

Abstract
In this review, we discuss the latest targeted enrichment methods and aspects of their utilization along with second-generation sequencing for complex genome analysis. In doing so, we provide an overview of issues involved in detecting genetic variation, for which targeted enrichment has become a powerful tool. We explain how targeted enrichment for next-generation sequencing has made great progress in terms of methodology, ease of use and applicability, but emphasize the remaining challenges such as the lack of even coverage across targeted regions. Costs are also considered versus the alternative of whole-genome sequencing which is becoming ever more affordable. We conclude that targeted enrichment is likely to be the most economical option for many years to come in a range of settings.
Keywords: targeted enrichment, next-generation sequencing, genome partitioning, exome, genetic variation
INTRODUCTION
Next-generation sequencing (NGS) [1, 2] is now a major driver in genetics research, providing a powerful way to study DNA or RNA samples. New and improved methods and protocols have been developed to support a diverse range of applications, including the analysis of genetic variation. As part of this, methods have been developed that aim to achieve ‘targeted enrichment’ of genome subregions [3, 4], also sometimes referred to as ‘genome partitioning’. Strategies for direct selection of genomic regions were already developed in anticipation of the introduction of NGS [5, 6]. By selective recover and subsequent sequencing of genomic loci of interest, costs and efforts can be reduced significantly compared with whole-genome sequencing.
Targeted enrichment can be useful in a number of situations where particular portions of a whole genome need to be analyzed [7]. Efficient sequencing of the complete ‘exome’ (all transcribed sequences) represents a major current application, but researchers are also focusing their experiments on far smaller sets of genes or genomic regions potentially being implicated in complex diseases [e.g. derived from genome-wide association studies (GWAS)], pharmacogenetics, pathway analysis and so on [1, 8, 9]. For identifying monogenetic diseases, exome sequencing can be a powerful tool [10]. Across all these areas of study, a typical objective is the analysis of genetic variation within defined cohorts and populations.
Targeted enrichment techniques can be characterized via a range of technical considerations related to their performance and ease of use, but the practical importance of any one parameter may vary depending on the methodological approach applied and the scientific question being asked. Arguably, the most important features of a method, which in turn reflect the biggest challenges in targeted enrichment, include: enrichment factor, ratio of sequence reads on/off target region (specificity), coverage (read depth), evenness of coverage across the target region, method reproducibility, required amount of input DNA and overall cost per target base of useful sequence data.
Within this review, we compare and contrast the most commonly used techniques for targeted enrichment of nucleic acids for NGS analysis. Additionally, we consider issues around the use of such methods for the detection of genetic variation, and some general points regarding the design of the target region, input DNA sample preparation and the output analysis.
ENRICHMENT TECHNIQUES
Current techniques for targeted enrichment can be categorized according to the nature of their core reaction principle (Figure 1):
‘Hybrid capture’: wherein nucleic acid strands derived from the input sample are hybridized specifically to preprepared DNA fragments complementary to the targeted regions of interest, either in solution or on a solid support, so that one can physically capture and isolate the sequences of interest;
‘Selective circularization’: also called molecular inversion probes (MIPs), gap-fill padlock probes and selector probes, wherein single-stranded DNA circles that include target region sequences are formed (by gap-filling and ligation chemistries) in a highly specific manner, creating structures with common DNA elements that are then used for selective amplification of the targeted regions of interest;
PCR amplification: wherein polymerase chain reaction (PCR) is directed toward the targeted regions of interest by conducting multiple long-range PCRs in parallel, a limited number of standard multiplex PCRs or highly multiplexed PCR methods that amplify very large numbers of short fragments.
Figure 1:

Commonly used targeted enrichment techniques. (1) Hybrid capture targeted enrichment either on solid support-like microarrays (a) or in solution (b). A shot-gun fragment library is prepared and hybridized against a library containing the target sequence. After hybridization (and bead coupling) nontarget sequences are washed away, the enriched sample can be eluted and further processed for sequencing. (2) Enrichment by MIPs which are composed of a universal sequence (blue) flanked by target-specific sequences. MIPs are hybridized to the region of interest, followed by a gap filling reaction and ligation to produce closed circles. The classical MIPs are hybridized to mechanically sheared DNA (a), the Selector Probe technique uses a restriction enzyme cocktail to fragment the DNA and the probes are adapted to the restriction pattern (b). (3) Targeted enrichment by differing PCR approaches. Typical PCR with single-tube per fragment assay (a), multiplex PCR assay with up to 50 fragments (b) and RainDance micro droplet PCR with up to 20 000 unique primer pairs (c) utilized for targeted enrichment.
Given the operational characteristics of these different targeted enrichment methods, they naturally vary in their suitability for different fields of application. For example, where many megabases needs to be analyzed (e.g. the exome), hybrid capture approaches are attractive as they can handle large target regions, even though they achieve suboptimal enrichment over the complete region of interest. In contrast, when small target regions need to be examined, especially in many samples, PCR-based approaches may be preferred as they enable a deep and even coverage over the region of interest, suitable for genetic variance analysis.
An overview of these different approaches is presented in Figure 1, and Table 1 lists the most common methods along with additional information.
