

Abstract
Background
There is growing evidence that exposure to small size particulate matter increases the risk of developing cardiovascular disease.
Methods
We investigated plaque progression and vasodilatory function in apolipoprotein E knockout (ApoE-/-) mice exposed to TiO2. ApoE-/- mice were intratracheally instilled (0.5 mg/kg bodyweight) with rutile fine TiO2 (fTiO2, 288 nm), photocatalytic 92/8 anatase/rutile TiO2 (pTiO2, 12 nm), or rutile nano TiO2 (nTiO2, 21.6 nm) at 26 and 2 hours before measurement of vasodilatory function in aorta segments mounted in myographs. The progression of atherosclerotic plaques in aorta was assessed in mice exposed to nanosized TiO2 (0.5 mg/kg bodyweight) once a week for 4 weeks. We measured mRNA levels of Mcp-1, Mip-2, Vcam-1, Icam-1 and Vegf in lung tissue to assess pulmonary inflammation and vascular function. TiO2-induced alterations in nitric oxide (NO) production were assessed in human umbilical vein endothelial cells (HUVECs).
Results
The exposure to nTiO2 was associated with a modest increase in plaque progression in aorta, whereas there were unaltered vasodilatory function and expression levels of Mcp-1, Mip-2, Vcam-1, Icam-1 and Vegf in lung tissue. The ApoE-/- mice exposed to fine and photocatalytic TiO2 had unaltered vasodilatory function and lung tissue inflammatory gene expression. The unaltered NO-dependent vasodilatory function was supported by observations in HUVECs where the NO production was only increased by exposure to nTiO2.
Conclusion
Repeated exposure to nanosized TiO2 particles was associated with modest plaque progression in ApoE-/- mice. There were no associations between the pulmonary TiO2 exposure and inflammation or vasodilatory dysfunction.
Introduction
The investigation of toxicological effects of nanoparticles is increasingly important due to their growing occupational use and presence as compounds in consumer products. The use of nanometer-size particles in paints can reduce the production costs by the addition of lower (mass) concentrations or add novel properties to the final product. TiO2 pigments are widely used by paint and plastic industries for whiteness and opacity. One of the major advantages of TiO2 is its resistance to discoloration by UV light. TiO2 particles are considered to have low toxicity to humans and animals [1]. However, it has been shown that decreasing particle size is associated with increased pro-inflammatory properties [2]. An important possible health risk of particle exposure is cardiovascular effects. It has been shown that exposure to combustion-based particles in ambient air is associated with progression of atherosclerosis, myocardial infarction and cardiovascular mortality in humans and the nanosized fraction is considered an important culprit [3-5]. The mechanisms of these effects are considered to involve oxidative stress and inflammation, vasomotor dysfunction, neuronal signalling and possible translocation of particles from the airways to the circulation [3]. Pulmonary exposure to carbon-based particles can accelerate plaques progression in animal models [6,7]. However, associations between exposure to TiO2 particles and progression of atherosclerosis have not been assessed, whereas pulmonary exposure has been associated with vasomotor dysfunction [8].
Endothelial cells have an important barrier function and produce factors that regulate vascular tone, cellular adhesion, smooth muscle cell proliferation, vessel wall inflammation, and thromboresistance [9,10]. In the functional endothelium, acetylcholine binds to muscarinic receptors on the luminal surface leading to nitric oxide (NO) production by the calcium dependent, constitutive isoform of NO synthase (eNOS) [9]. Impaired endothelium-dependent vasodilation is a central feature of endothelial dysfunction and is associated with increased risk of developing cardiovascular diseases [11]. Endothelial dysfunction is associated with increased levels of proinflammatory factors, such as adhesion molecules and chemoattractants [12], which can lead to atherosclerosis [9,11,13]. This is attributed to increased intravascular generation of reactive oxygen species (ROS) such as superoxide anion radicals, leading to reduced bioavailability of NO [12]. Superoxide dismutase (SOD) converts superoxide anion radicals to hydrogen peroxide, which may ameliorate the production of peroxynitrite [12]. Ex vivo exposure of isolated vessels to the exogenous SOD mimic, tempol, has been associated with improved endothelium-dependent vasodilation in arteries from hypertensive rats, most likely by reduced intracellular ROS production by tempol that diffuses freely across cell membranes [12,14].
Inflammation plays a key role in atherogenesis as well as destabilization of plaques [15]. According to the multistep theory, monocytes adhere to the endothelial cells by binding to intracellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1), and then migrate into the intima [16,17]. Monocyte chemoattractant protein-1 (MCP-1/CCL2) is a chemotactic cytokine produced by endothelial cells after exposure to cytokines and oxidized lipoproteins. MCP-1 plays an important role in the migration and activation of monocytes and T cells and regulates the proliferation of vascular smooth muscle cells [18]. Activated macrophages express proinflammatory cytokines including macrophage inflammatory protein 2 (MIP-2/CXCL2) [19]. This cytokine up-regulates the expression of ICAM-1 and VCAM-1 on the endothelial cell surface. Vascular endothelial growth factor (VEGF) stimulates the proliferation and growth of endothelial cells and additionally increases vascular permeability [20].
We hypothesised that pulmonary exposure to nanosized particles, compared to fine particle would evoke larger cardiovascular effects, whereas altered vasodilatory function would be blunted by ex vivo treatment with the superoxide dismutase mimic tempol. To this end we investigate plaque progression and vasodilatory function in dyslipidemic and atherosclerosis prone apolipoprotein E knockout (ApoE-/-) mice exposed by intratracheal instillation (i.t.) to three physicochemically different TiO2 particles including nanometer-size coated rutile TiO2 (nTiO2; 20.6 nm), nanometer-size and highly photocatalytic anatase-rich TiO2 (pTiO2; 12 nm), and sub-micrometer-size coated rutile TiO2 (fTiO2; 288 nm). Inflammation was evaluated by measuring mRNA levels of Mip2, Icam1, Vcam1, Mcp1, and Vegf in lung tissue. We also studied the NO production in human umbilical vein endothelial cells (HUVECs) exposed to the three TiO2 particles.
Results
Particle and exposure characterization
The particles had different crystalline form, crystallite size, surface area, coating and chemistry (table 1). The fTiO2 and nTiO2 were mainly rutile crystal structure. The fTiO2 particles were coated with Al2O3 (3.22 wt%) and polyol (1.3 wt%). The nTiO2 particles were coated with SiO2 (12.01 wt%), Al2O3 SiO2 (4.58 wt%), ZrO2 (1.17 wt%) and polylol (5.2 wt%) [21,22]. The pTiO2 had anatase crystal structure; it was uncoated, highly pure and delivered as a suspension in water (30 wt%).
Table 1.
Primary physicochemical characteristics of the particulate TiO2 materials and hydrodynamic sizes in exposure dispersions.
| Electron Microscopy images | Sample material | Product name | Phase(s) | Crystallite size for primary particles (nm) | Surface area BET (m2/g) |
Minor elements/surface coatings on the primary particles (wt%) | Particle size in exposure dispersions (nm ± SD) | |
|---|---|---|---|---|---|---|---|---|
| unfiltered | 3.0 μm filter | |||||||
 |
Fine TiO2 (fTiO2) | RDI-S | 99.5% rutile 0.5% anatase |
288 | 21 | Al2O: 3.22 P2O5: 0.12 ZrO2: 0.07 §Polyol: 1.3 |
3415.8 ± 228.3 |
560.9 ± 162.0 |
 |
Photocatalytic TiO2 (pTiO2) | VP Disp. W 2730 X | 7.8% rutile 92.2% anatase |
19 12 |
N.A. | Al2O3: 0.10 | NA | 2320.5 ± 272.6 |
 |
NanoTiO2 (nTiO2) | UV-Titan L181 | 100% rutile | 20.6 | 107.7 | Na2O: 0.60 SiO2: 12.01 Al2O3: 4.58 ZrO2: 1.17 §Polyol: 5.2 |
5223.5 ± 831.6 |
518.2 ± 118.2 |
§ Compound according to the manufacturer
Assessment of particle distribution in the lung of mice
We investigated the distribution of particles following i.t. instillation of Evans Blue, quantum dots and radioactively labelled gold nanoparticles in wild-type mice (Figure 1). The distribution of i.t. instilled particles was determined by three different test systems; this combination of the tests provides a reliable assessment of the distribution in whole lungs. The Evans blue staining served as real visual inspection of the instilled fluid, whereas the distribution of particles were visualised by quantum dots and radioactively labelled gold nanoparticles. Collectively, these results show that the i.t. instillation procedure yielded an even distribution in the lungs.
Figure 1.
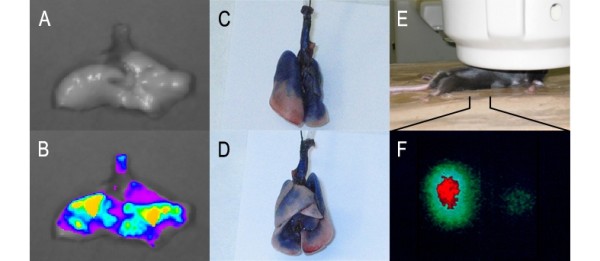
Distribution of particles after i.t. instillation in wild-type mice. Image A (front) and B (back) show the staining in the lung of mice after i.t. instillation of 1% Evans Blue solution (50 μl/mouse). Image C (no filter) and D (recorded as fluorescence passed through a 620 nm band pass filter) are images of the lung from a mouse after i.t. instillation of quantum dot QD621 solution (50 μl/mouse). Image E and F have been obtained in a single photon emission tomograph γ-camera in a mouse i.t. instilled with 18 nm nanogold particles.
Effect on vasodilatory response in aorta
Mice, aged 11-13 weeks, were exposed by i.t. instillation to either a control solution with 90% isotonic saline and 10% bronchoalveolar lavage (BAL) fluid, or particle (fTiO2, pTiO2, or nTiO2) suspended in 90% isotonic saline and 10% BAL fluid.
