

Abstract
A protein quality control system, consisting of molecular chaperones and proteases, controls the folding status of proteins and prevents the aggregation of misfolded proteins by either refolding or degrading aggregation-prone species. During severe stress conditions this protection system can be overwhelmed by high substrate load, resulting in the formation of protein aggregates. In such emergency situations, Hsp104/ClpB becomes a key player for cell survival, as it has the extraordinary capacity to rescue proteins from an aggregated state in cooperation with an Hsp70 chaperone system. The ring-forming Hsp104/ClpB chaperone belongs to the AAA+ protein superfamily, which in general drives the assembly and disassembly of protein complexes by ATP-dependent remodelling of protein substrates. A disaggregation activity was also recently attributed to other eubacterial AAA+ proteins, while such an activity has not yet been identified in mammalian cells. In this review, we report on new insights into the mechanism of protein disaggregation by AAA+ proteins, suggesting that these chaperones act as molecular crowbars or ratchets.
Review
Holding and folding: Molecular chaperones in protein quality control systems
The viability of all organisms crucially depends on the integrity and the correlated activity of its constituent proteins, stressing the fundamental importance of correctly folded proteins. While the information for a three-dimensional protein structure is provided by its corresponding primary amino acid sequence, the folding process is, however, sensitive to external parameters such as temperature, solvent properties or the concentration of macromolecules in solution. Given the high degree of molecular crowding in cells (340 gL-1 macromolecules) [1] and the exposure of hydrophobic residues during the de novo folding of newly synthesized polypeptides, proteins can be trapped during the folding process in misfolded intermediates that are prone to aggregation. In particular, large multidomain and oligomeric proteins encounter this problem, as they tend to follow a slow folding pathway. Even once a protein has achieved its correct conformation it is still subject to several forms of stress that interfere with its structural integrity. These include physical and chemical influences that can result in protein unfolding, such as changes in temperature or ionic strength, oxidative stress or the presence of heavy metals.
To cope with these stresses a group of highly conserved proteins has evolved, specialized in assisting the folding of nascent polypeptides and the refolding of misfolded proteins. This protein class was first discovered as part of the heat shock response as the gene expression of its members was stimulated by the exposure of cells to heat [2]. These effector molecules were therefore termed heat shock proteins (Hsp) and later on defined as molecular chaperones. Chaperones control the folding status of proteins and, together with ATP-dependent proteases, form the cellular protein quality control system (Fig. 1). Depending on their function, chaperones can be classified into "holder" or "folder" chaperones. Holder chaperones (e.g. small Hsps) can prevent the aggregation of misfolded protein species without mediating protein reactivation. Folder chaperones (e.g. Hsp60/GroEL, Hsp70/DnaK) in addition support substrate refolding (for review see [3]).
Figure 1.
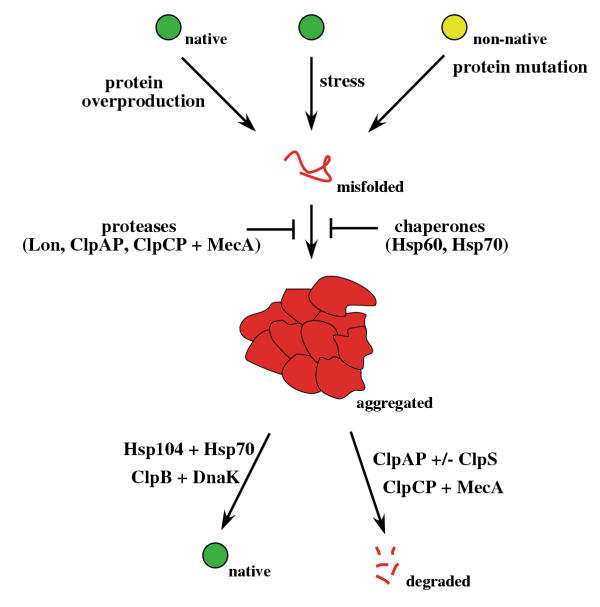
Prevention and reversion of protein aggregation by chaperones and proteases in eubacteria. Protein aggregation is caused by misfolded protein species and can be facilitated by overproduction of heterologous proteins, thermal or oxidative stress and protein mutations affecting protein folding. Formation of protein aggregates can be prevented either by Hsp60 or Hsp70-mediated refolding of aggregation-prone substrates or by removal of the misfolded protein species by proteolytic systems (Lon, ClpAP, ClpCP/MecA). High levels of misfolded proteins can overburden the capacity of chaperones and proteases, leading to protein aggregation. Aggregated proteins are rescued by the Hsp104/Hsp70 or the ClpB/DnaK bi-chaperone systems. Protein aggregates can also be solubilized by ClpCP/MecA or ClpAP (+/- ClpS) resulting in substrate hydrolysis.
However, the capacity of chaperones to protect misfolded proteins is limited. High levels of misfolded proteins can exceed the buffering capacity of chaperones, leading to protein aggregation. Protein aggregation is facilitated by partial unfolding during thermal or oxidative stress, by overproduction of heterologous proteins and/or by alterations in primary structure caused by mutation (Fig. 1). The terms "inclusion bodies" and "aggresomes" have been applied to intracellular foci into which aggregated proteins are sequestered [4]. The toxicity of protein aggregates is unclear. It is possible that the aggregates may disturb the fine-tuned folding equilibrium of cellular proteins by affecting their folding status or by sequestering molecular chaperones. Massive protein aggregation that occurs during severe stress conditions can therefore cause damage of cellular structures and metabolism, resulting in cell death. Since the ability of classical chaperones to rescue proteins from an aggregated state is limited, protein aggregates were once viewed as "dead ends" in the life cycle of proteins.
