

Abstract
This study examines the role of F-spondin, an extracellular matrix protein of osteoarthritic cartilage, during chondrocyte maturation in embryonic growth plate cartilage. In chick tibia, F-spondin expression localized to the hypertrophic and calcified zones of the growth plate. Functional studies using tibial organ cultures indicated that F-spondin inhibited (~35%, p =0.02), and antibodies to F-spondin increased (~30%, p <0.1) longitudinal limb growth relative to untreated controls. In cell cultures, induction of chondrocyte maturation, by retinoic acid (RA) or transforming growth factor (TGF)-β treatment led to a significant upregulation of F-spondin (p <0.05). F-spondin transfection increased mineral deposition, alkaline phosphatase (AP) and matrix metalloproteinase (MMP)-13 mRNA levels (p <0.05), and AP activity following RA stimulation, compared to mock transfected controls. Using AP as a differentiation marker we then investigated the mechanism of F-spondin promaturation effects. Blocking endogenous F-spondin via its thrombospondin (TSR) domain inhibited RA induced AP activity 40% compared to controls (p <0.05). The stimulatory effect of F-spondin on AP expression was also inhibited following depletion of TGF-β from culture supernatants. Our findings indicate that F-spondin is expressed in embryonic cartilage, where it has the capacity to enhance chondrocyte terminal differentiation and mineralization via interactions in its TSR domain and TGF-β dependent pathways.
Keywords: chondrocyte maturation, growth plate, hypertrophy, transforming growth factor (TGF)-β
Transient cartilage provides a platform for embryonic skeletal formation, postnatal bone growth and fracture repair.1 By contrast, articular and tracheal cartilage, under physiologic conditions, are permanent and serve a continuous function throughout life. In osteoarthritis (OA), however, many of the cellular processes observed in transient cartilage during endochondral ossification are recapitulated.2 Strikingly, during OA, articular chondrocytes synthesize markers of maturing chondrocytes such as type X collagen,3 MMP-13,4 osteopontin,5 and osteocalcin.6 In addition, osteoarthritic chondrocytes exhibit high alkaline phosphatase activity5 mineralize the extracellular matrix7, and die by apoptosis.8,9 Evidence from all these studies strongly supports the hypothesis that during osteoarthritis the articular chondrocyte switches its developmental program from a permanent cartilage cell to a transient cartilage one. Therefore, pathways controlling chondrocyte maturation and terminal differentiation are extremely important not only as targets for growth therapies but also for new treatments in OA. In this regard, developmental models of chondrocyte maturation and endochondral ossification are valuable tools for studying the function of OA-associated proteins.
We have previously reported upregulation of F-spondin in OA cartilage.10 F-spondin is an extracellular matrix, heparin-binding glycoprotein of the thrombospondin family, that regulates neuronal outgrowth in the embryonic floor plate.11 In OA cartilage explant cultures, F-spondin treatment was found to increase both type II collagen degradation and MMP-13 secretion, suggesting its ability to regulate articular chondrocyte metabolism. However, the role of F-spondin in normal cartilage biology, endochondral ossification and bone growth has not been described. In the present study, we examined F-spondin expression in growth plate cartilage and investigate its role in chondrocyte maturation and terminal differentiation using established developmental models.
MATERIALS AND METHODS
Immunohistochemistry of Embryonic Chick Tibia
Tibia from 18-day-old chick embryos were fixed in 10% formalin and decalcified in 4.1% disodium ETDA at 4°C for 2 weeks. After paraffin embedding and tissue processing, 5-μm-thick sections were cut, deparaffinized, rehydrated and stained with a polyclonal F-Spondin antibody (R4; 1:100 diln) and the Vectastain ABC kit (Vector Laboratories, Burlingame, CA) according to the manufacturer’s instructions. R4 and R1 F-spondin antibodies, with specificities to the Spondin and TSR domains respectively, were generously provided by Dr. Avihu Klar (Hebrew University, Hadassah Medical School, Jerusalem, Israel). Sections were counterstained with 1% alcian blue.