The endothelium-dependent vasodilation induced by acetylcholine showed an interaction between the treatment with particles and tempol (P < 0.05, ANOVA). The post-hoc analysis of the interaction showed that the tempol treatment was associated with a 45% (95% CI: 19-71%) reduction of the Emax value in animals i.t. instilled with control suspension. There were no differences in the Emax values between the particle-exposed mice and controls for the aorta segments that were not treated with tempol. However, the Emax value of tempol-treated vessels isolated from the animals exposed to pTiO2 particles was 71% (95% CI: 18 - 125%) higher than vessels isolated from mice exposed to the control suspension. There were no effects on EC50 values for the acetylcholine response (P = 0.83, single-factor effect of the particles) (table 2, Figure 2).
Table 2.
Endothelium dependent (acetylcholine: ACh, and calcitonin-gene related peptide: CGRP) and independent (nitroglycerin: NTG, and felodipine: FD) vasodilatory function in ApoE-/- mice after intratracheal instillation of TiO2 particles or control solution (90% saline and 10% BAL fluid).
| Exposure i.t. instillation | Drug | - tempol | + tempol | ||
|---|---|---|---|---|---|
| EC50 (nM) | Emax (%) | EC50 (nM) | Emax (%) | ||
| Control | ACh | 160.4 ± 65.0 | 56.7 ± 3.59 | 105.9 ± 35.6 | 31.2 ± 5.39* |
| fTiO2 | 207.7 ± 75.9 | 51.8 ± 4.98 | 107.5 ± 48.9 | 39.9 ± 6.23 | |
| pTiO2 | 222.3 ± 67.6 | 46.1 ± 3.92 | 49.3 ± 7.73 | 53.5 ± 7.31# | |
| nTiO2 | 101.7 ± 32.1 | 54.7 ± 4.99 | 64.2 ± 11.0 | 42.1 ± 7.43 | |
| Control | CGRP | 8.7 ± 2.1 | 87.9 ± 2.69 | 13.3 ± 2.73 | 72.4 ± 5.82* |
| fTiO2 | 11.6 ± 2.1 | 82.7 ± 3.23 | 15.2 ± 2.28 | 73.6 ± 3.23 | |
| pTiO2 | 9.4 ± 0.90 | 90.0 ± 1.57 | 11.1 ± 0.53 | 78.2 ± 3.34 | |
| nTiO2 | 14.8 ± 2.1 | 80.4 ± 6.20 | 14.5 ± 2.42 | 70.9 ± 6.96 | |
| Control | NTG | 23.8 ± 3.96 | 69.8 ± 3.41 | 68.7 ± 12.4* | 60.3 ± 2.36* |
| fTiO2 | 25.8 ± 4.63 | 66.5 ± 2.87 | 64.1 ± 16.9 | 60.3 ± 3.37 | |
| pTiO2 | 25.30 ± 8.26 | 69.8 ± 2.97 | 41.7 ± 5.3 | 65.6 ± 4.30 | |
| nTiO2 | 21.3 ± 4.43 | 67.3 ± 3.03 | 55.1 ± 20.9 | 64.4 ± 2.09 | |
| Control | FD | 10.4 ± 2.70 | 84.7 ± 3.74 | 7.57 ± 1.58 | 85.9 ± 2.69 |
| fTiO2 | 6.67 ± 1.05 | 84.6 ± 4.62 | 5.96 ± 0.94 | 89.7 ± 2.36 | |
| pTiO2 | 6.55 ± 0.90 | 84.8 ± 1.87 | 5.26 ± 1.04 | 86.1 ± 1.81 | |
| nTiO2 | 7.82 ± 1.79 | 90.6 ± 2.70 | 8.58 ± 2.95 | 83.5 ± 2.83 | |
All measurements were done in both absence and ex vivo presence of the SOD mimic, tempol. Data are expressed as means ± SEM (n = 10-11).
# Interaction between the treatment with particles and tempol (P < 0.05, ANOVA). The post-hoc analysis showed lower Emax values in animals intratracheally instilled with control solution, whereas there are higher Emax values in ex vivo tempol treated vessels of the animals exposed to pTiO2 particles. *P < 0.05 (ANOVA) compared to Emax values in segments that were not treated with tempol.
Figure 2.

Endothelium-dependent acetylcholine (ACh)-induced vasodilation of aorta segments from ApoE-/- mice exposed by intratracheal instillation to control solution containing 90% saline and 10% BAL fluid (black), nTiO2 (green), fTiO2 (blue), or pTiO2 (red). The measurements are performed in A) absence or B) ex vivo presence of the SOD mimic, tempol. The response is expressed as the percent vasodilation of the pre-contraction tension produced by PGF2α. Each point on the curves represents the cumulative response at each concentration of the vasodilator. Data are expressed as means ± SEM (n = 10-11). * P < 0.05 (ANOVA) compared with Emax in segments that were not treated with tempol. # P < 0.05 (ANOVA) compared with Emax values in tempol-treated aorta segments of control mice.
The vasodilation was also assessed by stimulation with calcitonin-gene related peptide (CGRP) that activates CGRP receptors on aortic smooth muscle cells and endothelial cells [23]. There was no difference in the CGRP-mediated vasodilation between the particle-exposed mice and controls. The effect of CGRP showed an 18% (95% CI: 3.0 - 33%) reduction of the maximal response (Emax) by ex vivo treatment with tempol, whereas there was no difference in terms of EC50 values (table 2, Figure 3).
Figure 3.

Calcitonic-gene related peptide (CGRP)-induced vasodilation of aorta segments from ApoE-/- mice exposed by intratracheal instillation to control solution consisting of 90% saline and 10% BAL fluid (black), nTiO2 (green), fTiO2 (blue), or pTiO2 (red). The measurements are performed in A) absence or B) ex vivo presence of the SOD mimic, tempol. The response is expressed as the percent vasodilation of the precontraction tension produced by PGF2α. Each point on the curves represents the cumulative response at each concentration of the vasodilator. Data are expressed as means ± SEM (n = 10-11). * P < 0.05 (ANOVA) compared with Emax values in tempol-treated aorta segments of control mice.
The endothelium-independent vasodilation was investigated as the vasodilatory response of aorta segments to the NO-donor nitroglycerin (NTG) or felodipine (FD; blocks the voltage-dependent calcium channels). The vasodilatory response to NTG indicated no difference between the groups of exposed animals, whereas the ex vivo tempol treatment was associated with 14% (95% CI: 1.1 - 26%) lower Emax value and 3.2 (95% CI: 1.8 - 5.6) fold higher EC50 value than the segments that were not treated with tempol (table 2, Figure 4).
Figure 4.

Endothelium-independent nitroglycerine (NTG)-induced vasodilation of aorta segments from ApoE-/- mice by intratracheal instillation to control solution consisting of 90% saline and 10% BAL fluid (black), nTiO2 (green), fTiO2 (blue), or pTiO2 (red). The measurements are performed in A) absence or B) ex vivo presence of the SOD mimic, tempol. The response is expressed as the percent vasodilation of the pre-contraction tension produced by PGF2α. Each point on the curves represents the cumulative response at each concentration of the vasodilator. Data are expressed as means ± SEM (n = 10-11).* P < 0.05 (ANOVA) compared with Emax or EC50 values in aorta segments of control mice.
The vasoactive response to the calcium channel antagonist FD was not affected by the in vivo exposure to any particle as compared to the control suspension and there was no difference in the vasodilation in the presence or absence of tempol (table 2, Figure 5).
Figure 5.

Felodipine (FD)-induced vasodilation of aorta segments from ApoE-/- mice exposed by intratracheal instillation to control solution consisting of 90% saline and 10% BAL fluid (black), nTiO2 (green), fTiO2 (blue), or pTiO2 (red). The measurements are performed in A) absence or B) ex vivo presence of the SOD mimic, tempol. The response is expressed as the percent vasodilation of the pre-contraction tension produced by PGF2α. Each point on the curves represents the cumulative response at each concentration of the vasodilator. Data are expressed as means ± SEM (n = 10-11).
The addition of the nitric oxide synthase inhibitor N'-monomethyl L-arginine (L-NMMA) increased the vasocontraction in the pre-constricted aorta segments, whereas there was no difference in regard to either exposure of animals to particles or ex vivo presence of tempol (table 3).
Table 3.
Excess vasocontraction (in %) induced by N'-monomethyl L-arginine (L-NMMA, 0.10 mM) after PGF2α precontraction (1 μM) in aorta segments from ApoE-/- mice after intratracheal instillation of TiO2 particles or control solution (90% saline and 10% BAL fluid).
| Exposure i.t. instillation | Drug | - tempol | + tempol |
|---|---|---|---|
| Control | L-NMMA | 17.0 ± 2.84 | 11.3 ± 1.08 |
| fTiO2 | 10.2 ± 1.49 | 14.3 ± 2.62 | |
| pTiO2 | 14.4 ± 2.89 | 13.2 ± 2.21 | |
| nTiO2 | 12.4 ± 1.65 | 15.9 ± 3.67 |
The measurements were done in both absence and ex vivo presence of the SOD mimic, tempol. Data are expressed as means ± SEM (n = 10-11).
Plaque progression
Figure 6 depicts the data from the assessment of plaque progression in whole aorta tissue of mice exposed to 0.5 mg/kg of nTiO2 by i.t. instillation once a week for four weeks giving a total dose of 2 mg/kg, followed by a five week particle-free period before sacrifice. We could not detect any plaques in two mice from each group. The plaque area was statistically significantly higher in the nTiO2 exposed mice (5.5 ± 1.2%) compared to the controls exposed to a solution with 90% saline and 10% BAL fluid (4.1 ± 0.8%; P = 0.018, Student's t-test). Inclusion of mice, in which we could not detect plaques in the aorta, in the statistical analysis showed no statistically significant effect (P = 0.096, Mann-Whitney U-test). We regard these results as showing a modestly increased plaque progression in the mice that were exposed to nTiO2.