The discovery of Hsp104: a chaperone with unique activities
In 1990 Susan Lindquist and co-workers discovered a new Hsp gene of Saccharomyces cerevisiae, termed HSP104, and its involvement in induced thermotolerance [5]. It was shown that upon mild pre-heat treatment (37°C) S. cerevisiae cells could subsequently survive extreme temperatures (50°C). In contrast, Δhsp104 mutant cells did not exhibit an adaptation to extreme temperatures and instead died with a rate up to 1000-fold faster than wild type cells. This defect could be entirely rescued through transformation of the HSP104 wild type gene, proving the dependence of thermotolerance development on Hsp104. It could be further demonstrated that Hsp104 is required for tolerance to many different forms of stress such as ethanol, arsenite or long-term storage of the yeast cells at low temperatures [6]. Even more striking was the finding that large electron-dense aggregates that formed after heat shock in the nucleus and the cytoplasm of S. cerevisiae were the main target of Hsp104 action [7]. S. cerevisiae cells expressing Hsp104 were able to resolubilize these aggregates, thus regaining the function of the affected proteins as demonstrated for the model substrate luciferase. Importantly Hsp104 was unable to prevent the aggregation of chemically denatured luciferase, differentiating it from classical chaperones [7]. These findings opened a new chapter in the understanding of molecular chaperones as their activities had been so far believed to be restricted to preventing the aggregation of misfolded protein species. The more defensive action of the classical holder and folder chaperones was now complemented by Hsp104's ability to act on aggregated proteins.
This extraordinary activity explains the cell's capacity to offensively cope with severe stress conditions that lead to protein aggregation. The unique function of Hsp104 is conserved in eubacteria, plants and mitochondria by its homologues ClpB, Hsp101 and Hsp78, which each are essential for thermotolerance and protein disaggregation [8-13]. The ability of these organisms and organelles to adapt to severe stress conditions relies on the activity of Hsp104/ClpB to reverse protein aggregation.
From a subfamily of heat shock proteins to a superfamily of AAA+ proteins
Through sequence alignments Hsp104/ClpB could be first assigned to the conserved Hsp100/Clp protein family, a group of ring-forming ATPases [14]. Neuwald and colleagues found that Hsp100/Clp proteins and AAA proteins (ATPase associated with a variety of cellular activities) share conserved motifs involved in ATP binding and hydrolysis and they combined both protein classes to generate a new protein superfamily: the AAA+ proteins [15]. Generally, AAA+ proteins remodel target substrates in an ATP-dependent manner, an activity that is of central importance for a large number of cellular processes [16,17]. AAA+ proteins share a common ATPase domain with a conserved sequence of 230–250 amino acid residues, termed the AAA domain. Each AAA domain consists of a core ATPase domain, containing the classical Walker A and B motifs, and a C-terminal α-helical domain. The structural features of this superfamily were confirmed by structure determinations of AAA domains of various proteins [18-24]. AAA domains mediate the oligomerization of AAA+ proteins, ATP binding and ATP hydrolysis. Depending on the number of AAAdomains, AAA+ proteins can be divided into two subgroups. Class I AAA+ proteins consist of two AAA domains, whereas class II members possess only one [14].
Hsp104/ClpB belongs to class I and the functionality of each of its AAA domains (AAA-1 and AAA-2) is crucial for efficient protein disaggregation (Fig. 2A) [25-29]. The recently published crystal structure of Thermus thermophilus ClpB in complex with AMPPNP shows that the structure and overall topology of the AAA domains is similar to that of E. coli ClpA (Fig. 2B) [18,24]. In a hexameric ClpB model based on single particle reconstitution by electron microscopy, the AAA domains are oriented in a head-to-tail arrangement and AAA-1 is located directly above AAA-2 of the same subunit [24]. AMPPNP is bound at the interface of neighbouring subunits in the oligomeric model and a conserved arginine residue, termed "arginine finger", is contacting the γ-phosphate from an adjacent ClpB molecule. This interaction serves to sense the state of bound nucleotide in the neighbouring subunit and can therefore explain the observed cooperativity in ATP hydrolysis within the Hsp104 and T. thermophilus ClpB oligomer [30,31]. E. coli ClpB mutants with mutational alterations of the "arginine finger" in either the first or the second AAA domain are deficient in protein disaggregation, demonstrating that this motif in each of the domains is important for ClpB function [26].
Figure 2.