Mouse Tibia Organ Culture
All animal experiments were conducted according to the guidelines of the Institutional Animal Care and Use Committee (IACUC) of New York University. CD1 timed-pregnant mice (Charles River Laboratories, Wilmington, MA) were euthanized, and tibiae isolated from E15.5 embryos using a stereomicroscope (Nikon Instruments, Melville, NY). Tibiae were then measured placed in 24-well plates and treated with either 0.5 μg/ml F-spondin recombinant protein (R&D Systems, Minneapolis, MN) or F-spondin neutralizing antibody (R1; 1:100 dilution). Media was changed every 24 h and tibial length measured again at the end of 1 week. The results were expressed as percentage change in length relative to day 1. After treatment some specimens were either fixed with glycerol/ethanol and placed in a staining solution (0.05% Alizarin red, 0.015% Alcian blue, 5% acetic acid in 70% ethanol) or processed for sectioning and stained with haemotoxylin and eosin.
In Vitro Chondrocyte Maturation Experiments
For monolayer experiments, chondrocytes were isolated from the upper portion of 14-day-old chick embryo sterna and cultured in DMEM with 10% FBS and hyaluronidase (4 U/ml) as previously described.12 At confluence, differentiation was induced by daily treatment with 10–100 nM all-trans retinoic acid (RA). To examine the effect of F-spondin overexpression, cultures were transfected at 70–80% confluence, with F-spondin or pcDNA3 control plasmids (Invitrogen, Carlsbad, CA) using GenePorter transfection reagent (Genlantis, San Diego, CA). Differentiation was induced the following day with 100 nM RA. For F-spondin inhibition studies, domain specific antibodies R1 or R4 were added to chondrocyte cultures (1:100 dilution), every other day, 1 h prior to RA treatment. To study the effect of TGF-β depletion, chondrocytes were transfected with pcDNA3 or pFS1 and supernatants harvested after 48 h. Conditioned supernatants were then incubated with 5 μg/ml anti-TGF-β antibody (R&D Systems) for 30 min at room temperature and added to freshly seeded chondrocytes in the presence of RA. After 24 h incubation, cells were harvested for determination of mRNA levels by RT-qPCR.
Serum-free alginate bead chondrocyte cultures were also employed to investigate F-spondin and TGF-β effects. Upper sternal chondrocytes were isolated from day 17 chick embryos and seeded in alginate beads at 5 × 106 cells/ml, as previously described.13 Beads were cultured in serum-free DMEM and ascorbate and stimulated with 0.5–5 ng/ml TGF-β2 (R&D systems) after 5 days, for 24 h. After treatment, RNA was harvested by resuspension in EDTA and lysis in Trizol® reagent (Life Technologies, Gaithersburg, MD).
Alkaline Phosphatase Activity Determination
To measure levels of activity of alkaline phosphatase, cells were harvested in 0.1% Triton X-100 mixed with 7.5 mM p-nitrophenylphosphate. Changes in absorbance were measured spectrophotometrically as previously described.14
Gene Expression Analyses
Different regions of chick tibial cartilage (E18) were micro-dissected, frozen in liquid nitrogen and crushed. Total RNA was extracted using Trizol® reagent and RNA was purified using a RNA micro kit (Qiagen, Inc., Chatsworth, CA) according to the RNeasy cleanup protocol. For chondrocyte cultures RNA was purified using Qiagen RNeasy mini columns. One-step RT-PCR was performed using 20 ng total RNA and QuantiTect SYBR Green RT-PCR reagents, a DNA Engine Optican2 system (Roche Molecular Systems, Pleasanton, CA), and primers specific for chick genes as previously described.15 In all studies, experiments were repeated 3–4 times and the mean and standard deviation were determined. Significant differences between test groups and controls were assessed by ANOVA or student’s t test. Significance was set at p <0.05.
