray DA, Nehrke KW, Brookes PS. A Novel Mitochondrial KATP Channel Assay. Circ Res. 2010;106 (7):1190–1196. doi: 10.1161/CIRCRESAHA.109.215400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Hekimi S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 2010;8 (12):e1000556. doi: 10.1371/journal.pbio.1000556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Shao Z, Zhai Z, Shen C, Powell-Coffman JA. The HIF-1 hypoxia-inducible factor modulates lifespan in C. elegans. PLoS One. 2009;4 (7):e6348. doi: 10.1371/journal.pone.0006348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao ZQ, Corvera JS, Halkos ME, Kerendi F, Wang NP, Guyton RA, Vinten-Johansen J. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2003;285 (2):H579–H588. doi: 10.1152/ajpheart.01064.2002. [DOI] [PubMed] [Google Scholar]