Figure 6.

Plaque area in whole aorta from 20-21 weeks old ApoE-/- mice on a regular diet and exposed to control solution (90% saline and 10% BAL fluid) or nTiO2. The plaque areas were determined in the mice after an exposure period of nTiO2 (20.6 nm) or control solution once a week by intratracheal instillation for four weeks, followed by a period of five weeks without exposure to particles. The data are expressed as means ± SEM (n = 8). We could not detect plaques in two mice from each group and these are not shown in the figure. * denotes a significant effect on plaque progression in exposed mice vs. the control group.
Levels of mRNA of Mip-2, Mcp-1, Icam-1, Vcam-1 and Vegf in lung tissue
The mRNA levels of Mip-2, Mcp-1, Icam-1, Vcam-1, and Vegf in the lung tissue of mice exposed twice (26 and 2 hours before sacrifice) to nTiO2, pTiO2, or fTiO2 are reported in table 4. There was no difference in the mRNA levels between the control group and particle exposed mice. The results from the study on repeated exposure to nTiO2 (sacrificed five weeks after the last exposure) are shown in table 5. This exposure was also associated with unaltered mRNA levels of Mip-2, Mcp-1, Icam-1, Vcam-1, and Vegf in the lung tissue.
Table 4.
Inflammatory markers measured in mice having received two intratracheal instillations of either control bronchoalveolar lavage (BAL) suspension or 0.5 mg/(kg bodyweight) of one of the particle suspensions; fTiO2, pTiO2, or nTiO2, separated by 24 hours (n = 10-11).
| Exposure i.t. instillation | mRNA expression level (Median (quartile 25-75%)) | ||||
|---|---|---|---|---|---|
| Mcp-1 (*10-9) | Mip-2 (*10-9) | Icam-1 (*10-6) | Vcam-1 (*10-6) | Vegf (*10-6) | |
| Control | 54.5 (27.7-89.3) | 84.5 (41.5-150.6) | 40.8 (27.4-70.2) | 0.62 (0.32-0.88) | 96.9 (59.3-271.2) |
| fTiO2 | 24.4 (9.9-38.9) | 71.9 (35.2-111.6) | 32.8 (29.0-54.4) | 0.34 (0.19-0.60) | 99.7 (74.2-109.4) |
| pTiO2 | 37.1 (24.3-52.8) | 77.8 (36.2-140.0) | 30.3 (25.7-55.2) | 0.38 (0.19-1.04) | 105.1 (90.3-168.0) |
| nTiO2 | 27.3 (20.7-64.9) | 49.1 (39.0-237.9) | 38.9 (18.3-66.8) | 0.36 (0.27-0.63) | 80.1 (47.0-144.1) |
Expression of mRNA is normalised to 18S rRNA.
Table 5.
Inflammatory markers measured in mice exposed to nTiO2 (0.5 mg/kg) or control bronchoalveolar lavage (BAL) suspension by intratracheal instillation once a week for four weeks and sacrificed 5 weeks later (n = 10).
| Exposure i.t. instillation | mRNA expression level (Median (quartile 25-75%)) | ||||
|---|---|---|---|---|---|
| Mcp-1 (*10-9) | Mip-2 (*10-9) | Icam-1 (*10-6) | Vcam-1 (*10-6) | Vegf (*10-6) | |
| Control | 141.9 (98.3-166.5) | 43.2 (39.3-53.0) | 21.8 (17.4-39.0) | 0.30 (0.13-0.50) | 0.70 (0.45-1.18) |
| nTiO2 | 265.9 (80.6-359.5) | 53.3 (25.4-140.2) | 38.9 (18.6-56.9) | 0.40 (0.23-0.83) | 0.90 (0.78-1.38) |
Expression of mRNA is normalised to 18S rRNA.
Assessment of NO response in HUVECs
The exposure to nTiO2 particles was associated with a 32% (95% CI: 23% - 42%) increased level of NO in HUVEC cultures treated with 100 μg/ml (P < 0.05, ANOVA) (Figure 7). The NO levels were unaltered by co-incubation with diphenyleneiodonium chloride (DPI), which is a non-selective inhibitor of NO synthase activity. This abolished the particle-induced NO production in cells incubated with nTiO2 (P < 0.001, ANOVA). The incubation with fTiO2 or pTiO2 did not increase the level of NO (P < 0.01 and P < 0.05 for cultures treated with fTiO2 and pTiO2, respectively).
Figure 7.

Nitric oxide (NO) production in human umbilical vein endothelial cells (HUVECs) exposed to three physicochemically different TiO2 particles and compared to control cells. The NO production was measured in absence or presence of the NO synthase inhibitor diphenyleneiodonium chloride (DPI). Values are means ± SEM. * P < 0.05 control versus nTiO2. # P < 0.05 significant treatment effect within group.
We assessed a possible NO scavenging effect of tempol in HUVECs exposed to 3-morpholinosydnonimine (SIN-1) (Figure 8) or NTG. There was a strong single-factor effect of the SIN-1 exposure, showing increased NO production at all concentrations as compared to the unexposed cells (P < 0.001, single-factor effect of SIN-1). The concentrations of SIN-1 were relatively large and there were more than 60-fold larger NO levels compared to the unexposed cells. This probably explains the flat concentration-response relationship. A wider concentrations span of SIN-1 (e.g. ten-fold rather than two-fold dilutions) might have yielded a clearer concentration-response relationship. The SIN-1 treated cultures had higher levels of NO in the presence of tempol (P < 0.01, single-factor effect of tempol). These results indicate that tempol preferentially removes superoxide anion radicals and therefore increases the level of NO in the HUVECs. In a different experiment we treated HUVECs with 0.55 mM NTG in the absence or presence of 1 mM tempol. HUVECs that were treated with NTG, in the absence of tempol, had a 1.45 (95% CI: 1.38-1.51) fold larger NO level as compared to the unexposed control. In the presence of tempol, the NTG treatment increased the NO level by 1.47 (95% CI: 1.34-1.60) fold. NTG was a less potent NO donor than SIN-1, which is clearly observed by the lower production of NO in our experiment, although it was increased relative to the control (P < 0.001, single-factor effect of NTG). The presence of tempol did not affect the NTG-induced NO production in HUVECs (P = 0.16, single-factor effect of tempol). This is possibly because the spontaneously generated superoxide anion radicals in HUVECs are efficiently removed by endogenous SOD.
Figure 8.

Nitric oxide (NO) production in human umbilical vein endothelial cells (HUVECs) exposed to the NO donor SIN-1 for a total of three hours in the absence or presence of 1 mM tempol. Values are means ± SEM. * P < 0.05 SIN-1 versus unexposed cells. # P < 0.05 for difference between SIN-1 generated NO in the absence or presence of tempol.
Discussion
In this study we show that four repeated i.t. instillations of nTiO2 were associated with a modest increase in plaque progression in the aorta of ApoE-/- mice, whereas there was little pulmonary inflammation and only minor effects on vasodilatory function of endothelial and smooth muscle cells in segments after two instillations. Ex vivo addition of the SOD mimic agent, tempol, reduced the endothelium-dependent vasodilatory function in aorta segments from unexposed mice, whereas tempol increased this function in segments from mice exposed to pTiO2. The minimal effect of pulmonary TiO2 exposure on vasodilatory function was supported by minimal effects on NO production in HUVECs.
The endothelium and its product NO are key regulators of vascular function. Reduced bioavailability of NO is involved in the initiation and progression of atherosclerosis. The particle exposure of HUVECs showed an increase in the level of NO when the cells were exposed to nTiO2. This increase was abolished after addition of DPI, which is a non-selective inhibitor of NO synthase activity in the concentration that we have used. The addition of DPI also blocked the accumulation of NO in HUVECs that were not exposed to particles, indicating that the basal NO production originated from NO-generating enzymes. DPI also inhibits NAD(P)H oxidase, but this probably increases the cellular NO level because of lower possibility of reaction with superoxide anion radicals. iNOS enzymes are expressed during inflammatory conditions, but the one-hour incubation time in our study is probably too short to be associated with upregulation of iNOS. Collectively, the results suggest that the particle-derived NO production originated from eNOS activity.
We investigated the plaque progression in ApoE-/- mice exposed to nTiO2 once a week for four weeks, followed by a period of five weeks before sacrifice. This particular sample was chosen because previous in vitro studies had indicated inflammatory response. Furthermore, Rossi et al. showed that a silica-coated TiO2 material (similar to nTiO2) was the only among several TiO2 materials that gave pulmonary inflammation [24]. The protocol used in the present study was similar to the one used for assessment of plaque progression by i.t. instillation of single-walled carbon nanotubes (SWCNT), which had been associated with slightly larger plaque area (9%) in the aorta compared with controls (5.5%) in mice on a high-fat diet [25]. We observed only a modest increase in plaque progression in the aorta of nTiO2 exposed mice compared to the control group. This is to the best of our knowledge the first report on plaque progression enhanced by i.t. instillation of nanosized TiO2 particles. This observation could be further supported by histology of key sites in the vascular tree or staining of lipids to show fatty streaks/plaques. The exposure was not associated with increased expression of Mcp-1 or Mip-2 or altered vascular response in terms of Icam-1, Vcam-1 or Vegf mRNA expression levels in the lungs at the time of sacrifice. The mice in our study were fed a regular diet, which might have been associated with slow plaque progression. This is supported by observations that i.t. instillation of carbon black particles was associated with accelerated plaque progression (9.1%) compared to controls (5.5%) in LDLr-/- mice on a high-fat diet, whereas the same exposure was statistically non-significantly increased from 2.2% (controls) to 3.0% (exposed) in mice on a regular diet [6]. Similar findings have been observed in ApoE-/- mice exposed by inhalation to concentrated ambient air particles where the progression of atherosclerosis was largest in mice on a high-fat diet [26,27]. The plaque progression in mice on a low-fat diet was statistically non-significantly increased in the aorta from 13.2% (filtered air) to19.2% (concentrated ambient air particles) [26] and 10.4% (filtered air) to 14.2% (concentrated ambient air particles) after six months exposure [27]. It is possible that a high-fat diet and a longer exposure period would have rendered the mice more susceptible to particle-induced plaque progression. The mechanisms leading to accelerated plaque progression by inhalation of concentrated ambient air particles involves pro-oxidant and inflammatory milieu in the vasculature, which seems to occur in animals without pulmonary inflammation [28]. It has been shown that accelerated progression of atherosclerosis in ApoE-/- mice on a regular diet occurred concomitantly with loss of HDL anti-inflammatory property and systemic inflammation (assessed as elevated hepatic lipid peroxidation) after inhalation of fine or ultrafine concentrated ambient air particles at a concentration that did not generate pulmonary inflammation [29]. Other studies of exposure to concentrated ambient air particles in ApoE-/- mice have shown evidence of NAD(P)H oxidase dependent generation of reactive oxygen species, expression of iNOS and nitrotyrosine in aorta tissue [30].