RESULTS
Localized Expression of F-Spondin in Embryonic Growth Plate Cartilage
Our previous work has identified F-spondin as a marker of osteoarthritic cartilage.10 In this study, we investigated whether F-spondin is also expressed in maturing chondrocytes during endochondral bone development. Immunohistochemical staining identified cell-associated expression of F-spondin which was restricted to the hypertrophic and mineralized regions of growth plate cartilage of chick embryonic tibia (Fig. 1A). Expression appeared to correlate with increasing maturation; greater numbers of F-spondin positive chondrocytes were present in late-stage mineralized cartilage compared with early, hypertrophic cartilage (Fig. 1A). To confirm expression of F-spondin as a marker of chondrocyte maturation, we isolated the proliferative, hypertrophic and mineralized regions of tibial cartilage by micro-dissection and performed RT-PCR on mRNA extracted from the different regions. Figure 1B shows the relative expression levels of F-spondin within each region, in parallel with established hypertrophic markers. As expected, type II collagen expression peaked in the proliferative region, while type X collagen was highest in the hypertrophic region. F-spondin mRNA levels, along with MMP13, were highest within the calcified region. Together these findings indicate that F-spondin is a marker of hypertrophic and calcified cartilage.
Figure 1.
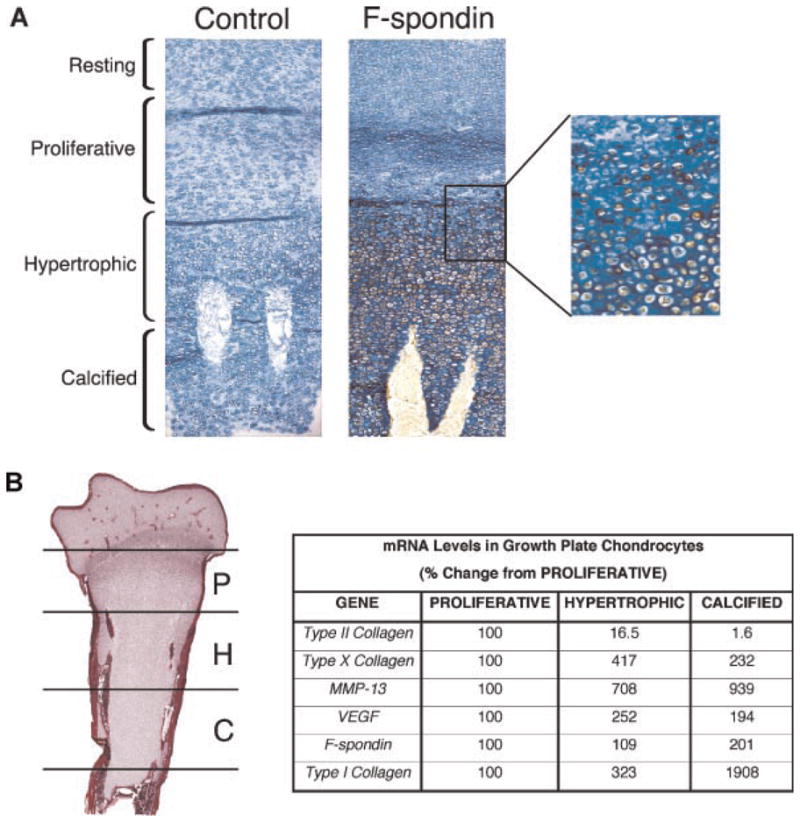
F-spondin is expressed in embryonic growth plate cartilage. (A) Immunohistochemical staining of chick tibia with pre-immune serum (left panel) or F-spondin antibody (right panel). Sections were counterstained with alcian blue. Brown color denotes positive staining for F-spondin. Magnification 200×. Boxed region shows detail of transition from proliferative to hypertrophic region. Magnification 400×. (B) Relative gene expression of F-spondin and chondrocyte markers in proliferative (P), hypertrophic (H), and calcified (C) cartilage regions. Results show percentage change in mRNA levels relative to proliferative region.