Table 1:
Currently employed targeted enrichment techniques
| Enrichment technique | Vendor | Features | Pros | Cons | Number of loci (target size) | Library prep for NGS |
|---|---|---|---|---|---|---|
| Hybrid capture | ||||||
| Solid support | Agilent SureSelect, Roche NimbleGen SeqCap EZ | Medium to large target regions, custom and preconfigured target regions (i.e. whole exome), Multiplexing possible, ready to use kits | Ease of production, large target sets | Large amount of input DNA, high-tech equipment (3–10 µg) | 104–106 (1–50 Mb) | Before enrichment |
| In solution | Agilent SureSelect, FlexGen FleXelect, MYcroarray MYselect, Roche NimbleGen SeqCap EZ | Ease of use, small amount of input DNA (<1–3 µg) | ||||
| Circularization | ||||||
| Molecular inversion probes | No dedicated instruments, high specificity, input DNA (<1 µg) | 102–104 (0.1–5 Mb) | During enrichment (incorporated into hybridization probes | |||
| Selector probes | HaloGenomics | Custom kits and clinically relevant panel kits | exome kit not available yet | 10–200 (0.1–1.5 Mb) | ||
| PCR | ||||||
| Long range | Invitrogen SequalPrep, Qiagen SeqTarget system | Smaller target regions, coverage by tiling | Relatively easy to set up and automatable, even coverage | PCR conditions largely influence effectiveness, >10 µg DNA for Large sets | 102–104 (0.1–5 Mb) | After enrichment |
| Multiplex | Multiplicom, Fluidigm | Smaller target regions, coverage by tiling, multiplex PCR of 150–200 amplicons (150–450 bp) | Easy to perform, reasonably economical in terms of, even coverage | |||
| Micro droplet | RainDance | Smaller target regions, coverage by tiling, micro droplet PCR of up to 20 000 amplicons (150–1500 bp) | Even coverage, low input DNA | Relatively expensive, specialist equipment | 103–104 (up to 10 Mb) |
All major targeted enrichment techniques show relative pros and cons.
Basic considerations for targeted enrichment experiments
The design of a targeted enrichment experiment begins with a general consideration of the target region of interest. In particular, a major obstacle for targeted enrichment is posed by repeating elements, including interspersed and tandem repeats as well as elements such as pseudogenes located within and outside the region of interest. Exclusion of repeat masked elements [11] from the targeted region is a straightforward and efficient way to reduce the recovery of undesirable products due to repeats. Furthermore, at extreme values (<25% or >65%), the guanine-cytosine (GC) content of the target region has a considerable impact on the evenness and efficiency of the enrichment [12]. This can adversely affect the enrichment of the 5′-UTR/promoter region and the first exon of genes, which are often GC rich [13]. Therefore, expectations regarding the outcome of the experiment require careful evaluation in terms of the precise target region in conjunction with the appropriate enrichment method.
The performance of a targeted enrichment experiment will also depend upon the mode and quality of processing of the input DNA sample. Having sufficient high-quality DNA is key for any further downstream handling. When limited genomic DNA is available, whole-genome amplification (WGA) is usually applied. Since WGA produces only a representation and not a replica of the genome, a bias is assumed to be introduced though the impact of this on the final results can be compensated for, to a degree by identically manipulating control samples [14].
All three major targeted enrichment techniques (hybrid capture, circularization and PCR) differ in terms of sample library preparation workflow enabling sequencing on any of the current NGS instruments (e.g. Illumina, Roche 454 and SOLiD). Enrichment by hybrid selection relies on short fragment library preparations (typically range from 100 to 250 bp) which are generated before hybridization to the synthetic library comprising the target region. In contrast, enrichment by PCR is performed directly on genomic DNA and thereafter are the library primers for sequencing added. Enrichment by circularization offers the easiest library preparation for NGS because the sequencing primers can be added to the circularization probe, thus eliminating the need for any further library preparation steps. Sequencing can be performed either as single read or paired-end reads of the fragment library. In general, mate–pair libraries are not used for hybridization-based targeted enrichments due to the extra complications this implies in terms of target region design.
In general, a single NGS run produces enough reads to sequence several samples enriched by one of the mentioned methods. Therefore, pooling strategies and indexing approaches are a practical way to reduce the per sample cost. Depending on the method used for targeted enrichment, different multiplexing strategies can be envisaged that enable multiplexing in different stages of the enrichment process: before, during and after the enrichment. For targeted enrichment by hybrid capture, indexing of the sample is usually performed after the enrichment but to reduce the number of enrichment reactions, the sample libraries can alternatively be indexed during the library preparations and then pooled for enrichment [15]. Enrichment by PCR and circularization offers indexing during the enrichment by using bar-coded primers in the product amplification steps [16]. Furthermore, two multiplexing strategies can be combined in a single experiment. First, multiple samples can be enriched as a pool, with each harboring a unique pre-added bar-code. Then second, another bar-coding procedure can be applied postenrichment, to each of these pools, giving rise to a highly multiplexed final pool. If such extensive multiplexing is used, great care must be taken to normalize the amount of each sample within the pool to achieve sufficiently even representation over all samples in the final set of sequence reads. In addition, highly complex pooling strategies also imply far greater challenges when it comes to deconvoluting the final sequence data back into the original samples.
The task of designing the target region is relatively straightforward, and this can be managed with web-based tools offered by UCSC, Ensembl/BioMart, etc. and spreadsheet calculations (e.g. Excel) on a personal computer. Web-based tools like MOPeD offer a more user-friendly approach for oligoncleotide probe design [17]. Far more difficult, however, is the final sequence output analysis, which needs dedicated computer hardware and software. Fortunately, great progress has recently been made in read mapping and parameter selection for this process, leading to more consistent and higher quality final results [18]. Reads generated by hybrid selection will always tend to extend into sequences beyond the target region and the longer the fragment library is, the more of these ‘near target’ sequences will be recovered. Therefore, read mapping must start with a basic decision regarding the precise definition of the on/off target boundaries, as this parameter is used for counting on/off target reads and so influences the number of sequence reads considered as on target. This problem is not so critical for enrichments based on PCR and circularization as these methods do not suffer from ‘near target’ products. Another major consideration in data analysis is the coverage needed to reliably identify sequence variants, e.g. single nucleotide polymorphisms (SNP). This depends on multiple factors such as the nature of the region of interest in question, the method used for targeted enrichment. In different reports, it has ranged from 8x coverage [19], which was the minimum coverage for reliable SNP calling and up to 200x coverage [20], in this case the total average coverage for the targeted region.