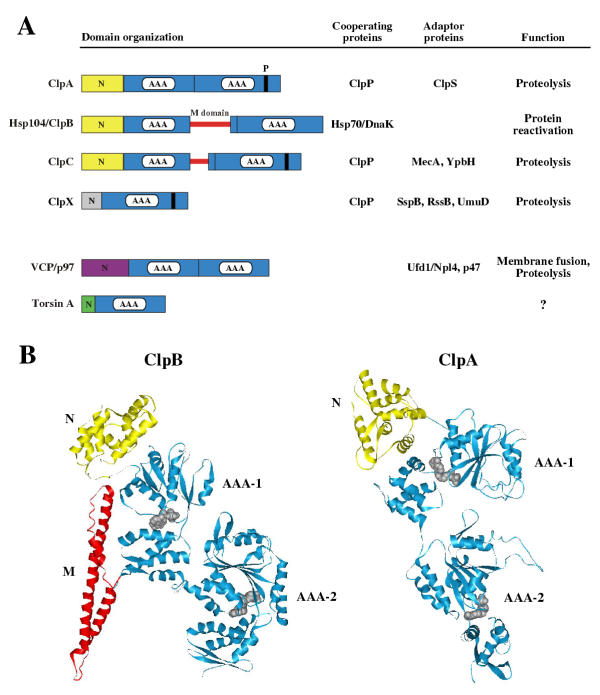
Structural features of AAA+ proteins. (A) Domain organization and cooperating proteins of AAA+ chaperones. N-terminal domains of AAA+ proteins are diverse and are in most cases involved in substrate recognition, either directly or indirectly by serving as a binding platform for adaptor proteins. In the case of Torsin A, the N-domain represents a membrane-spanning region. The first AAA domains of ClpB and ClpC are interrupted by a middle domain (M-domain) of variable length. ClpP-association of ClpA, ClpC and ClpX is mediated by a conserved docking site (P). Adaptor proteins, cooperating proteins and functions of the corresponding AAA+ proteins are given. The ClpX adaptor proteins SspB, RssB and UmuD target SsrA-tagged proteins, the alternative sigma factor σS and UmuD' for degradation by ClpXP, respectively [90-93]. (B) Structure of monomeric T. thermophilus ClpB and E. coli ClpA. The colour code of the individual domains is equivalent to (A). AMPPNP (ClpB) or ADP (ClpA) is shown as CPK model in grey.
Hsp104/ClpB: an AAA+ protein with unique features
In addition to their AAA domains AAA+ proteins also contain variable regions at their N-termini. N-domains of Hsp104/ClpB are homologous to those of ClpA and ClpC and form independent structural domains that are connected via flexible linkers to AAA-1 (Fig. 2A/2B) [24,32]. Comparison of the individual ClpB molecules present in the asymmetric ClpB crystal unit revealed that the N-domains are highly mobile and possess a large degree of rotational freedom [24]. The function of the N-domains in ClpB-mediated protein disaggregation is still enigmatic. While ClpB variants carrying point mutations in the N-domain exhibited a reduced disaggregation activity, ClpB deletion variants from various sources missing the entire N-domain were as active as the full-length proteins [26,32-35]. The existence of ClpB homologues lacking the N-domain in Mycoplasma sp. also argues against an essential function of N-domains in the disaggregation process [36,37]. Therefore N-domains of ClpB may rather be involved in other more specialized ClpB functions that are distinct from protein disaggregation.
The most striking feature of Hsp104/ClpB is the presence of a middle domain (M-domain) that is absent in ClpA, the closest homologue of ClpB in E. coli (Fig. 2A/2B). While not directly involved in Hsp104/ClpB oligomerization, the M-domains possess an essential function in protein disaggregation [26,38]. The dependence of the disaggregation reaction on Hsp104/ClpB in vivo underlines the crucial role of this unique domain in the solubilization process. A major step in understanding the M-domain function was achieved by structure determination of Thermus thermophilus ClpB [24]. The M-domain forms a large coiled-coil structure that resembles a two-bladed propeller (Fig. 2B). In a hexameric ClpB model, the M-domains are located adjacent to AAA-1 on the outer surface of the molecule [24]. Interestingly, the M-domains exhibited a high degree of mobility as revealed by superposition of the individual ClpB molecules present in the asymmetric ClpB crystal unit. M-domain mobility was demonstrated to be crucial for protein disaggregation, as inhibition of M-domain motion caused a loss of ClpB chaperone activity without affecting ClpB oligomerization and ATPase activity [24]. These findings led to the suggestion that the M-domains may serve as molecular crowbars that modulate the structure of protein aggregates (see below). Multiple contacts between the M-domain and exposed peptide segments on the aggregate surface may also serve as a scanning device for aggregated proteins. Direct evidence for interactions between M-domains and aggregated proteins is still missing and such binding might be difficult to detect given the often transient nature of AAA+ protein-substrate interaction.
In contrast to many other prokaryotic AAA+ proteins that are key players in the cellular protein quality control system, ClpB is not involved in proteolysis. AAA+ proteins that are implicated in proteolysis usually associate with peptidases to form proteolytic systems that resemble the eukaryotic proteasome. The AAA+ subunits of these protease complexes function as regulatory components since they control the access of substrates to the proteolytic chamber of the associated peptidase. The association of the AAA+ component (e.g. ClpA, ClpC, ClpX) with the peptidase (e.g. ClpP) is mediated by a conserved docking site (Fig. 2A). The signature motif (LIV-G-FL) is present in the AAA domains of all ClpP-interacting AAA+ proteins and is located distal to their substrate binding site [39]. Importantly, ClpB is lacking the signature motif and, as a consequence, ClpB is not involved in proteolysis, yet is able to act in protein reactivation.
Crowbars and ratchets: potential activities of Hsp104/ClpB
Which specific activities enable Hsp104/ClpB to reverse protein disaggregation? The unique function of Hsp104/ClpB combined with the determined characteristics of the Hsp104/ClpB-specific M-domain led to the suggestion of a crowbar activity. This model suggests that movements of the M-domains of Hsp104/ClpB cause changes in the aggregate structure and/or a conversion of large protein aggregates into smaller species (Fig. 3) [24,40,41]. Fragmentation of large aggregates by the postulated crowbar mechanism has not yet been demonstrated as Hsp104/ClpB-mediated changes in the aggregate might be rather transient in the absence of additional chaperone systems.
Figure 3.