F-Spondin Regulates Tibial Growth Ex Vivo
Embryonic mouse tibia (E15.5) were isolated and cultured ex vivo to investigate the role of F-spondin on endochondral bone growth. Relative to untreated controls, F-spondin treatment for 1 week (0.5 μg/ml) inhibited tibial growth approximately 37 % (p =0.02; Fig. 2B). Morphologically, treated limbs also appeared more curved, with greater width (5–10%) in the epiphyseal regions (Fig. 2B). Accordingly, blocking F-spondin via antibody treatment led to the opposite effects: Limb growth was increased approximately 32% (p =0.008; Fig. 2B), and the limbs appeared thinner and straighter compared to untreated controls (Fig. 2A). Histological examination of F-spondin treated limbs revealed increased numbers of hypertrophic chondrocytes in the growth plate cartilage adjacent to the mineralized core (Fig. 2C). Conversely, inhibition of F-spondin by antibody treatment caused a reduction in the number of hypertrophic chondrocytes within the same region. Based on these observations we hypothesized that F-spondin functions to regulate chondrocyte terminal differentiation within the growth plate.
Figure 2.
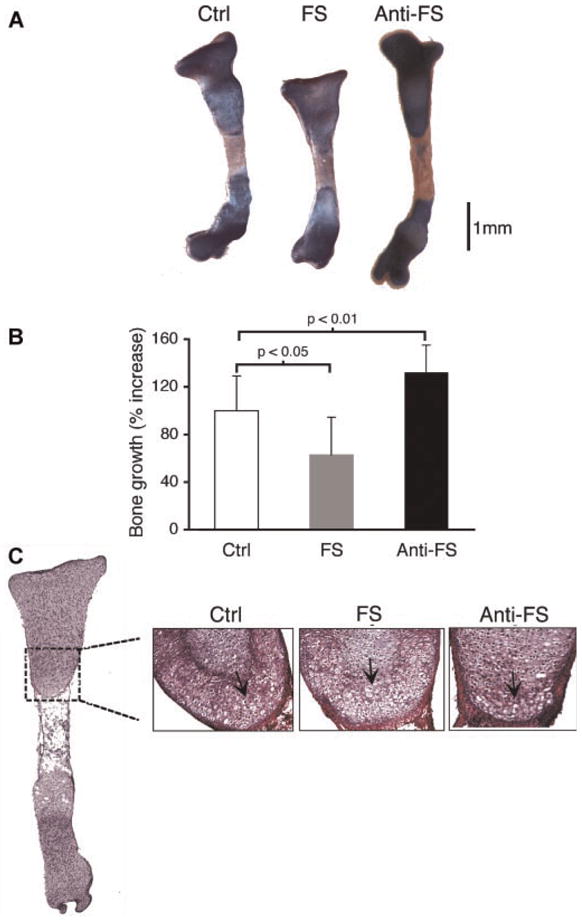
F-Spondin regulates bone growth and morphology in mouse tibia cultures. Tibia were treated with F-spondin (0.5 μg/ml) or a TSR-domain specific antibody for 7 days. (A) Representative images of whole tibia stained with alcian blue (proteoglycans) and alizarin red (mineral). (B) Growth is expressed as percent of original length at day 0. Average growth for control cultures was set to 100%. (C) H&E stained sections of corresponding limbs showing hypertrophic chondrocytes in the diaphysis (arrows).
F-spondin Expression Increases during Maturation of Chick Sternal Chondrocytes In Vitro
To further investigate F-spondin cellular effects we used an in vitro model of chondrocyte maturation, in which sternal chondrocytes mimic growth plate chondrocyte maturation in response to RA treatment. Figure 3A shows a dose dependant increase in AP activity (red staining) in response to RA consistent with previous observations.14 Gene expression analysis revealed a marked downregulation of type II collagen, accompanied by a pronounced upregulation of type I collagen in response to RA (Fig. 3B). Corroborating our observations in the growth plate, late-stage maturation markers such as MMP-13 and VEGF increased with RA dose, while type X collagen, a marker of early chondrocyte hypertrophy, decreased. Thus, this model appears to recapitulate the transition of growth plate cartilage from hypertrophic to calcified (Fig. 1B). Accordingly, F-spondin expression also increased with RA dose, and was approximately threefold higher than controls in the presence of 100 nM RA (p =0.04; Fig. 3B).