Enrichment by hybrid capture
Enrichment by hybrid capture (Figure 1.1a and b) builds on know-how developed over the decade or more of microarray research that preceded the NGS age [21, 22]. The hybrid capture principle is based upon the hybridization of a selection ‘library’ of very many fragments of DNA or RNA representing the target region against a shotgun library of DNA fragments from the genome sample to be enriched. Two alternative strategies are used to perform the hybrid capture: (i) reactions in solution [4] and (ii) reactions on a solid support [3]. Each of these two approaches brings different advantages, as listed in Table 1.
Selection libraries for hybrid capture are typically produced by oligonucleotide synthesis upon microarrays, with lengths ranging from ~60 to ~180 bases. These microarrays can be used directly to perform the hybrid capture reaction (i.e. surface phase methods), or the oligonucleotide pool can be harvested from the array and used for an in-solution targeted enrichment (i.e. solution phase methods). The detached oligonucleotide pool enables versatile downstream processing: if universal 5′- and 3′-end sequences are included in the design of the oligonucleotides, the pool can be reamplified by PCR and used to process many genomic samples. Furthermore, it is possible to introduce T7/SP6 transcription start sites via these PCRs [23], so that the pool can be transcribed into RNA before being used in an enrichment experiment.
Recently, an increasing number of protocols and vendors have begun offering out of the box solutions for hybrid capture, meaning, the researcher need not do development work but merely choose between a preset targeted enrichment regions (e.g. whole exome) or specify their own custom enrichment region. Example vendors include: Agilent (SureSelect product), NimbleGen (SeqCap EZ product), Flexgen and MYcroArray. Alternatively, a more cost efficient option compared with buying a complete kit involves ordering a synthetic bait library, reamplifying this by PCR [24], optionally transcribing this into RNA and undertaking a do-it-yourself enrichment experiment based upon published protocols.
Enrichment by circularization
Enrichment by DNA fragment circularization is based upon the principle of selector probes [6, 25] and gap-fill padlock or MIPs [26]. This approach differs significantly compared with the aforementioned hybrid capture method. Most notably, it is greatly superior in terms of specificity, but far less amenable to multiple sample co-processing in a single reaction. Each probe used for enrichment by circularization comprises a single-stranded DNA oligonucleotide that at its ends contains two sequences that are complementary to noncontiguous stretches of a target genomic fragment, but in reversed linear order. Specific hybridization between such probes and their cognate target genomic fragments generates bipartite circular DNA structures. These are then converted to closed single-stranded circles by gap filling and ligation reactions (Figure 1.2). A rolling circle amplification step or a PCR directed toward sequences present in the common region of all the circles is then finally applied to amplify the target regions (circularized sequences) to generate an NGS library.
Variations on this basic method concept exist, in particular with regard to the differences in sample material preparation and downstream processing for NGS library preparation. In the gap-fill padlock or MIPs implementation (Figure 1.2a), the sample DNA is fragmented by shearing and used in the bipartite circular structure to provide a template for the probe DNA to be extended by gap filling and converted to a closed circle. In this incarnation, the design of the MIPs merely has to consider the uniqueness of each target region fragment and the most suitable hybridization conditions. In contrast, a more elaborated design is offered by the ‘Selector Probe’ technique [6, 27]. Here the genomic DNA is fragmented in a controlled manner by means of a cocktail of restriction enzymes, and the selector probes are designed to accommodate the restriction pattern of the target region. The ends of each genomic DNA thus become adjacently positioned in the bipartite circles, enabling them to be gap filled and ligated into closed single-stranded circles (Figure 1.2b).
A particularly appealing feature of enrichment by circularization with MIPs and selectors is their ‘library free’ nature [28]. Since MIPs and selectors comprise a target-specific 5′- and 3′-end with a common central linker, the sequencing primer information for NGS applications can be directly included into this common linker. Burdensome NGS library preparations are therefore not required, reducing processing time markedly.
Enrichment by PCR
Enrichment by PCR (Figures 1.3a–c) is in terms of methodology, a more straightforward method compared with the other genome partioning techniques. It takes advantage of the great power of PCR to enrich genome regions from small amounts of target material. Just as for circularization methods, if the PCR product sizes fall within the sequencing length of the applied NGS platform (maximum read length for SOLiD: 110 bp, Illumina: 240 bp and 454: 1000 bp) PCR-based enrichment can allow one to bypass the need for shot-gun library preparation by using suitably 5′-tailed primers in the final amplification steps.
The main downside of the method is that it does not scale easily, in any format, to enable the targeting of very large genome subregions or many DNA samples. To use this method effectively, any significant extent of parallelized singleplex or multiplex PCR would need to be supported by the use of automated robotics, individual PCR amplicons (or multiplex products) need to be carefully normalized to equivalent molarities when pooling in advance of NGS (so that the final coverage of the total region of interest is as even as possible), and the amount of DNA material a study requires can be substantial as this requirement grows linearly with the number of utilized PCR reactions. But if the target region is small, PCR can be the method of choice. For example, a target region of 50–100 kb or so, could be spanned by a handful of long-range PCRs each of 5–10 kb [29], or by tiling a few hundred shorter PCRs and using microtiter plates and robotics, or by one or other approaches toward PCR multiplexing [30, 31].