We a priori hypothesized that excess generation of superoxide anion radicals would be implicated in dysfunction of vasomotor response. Thus, we measured the vasomotor function with and without the SOD mimic, tempol. The results from our investigation do not indicate any alterations of the vasomotor function related to particle exposure. We obtained full concentration-effect curves for all included vasodilators, indicating that we have reliable measurements of these responses covering a wide range of concentrations. In addition, aorta segments treated with L-NMMA had increased vasocontraction, whereas there was no difference between vessels from control and particle-exposed mice, indicating that spontaneous vasocontraction did not bias the vasodilatory response. The largest effect on vasomotor function was observed in the measurement of the endothelium-dependent vasodilation, where the exposure to TiO2 was associated with a statistically non-significant reduction of the maximal acetylcholine response. Mice exposed to the small anatase pTiO2 had lower Emax (46.1% ± 3.9) than the large rutile fTiO2 (Emax 51.8% ± 5.0) and small rutile nTiO2 (Emax 54.7% ± 5.0), whereas the mice exposed to the control solution had the largest Emax value (56.7% ± 3.6). This is in accordance with an earlier study showing no effect on acetylcholine-mediated vasodilation in pulmonary arteries of rats exposed to ultrafine and fine TiO2 particles (15 and 140 nm, respectively), whereas the same experiment showed a transient endothelial dysfunction in rats exposed to SRM1648 urban dust [31]. Results from another experiment showed unaltered acetylcholine-induced vasodilation in isolated aorta segments exposed to TiO2, further supporting the lack of effect in our study [32]. However, these results are in contrast to data obtained from a different experimental model where the vasomotor function was measured in spinotrapezius arteries following intraluminal infusion of the calcium ionophore A23187. It was demonstrated that spinotrapezius muscle arterioles had unaltered responsiveness to NO, whereas the endothelial dysfunction depended on the NO bioavailability [33]. Several studies by the same group have shown that both inhalation and i.t. instillation of fine and ultrafine TiO2 were associated with reduced or abolished endothelium-dependent vasodilation in spinotrapezius muscle arteries of normal rats [33-36]. These observations were later extended to coronary arterioles that displayed marked endothelial dysfunction in terms of altered vasodilatory response to shear stress, acetylcholine and the Ca2+ ionophore A23187, whereas the endothelium-independent vasodilatory response to sodium nitroprusside was unaffected [37]. Interestingly, these authors have also reported that the L-NMMA treatment of coronary arterioles led to lower additional increase in vasocontraction in nanosized TiO2-exposed rats [38]. The discrepancy between our results and those obtained by the intraluminal infusion of vasodilators might be explained by differences in experimental models, the mode of particle exposure, type of TiO2 particles, or species. It has been shown that inhalation of fine or nanosized TiO2 was associated with more severe pulmonary inflammation in rats compared to mice and hamsters under conditions where the lung TiO2 burdens were equivalent [39,40]. However, we have previously shown that ApoE-/- mice had more pulmonary inflammation than wild type mice after i.t. instillation of nanosized carbon black and there was vasomotor dysfunction in the exposed ApoE-/- mice as well [41,42].
Most investigations of the effect of tempol have been carried out in animal models of disease, such as hypertension or substantial burdens of atherosclerotic plaques, including studies in old ApoE-/- mice showing that treatments with tempol and other SOD-mimicking agents increases the vasodilatory response to acetylcholine [12,43,44]. Tempol has been found to be ineffective as an antihypertensive agent in animal models that are not associated with elevated oxidative stress [45]. We used 12-13 weeks old ApoE-/- mice for the vasomotor function experiments, which have the same vasodilatory function as wild type mice if they are not exposed to particles, whereas aged ApoE-/- mice are shown to have substantially decreased endothelium-dependent vasodilation in aorta vessels [46,47]. It has been shown that tempol decreased acetylcholine-induced vasodilation in normal rabbits and rats [48,49], whereas it restored acetylcholine-mediated vasodilation in aorta of rabbits pre-treated with an inhibitor of endogenous SOD (diethyldithiocarbamate) and the hypoxanthine/xanthine oxidase superoxide generating system [48]. Similarly, the SOD-mimicking compound (M40430) improved the acetylcholine-mediated vasodilation in ApoE-/- mice with compromised vascular function, whereas it was associated with a statistically non-significant 17% decrease in acetylcholine-mediated vasodilation in vessels from wild type mice [50]. The mechanism of endothelial dysfunction in ApoE-/- mice involves both decreased bioavailability of NO caused by superoxide anion radicals and degradation of the eNOS cofactor tetrahydrobiopterin [51]. The results from the present study indicate that tempol eliminates vasodilator compounds and/or augments the vascular tone, as have been shown in earlier studies [48,52]. Our results in SIN-1 exposed HUVECs indicate that tempol increased the NO levels by removing superoxide anion radicals, which is in accordance with previously reported observations [53]. This effect of tempol was not observed in NTG exposed HUVECs, which could be because the endogeneous level of SOD efficiently removes the cellular production of superoxide anion radicals. It is possible that exposure to a pure NO donor (e.g. NONOate) and/or inhibition of the endogenous SOD activity would show a clearer effect of tempol. In addition, tempol converts superoxide anion radicals to hydrogen peroxide that has been shown to mediate vasoconstriction in aorta tissue [12,54]. This effect of hydrogen peroxide would be associated with reduced vasodilatory response in our experiment. It seems that the tempol treatment has different effect in healthy and aged/diseased vessels. This possibly limits the extrapolation of our results to effects of tempol-generated hydrogen peroxide in aged/diseased vessels and firm conclusions about the possible role of hydrogen peroxide may require co-incubation with compounds that removes it (e.g. catalase).
Our investigation showed no effect of pulmonary exposure of TiO2 particle with different physicochemical characteristics on the vasodilatory function in aorta or mRNA expression of genes relevant in the inflammatory response in the lung, whereas the effect of one form on plaques progression was modest. Therefore, the outcome of the investigation is not optimal for assessment of the importance of particle size or composition or dose. We used mass doses and exposure protocols of TiO2 in ApoE-/- mice similar to what had shown significant effect for carbon black and SWCNT in terms of vasodilation function and plaque progression, respectively [25,42]. However, a direct dose comparison as mass burden or even surface area, between TiO2 particles and SWCNT does not take the overt differences in structure and reactivity into account. The latter exposure was associated with pulmonary inflammatory reaction, systemic oxidative stress and mitochondrial dysfunction, whereas minimal systemic inflammation was detected [25]. TiO2 and carbon black are regarded as "low-solubility low-toxicity" particles with similar inflammogenic potency per instilled surface area [55,56]. Indeed, we also found little pulmonary inflammation at doses that were four times larger than the individual doses of fTiO2 and nTiO2, whereas there was a linear relationship between the surface area and inflammation in relation to fine and nanosized carbon black after a single large-dose exposure [22]. The fTiO2 and nTiO2 samples in our study also had rutile crystal structure and surface coating, whereas pTiO2 was an uncoated mixture of anatase and rutile structure. It has been argued that the anatase crystal structure of TiO2 might be more reactive and cytotoxic as compared to rutile crystal structure [57]. In addition, the dispersion protocols might have an influence; some studies on TiO2 use dispersions in saline solution [36]. We used BAL fluid, which provides a well dispersed suspension of nanosized TiO2 and it has been shown to cause pulmonary inflammation after i.t. instillation in rats, whereas poorly dispersed nanosized TiO2 suspensions in saline had similar inflammogenic potency as fine TiO2 particles in rats after i.t. instillation [58,59]. However, suspensions in BAL fluid may also lead to formation of a protein corona on the particles, which may change the particle surface chemistry, although we cannot predict whether our TiO2 particles would be less potent by coating with proteins.
Conclusions
We show for the first time that pulmonary exposure to nanosized TiO2 is associated with a modest increase in plaque progression. On the other hand, three physicochemically different TiO2 nanomaterials (including nanosized TiO2) have virtually no effect on vasodilation induced from either endothelial or smooth muscle cells at exposure levels that were not associated with increased Mcp-1 and Mip-2 expression.
Materials and methods
Particles
The following materials were used in this study: fine TiO2 (fTiO2, RDI-S from Kemira Pigments, Finland) and photocatalytic TiO2 (pTiO2; W2730X from Kemira Pigments, Finland) both delivered by Beck and Joergensen A/S, Denmark, and nano TiO2 (nTiO2, UV-Titan L181 from Degussa, Germany) delivered by Boesens Fabrikker ApS, Denmark. A physicochemical characterization has been reported in detail elsewhere [21,22,60].