Figure 3.

F-Spondin is upregulated during maturation of chick chondrocytes. Upper sternal chondrocytes were exposed to RA for 5 days in culture. (A) Alkaline phosphatase staining indicating dose-dependent maturation of chick chondrocytes in response to RA. (B) Relative expression levels of chondrocyte maturation markers harvested from parallel cultures. Results are shown as percent change from RA 0.
F-Spondin Enhances Expression of Chondrocyte Maturation Markers
We next examined the effects of F-spondin on maturation of chick sternal chondrocytes in vitro. F-spondin overexpression in RA treated chondrocytes caused a moderate increase in mineral accumulation, detectable by Von Kossa staining, relative to controls (Fig. 4A). In parallel cultures, F-spondin induced a 5-fold increase in MMP-13 and a 14-fold increase in AP expression, relative to pcDNA3-transfected controls (Fig. 4B). F-spondin had no effect on type X collagen and runx-2. Interestingly, in the absence of RA, F-spondin did not change the expression of these maturation markers (data not shown). These results indicate that in the presence of RA, F-spondin enhances terminal differentiation and mineralization, but not induction of hypertrophy, of chick sternal chondrocytes.
Figure 4.

F-spondin enhances RA-induced chondrocyte maturation of chick chondrocytes. Upper sternal chondrocytes were transfected with full length F-spondin cDNA (FS), or pcDNA3 control vector (Ctrl) and treated with or without RA (100 nM). (A) Von Kossa staining of mineral accumulation after 7 days of RA treatment (B) Gene expression levels 5 days after transfection determined by RT-qPCR. Values are expressed relative to pcDNA transfected controls (=1). *p <0.05 versus vector control.
F-Spondin Induces AP Activity Via its TSR Domain
Using AP activity as a quantitative measure of chondrocyte maturation, we investigated the role of the F-spondin TSR domain in this process by transient transfection of cDNA constructs and culture with domain specific antibodies (Fig. 5A). Overexpression of full-length F-spondin cDNA (FS1) increased AP activity approximately threefold compared to control (Fig. 5B). In the absence of RA, F-spondin had no effect. Overexpression of an F-spondin cDNA construct containing a deletion of the TSR domain (FS6) did not elicit this stimulatory effect (Fig. 5A and B). Blocking endogenous F-spondin activity, by culturing cells in the presence of domain specific antibodies also inhibited AP activity (Fig. 5C). The level of inhibition was dependent upon RA dose and antibody specificity. At higher doses of RA (35 and 100 nm), blocking the TSR domain caused a ~40–50% inhibition of AP activity (p <0.02 vs. ctrl; Fig. 5C), while targeting the N-terminal spondin domain had no inhibitory effect (Fig. 5C). These findings suggest that F-spondin induction of AP during chondrocyte maturation is mediated via its TSR domain.
Figure 5.

F-spondin regulation of chondrocyte maturation requires the TSR domain. (A) Schematic representation of F-spondin protein domains and plasmid constructs encoding either full length (FS1) or TSR domain truncated (FS6) cDNAs. Relative AP activity 5 days after transfection with FS constructs (B) or following treatment with F-spondin antibodies (C). *p <0.05 versus RA control.