Long-range PCR is the most commonly applied approach and it is reasonably straightforward to accomplish. Many vendors now offer specially formulated kits (e.g. Invitrogen SequalPrep, Qiagen SeqTarget) that can amplify fragments of up to 20 kb in length. And obviously, this approach is fully compatible with automation. Long-range PCR products do, however, have to be cleaned, pooled and processed for shot-gun library preparation so that they are ready for analysis by NGS.
To increase the throughput of PCR by keeping the number of PCR reactions as low as possible, there is the alternative of multiplex PCR (Figure 1.3b). Given careful primer design and reaction optimization, several dozen primer pairs can be used together effectively in a multiplexing reaction [32]. Indeed, software specifically created to help with multiplex PCR assay design is available [33]. Then, by running many such reactions in parallel, many hundred different DNA fragments can be amplified. An alternative method that is commercially available from Fluidigm (Table 1), uses a microfluidics PCR chip to conduct several thousand singleplex PCRs in parallel.
Yet, another strikingly elegant method is the micro-droplet PCR technology developed by Raindance [34, 35]. Here, two libraries of lipid encapsulated water droplets are prepared—one in which each droplet contains a small amount of the test sample DNA and the other comprising droplets that harbor distinct pairs of primers. These two libraries are then merged (respective droplet pairs are fused together) to generate a highly multiplexed total emulsion PCR wherein each reaction is actually isolated from all others in its own fused droplet (Figure 1.3c). Using this technology, up to 20 000 primer pairs can be used effectively in parallel in a single tube.
Overall, one can draw the following conclusions from a comparison of the currently used enrichment techniques shown in Table 1: (i) that hybrid capture has its main advantages for medium to large target regions (10–50 Mb) in contrast to the other two approaches which typically only target small regions within the kilo base pairs and low mega base pairs range. The ability to enrich for mega base pair-sized targets is particularly advantageous in research studies where typically whole exomes or many genes are involved. Especially for clinical applications, this may be relevant for oncological applications where one would expect to sequence 100–1000's of genes. (ii) The advantage of PCR and circularization-based methods is that they achieve very high enrichment factors and few off-target reads, but only for small target regions. This is more suited to clinical genetics where typically only a few critical loci need to be assessed.
Descriptive metrics for targeted DNA enrichment experiments
To allow meaningful comparison of enrichment methods and experiments that employ them, and to rationally decide which technologies are most suitable when designing a research project, it is important that an objective set of descriptive metrics are defined and then widely used when reporting enrichment datasets. A series of metrics need to be considered, and the importance of each can be weighted according to specific needs and objectives of any experiment. A proposal for such a set of metrics is soon to be published, and it contains the following (Nilsson et al., manuscript in preparation):
Region of interest (size): ROI;
Average read depth (in ROI): D;
Fraction of ROI sufficiently covered (at a specified D): F;
Specificity (fraction of reads in ROI): S;
Enrichment Factor (D for ROI versus D for rest of genome): EF;
Evenness (lack of bias): E and
Weight (input DNA requirement): W.
A theoretical examination of how a method's innate enrichment capability and the size of the targeted region work together to determine other parameters (such as specificity and read depth) can be very instructive when choosing an enrichment method for a particular application. This is illustrated in Figure 2. For example, given a method's specific enrichment factor and knowledge about the size of the region of interest, the corresponding sequencing effort can be estimated for a given desired specificity (percent of sequences on target). Similarly, for a given region of interest and a minimum desired specificity, the necessary enrichment factor capabilities can be calculated.
Figure 2:

Comparison of enrichment factor calculations on sequencing depth and percent on target sequences for different target region sizes employed for targeted enrichment. Calculations were performed as follows:  [52]
[52] 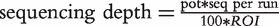 EF, enrichment factor; ROI, region of interest in kb; genome, genome size in kb; pot, percent on target sequences; seq per run, assumed sequences per run in kb.
EF, enrichment factor; ROI, region of interest in kb; genome, genome size in kb; pot, percent on target sequences; seq per run, assumed sequences per run in kb.
Finally, the specific per sample costs for a targeted enrichment is useful to consider. To make costs comparable, either for different target region sizes or across different methods, the costs can be normalized as costs per base pair. Costs also change with time and as technologies improve, and so at some stage the overall price of any particular experiment (i.e. targeted enrichment plus sequencing costs) will not be cheaper than the alternative of whole-genome sequencing combined with in silico-based isolation of the region of interest.
DISCOVERY OF GENETIC VARIATION
To investigate genetic variation by NGS, many DNA samples need to be tested. To reduce the cost of such studies, researchers typically focus their attention on genome subregions of particular interest, and this implies a major role for targeted enrichment in such undertakings. A set of concerns then arises regarding the accuracy of variation discovery within NGS data obtained from DNA that has been subjected to one or other enrichment methods. Other questions, such as whether the input genomic DNA was also preamplified by WGA, whether sample pooling or multiplexing was applied and whether proper experimental controls were employed, also come into play. Currently, however, the field is lacking a complete understanding of all the issues and influences relevant to these important questions. For these reasons, it is critical that thorough downstream validation experiments are performed, using independent experimental approaches.