Material and Exposure Characterization
The phase composition and crystallite sizes were determined by Monochromated CuKα1 (1.540598 Å) X-ray diffraction using a Bruker D8 Advance X-ray diffractometer equipped with a Lynxeye CCD detector (Bruker AXS Inc., Madison, WI 53711-5373, USA). Electron microscopy imaging was completed on lacey carbon-coated Cu TEM-grids using a 200 kV Transmission Electron Microscope (TEM) (Tecnai G20, FEI Company, Hillsboro, Oregon, USA) and a Quanta 200 FEG MKII Scanning Electron Microscope [21]. Specific surface area was determined according to DIN ISO 9277 on a Quantachrome Autosorp-1 (Quantachrome GmbH & Co. KG, Odelzhausen, Germany) using multipoint Brunauer, Emmett, and Teller (BET) nitrogen adsorption method after 1 hour degassing at 300°C as a commercial service by Quantachrome GmbH & Co. KG. Elemental composition was analysed by X-ray Fluorescence analysis on a Philips PW-2400 spectrometer as a commercial service by the Department of Earth Sciences, University of Aarhus, Denmark. The organic content was determined indirectly from loss on ignition.
The size distribution of the particle suspensions used for the animal exposures was characterized by dynamic light scattering (DLS) analysis in a Nano Zetasizer (Malvern Instruments, UK). Particle dispersions (0.05 mg/kg bodyweight) were made as described above. Dispersion viscosities were determined using a SV-10 Vibro Viscometer (A&D Company Ltd., Japan). Particle size distributions were measured in disposable polystyrene cuvettes containing 150 μl sample. The optical data were recorded and calculated for both normal and high resolution size distribution using the Dispersion Technology Software v. 5.0 (Malvern Instruments). Particle suspensions were analysed unfiltered and following filtration (3.0 μm filter) and compared with unfiltered BAL solution. Six measurements, consisting of 12-16 scans, were conducted for unfiltered or filtered sample.
Nitric oxide production in human Umbilical Vein Endothelial Cells (HUVEC)
HUVECs were purchased from Cell Applications (San Diego, CA). The cells were cultured in T75 flasks in Endothelial Cell Growth Medium Kit (Cell Applications, San Diego, CA, USA). Cell cultures were incubated at 37°C in 5% CO2-95% air gas mixtures. Media were changed 24-36 hours after seeding and cells were grown to confluence. The cells were used between passages 2-6 because they have morphologic and phenotypic characteristics of endothelial cells.
We used the fluorimetric NOS Detection System DAF-2 DA (Sigma-Aldrich, Schnelldorf, Germany) for the measurement of NO production. The DAF-2 DA probe penetrates cells rapidly where it is hydrolyzed by intracellular esterase to DAF-2 that can react with NO to the fluorescent triazolofluorescein. HUVECs were trypsinated, transferred to a black clear bottom 96-well MT-plate (5*105 cells/well) and allowed to attach overnight. Particles were added in triplicate in four different concentrations (0.1, 10, 50, and 100 μg/ml) and the cells were incubated at 37°C for 1 hour. The cells were washed briefly and then treated with a reaction mixture, consisting of arginine, DAF-2 DA and Reaction Buffer. The NO production measurement was carried out in the absence or presence of the NOS inhibitor DPI (2 μM). The plates were incubated for 2 hours in the dark before the measurement of triazolofluorescein (excitation filter of 492 nm and an emission filter of 515 nm). All necessary controls, both negative and positive, were included (non-induced, cell-free, without dye, and TNF-α induced with and without DPI).
We investigated whether or not tempol was able to scavenge NO in HUVECs treated with SIN-1 (Sigma-Aldrich, Schnelldorf, Germany) or NTG. The HUVECs were exposed to SIN-1 in absence or presence of tempol (1 mM). We used the same concentration of tempol in this experiment as the concentration in the aorta rings. SIN-1 generates superoxide anion radicals and NO by spontaneous decomposition [53]. It is therefore possible to test the specificity of tempol on NO in our experimental setup. In a different experiment we treated HUVECs with 0.55 mM NTG. The level of NO in HUVECs was determined DAF-2 DA assay.
Animals
Eleven weeks old female ApoE-/- (C57BL/6-Apoe tm1) mice were obtained from Taconic MB (Ejby, Denmark) and acclimatized before entering the experiments. The mice were housed in a temperature- (22-24°C) and moisture- (40-70%) controlled room with a 12:12 h light-dark cycle. All mice were given free access to tap water and standard mouse chow (Standard Altromin no. 1314, Lage, Germany) during acclimatization and housing periods. The experiments were approved by the Danish "Animal Experimental Inspectorate" and carried out following their guidelines for ethical conduct and care when using animals in research.
Instillation exposure of mice
We assessed the distribution of particles by our i.t. instillation procedure in wild-type mice. This was assessed by i.t. instillation of 1% Evans Blue solution (50 μl/mouse). The lungs were subsequently dried and inflated. In another experiment we instilled 50 μl/mouse of quantum dots (ADSQD621, American Dye Source Inc., Quebec, Canada) and obtained images in a Unit-One dark box using a super sensitive camera (IXON EM+ DU-897 BI, Andor Technologies). The lungs were illuminated through a 600 nm short wave pass filter and the image was obtained through a 620 nm band pass filter. In a third experiment we instilled 40 μl/mouse of radioactive gold particles (18 nm) generated by neutron activation. The images were obtained for 40 min in a single photo emission computed tomography γ-camera (Prism 2000, Phillips) equipped with a pinhole collimator and adjusted for γ-energy of 4000 keV of 198Au.
The particles were suspended by sonication in a solution containing 90% sterile, isotonic saline and 10% BAL fluid. The latter was prepared by flushing the lungs of unexposed female ApoE-/- mice twice with 0.6 ml isotonic saline. The particle suspensions were sonicated on ice using a Branson Sonifier S-450D (Branson Ultrasonics Corp., Danbury, CT, USA) equipped with a disruptor horn (Model number: 101-147-037). Total sonication time was 16 min, with alternating 10 s pulses ON and 10 s pauses and with amplitude of 10%. Control solutions contained 90% sterile, isotonic saline and 10% BAL fluid from ApoE-/- mice. The solutions were divided in aliquots and immediately frozen at -80°C until use. Prior to use, the solutions were thawed at room temperature and vigorously vortexed. We have previously observed that i.t. instillation of saline with 10% BAL fluid was associated with a slightly increased influx of neutrophils, which we have attributed to the instillation procedure rather than the content of BAL fluid [41]. We have not assessed the effect of BAL fluid in regard to vascular endpoints because all the mice in our experiment received vehicle with BAL fluid and the results cannot be biased because of that.
For the assessment of vasodilatory function, four groups of 10-11 mice aged 11-12 weeks received two i.t. instillations of either control solution (90% saline and 10% BAL fluid) or 0.5 mg/(kg bodyweight) of one of the particle suspensions; fTiO2, pTiO2, or nTiO2, separated by 24 hours. We used this dose and exposure period because we have previously observed that exposure to nanosized carbon black was associated with endothelium-dependent vasorelaxation by an identical protocol in ApoE-/- mice [42]. The mice were anesthetized using Hypnorm® (fentanyl citrate 0.315 mg/ml and fluanisone 10 mg/ml from Janssen Pharma) and Dormicum® (Midazolam 5 mg/mL from Roche), both mixed with equal volume sterile water. A volume of 0.2 ml was injected subcutaneously in the neck of each mouse before each instillation. The sedated mice were kept on 37°C heating plates prior and subsequent to instillation until recovery from anaesthesia. Two hours after the second dose the mice were sacrificed. To minimize day to day variation, 2-3 particle suspensions were instilled on each exposure day and a control mice was furthermore included each day.
For assessment of effects on plaque progression two groups of 10 mice received 0.5 mg/kg nTiO2 or control solution (90% saline and 10% BAL fluid) by i.t. instillation once a week for four weeks and sacrificed 5 weeks later. We used this dose and exposure period because it was similar to a study on SWCNT [25], which we used as a reference condition although we kept the mice on regular chow. We did not use a high-fat diet because we wanted to compare the results from plaque progression experiment with the assessment of vasodilatory function that was carried out on mice fed a regular chow. The plaque progression was not assessed in mice exposed to fTiO2 and pTiO2 because there was little effect on atherosclerosis in our preliminary experiment with nTiO2.
During instillation the mouse was placed supine on an approximately 40 degree slope, where it was held in a favourable position by its front teeth. A light was gently touching the larynx to allow intubation of the trachea with a 24 gauge BD Insyte catheter (Ref: 381212, Becton Dickinson, Denmark) with a shortened needle. The correct placement was verified by a highly sensitive pressure transducer confirming breath and thereby the right placement in the trachea. A volume of 50 μl particle suspension followed by 150 μl air was instilled and the catheter was quickly removed. To ensure that the particle suspension remained in the lungs, the mouse was placed vertically with its head up for 30-60 s.
Isolation of organs
After the mice were sacrificed the tissues, except the heart and aorta, were immediately removed, quickly frozen in liquid nitrogen, and stored at -80°C until further analysis. The heart and aorta were carefully removed from the animal, stored at ice cold physiological saline solution (PSS: 119 mM NaCl, 25 mM NaHCO3, 4.7 mM KCl, 1.18 mM KH2PO4, 1.17 mM MgSO4 7H2O, 1.5 mM CaCl2 2H2O, 0.027 mM ethylene diamine tetraacetic acid and 5.5 mM glucose, pH = 7.4) and dissected free of connective tissue under a light microscope to further use.