Promaturation Effects of F-Spondin Are Dependent on TGF-β
Since the TSR domain of F-spondin harbors conserved TGF-β binding motifs,10 we proposed that F-spondin-mediated enhancement of chondrocyte maturation similarly occurs via TGF-β. Consistent with this hypothesis, treatment of embryonic chick chondrocytes with TGF-β2 for 24 h in a serum-free alginate culture system led to dose dependent increases in expression of hypertrophic markers AP (~1.3- to 4.3-fold) and MMP-13 (~3.5- to 30-fold), as well as F-spondin (~2.4 to 5-fold) (Fig. 6A). This finding indicates that TGF-β2 can act as an inducer of chondrocyte maturation, and may provide a mechanism for the promaturation effects of F-spondin. To determine whether TGF-β is required for F-spondin induction of AP, we transfected chick chondrocytes with FS1 or control vector and harvested conditioned media supernatants after 48 h. Supernatants were then depleted of TGF-β by incubation with anti-TGF-β antibody and added to separate cultures of chick chondrocytes in the presence of retinoic acid for 24 h. Chondrocytes treated with conditioned media generated from FS1 transfected cultures demonstrated a twofold increase in AP gene expression compared to those receiving media from control vector transfected cells (open bars; Fig. 6B). Immunodepletion of TGF-β in the conditioned media of both control and FS1 transduced cultures led to a marked reduction in AP expression (gray bars; Fig. 6B). Incubation of conditioned media with control IgG antibody had no effect on AP gene expression (data not shown). Since TGF-β depletion of the conditioned media of FS1 transfected cultures completely abolished its stimulatory effect on AP gene expression, our results suggest that TGF-β activation is required for F-spondin mediated induction of AP.
Figure 6.

TGF-β enhances chondrocyte maturation and is required for F-spondin mediated induction of AP expression. (A) Relative gene expression of chondrocyte maturation markers following 24 h treatment with TGF-β2. Values are expressed relative to untreated controls. (B) Effect of TGF-β depletion on F-spondin-mediated induction of AP gene expression. The conditioned media of FS1 or pcDNA3 (Vec Ctrl) transfected chondrocytes was harvested and immunodepleted of TGF-β or left untreated, and added to separate cultures in the presence of RA. Graph shows relative AP gene expression after 24 h treatment. Values represent mean expression levels of triplicate samples relative to untreated supernatants from control vector cultures (=1).
DISCUSSION
In the present study we employed developmental models of chondrocyte maturation to investigate the role of F-spondin on chondrocyte phenotype and function. Our findings show that F-spondin is a marker of hypertrophic and mineralized cartilage. Functional studies indicate that F-spondin regulates endochondral bone growth and promotes chondrocyte terminal differentiation and mineralization in organ and cell cultures, respectively.
We propose that inhibition of limb growth occurs due to premature terminal differentiation and mineralization, at the expense of cell proliferation and hypertrophy. We are performing F-spondin gene deletion studies to confirm whether this hypothesis is correct. Interestingly, other TSR (thrombospondin) type I class molecule family members have been shown to play a role in normal skeletal development. Mutations in the cartilage oligo-meric matrix protein (COMP) have been reported to cause pseudoachondroplasias, a well described form of dwarfism also associated with severe osteoarthritis requiring joint replacement.16,17 Murine models of pseudoachondroplasia, generated by engineering mutations in both globular and thrombospondin domains of COMP, exhibit short limb dwarfism, and growth plate disorganization suggesting a deregulation of chondrocyte terminal differentiation.18,19
Our findings suggest that F-spondin enhances terminal differentiation of chondrocytes, characterized by upregulation of AP and MMP-13 (Figs. 4 and 5). Interestingly, both markers have been shown to be induced by F-spondin in cementoblasts, specialized cells that deposit a calcified matrix of cementum at the roots of teeth.20 F-spondin induction of AP activity was also found to be TSR domain dependent (Fig. 5). In OA chondrocytes, we have observed TSR-dependent induction of PGE2.10 This region also harbors a highly conserved latency associated peptide (LAP)-binding KFRK motif, which may modulate TGF-β activity via binding to the latent complex in a manner analogous to thrombospondin-1.21 Indeed, addition of F-spondin to cartilage explant cultures was found to increase active TGF-β levels in culture supernatants and regulate synthetic activity.10 It is well established that TGF-β effects on chondrocyte maturation depend on the differentiation state of its target cell population.22,23 While TGF-β is generally regarded as an inhibitor of chondrocyte hypertrophy, MMP-13 dependent increased TGF-β activation has also been reported in chick embryonic chondrocytes undergoing terminal differentiation.13 Late hypertrophic chondrocytes produce more TGF-β than early hypertrophic chondrocytes and the percentage of TGF-β that is activated increases as the chondrocyte nears terminal differentiation.13,24 In this study, we have shown that addition of exogenous recombinant TGF-β increases the expression of MMP-13 and AP as well as F-spondin. Thus, late-stage hypertrophic chondrocytes may respond to activated TGF-β by further progression toward terminal differentiation. Indeed, our data suggest that F-spondin is part of a positive feedback loop that accelerates this process, ensuring terminal differentiation of hypertrophic chondrocytes and subsequent endochondral ossification via induction of MMP-13 and further activation of TGF-β (Fig. 7).