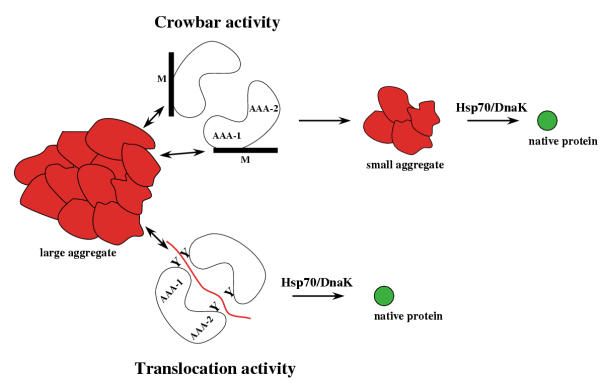
Potential mechanisms of protein disaggregation by Hsp104/ClpB. Hsp104/ClpB may break large aggregates into smaller species via a crowbar activity, mediated by movements of M-domains. Small-sized aggregates serve as substrates for Hsp70/DnaK in a sequential reaction. Hsp104/ClpB may also extract unfolded proteins from an aggregate via a translocation activity with major contributions of pore-located aromatic residues (Y). Hsp70/DnaK may directly take over the translocated polypeptide, thereby preventing the re-aggregation of extracted proteins and ensuring substrate refolding. Both suggested Hsp104/ClpB activities are not mutually exclusive, but rather may act sequentially or in concert.
In the case of peptidase-associated AAA+ proteins, a ratchet mechanism that mediates the unfolding and translocation of bound substrates has been proposed [42]. Key players of this postulated activity are conserved aromatic residues that are located at the central pore of AAA+ proteins. These aromates may serve as molecular clamps by binding and releasing substrates, thereby preventing the backsliding of polypeptides during the translocation process. It has been further suggested that nucleotide-dependent conformational changes of these residues directly couple substrate translocation to substrate unfolding [42]. Consistent with such an activity, the corresponding aromatic residues in HslU and FtsH have been shown to be crucial for substrate degradation [43,44]. Interestingly, the aromatic residues and their relative positioning are conserved in both AAA domains of Hsp104/ClpB [41]. Furthermore, these residues appear to be located on mobile loops, as the corresponding regions could not be resolved in the ClpB crystal structure [24]. These observations suggest that Hsp104/ClpB may, like peptidase-associated AAA+ proteins, exhibit a translocation activity that could mediate the extraction of an unfolded polypeptide from the aggregate (Fig. 3). A translocation activity of Hsp104/ClpB as part of the protein disaggregation reaction and a role of the aromatic residues in such an activity still needs to be investigated. It is important to note that the potential crowbar and translocation activities of Hsp104/ClpB are not mutually exclusive but rather may act sequentially or in concert, resulting in efficient protein disaggregation.
The missing partner: Hsp70 systems cooperate with Hsp104/ClpB in reversing protein aggregation
While the dependence of protein disaggregation in vivo on Hsp104/ClpB could be established for various organisms, the reconstitution of this activity in vitro failed for a long time. Incubation of aggregated proteins with either Hsp104 or ClpB alone did not result in aggregate solubilization. A breakthrough was made by Glover and Lindquist, when they demonstrated that Hsp104 needs the assistance of an Hsp70 system to reverse protein aggregation [40]. The disaggregation activity of this novel bi-chaperone system could be subsequently established for the E. coli and T. thermophilus chaperone homologues [45-47]. The essential role of the Hsp70 chaperone system in protein disaggregation was also confirmed in vivo, as E. coli ΔdnaK mutant cells could no longer solubilize aggregated proteins formed during heat shock [8].
The precise mechanism of Hsp104/Hsp70 or ClpB/DnaK cooperation during protein disaggregation is unknown; however there are two working models. The first model proposes a sequential action of both chaperones [45]. Hsp70/DnaK are suggested to act on small-sized protein aggregates, generated by the postulated crowbar activity of ClpB (Fig. 3). This model is supported by the findings that Hsp70/DnaK are able to solubilize small protein aggregates in the absence of Hsp104/ClpB [40,48].
The second model relies on the observation that cooperation of the two chaperone systems exists only between proteins of the same species [40,49]. Thus, mixtures of S. cerevisiae Hsp104 and E. coli DnaK (and vice versa) do not function in protein disaggregation. Interestingly, species-specificity can be directly attributed to the Hsp70 chaperones as the Hsp40 co-chaperones (Ydj1, DnaJ) of the individual species can be exchanged without loosing disaggregation activity. These findings suggest that the solubilization of protein aggregates is achieved by the simultaneous action of a Hsp104/Hsp70 or a ClpB/DnaK complex. The role of Hsp70/DnaK in such a potential complex is still unclear. Hsp70/DnaK might be necessary to accept translocated substrates directly from Hsp104/ClpB, ensuring efficient substrate translocation and refolding (Fig. 3). Such an activity would be reminiscent to the function of AAA+ proteins that are associated with peptidases, but links Hsp104/ClpB activity to protein refolding instead of substrate degradation. Direct evidence in support of either model is still missing. Clearly, the determination of Hsp70/DnaK positioning in the disaggregation process will help to clarify the mechanism of the Hsp104/ClpB-mediated solubilization process.