Vasodilatory function
Four aorta segments from each mouse were mounted on steel pins with a diameter 150 μm in the organ chambers of the myograph (multi Myograph 610 M from Danish Myo Technology, Aarhus, Denmark) containing 5 ml cold oxygenated PSS continually perfused with a 95% O2 and 5% CO2 gas mixture. The distal region of the thoracic aorta was purposely selected to avoid lesions in the segments used for the contractility studies. Each myograph was connected to a computer and the data were collected by the software Myodaq (Danish Myo Technology, Aarhus, Denmark).
The experimental procedure has previously been described in detail [47]. In brief, the temperature in the organ baths were slowly raised to 37°C and the segments were allowed to equilibrate for 30 min. A standard normalization procedure was performed by which the internal lumen diameter of each aortic segment was determined. This procedure enables us to obtain an optimal active tension development in the aortic segments. Preliminary to the specific experiments, it was verified that the aorta segments were variable and that contraction was reproducible. This was done by substituting the PSS in each organ bath with 5 ml warm, oxygenated 125 mM K+-PSS (119 mM KCL, 25 mM NaHCO3, 4.7 mM KCL, 1.18 mM KH2PO4, 1.17 mM MgSO4 7H2O, 1.5 mM CaCl2 2H2O, 0.027 mM ethylene diamine tetraacetic acid and 5.5 mM glucose, pH = 7.4) followed by 4 times of wash with PSS. This procedure was repeated for a total of three to four times. The K+-PSS treatment furthermore ensured that the sympathetic nerve endings were depleted for neurotransmitters. The segments were pre-contracted PGF2α (1 μM) before the response to the vasodilators was assessed.
The endothelial function was analyzed by the following vasodilators; acetylcholine (ACh; 10-10-10-5 M) and calcitonin-gene related peptide (CGRP; 10-11-3*10-7 M). Acetylcholine-induced vasodilation is endothelium dependent as the dilatory effect is exerted via binding to muscarinic receptors (M3) on the endothelial cells, whereas CGRP-induced vasodilation is only partly mediated through the endothelium [23]. Furthermore, the endothelium-independent vasomotor response was investigated by using the NO-donor NTG (0-10-3*10-5 M), and by using FD (10-13-10-6 M), which blocks the voltage-dependent calcium channels on smooth muscle cells.
We tested the effect of particle exposure on vasocontraction in vessels that were treated with L-NMMA (10-4 M), which is a non-selective inhibitor of NOS. In addition, all measurements were done in the absence and presence of tempol (1 mM).
The values of the maximal steady state contraction or dilation (Emax) and the concentration at which half the Emax was obtained (EC50) were calculated using the GraphPad Prism version 4 (San Diego, CA, USA). Nonlinear regression analysis with equation for sigmoid concentration-response (with variable slope) was used. The results are expressed as the means ± standard error of the mean (S.E.M.).
Plaque area assessment
The area of atherosclerotic plaques was determined in the aorta of mice five weeks after four weekly exposures. The entire aorta from the junction with the heart to the iliac bifurcation was placed in a flat bed scanner. The plaques were observed as whitish spots against a black background (Figure 9). The fraction of area of the total aortic surface that contained plaques was determined on a digital microscope image (Digital Imaging Solutions; analySIS® getIT!) by means of the computer software Image J and calculated as % plaques proportional to total aorta area.
Figure 9.

Representative images of whole aorta tissue from the junction with the heart to the iliac bifurcation from a mouse with undetectable plaque (A) or detectable (B) plaque areas seen as white spots.
Analysis of mRNA expression levels of Mcp-1, Mip-2, Icam-1, Vcam-1, and Vegf in lungs from ApoE-/- mice
We have previously found close correspondence between Mcp-1 and Mip-2 mRNA levels in the lung and neutrophil infiltration in BAL fluid in mice exposed to different nanosized particles [41,61,62]. We did not analyse the cell content of cells in the BAL fluid because we did not want to influence the endothelium by the lung infusion and anaesthetic administration that are parts of the BAL fluid collection procedure. We have measured mRNA levels in the lung tissue for Mcp-1 and Mip-2 as markers of pulmonary inflammation.
Mcp-1, Mip-2, Vcam-1, Icam-1, and Vegf gene expression was determined using Real-Time Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR). Quantification of the target mRNA was done relative to reference 18S RNA using the relative 2ΔCt method [63]. Total RNA was purified from lung tissue using the TRIzol® Reagent method (Invitrogen A/S, Taastrup, Denmark) and DNase treated as described by the manufacturer (Promega Corporation, Madison, WI) prior to RT-PCR. For cDNA synthesis, less than 1 μg of RNA was used as recommended by Applied Biosystems.
For quantification of the mRNA levels, we used probe and primer solutions (Applied Biosystems,, Foster City, CA, USA) as follows: Mcp-1 (AssayID. Mm99999056_m1), Mip-2 (AssayID. Mm00436450_m1), Vcam-1 (AssayID. Mm01320973_m1), Icam-1 (AssayID. Mm00516024_g1), Vegf (AssayID. Mm01281449_m1), and 18S rRNA (TaqMan® Ribosomal RNA control reagents: VIC™ Probe). The mRNA levels were quantified in separate wells.
The PCR reactions were performed in triplicate on the Applied Biosystems 7900HT Fast Real-Time PCR System in 10 μl reactions. Four μl of the cDNA preparation was mixed with 98 μl Mastermix (Applied Biosystems, Foster City, CA, USA) and sterile water was added to a final volume of 185 μl. Aliquots of 32 μl were mixed with the respective probe and primer mix solutions. The final concentration of primers and probes were 900 nM and 200 nM, respectively.
Data analysis
The results are expressed as the means ± standard error of the mean (S.E.M.). Graphs are composed using Graph-Pad Prism by Intuitive Software for Science (San Diego, CA, USA, http://www.graphpad.com). Nonlinear regression analysis with equation for sigmoid concentration-response (with variable slope) was used.
Statistics
The data on the production of NO by HUVEC were analyzed by three-factor ANOVA test with the concentration, type of particle and ex vivo treatment with DPI as categorical variables. The data on vasodilatory function endpoints were analysed by two-factor ANOVA test with the particle and ex vivo treatment with tempol as categorical variables. The statistical significance of the tempol treatment is reported as single-factor effects. The mRNA expressions in lung tissue were analyzed by Kruskal-Wallis or Mann-Whitney U-test because of unequal variance between the groups. All tests were accepted as statistically significant at 5% level. The statistical analysis was performed in Statistica version 5.5 (StatSoft Inc., Tulsa, OK, USA).
Abbreviations
ACh: acetylcholine; apoE: apolipoprotein E; CGRP: calcitonin-gene related peptide; eNOS: endothelial nitric oxide synthase; FD: felodipine; fTiO2: fine titanium dioxide; ICAM-1: intracellular adhesion molecule 1; L-NMMA: N'-monomethyl L-arginine; MCP-1: monocyte chemoattractant protein-1; MIP2: macrophage inflammatory protein 2; NO: nitric oxide; nTiO2: nano titanium dioxide; NTG: nitroglycerin; pTiO2: photocatalytic titanium dioxide; PSS: physiological saline solution; SIN-1: 3-morpholinosydnonimine; SOD: superoxide dismutase; SWCNT: single-walled carbon nanotubes; VCAM-1: vascular cellular adhesion molecule 1; VEGF: vascular endothelial growth factor
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
PM, SL, ATS, HW, and UV conceived the study. LM, PM, SL, HW, and UV designed the experiments. LM made the experiments on vasomotor function, assisted and supervised by MS. KAJ made the characterization experiments. NRJ made the experiments on the assessment of particle distribution in mice lungs. LM made the draft of the manuscript, which was revised critically by all authors, who have read and approved the manuscript.
Contributor Information
Lone Mikkelsen, Email: lomi@sund.ku.dk.
Majid Sheykhzade, Email: mash@farma.ku.dk.
Keld A Jensen, Email: kaj@nrcwe.dk.
Anne T Saber, Email: ats@nrcwe.dk.
Nicklas R Jacobsen, Email: nrj@nrcwe.dk.
Ulla Vogel, Email: ubv@nrcwe.dk.
Håkan Wallin, Email: hwa@nrcwe.dk.
Steffen Loft, Email: stl@sund.ku.dk.
Peter Møller, Email: pemo@sund.ku.dk.
Acknowledgements
The technical assistance from Julie Hansen and Michael Guldbrandsen as well as XRD-analysis from Renie Birkedal at NRCWE is gratefully acknowledged. The experiments were approved by the Danish "Animal Experimental Inspectorate" and carried out following their guidelines for ethical conduct and care when using animals in research. Thanks to Manuela Semmler-Behnke and Wolfgang Kreyling (GSF, Münich, Germany) for help with the nanogold instillation experiment. The work was part of the Nanokem project and supported by The Danish Working Environment Research Fund (Nanokem, Grant No. 20060068816). The Danish Association for the Paint and Lacquer Industry provided the tested particle samples. Electron Microscopy was conducted at the Center for Electron Nanoscopy (DTU CEN) funded by the A.P. Møller and Chastine Mc-Kinney Møller Foundation, the Center for Electron Nanoscopy (DTU CEN).