Another dimension to the problem of reliably discovering sequence variation, and one where there is perhaps a little more clarity, is the impact of different software and algorithm choices used for primary sequence data analysis (e.g. the choice of suitable genome alignment tool, filter parameters for the analysis, coverage thresholds at intended bases). It has been shown that the detection of variants depends strongly on the particular software tools employed [36]. Indeed, because current alignment and analytical tools perform so heterogeneously, the 1000 Genomes Project Consortium [37] decided to avoid calling novel SNPs unless they were discovered by at least two independent analytical pipelines. In general, unified analysis workflows can and must be developed [38] to enable the combination and processing of data produced from different machines/approaches, to at least minimize instrument-specific biases and errors that otherwise detract from making high-confidence variant base calls.
Whatever mapping and analysis approach is applied, sufficient coverage on a single base resolution ranging from 20 to 50x is usually deemed necessary for reliable detection of sequence variation [39–42]. In one simulation study, the SNP discovery performances of two NGS platforms in a specific disease gene were shown to fall rapidly when the coverage depth was below 40x [43]. In addition, all called variants should ideally be supported by data from both read orientations (forward and reverse). Some researchers further insist on obtaining at least three reads from both the forward and the reverse DNA strands (double-stranded coverage) for any nonreference base before it is called [20]. Such stringent quality control practices are surely needed to minimize error rates and the impact of random sampling variance, so that true variations and sequencing artifacts can be resolved and homozygous and heterozygous genotypes at sites of variation reliably scored.
Deep coverage alone seems not, by itself, to always be sufficient for accurate variation discovery. For example, a naïve Bayesian model for SNP calling—even with deep coverage—can lead to considerable false positive rates [38]. Thus, other stringent filtering parameters should also be applied, such as filtering out SNP calls that occur at positions with too great a coverage [44], e.g. on positions where massive pile-ups are found which are either sequencing or mapping artifacts. Increasing the number of sequenced samples (individually or multiplexed) may also result in more power to confidently call variations [45]. For instance, applying an index-based multiplexed targeted sequencing approach would remove run-to-run biases and in turn facilitate calculating error estimates for genetic polymorphism detection [46]. Computing inter-sample concordance rates at each base provides yet another way to highlight sequencing errors. Sometimes, manual read inspection is necessary to refine SNP calls, but this is time consuming unless it can be partially automated. Other useful strategies include applying index-based sample multiplexing, processing controls of known sequence (e.g. HapMap DNAs) and testing parent–child trios. These ‘multisample’ approaches allow one to estimate genotype concordance rates, detect Mendelian errors and measure allelic bias at heterozygous sites. This latter problem (systematic distortions in the recovery of one nucleotide allele over another) could be due to a bias in the targeted enrichment process, in the preparation and amplification of the sequencing library, or during sequencing or postsequencing analysis [47].
CHALLENGES AND FUTURE PERSPECTIVES
The main reason that targeted enrichment has been developed as an adjunct for NGS in recent years is that it was needed to make extensive sequencing affordable for subregions of complex genomes. The alternative of fully sequencing many complete genomes to high average coverage (~30x or higher) to enable things like genetic variation analysis, was simply not affordable. Another reason for assaying, e.g. exome rather than whole-genome sequencing is the simpler data interpretation of the former. This is a crucial consideration as it is generally much more challenging to find the functional impact of variants in noncoding regions. A comparison of today's costs for whole-genome sequencing and targeted enrichment is show in Figure 3.
Figure 3:

Sequencing costs for short read next-generation sequencing. Since introduction in early 2008, costs dropped radically and are represented in a straight line on a logarithmic scale. The cost differential between sequencing, e.g. a full human genome or a human exome (plotted with already doubled coverage) is the cost that can be spent for targeted enrichment. Therefore, targeted enrichment is still an overall cost-efficient method, if costs for targeted enrichment stays within this cost space, this especially holds true for small target regions. Data adapted from NHGRI [51].
Current targeted enrichment methods are not yet optimal, and must be improved if they are to be relevant for a long time to come. One fundamental problem is the lack of evenness of coverage [48], which is especially troublesome if the results are intended for diagnostic purposes. Poor evenness across regions with differing percentages of GC bases is a general problem for NGS itself [2], which directly translates into lower coverage of promoter regions and the first exon of genes as these are often GC rich. Such problems are exacerbated by GC content and other biases suffered by enrichment technologies. Therefore, for reliable results, a high coverage is invaluable—but current methods for targeting several mega base pairs might only return 60–80% of the ROI at a read depth of over 40x, and 80–90% at around 20x coverage.
The comparison of different genome partitioning methods in Figure 4 gives a real-world indication of how very divergent the results of the available methods can be. Even for the same genetic locus, processed by the same people in the same laboratory, the different enrichment methods produce very different average coverage, evenness and specificity. All four hybrid capture methods, including three solution phase methods (home made, Flexelect, SureSelect) and one solid phase method (NimbleGen) show considerable fluctuation in coverage over the targeted region of interest. Depending on the length of fragment library, off-target sequences protrude more or less into genomic regions adjacent to the target region. In comparison, the SelectorProbe enrichment shows a more even coverage for the targeted region and fluctuations in coverage are due to the number of hybridization probes designed. The PCR-based enrichment (RainDance) results in the most even coverage across the targeted region, but this is flanked by the typical high coverage reads for the primer pair used for enrichment.