Figure 7.

Proposed role of F-spondin in chondrocyte terminal differentiation. F-spondin expression increases during RA- and TGF-β-induced chondrocyte maturation. This protein, through activation of TGF-βinduces the expression of important maturation markers such as AP and MMP13, creating a positive feedback loop that accelerates chondrocyte terminal differentiation.
Acknowledgments
This work was supported by grants from the Arthritis Foundation, New York Chapter, NIH (R01-AR054817), and NIDCR (K08DE017426).
References
- 1.Iwamoto M, Higuchi Y, Enomoto-Iwamoto M, et al. The role of ERG (ets related gene) in cartilage development. Osteoarthr Cartilage. 2001;9(Suppl A):S41–S47. doi: 10.1053/joca.2001.0443. [DOI] [PubMed] [Google Scholar]
- 2.Aigner T, Gerwin N. Growth plate cartilage as developmental model in osteoarthritis research—Potentials and limitations. Curr Drug Targets. 2007;8:377–385. doi: 10.2174/138945007779940052. [DOI] [PubMed] [Google Scholar]
- 3.Von der Mark K, Kirsch T, Nerlich A, et al. Type X collagen synthesis in human osteoarthritic cartilage. Indication of chondrocyte hypertrophy. Arthritis Rheum. 1992;35:806–811. doi: 10.1002/art.1780350715. [DOI] [PubMed] [Google Scholar]
- 4.Fernandes JC, Martel-Pelletier J, Lascau-Coman V, et al. Collagenase-1 and collagenase-3 synthesis in normal and early experimental osteoarthritic canine cartilage: An immunohistochemical study. J Rheumatol. 1998;25:1585–1594. [PubMed] [Google Scholar]
- 5.Pullig O, Weseloh G, Gauer S, et al. Osteopontin is expressed by adult human osteoarthritic chondrocytes: Protein and mRNA analysis of normal and osteoarthritic cartilage. Matrix Biol. 2000;19:245–255. doi: 10.1016/s0945-053x(00)00068-8. [DOI] [PubMed] [Google Scholar]
- 6.Pullig O, Weseloh G, Ronneberger D, et al. Chondrocyte differentiation in human osteoarthritis: Expression of osteocalcin in normal and osteoarthritic cartilage and bone. Calcif Tissue Int. 2000;67:230–240. doi: 10.1007/s002230001108. [DOI] [PubMed] [Google Scholar]
- 7.Hashimoto S, Ochs RL, Rosen F, et al. Chondrocyte-derived apoptotic bodies and calcification of articular cartilage. Proc Natl Acad USA. 1998;95:3094–3099. doi: 10.1073/pnas.95.6.3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blanco FJ, Ochs RL, Schwarz H, et al. Chondrocyte apoptosis induced by nitric oxide. Am J Path. 1995;146:75–85. [PMC free article] [PubMed] [Google Scholar]
- 9.Aigner T, Kurz B, Fukui N, et al. Roles of chondrocytes in the pathogenesis of osteoarthritis. Curr Opin Rheumatol. 2002;14:578–584. doi: 10.1097/00002281-200209000-00018. [DOI] [PubMed] [Google Scholar]
- 10.Attur MG, Palmer GD, Al-Mussawir HE, et al. F-Spondin, a neuroregulatory protein, is up-regulated in osteoarthritis and regulates cartilage metabolism via TGF-beta activation. FASEB J. 2009;23:79–89. doi: 10.1096/fj.08-114363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klar A, Baldassare M, Jessell TM. F-Spondin: A gene expressed at high levels in the floor plate encodes a secreted protein that promotes neural cell adhesion and neurite extension. Cell. 1992;69:95–110. doi: 10.1016/0092-8674(92)90121-r. [DOI] [PubMed] [Google Scholar]