References
- Zhang DD, Hartsky MA, Warheit DB. Time course of quartz and TiO(2) particle-induced pulmonary inflammation and neutrophil apoptotic responses in rats. Exp Lung Res. 2002;28:641–670. doi: 10.1080/01902140260426742. [DOI] [PubMed] [Google Scholar]
- Renwick LC, Brown D, Clouter A, Donaldson K. Increased inflammation and altered macrophage chemotactic responses caused by two ultrafine particle types. Occup Environ Med. 2004;61:442–447. doi: 10.1136/oem.2003.008227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brook RD, Rajagopalan S, Pope CA III, Brook JR, Bhatnagar A, Diez-Roux AV, Holguin F, Hong Y, Luepker RV, Mittleman MA, Peters A, Siscovick D, Smith SC Jr, Whitsel L, Kaufman JD. Particulate matter air pollution and cardiovascular disease: An update to the scientific statement from the American Heart Association. Circulation. 2010;121:2331–2378. doi: 10.1161/CIR.0b013e3181dbece1. [DOI] [PubMed] [Google Scholar]
- Mills NL, Tornqvist H, Robinson SD, Gonzalez M, Darnley K, MacNee W, Boon NA, Donaldson K, Blomberg A, Sandstrom T, Newby DE. Diesel exhaust inhalation causes vascular dysfunction and impaired endogenous fibrinolysis. Circulation. 2005;112:3930–3936. doi: 10.1161/CIRCULATIONAHA.105.588962. [DOI] [PubMed] [Google Scholar]
- Seaton A, Tran L, Aitken R, Donaldson K. Nanoparticles, human health hazard and regulation. J R Soc Interface. 2010;7(Suppl 1):S119–S129. doi: 10.1098/rsif.2009.0252.focus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa Y, Hiura Y, Murayama T, Yokode M, Iwai N. Nano-sized carbon black exposure exacerbates atherosclerosis in LDL-receptor knockout mice. Circ J. 2007;71:1157–1161. doi: 10.1253/circj.71.1157. [DOI] [PubMed] [Google Scholar]
- Suwa T, Hogg JC, Quinlan KB, Ohgami A, Vincent R, van Eeden SF. Particulate air pollution induces progression of atherosclerosis. J Am Coll Cardiol. 2002;39:935–942. doi: 10.1016/S0735-1097(02)01715-1. [DOI] [PubMed] [Google Scholar]
- Møller P, Mikkelsen L, Vesterdal LK, Folkmann JK, Forchhammer L, Roursgaard M, Danielsen PH, Loft S. Hazard identification of particulate matter on vasomotor dysfunction and progression of atherosclerosis. Crit Rev Toxicol. 2011;41:339–368. doi: 10.3109/10408444.2010.533152. [DOI] [PubMed] [Google Scholar]
- Vanhoutte PM. Endothelial dysfunction: the first step toward coronary arteriosclerosis. Circ J. 2009;73:595–601. doi: 10.1253/circj.CJ-08-1169. [DOI] [PubMed] [Google Scholar]
- Deanfield JE, Halcox JP, Rabelink TJ. Endothelial function and dysfunction: testing and clinical relevance. Circulation. 2007;115:1285–1295. doi: 10.1161/CIRCULATIONAHA.106.652859. [DOI] [PubMed] [Google Scholar]
- Bonetti PO, Lerman LO, Lerman A. Endothelial dysfunction: a marker of atherosclerotic risk. Arterioscler Thromb Vasc Biol. 2003;23:168–175. doi: 10.1161/01.ATV.0000051384.43104.FC. [DOI] [PubMed] [Google Scholar]
- Simonsen U, Rodriguez-Rodriguez R, Dalsgaard T, Buus NH, Stankevicius E. Novel approaches to improving endothelium-dependent nitric oxide-mediated vasodilatation. Pharmacol Rep. 2009;61:105–115. doi: 10.1016/s1734-1140(09)70012-x. [DOI] [PubMed] [Google Scholar]
- Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- Chen Y, Pearlman A, Luo Z, Wilcox CS. Hydrogen peroxide mediates a transient vasorelaxation with tempol during oxidative stress. Am J Physiol Heart Circ Physiol. 2007;293:H2085–H2092. doi: 10.1152/ajpheart.00968.2006. [DOI] [PubMed] [Google Scholar]
- Stoll G, Bendszus M. Inflammation and atherosclerosis: novel insights into plaque formation and destabilization. Stroke. 2006;37:1923–1932. doi: 10.1161/01.STR.0000226901.34927.10. [DOI] [PubMed] [Google Scholar]
- Butcher EC. Leukocyte-endothelial cell recognition: three (or more) steps to specificity and diversity. Cell. 1991;67:1033–1036. doi: 10.1016/0092-8674(91)90279-8. [DOI] [PubMed] [Google Scholar]
- Ashida N, Arai H, Yamasaki M, Kita T. Distinct signaling pathways for MCP-1-dependent integrin activation and chemotaxis. J Biol Chem. 2001;276:16555–16560. doi: 10.1074/jbc.M009068200. [DOI] [PubMed] [Google Scholar]
- Papayianni A, Alexopoulos E, Giamalis P, Gionanlis L, Belechri AM, Koukoudis P, Memmos D. Circulating levels of ICAM-1, VCAM-1, and MCP-1 are increased in haemodialysis patients: association with inflammation, dyslipidaemia, and vascular events. Nephrol Dial Transplant. 2002;17:435–441. doi: 10.1093/ndt/17.3.435. [DOI] [PubMed] [Google Scholar]
- Wiesner P, Choi SH, Almazan F, Benner C, Huang W, Diehl CJ, Gonen A, Butler S, Witztum JL, Glass CK, Miller YI. Low doses of lipopolysaccharide and minimally oxidized low-density lipoprotein cooperatively activate macrophages via nuclear factor kappa B and activator protein-1: possible mechanism for acceleration of atherosclerosis by subclinical endotoxemia. Circ Res. 2001;107:56–65. doi: 10.1161/CIRCRESAHA.110.218420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm PW, Slart RH, Zeebregts CJ, Hillebrands JL, Tio RA. Atherosclerotic plaque development and instability: a dual role for VEGF. Ann Med. 2011;41:257–264. doi: 10.3109/17435390.2011.587900. [DOI] [PubMed] [Google Scholar]
- Hougaard KS, Jackson P, Jensen KA, Sloth JJ, Loschner K, Larsen EH, Birkedal RK, Vibenholt A, Boisen AM, Wallin H, Vogel U. Effects of prenatal exposure to surface-coated nanosized titanium dioxide (UV-Titan). A study in mice. Part Fibre Toxicol. 2010;7:16. doi: 10.1186/1743-8977-7-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saber AT, Jensen KA, Jacobsen NR, Birkedal RK, Mikkelsen L, Møller P, Loft S, Wallin H, Vogel U. Inflammatory and genotoxic effects of nanoparticles designed for inclusion in paints and lacquers. Nanotoxicology. 2010. in press . [DOI] [PubMed]
- Chan SL, Fiscus RR. Vasorelaxant response to isoprenaline, nitric oxide donor, calcitonin gene-related peptide and vasoactive intestinal peptide in aortic rings of adult C57BL/6J mice. Eur J Pharmacol. 2001;431:229–236. doi: 10.1016/S0014-2999(01)01418-2. [DOI] [PubMed] [Google Scholar]
- Rossi EM, Pylkkanen L, Koivisto AJ, Vippola M, Jensen KA, Miettinen M, Sirola K, Nykasenoja H, Karisola P, Stjernvall T, Vanhala E, Kiilunen M, Pasanen P, Makinen M, Hameri K, Joutsensaari J, Tuomi T, Jokiniemi J, Wolff H, Savolainen K, Matikainen S, Alenius H. Airway exposure to silica-coated TiO2 nanoparticles induces pulmonary neutrophilia in mice. Toxicol Sci. 2010;113:422–433. doi: 10.1093/toxsci/kfp254. [DOI] [PubMed] [Google Scholar]
- Li Z, Hulderman T, Salmen R, Chapman R, Leonard SS, Young SH, Shvedova A, Simeonova PP. Cardiovascular effects of pulmonary exposure to single-wall carbon nanotubes. Environ Health Perspect. 2007;115:377–382. doi: 10.1289/ehp.9688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Wang A, Jin X, Natanzon A, Duquaine D, Brook RD, Aguinaldo JG, Fayad ZA, Fuster V, Lippmann M, Chen LC, Rajagopalan S. Long-term air pollution exposure and acceleration of atherosclerosis and vascular inflammation in an animal model. JAMA. 2005;294:3003–3010. doi: 10.1001/jama.294.23.3003. [DOI] [PubMed] [Google Scholar]
- Sun Q, Yue P, Kirk RI, Wang A, Moatti D, Jin X, Lu B, Schecter AD, Lippmann M, Gordon T, Chen LC, Rajagopalan S. Ambient air particulate matter exposure and tissue factor expression in atherosclerosis. Inhal Toxicol. 2008;20:127–137. doi: 10.1080/08958370701821482. [DOI] [PubMed] [Google Scholar]
- Araujo JA, Nel AE. Particulate matter and atherosclerosis: role of particle size, composition and oxidative stress. Part Fibre Toxicol. 2009;6:24. doi: 10.1186/1743-8977-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araujo JA, Barajas B, Kleinman M, Wang X, Bennett BJ, Gong KW, Navab M, Harkema J, Sioutas C, Lusis AJ, Nel AE. Ambient particulate pollutants in the ultrafine range promote early atherosclerosis and systemic oxidative stress. Circ Res. 2008;102:589–596. doi: 10.1161/CIRCRESAHA.107.164970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying Z, Kampfrath T, Thurston G, Farrar B, Lippmann M, Wang A, Sun Q, Chen LC, Rajagopalan S. Ambient particulates alter vascular function through induction of reactive oxygen and nitrogen species. Toxicol Sci. 2009;111:80–88. doi: 10.1093/toxsci/kfp004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtois A, Andujar P, Ladeiro Y, Baudrimont I, Delannoy E, Leblais V, Begueret H, Galland MA, Brochard P, Marano F, Marthan R, Muller B. Impairment of NO-dependent relaxation in intralobar pulmonary arteries: comparison of urban particulate matter and manufactured nanoparticles. Environ Health Perspect. 2008;116:1294–1299. doi: 10.1289/ehp.11021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knaapen AM, den Hartog GJ, Bast A, Borm PJ. Ambient particulate matter induces relaxation of rat aortic rings in vitro. Hum Exp Toxicol. 2001;20:259–265. doi: 10.1191/096032701678227677. [DOI] [PubMed] [Google Scholar]