Figure 4:

Comparison of different enrichment techniques for regions of interest. The graph shows enrichments for the genes CTNNB1 (1), PT53 (2) and ADAM10 (3) converted to wiggle format on the UCSC genome browser. For every targeted enrichment experiment, the upper graph indicates the coverage obtained after data analysis, the corresponding bar line below specifies the region of interest used for probe design. Enrichment was performed with hybridization-based techniques either in solution (home made, blue; SureSelect whole exome, magenta; FlexGen, green) or on solid support (NimbleGen, yellow), with a PCR-based approach (RainDance, orange) and a circularization method (SelectorProbes, light blue).
For an improved understanding of many single gene disorders, targeted enrichment can help produce a catalog of rare causative mutations by deeply sequencing genomic loci of a large number of patients. The analysis of genetic variation in complex disease is not necessarily limited to human DNA but can also be applied in other health-relevant fields such as microbiology [49]. In principle, targeted enrichment in conjunction with NGS provides emerging possibilities in many areas relying on molecular-based technologies ranging from microbial testing to diagnostics [50].
Still, clinical diagnostic applications of sequencing where specific clinical questions need to be answered might favor analysis of only the relevant loci at high coverage. This has a number of advantages. First, a highly accurate answer is provided, which is required when clinicians take decisions about supplying or withholding expensive targeted biological drugs to, for instance, cancer patients. Second, a targeted sequencing approach has the advantage of focusing directly to the region of interest and therefore omitting not directly relevant genomic information. Third, an important point to consider is regulatory approval of further sequencing-based diagnostic tests. Given that regulatory approval is supplied for a dedicated and specific test that addresses a specific question, a targeted sequencing approach might be more acceptable to regulatory agencies. Hence, ultimately the adoption of enrichment methods in the sequencing field may evolve differently in the research and diagnostics fields. Indeed, the future use of sequencing for diagnostics may naturally move toward a ‘single cartridge per patient’ approach, as is the current practice for other types of molecular diagnostics.
Looking to the future, whole-genome sequencing will continue to become cheaper, simpler, and faster. This will steadily erode the rationale for using targeted enrichment rather than directly sequencing the complete genome and bioinformatically extracting the sequences of interest. The long term utility of targeted enrichment will depend increasingly on progress toward evenness and enrichment power improvements (to increase the value of the data), and also on new and better strategies for sample multiplexing and pooling (to bring down the per sample cost).
In conclusion, with cheap 3rd generation sequencing on the horizon, and with improvements in targeted enrichment still occurring, the field of targeted enrichment has not yet lost its raison d'être. Current international large-scale sequencing projects like the 1000 Genomes Project [37] also rely on targeted enrichment for NGS besides whole-genome sequencing because, the upfront expenses in sample preparation are more than reimbursed by a significantly reduced total sequencing demand and reduced downstream processing in terms of data analysis and storage for generating high coverage sequence data.
Key Points.
Discussion of current targeted enrichment methods.
Use of targeted enrichment in the context of analyzing complex genomes.
Detecting genetic variation by targeted enrichment.
Considerations in terms of methodology, applicability and descriptive metrics.
Challenges and future perspectives of targeted enrichment.
FUNDING
The research leading to these results has received funding from the European Union's Seventh Framework Program [FP7/2007-2013, under grant no. 201418 (READNA) and no. 262055 (ESGI)]; the German Ministry for Education and Research (BMBF, grant no. 0315082); the Max Planck Society.
Biographies
Florian Mertes studied biotechnology and earned a Doctorate from the Technical University Berlin. Currently, he is a postdoctoral researcher focusing on applied research to develop test/screening assays based on high-throughput technologies, using both PCR and next-generation sequencing.
Abdou ElSharawy is a postdoctoral researcher (University of Kiel, CAU, Germany), and lecturer of Biochemistry and Cell Molecular Biology (Manusoura University, Egypt). He focuses on disease-associated mutations and miRNAs, allele-dependent RNA splicing, and high-throughput targeted, whole exome, and genome sequencing.
Sascha Sauer is a research group leader at the Max Planck Institute for Molecular Genetics, and coordinates the European Sequencing and Genotyping Infrastructure.
Joop M.L.M. van Helvoort is CSO at FlexGen. He received his PhD at the University of Amsterdam. His expertise is in microarray applications currently focusing on target enrichment.
P.J. van der Zaag is with Philips Research, Eindhoven, The Netherlands. He holds a doctorate in physics from Leiden University. At Philips, he has worked on a number of topics related to microsystems and nanotechnology, lately in the field of nanobiotechnology.
Andre Franke is a biologist by training and currently holds an endowment professorship for Molecular Medicine at the Christian-Albrechts-University of Kiel in Germany and is guest professor in Oslo (Norway).
Mats Nilsson is Professor of Molecular Diagnostics at the Department of Immunology, Genetics, and Pathology, Uppsala University, Sweden. He has pioneered a number of molecular analysis technologies for multiplexed targeted analyses of genes.
Hans Lehrach is Director at the Max Planck Institute for Molecular Genetics. His expertise lies in genetics, genomics, systems biology, and personalized medicine. Highlights include key involvement in several large-scale genome sequencing projects.
Anthony J Brookes is a Professor of Bioinformatics and Genomics at the University of Leicester (UK) where he runs a research team and several international projects in method development and informatics for DNA analysis through to healthcare.