Protein disaggregation by other AAA+ proteins
While the solubilization of aggregated proteins in E. coli cells is dominated by ClpB, a disaggregation activity could be also assigned to other AAA+ proteins (Fig. 1). E. coli ClpA can, in complex with the peptidase ClpP, solubilize heat-aggregated MDH and firefly luciferase in vitro [50]. ClpA is homologous to ClpB, but is missing the ClpB-specific middle domain (Fig. 2A/2B). The ability of ClpAP to degrade aggregated proteins implies that protein disaggregation can, in principal, be achieved by a "translocation only" mechanism. The disaggregation activity of ClpAP is further stimulated in the presence of ClpS, a ClpA-specific adaptor protein [50]. Adaptor proteins increase the substrate specificity of AAA+ proteins in general by trans-targeting specific substrates to their AAA+ partner proteins. Interestingly, ClpS has dual activity, as it not only increases protein disaggregation by ClpAP, but also prevents the degradation of SsrA-tagged substrates and ClpA itself by ClpAP [50]. The influence of ClpS on protein disaggregation by ClpAP can be explained by improved targeting of the proteolytic system to aggregated proteins. ClpS may also exert an indirect effect as it protects ClpA from autodegradation, thereby ensuring constant ClpA levels throughout the disaggregation reaction.
ClpS activity is achieved by binding to the N-domain of ClpA [50]. The activity of ClpS is restricted to ClpA, as it does not interact with ClpB, despite the overall similarity between the N-domains of both AAA+ proteins. The solved crystal structure of ClpS in complex with the N-domain of ClpA provides a structural rationale for the binding specificity of ClpS. Thus conserved residues of the N-domain of ClpA, which make crucial contacts to ClpS, are missing in the N-domain of ClpB [51]. While efficient in degrading aggregated proteins in vitro, ClpAPS cannot replace ClpB function in vivo [8,50]. Protein aggregates generated upon heat shock in vivo may be different in structure and complexity to those formed by thermolabile model substrates in vitro. The true nature of ClpAPS substrates and the in vivo function of this proteolytic system remain unknown.
A disaggregation activity towards aggregated λ O protein has been described for the AAA+ protein ClpX from E. coli [52]. ClpX belongs to the class II AAA+ proteins, as it consists of only one AAA domain (Fig. 2A). While efficient in disaggregating λ O protein, ClpXP fails to solubilize other aggregated model substrates, including Malate Dehydrognease (MDH) and luciferase (A. Mogk and B. Bukau, unpublished results). One explanation for these controversial results may be the size of the tested protein aggregates. Generated λ O aggregates, in contrast to aggregated MDH, can still be recovered by gelfiltration chromatography, indicating protein aggregates of smaller size. Since ClpA and ClpB can solubilize large-sized protein aggregates (e.g. MDH aggregates) in vitro, it is tempting to speculate that the solubilization of large protein aggregates may require a stronger force and therefore the presence of two AAA domains. Consistent with this hypothesis, other class II AAA+ proteins including HslU and Lon also do not exhibit a general disaggregation activity in vitro (A. Mogk and B. Bukau, unpublished results).
In Bacillus subtilis, the gram-positive model organism, ClpA and ClpB are missing. Instead B. subtilis encodes for the AAA+ protein ClpC, which resembles ClpA and ClpB, as it possesses an M-domain of reduced size and a ClpP docking site (Fig. 2A). ClpC was first identified together with its adaptor protein MecA in a genetic screen for repressors of competence development, the ability of cells to take up foreign DNA [53]. In this regulatory pathway ClpC acts together with MecA to target the master regulator of competence development (ComK) for degradation by the ClpCP proteolytic system [54,55]. ClpC also functions as a general stress protein. Its expression is heat shock regulated and B. subtilis ΔclpC mutant cells are thermosensitive and accumulate deposits of misfolded proteins [56-58]. Furthermore, ClpC, like ClpP, localizes to protein aggregates in heat-shocked B. subtilis cells and to inclusion bodies formed by overexpressed insoluble heterologous proteins [58,59]. These data suggest that ClpC might replace ClpB function in the gram-positive counterpart of E. coli, although it rather links protein disaggregation to substrate degradation instead of substrate reactivation.
A protein disaggregation activity of ClpC could be recently reconstituted in vitro [60]. Surprisingly, ClpCP activity was strictly dependent on the adaptor protein MecA, regardless of the nature of substrate. Thus ClpCP-mediated degradation of the specific substrate ComK, as well as hydrolysis of aggregated model substrates and unfolded α-casein, occurred only in presence of MecA. Further analysis revealed that MecA has a dual functions: it delivers substrates to ClpC and concurrently stimulates the ATPase activity of the AAA+ protein [60]. This activity is reflected in the domain organization of the adaptor protein: the interaction with the ClpC partner protein is mediated via a C-terminal domain, while substrates are recognized via its N-domain [61]. Interestingly B. subtilis also encodes a parologue of MecA: YpbH. YpbH can, at least in vitro, take over MecA function and enable ClpCP to degrade aggregated proteins [60]. Since B. subtilis cells lacking MecA and YpbH function are, in contrast to ΔclpC mutant cells, not thermosensitive, it is tempting to speculate that ClpC activity in vivo is further controlled by other yet to be identified adaptor proteins.
Protein aggregation and neurodegenerative diseases: a connection to AAA+ proteins?