- Nurkiewicz TR, Porter DW, Hubbs AF, Stone S, Chen BT, Frazer DG, Boegehold MA, Castranova V. Pulmonary nanoparticle exposure disrupts systemic microvascular nitric oxide signaling. Toxicol Sci. 2009;110:191–203. doi: 10.1093/toxsci/kfp051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurkiewicz TR, Porter DW, Hubbs AF, Cumpston JL, Chen BT, Frazer DG, Castranova V. Nanoparticle inhalation augments particle-dependent systemic microvascular dysfunction. Part Fibre Toxicol. 2008;5:1. doi: 10.1186/1743-8977-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurkiewicz TR, Porter DW, Barger M, Castranova V, Boegehold MA. Particulate matter exposure impairs systemic microvascular endothelium-dependent dilation. Environ Health Perspect. 2004;112:1299–1306. doi: 10.1289/ehp.7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurkiewicz TR, Porter DW, Barger M, Millecchia L, Rao KM, Marvar PJ, Hubbs AF, Castranova V, Boegehold MA. Systemic microvascular dysfunction and inflammation after pulmonary particulate matter exposure. Environ Health Perspect. 2006;114:412–419. doi: 10.1289/ehp.114-a412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBlanc AJ, Cumpston JL, Chen BT, Frazer D, Castranova V, Nurkiewicz TR. Nanoparticle inhalation impairs endothelium-dependent vasodilation in subepicardial arterioles. J Toxicol Environ Health A. 2009;72:1576–1584. doi: 10.1080/15287390903232467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBlanc AJ, Moseley AM, Chen BT, Frazer D, Castranova V, Nurkiewicz TR. Nanoparticle inhalation impairs coronary microvascular reactivity via a local reactive oxygen species-dependent mechanism. Cardiovasc Toxicol. 2010;10:27–36. doi: 10.1007/s12012-009-9060-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermudez E, Mangum JB, Wong BA, Asgharian B, Hext PM, Warheit DB, Everitt JI. Pulmonary responses of mice, rats, and hamsters to subchronic inhalation of ultrafine titanium dioxide particles. Toxicol Sci. 2004;77:347–357. doi: 10.1093/toxsci/kfh019. [DOI] [PubMed] [Google Scholar]
- Bermudez E, Mangum JB, Asgharian B, Wong BA, Reverdy EE, Janszen DB, Hext PM, Warheit DB, Everitt JI. Long-term pulmonary responses of three laboratory rodent species to subchronic inhalation of pigmentary titanium dioxide particles. Toxicol Sci. 2002;70:86–97. doi: 10.1093/toxsci/70.1.86. [DOI] [PubMed] [Google Scholar]
- Jacobsen NR, Møller P, Jensen KA, Vogel U, Ladefoged O, Loft S, Wallin H. Lung inflammation and genotoxicity following pulmonary exposure to nanoparticles in ApoE-/- mice. Part Fibre Toxicol. 2009;6:2. doi: 10.1186/1743-8977-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vesterdal LK, Folkmann JK, Jacobsen NR, Sheykhzade M, Wallin H, Loft S, Møller P. Pulmonary exposure to carbon black nanoparticles and vascular effects. Part Fibre Toxicol. 2010;7:33. doi: 10.1186/1743-8977-7-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d'Uscio LV, Baker TA, Mantilla CB, Smith L, Weiler D, Sieck GC, Katusic ZS. Mechanism of endothelial dysfunction in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2001;21:1017–1022. doi: 10.1161/01.ATV.21.6.1017. [DOI] [PubMed] [Google Scholar]
- Kitayama J, Faraci FM, Lentz SR, Heistad DD. Cerebral vascular dysfunction during hypercholesterolemia. Stroke. 2007;38:2136–2141. doi: 10.1161/STROKEAHA.107.481879. [DOI] [PubMed] [Google Scholar]
- Wilcox CS, Pearlman A. Chemistry and antihypertensive effects of tempol and other nitroxides. Pharmacol Rev. 2008;60:418–469. doi: 10.1124/pr.108.000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen CS, Sheykhzade M, Møller P, Folkmann JK, Amtorp O, Jonassen T, Loft S. Diesel exhaust particles induce endothelial dysfunction in apoE-/- mice. Toxicol Appl Pharmacol. 2007;219:24–32. doi: 10.1016/j.taap.2006.10.032. [DOI] [PubMed] [Google Scholar]
- Vesterdal LK, Folkmann JK, Jacobsen NR, Sheykhzade M, Wallin H, Loft S, Møller P. Modest vasomotor dysfunction induced by low doses of C60 fullerenes in apolipoprotein E knockout mice with different degree of atherosclerosis. Part Fibre Toxicol. 2009;6:5. doi: 10.1186/1743-8977-6-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie A, Martin W. Loss of endothelium-derived nitric oxide in rabbit aorta by oxidant stress: restoration by superoxide dismutase mimetics. Br J Pharmacol. 1998;124:719–728. doi: 10.1038/sj.bjp.0701899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preti SC, da Cunha V, Vassallo DV, Stefanon I. The superoxide dismutase mimetic, tempol, reduces the bioavailability of nitric oxide and does not alter L-NAME-induced hypertension in rats. Basic Clin Pharmacol Toxicol. 2005;97:29–34. doi: 10.1111/j.1742-7843.2005.pto_97105.x. [DOI] [PubMed] [Google Scholar]
- Jiang F, Guo Y, Salvemini D, Dusting GJ. Superoxide dismutase mimetic M40403 improves endothelial function in apolipoprotein(E)-deficient mice. Br J Pharmacol. 2003;139:1127–1134. doi: 10.1038/sj.bjp.0705354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laursen JB, Somers M, Kurz S, McCann L, Warnholtz A, Freeman BA, Tarpey M, Fukai T, Harrison DG. Endothelial regulation of vasomotion in apoE-deficient mice: implications for interactions between peroxynitrite and tetrahydrobiopterin. Circulation. 2001;103:1282–1288. doi: 10.1161/01.cir.103.9.1282. [DOI] [PubMed] [Google Scholar]
- MacKenzie A, Filippini S, Martin W. Effects of superoxide dismutase mimetics on the activity of nitric oxide in rat aorta. Br J Pharmacol. 1999;127:1159–1164. doi: 10.1038/sj.bjp.0702670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zollner S, Haseloff RF, Kirilyuk IA, Blasig IE, Rubanyi GM. Nitroxides increase the detectable amount of nitric oxide released from endothelial cells. J Biol Chem. 1997;272:23076–23080. doi: 10.1074/jbc.272.37.23076. [DOI] [PubMed] [Google Scholar]
- Ardanaz N, Beierwaltes WH, Pagano PJ. Distinct hydrogen peroxide-induced constriction in multiple mouse arteries: potential influence of vascular polarization. Pharmacol Rep. 2008;60:61–67. [PubMed] [Google Scholar]
- Sager TM, Castranova V. Surface area of particle administered versus mass determining the pulmonary toxicity of ultrafine and fine carbon black: comparison to ultrafine titanium dioxide. Part Fibre Toxicol. 2009;6:15. doi: 10.1186/1743-8977-6-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffin R, Tran L, Brown V, Stone V, Donaldson K. Proinflammogenic effects of low-toxicity and metal nanoparticles in vivo and in vitro: Highlighting the role of particle surface are and surface reactivity. Inhal Toxicol. 2007;19:849–856. doi: 10.1080/08958370701479323. [DOI] [PubMed] [Google Scholar]
- Sayes CM, Wahi R, Kurian PA, Liu Y, West JL, Ausman KD, Warheit DB, Colvin VL. Correlating nanoscale titania structure with toxicity: A cytotoxic and inflammatory response study with human dermal fibroblasts and human lung epithelial cells. Toxicol Sci. 2006;92:174–185. doi: 10.1093/toxsci/kfj197. [DOI] [PubMed] [Google Scholar]
- Warheit DB, Webb TR, Sayes CM, Colvin VL, Reed KL. Pulmonary instillation studies with nanoscale TiO2 rods and dots in rats: Toxicity is not dependent upon particle size and surface area. Toxicol Sci. 2006;91:227–236. doi: 10.1093/toxsci/kfj140. [DOI] [PubMed] [Google Scholar]
- Warheit DB, Webb TR, Reed KL, Frerichs S, Sayes CM. Pulmonary toxicity study in rats with three forms of ultrafine-TiO2 particles: Differential responses related to surface properties. Toxicology. 2007;230:90–104. doi: 10.1016/j.tox.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Larsen ST, Roursgaard M, Jensen KA, Nielsen GD. Nano titanium dioxide particles promote allergic sensitization and lung inflammation in mice. Basic Clin Pharmacol Toxicol. 2010;106:114–117. doi: 10.1111/j.1742-7843.2009.00473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saber AT, Jacobsen NR, Bornholdt J, Kjaer SL, Dybdahl M, Risom L, Loft S, Vogel U, Wallin H. Cytokine expression in mice exposed to diesel exhaust particles by inhalation. Role of tumor necrosis factor. Part Fibre Toxicol. 2006;3:4. doi: 10.1186/1743-8977-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saber AT, Halappanavar S, Folkmann JK, Bornholdt J, Boisen AM, Møller P, Williams A, Yauk C, Vogel U, Loft S, Wallin H. Lack of acute phase response in the livers of mice exposed to diesel exhaust particles or carbon black by inhalation. Part Fibre Toxicol. 2009;6:12. doi: 10.1186/1743-8977-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risom L, Møller P, Dybdahl M, Vogel U, Wallin H, Loft S. Dietary exposure to diesel exhaust particles and oxidatively damaged DNA in young oxoguanine DNA glycosylase 1 deficient mice. Toxicol Lett. 2007;175:16–23. doi: 10.1016/j.toxlet.2007.09.003. [DOI] [PubMed] [Google Scholar]