References
- 1.Fuller CW, Middendorf LR, Benner SA, et al. The challenges of sequencing by synthesis. Nat Biotechnol. 2009;27(11):1013–23. doi: 10.1038/nbt.1585. [DOI] [PubMed] [Google Scholar]
- 2.Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet. 2010;11(1):31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 3.Albert TJ, Molla MN, Muzny DM, et al. Direct selection of human genomic loci by microarray hybridization. Nat Methods. 2007;4(11):903–5. doi: 10.1038/nmeth1111. [DOI] [PubMed] [Google Scholar]
- 4.Gnirke A, Melnikov A, Maguire J, et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat Biotechnol. 2009;27(2):182–9. doi: 10.1038/nbt.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bashiardes S, Veile R, Helms C, et al. Direct genomic selection. Nat Methods. 2005;2(1):63–9. doi: 10.1038/nmeth0105-63. [DOI] [PubMed] [Google Scholar]
- 6.Dahl F, Gullberg M, Stenberg J, et al. Multiplex amplification enabled by selective circularization of large sets of genomic DNA fragments. Nucleic Acids Res. 2005;33(8):e71. doi: 10.1093/nar/gni070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Summerer D. Enabling technologies of genomic-scale sequence enrichment for targeted high-throughput sequencing. Genomics. 2009;94(6):363–8. doi: 10.1016/j.ygeno.2009.08.012. [DOI] [PubMed] [Google Scholar]
- 8.Hoischen A, Gilissen C, Arts P, et al. Massively parallel sequencing of ataxia genes after array-based enrichment. Hum Mutat. 2010;31(4):494–9. doi: 10.1002/humu.21221. [DOI] [PubMed] [Google Scholar]
- 9.Li YR, Vinckenbosch N, Tian G, et al. Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nat Genet. 2010;42(11):969–82. doi: 10.1038/ng.680. [DOI] [PubMed] [Google Scholar]
- 10.Krawitz PM, Schweiger MR, Rodelsperger C, et al. Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome. Nat Genet. 2010;42(10):827–9. doi: 10.1038/ng.653. [DOI] [PubMed] [Google Scholar]
- 11.Smit A, Hubley R, Green P. RepeatMasker Open-3.0. http://www.repeatmasker.org, 1996-2010 (27 June 2011, date last accessed) [Google Scholar]
- 12.Tewhey R, Nakano M, Wang X, et al. Enrichment of sequencing targets from the human genome by solution hybridization. Genome Biol. 2009;10(10):R116. doi: 10.1186/gb-2009-10-10-r116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Antequera F, Bird A. Number of CpG islands and genes in human and mouse. Proc Natl Acad Sci USA. 1993;90(24):11995–9. doi: 10.1073/pnas.90.24.11995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marusyk A, Polyak K. Tumor heterogeneity: Causes and consequences. BBA-Rev Cancer. 2010;1805(1):105–117. doi: 10.1016/j.bbcan.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cummings N, King R, Rickers A, et al. Combining target enrichment with barcode multiplexing for high throughput SNP discovery. BMC Genomics. 2010;11:641. doi: 10.1186/1471-2164-11-641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith AM, Heisler LE, St Onge RP, et al. Highly-multiplexed barcode sequencing: an efficient method for parallel analysis of pooled samples. Nucleic Acids Res. 2010;38(13):e142. doi: 10.1093/nar/gkq368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patel VC, Mondal K, Shetty AC, et al. Microarray oligonucleotide probe designer (MOPeD): A web service. Open Access Bioinformatics. 2010;2(2010):145–55. doi: 10.2147/OAB.S13741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H, Homer N. A survey of sequence alignment algorithms for next-generation sequencing. Brief Bioinform. 2010;11(5):473–83. doi: 10.1093/bib/bbq015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kenny EM, Cormican P, Gilks WP, et al. Multiplex target enrichment using DNA indexing for ultra-high throughput SNP detection. DNA Res. 2011;18(1):31–8. doi: 10.1093/dnares/dsq029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mokry M, Feitsma H, Nijman IJ, et al. Accurate SNP and mutation detection by targeted custom microarray-based genomic enrichment of short-fragment sequencing libraries. Nucleic Acids Res. 2010;38(10):e116. doi: 10.1093/nar/gkq072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schena M, Heller RA, Theriault TP, et al. Microarrays: biotechnology's discovery platform for functional genomics. Trends Biotechnol. 1998;16(7):301–6. doi: 10.1016/s0167-7799(98)01219-0. [DOI] [PubMed] [Google Scholar]
- 22.Sauer S, Lange BM, Gobom J, et al. Miniaturization in functional genomics and proteomics. Nat Rev Genet. 2005;6(6):465–76. doi: 10.1038/nrg1618. [DOI] [PubMed] [Google Scholar]
- 23.Weier HU, Rosette C. Generation of clonal DNA templates for in vitro transcription without plasmid purification. Biotechniques. 1990;8(3):252–7. [PubMed] [Google Scholar]
- 24.Williams R, Peisajovich SG, Miller OJ, et al. Amplification of complex gene libraries by emulsion PCR. Nat Methods. 2006;3(7):545–50. doi: 10.1038/nmeth896. [DOI] [PubMed] [Google Scholar]
- 25.Dahl F, Stenberg J, Fredriksson S, et al. Multigene amplification and massively parallel sequencing for cancer mutation discovery. Proc Natl Acad Sci USA. 2007;104(22):9387–92. doi: 10.1073/pnas.0702165104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Porreca GJ, Zhang K, Li JB, et al. Multiplex amplification of large sets of human exons. Nat Methods. 2007;4(11):931–6. doi: 10.1038/nmeth1110. [DOI] [PubMed] [Google Scholar]