Protein misfolding and aggregation is thought to be central in the pathogenesis of several neurodegenerative diseases including Parkinson's disease, polyglutamine diseases and prion diseases. In contrast to stress-generated amorphous aggregates, disease-related protein aggregation leads to the formation of amyloid fibrils. Here we will concentrate on polyglutamine (polyQ) diseases and their potential connection to AAA+ proteins. PolyQ diseases (e.g. Huntington's disease) are caused by trinucleotide (CAG) repeat expansions. The genes responsible for these diseases are functionally unrelated and the only known common feature is a CAG trinucleotide repeat in each gene's coding region, resulting in a polyQ tract in the disease protein. Expansion of this polyQ tract results in aggregation of the corresponding proteins that is coupled to neurodegeneration [62]. It has been proposed that proteins with long polyglutamine tracts misfold and aggregate as antiparallel β strands [63]. PolyQ-dependent protein aggregation has now been demonstrated in many different model systems, including S. cerevisiae [64], mammalian cell-culture systems [65] and transgenic animals [66-68]. Regulated expression of huntingtin (Htt) exon 1 protein, which aggregates and causes toxicity in Huntington's disease, resulted in polyQ inclusions and neurodegeneration in a transgenic mouse model [69]. Surprisingly, when the expression of Htt exon 1 protein was switched off after intracellular inclusion had been formed, neurological symptoms and polyQ aggregates disappeared [69]. These findings suggest the existence of a disaggregation activity in mammalian cells and could point to a potential involvement of AAA+ proteins in polyQ aggregation.
A direct connection between AAA+ proteins and polyQ-dependent protein aggregation could first be demonstrated by Lindquist and co-workers. They established a model system in S. cerevisiae to investigate the influence of chaperones on polyQ-induced protein aggregation by fusing the N-terminal region of Htt protein, carrying polyQ tracts of variable length, to GFP [64]. Only fusion proteins with long polyQ tracts (Q72, Q103) formed cytoplasmic protein aggregates. Importantly, deletion of HSP104 completely blocked the formation of polyQ-aggregates and overproduction of Hsp104 strongly reduced polyQ aggregation. Similar findings were subsequently reported when C. elegans was used as model system: expression of GFP-polyQ82 led to protein aggregation and retarded growth rates and both effects were reversed upon co-expression of Hsp104 [68]. These findings are reminiscent to the influence of Hsp104 on [PSI+] propagation in S. cerevisiae, a self-perpetuating aggregate of Sup35 (for review see [70]). [PSI+] propagation strictly depends on Hsp104 as loss of Hsp104 function or Hsp104 overproduction lead to curing of [PSI+] [71]. It is proposed that the fragmentation of Sup35 fibers into shorter polymers by Hsp104 is essential for their inheritance [72]. Overproduction of Hsp104 may therefore cause too excessive fiber fragmentation whereas missing Hsp104 function will lead to the formation of large Sup35 aggregates, resulting in [PSI+] loss in either case. Given the dependence of polyQ aggregation on Hsp104 in yeast, it is tempting to speculate that the formation and reversion of yeast prion Sup35 and polyQ aggregates may follow a common principle and that proteins with disaggregation activities might be involved in the development of neurodegenerative diseases in mammalian cells.
A mammalian counterpart of Hsp104 has not been identified yet, however, several paralogues have been described which may reverse protein aggregation. To investigate the potential involvement of chaperones in the propagation of neurodegenerative diseases, many laboratories tested for the presence of chaperones in polyQ inclusions or Lewy bodies, which are deposits of α-synuclein. Besides Hsp70 and Hsp40 chaperones, several AAA+ proteins were detected in such neuronal inclusions, including VCP/p97, TorsinA and the 19S proteasomal complexes. Since the role of Hsp70/Hsp40 proteins in pathogenesis has been extensively described by others (for review see [73]), we will concentrate on recent findings on AAA+ proteins in this process.
VCP/p97 (valosin-containing protein), the mammalian homologue of yeast Cdc48, possess two AAA domains and is a prime candidate for representing a disaggregation activity in mammalian cells (Fig. 2A). VCP/p97 is involved in a variety of cellular processes, including membrane fusion, protein disassembly and ubiquitin-dependent degradation (for review see [74]). VCP/p97 was shown to co-localize with polyQ-aggregates in cultured cells and with Lewy bodies in patient samples [75]. Importantly, loss of function mutants of VCP/p97 could suppress eye degeneration in transgenic flies, caused by targeted expression of polyQ proteins in the Drosophila compound eye [76]. In contrast, co-expression of VCP/p97 with polyQ-containing proteins enhanced polyQ toxicity in the same study. These recent findings demonstrate a direct influence of VCP/p97 on polyQ toxicity. Since the curing of polyQ toxicity by VCP/p97 mutations was not directly coupled to a loss of polyQ aggregates, it is unclear whether VCP/p97 acted on polyQ aggregates or influenced the aggregation process or toxicity of intermediate species.
Torsin A is the second mammalian AAA+ protein that may exhibit a disaggregation activity. TorsinA is the causative protein in the neurologic disease torsin dystonia, a movement disorder [77]. TorsinA is an ER luminal protein with a signal sequence and a short membrane-spanning domain at the N-terminus (Fig. 2A) [78-80]. The C-terminal AAA domain of TorsinA exhibits greatest homology to Hsp104/ClpB. TorsinA was found to accumulate with α-synuclein in Lewy bodies and to localize to sites of polyQ-dependent protein aggregation in C. elegans [81-83]. Interestingly, overexpression of TorsinA could suppress α-synuclein aggregation in a cell culture model, whereas a mutant TorsinA derivative had no effect [81]. Similarly, overexpression of Torsin proteins reduced the number and size of GFP-polyQ82 containing aggregates in C. elegans, while a mutant Torsin variant did not [83].