- 12.Teixeira CC, Ischiropoulos H, Leboy PS, et al. Nitric oxide-Nitric oxide synthase regulates key maturational events during chondrocyte terminal differentiation. Bone. 2005;37(1):37–45. doi: 10.1016/j.bone.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 13.D’Angelo M, Sarment DP, Billings PC, et al. Activation of transforming growth factor beta in chondrocytes undergoing endochondral ossification. J Bone Miner Res. 2001;16:2339–2347. doi: 10.1359/jbmr.2001.16.12.2339. [DOI] [PubMed] [Google Scholar]
- 14.Teixeira CC, Hatori M, Leboy PS, et al. A rapid and ultrasensitive method for measurement of DNA, calcium and protein content, and alkaline phosphatase activity of chondrocyte cultures. Calcif Tissue Int. 1995;56:252–256. doi: 10.1007/BF00298620. [DOI] [PubMed] [Google Scholar]
- 15.Oliveira SM, Amaral IF, Barbosa MA, et al. Engineering endochondral bone: In vitro studies. Tissue Eng A. 2009;15:625–634. doi: 10.1089/ten.tea.2008.0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hecht JT, Nelson LD, Crowder E, et al. Mutations in exon 17B of cartilage oligomeric matrix protein (COMP) cause pseudoachondroplasia. Nat Genet. 1995;10:325–329. doi: 10.1038/ng0795-325. [DOI] [PubMed] [Google Scholar]
- 17.Briggs MD, Hoffman SM, King LM, et al. Pseudoachondroplasia and multiple epiphyseal dysplasia due to mutations in the cartilage oligomeric matrix protein gene. Nat Genet. 1995;10:330–336. doi: 10.1038/ng0795-330. [DOI] [PubMed] [Google Scholar]
- 18.Pirog-Garcia KA, Meadows RS, Knowles L, et al. Reduced cell proliferation and increased apoptosis are significant pathological mechanisms in a murine model of mild pseudoachondroplasia resulting from a mutation in the C-terminal domain of COMP. Hum Mol Genet. 2007;16:2072–2088. doi: 10.1093/hmg/ddm155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmitz M, Niehoff A, Miosge N, et al. Transgenic mice expressing D469Delta mutated cartilage oligomeric matrix protein (COMP) show growth plate abnormalities and sternal malformations. Matrix Biol. 2008;27:67–85. doi: 10.1016/j.matbio.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 20.Kitagawa M, Kudo Y, Iizuka S, et al. Effect of F-spondin on cementoblastic differentiation of human periodontal ligament cells. Biochem Biophys Res Commun. 2006;349:1050–1056. doi: 10.1016/j.bbrc.2006.08.142. [DOI] [PubMed] [Google Scholar]
- 21.Schultz-Cherry S, Lawler J, Murphy-Ullrich JE. The type 1 repeats of thrombospondin 1 activate latent transforming growth factor-beta. J Biol Chem. 1994;269:26783–26788. [PubMed] [Google Scholar]
- 22.Serra R, Chang C. TGF-beta signaling in human skeletal and patterning disorders. Birth Defects Res C Embryo Today. 2003;69:333–351. doi: 10.1002/bdrc.10023. [DOI] [PubMed] [Google Scholar]
- 23.Spagnoli A, O’Rear L, Chandler RL, et al. TGF-beta signaling is essential for joint morphogenesis. J Cell Biol. 2007;177:1105–1117. doi: 10.1083/jcb.200611031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.D’Angelo M, Pacifici M. Articular chondrocytes produce factors that inhibit maturation of sternal chondrocytes in serum-free agarose cultures: A TGF-beta independent process. J Bone Miner Res. 1997;12:1368–1377. doi: 10.1359/jbmr.1997.12.9.1368. [DOI] [PubMed] [Google Scholar]