- 27.Johansson H, Isaksson M, Sorqvist EF, et al. Targeted resequencing of candidate genes using selector probes. Nucleic Acids Res. 2011;39(2):e8. doi: 10.1093/nar/gkq1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Turner EH, Lee C, Ng SB, et al. Massively parallel exon capture and library-free resequencing across 16 genomes. Nat Methods. 2009;6(5):315–6. doi: 10.1038/nmeth.f.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harismendy O, Frazer K. Method for improving sequence coverage uniformity of targeted genomic intervals amplified by LR-PCR using Illumina GA sequencing-by-synthesis technology. Biotechniques. 2009;46(3):229–31. doi: 10.2144/000113082. [DOI] [PubMed] [Google Scholar]
- 30.Cronn R, Liston A, Parks M, et al. Multiplex sequencing of plant chloroplast genomes using Solexa sequencing-by-synthesis technology. Nucleic Acids Res. 2008;36(19):e122. doi: 10.1093/nar/gkn502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meuzelaar LS, Lancaster O, Pasche JP, et al. MegaPlex PCR: a strategy for multiplex amplification. Nat Methods. 2007;4(10):835–7. doi: 10.1038/nmeth1091. [DOI] [PubMed] [Google Scholar]
- 32.Shen Z, Qu W, Wang W, et al. MPprimer: a program for reliable multiplex PCR primer design. BMC Bioinformatics. 2010;11:143. doi: 10.1186/1471-2105-11-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holleley CE, Geerts PG. Multiplex Manager 1.0: a cross-platform computer program that plans and optimizes multiplex PCR. Biotechniques. 2009;46(7):511–7. doi: 10.2144/000113156. [DOI] [PubMed] [Google Scholar]
- 34.Mondal K, Shetty AC, Patel V, et al. Targeted sequencing of the human X chromosome exome. Genomics. 2011;98:260–5. doi: 10.1016/j.ygeno.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tewhey R, Warner JB, Nakano M, et al. Microdroplet-based PCR enrichment for large-scale targeted sequencing. Nat Biotechnol. 2009;27(11):1025–31. doi: 10.1038/nbt.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Margulies EH, Cooper GM, Asimenos G, et al. Analyses of deep mammalian sequence alignments and constraint predictions for 1% of the human genome. Genome Res. 2007;17(6):760–74. doi: 10.1101/gr.6034307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Durbin RM, Abecasis GR, Altshuler DL, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–73. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Depristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–8. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brockman W, Alvarez P, Young S, et al. Quality scores and SNP detection in sequencing-by-synthesis systems. Genome Res. 2008;18(5):763–70. doi: 10.1101/gr.070227.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chou LS, Liu CS, Boese B, et al. DNA sequence capture and enrichment by microarray followed by next-generation sequencing for targeted resequencing: neurofibromatosis type 1 gene as a model. Clin Chem. 2010;56(1):62–72. doi: 10.1373/clinchem.2009.132639. [DOI] [PubMed] [Google Scholar]
- 41.McKernan KJ, Peckham HE, Costa GL, et al. Sequence and structural variation in a human genome uncovered by short-read, massively parallel ligation sequencing using two-base encoding. Genome Res. 2009;19(9):1527–41. doi: 10.1101/gr.091868.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith DR, Quinlan AR, Peckham HE, et al. Rapid whole-genome mutational profiling using next-generation sequencing technologies. Genome Res. 2008;18(10):1638–42. doi: 10.1101/gr.077776.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Melum E, May S, Schilhabel MB, et al. SNP discovery performance of two second-generation sequencing platforms in the NOD2 gene region. Hum Mutat. 2010;31(7):875–85. doi: 10.1002/humu.21276. [DOI] [PubMed] [Google Scholar]
- 44.Wheeler DA, Srinivasan M, Egholm M, et al. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452(7189):872–6. doi: 10.1038/nature06884. [DOI] [PubMed] [Google Scholar]
- 45.Teer JK, Mullikin JC. Exome sequencing: the sweet spot before whole genomes. Hum Mol Genet. 2010;19(R2):R145–51. doi: 10.1093/hmg/ddq333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Craig DW, Pearson JV, Szelinger S, et al. Identification of genetic variants using bar-coded multiplexed sequencing. Nat Methods. 2008;5(10):887–93. doi: 10.1038/nmeth.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hedges DJ, Guettouche T, Yang S, et al. Comparison of three Targeted Enrichment strategies on the SOLiD sequencing platform. PLoS One. 2011;6(4):e18595. doi: 10.1371/journal.pone.0018595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harismendy O, Ng PC, Strausberg RL, et al. Evaluation of next generation sequencing platforms for population targeted sequencing studies. Genome Biol. 2009;10(3):R32. doi: 10.1186/gb-2009-10-3-r32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sauer S, Kliem M. Mass spectrometry tools for the classification and identification of bacteria. Nat Rev Microbiol. 2010;8(1):74–82. doi: 10.1038/nrmicro2243. [DOI] [PubMed] [Google Scholar]
- 50.Dahl A, Mertes F, Timmermann B, et al. The application of massively parallel sequencing technologies in diagnostics. F1000 Biol Rep. 2010;2:59. doi: 10.3410/B2-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wetterstrand KA. DNA Sequencing Costs: Data from the NHGRI Large-Scale Genome Sequencing Program. www.genome.gov/sequencingcosts (18 October 2011, date last accessed) [Google Scholar]
- 52.Okou DT, Locke AE, Steinberg KM, Hagen K, Athri P, Shetty AC, Patel V, Zwick ME. Combining microarray-based genomic selection (MGS) with the Illumina Genome Analyzer platform to sequence diploid target regions. Ann Hum Genet. 2009;73(Pt 5):502–13. doi: 10.1111/j.1469-1809.2009.00530.x. [DOI] [PMC free article] [PubMed] [Google Scholar]