PolyQ inclusions also stain positively for 20S and 19S proteasomal complexes [84]. The 19S cap of the proteasome is functionally equivalent to the AAA+ components of ATP-dependent proteolytic systems in eubacteria and contains a ring of six AAA+ proteins. The finding that the addition of proteasome inhibitors caused an increase in aggregate formation in transfected cells led to the suggestion that the proteasome may be involved in polyQ pathogenesis [85]. Other findings point to a rather indirect role of proteasomes in protein aggregation. A surprisingly high proportion (30%) of all newly synthesized proteins are apparently misfolded and degraded by proteasomes shortly after their synthesis [86]. Inhibition of the proteasome will therefore lead to an imbalance between the capacity of chaperones to prevent protein aggregation and the number of aggregation-prone protein species, resulting in increased protein aggregation. The role of proteasomes in protein aggregation may thereby be restricted to the degradation of misfolded substrates before they aggregate.
To summarize, recent findings point to a potential role of mammalian AAA+ proteins in the propagation of neurodegenerative diseases. Direct evidence for a disaggregation activity of AAA+ proteins as basis for such a function is still missing. The presence of AAA+ proteins in protein inclusions may also only indicate an unsuccessful attempt of these chaperones to prevent the aggregation of misfolded and still soluble intermediates. Similarly, reduction of protein aggregation by co-expression of AAA+ proteins in transfected cells may be caused by the interaction of AAA+ proteins with such non-native intermediates and not by the active solubilization of pre-existing protein aggregates. Furthermore in vitro and in vivo studies are required to investigate whether VCP/p97 or Torsin A fulfill a similar function in mammalian cells as ClpB/Hsp104 does in yeast and eubacteria.
Perspectives
The discovery of the substrate-rescuing disaggregation activity of Hsp104/ClpB revealed a new and highly remarkable chaperone function illuminating the fate of aggregated proteins. A disaggregation activity could now be attributed to various eubacterial AAA+ proteins. Interestingly, each AAA+ disaggregation system requires the assistance of different cooperating proteins. Future work will clarify how the various partner proteins affect the mechanism of the disaggregation reaction and will lead to a better understanding of AAA+ protein activity. So far no orthologues of Hsp104/ClpB are evident in animal genomes. However, recent studies on the development of conformational diseases suggest the existence of AAA+ proteins with disaggregation activities in mammalian cells. It will be important to obtain biochemical evidence for this activity and to identify proteins that cooperate in the solubilization process. Such proteins may then also serve as therapeutic targets, potentially offering new strategies in the treatment of neurodegenerative diseases.
The extraordinary chaperone activity of AAA+ proteins may also offer a new perspective in biotechnology. High-level expression of heterologous proteins in eubacteria often results in the deposition of the desired product in inclusion bodies. While this phenomenon is beneficial for initial purification steps, it is often difficult and inefficient to regain the functional protein. Therefore co-overexpression of chaperones and the protein of interest were performed in many studies to prevent inclusion body formation and to increase the yield of soluble, native client protein. While this approach has been successful in a number of cases, it failed in many others and as a consequence the applications of molecular chaperones rely on trial and error experiments [87]. The disaggregation activity of AAA+ proteins represents a new quality in the biotechnical application of molecular chaperones, since the co-overproduction of these systems allows transient aggregation of heterologous proteins. Indeed, overproduction of ClpB and the DnaK chaperone machinery completely solubilized the inclusion-body like aggregates formed in E. coli ΔrpoH mutant cells after heat stress [88]. Furthermore, Villaverde and co-workers could demonstrate that even the formation of inclusion bodies can be reversed in E. coli cells [89]. Inclusion body disintegration was achieved by simultaneously arresting protein synthesis and lowering the growth temperature. It is tempting to speculate that solubilization of inclusion bodies relies on the disaggregation activity of AAA+ proteins (e.g. ClpB/DnaK). Together these findings offer a new perspective to increase the solubility of a wide variety of heterologous proteins upon overproduction in eubacteria.
Acknowledgments
Acknowledgements
This work was supported by grants from the DFG (Leibnizprogramm Bu617/14-1 and Bu617/15-1) and the Fond der Chemischen Industrie to B.B. and A.M We are grateful to Renee Wegrzyn for critical reading of the manuscript.
Contributor Information
Jimena Weibezahn, Email: j.weibezahn@zmbh.uni-heidelberg.de.
Bernd Bukau, Email: bukau@zmbh.uni-heidelberg.de.
Axel Mogk, Email: a.mogk@zmbh.uni-heidelberg.de.
References
- Goodsell DS. Inside a living cell. TiBS. 1991;16:203–206. doi: 10.1016/0968-0004(91)90083-8. [DOI] [PubMed] [Google Scholar]
- Moran L, Mirault ME, Arrigo AP, Goldschmidt-Clermont M, Tissieres A. Heat shock Drosophila melanogaster induces the synthesis of new messenger RNAs and proteins. Philos Trans R Soc Lond B Biol Sci. 1978;283:391–406. doi: 10.1098/rstb.1978.0044. [DOI] [PubMed] [Google Scholar]
- Hartl FU, Hayer-Hartl M. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science. 2002;295:1852–1858. doi: 10.1126/science.1068408. [DOI] [PubMed] [Google Scholar]
- Kopito RR, Sitia R. Aggresomes and Russell bodies. Symptoms of cellular indigestion? EMBO Rep. 2000;1:225–231. doi: 10.1093/embo-reports/kvd052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez Y, Lindquist SL. HSP104 required for induced thermotolerance. Science. 1990;248:1112–1115. doi: 10.1126/science.2188365. [DOI] [PubMed] [Google Scholar]
- Sanchez Y,, Taulin J,, Borkovich KA, Lindquist S. Hsp104 is required for tolerance to many forms of stress. EMBO Journal. 1992;11:2357–2364. doi: 10.1002/j.1460-2075.1992.tb05295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsell DA, Kowal AS, Singer MA, Lindquist S. Protein disaggregation mediated by heat-shock protein Hsp104. Nature. 1994;372:475–478. doi: 10.1038/372475a0. [DOI] [PubMed] [Google Scholar]
- Mogk A, Tomoyasu T, Goloubinoff P, Rüdiger S, Röder D, Langen H, Bukau B. Identification of thermolabile E. coli proteins: prevention and reversion of aggregation by DnaK and ClpB. EMBO J. 1999;18:6934–6949. doi: 10.1093/emboj/18.24.6934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt M, Neupert W, Langer T. The molecular chaperone Hsp78 confers compartment-specific thermotolerance to mitochondria. J Cell Biol. 1996;134:1375–1386. doi: 10.1083/jcb.134.6.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queitsch C, Hong SW, Vierling E, Lindquist S. Heat shock protein 101 plays a crucial role in thermotolerance in Arabidopsis. Plant Cell. 2000;12:479–492. doi: 10.1105/tpc.12.4.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JG, Baneyx F. Roles of the Escherichia coli small heat shock proteins IbpA and IbpB in thermal stress management: comparison with ClpA, ClpB, and HtpG in vivo. J Bacteriol. 1998;180:5165–5172. doi: 10.1128/jb.180.19.5165-5172.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowska E, Kuczynska-Wisnik D, Skórko-Glonek J, Taylor A. Degradation by proteases Lon, Clp and HtrA, of Escherichia coli proteins aggregated in vivo by heat shock; HtrA protease action in vivo and in vitro. Mol Microbiol. 1996;22:555–571. doi: 10.1046/j.1365-2958.1996.1231493.x. [DOI] [PubMed] [Google Scholar]
- Hong SW, Vierling E. Mutants of Arabidopsis thaliana defective in the acquisition of tolerance to high temperature stress. Proc Natl Acad Sci U S A. 2000;97:4392–4397. doi: 10.1073/pnas.97.8.4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmer EC, Glover JR, Singer MA, Lindquist S. HSP100/Clp proteins: a common mechanism explains diverse functions. Trends Biochem Sci. 1996;21:289–296. doi: 10.1016/0968-0004(96)10038-4. [DOI] [PubMed] [Google Scholar]
- Neuwald AF, Aravind L, Spouge JL, Koonin EV. AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 1999;9:27–43. [PubMed] [Google Scholar]
- Ogura T, Wilkinson AJ. AAA+ superfamily ATPases: common structure--diverse function. Genes Cells. 2001;6:575–597. doi: 10.1046/j.1365-2443.2001.00447.x. [DOI] [PubMed] [Google Scholar]
- Vale RD. AAA proteins. Lords of the ring. J Cell Biol. 2000;150:F13–9. doi: 10.1083/jcb.150.1.F13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Maurizi MR, Esser L, Xia D. Crystal structure of ClpA, an Hsp100 chaperone and regulator of ClpAP protease. J Biol Chem. 2002;277:46743–46752. doi: 10.1074/jbc.M207796200. [DOI] [PubMed] [Google Scholar]
- Bochtler M, Hartmann C, Song HK, Bourenkov GP, Bartunik HD, Huber R. The structures of HsIU and the ATP-dependent protease HsIU-HsIV. Nature. 2000;403:800–805. doi: 10.1038/35001629. [DOI] [PubMed] [Google Scholar]
- Li J, Sha B. Crystal structure of E. coli Hsp100 ClpB nucleotide-binding domain 1 (NBD1) and mechanistic studies on ClpB ATPase activity. J Mol Biol. 2002;318:1127–1137. doi: 10.1016/S0022-2836(02)00188-2. [DOI] [PubMed] [Google Scholar]
- Yu RC, Hanson PI, Jahn R, Brunger AT. Structure of the ATP-dependent oligomerization domain of N-ethylmaleimide sensitive factor complexed with ATP. Nat Struct Biol. 1998;5:803–811. doi: 10.1038/1843. [DOI] [PubMed] [Google Scholar]
- Lenzen CU, Steinmann D, Whiteheart SW, Weis WI. Crystal structure of the hexamerization domain of N-ethylmaleimide-sensitive fusion protein. Cell. 1998;94:525–536. doi: 10.1016/s0092-8674(00)81593-7. [DOI] [PubMed] [Google Scholar]
- Zhang X, Shaw A, Bates PA, Newman RH, Gowen B, Orlova E, Gorman MA, Kondo H, Dokurno P, Lally J, Leonard G, Meyer H, van Heel M, Freemont PS. Structure of the AAA ATPase p97. Mol Cell. 2000;6:1473–1484. doi: 10.1016/s1097-2765(00)00143-x. [DOI] [PubMed] [Google Scholar]
- Lee S, Sowa ME, Watanabe Y, Sigler PB, Chiu W, Yoshida M, Tsai FT. The Structure of ClpB. A Molecular Chaperone that Rescues Proteins from an Aggregated State. Cell. 2003;115:229–240. doi: 10.1016/s0092-8674(03)00807-9. [DOI] [PubMed] [Google Scholar]
- Watanabe YH, Motohashi K, Yoshida M. Roles of the two ATP binding sites of ClpB from Thermus thermophilus. J Biol Chem. 2002;277:5804–5809. doi: 10.1074/jbc.M109349200. [DOI] [PubMed] [Google Scholar]
- Mogk A, Schlieker C, Strub C, Rist W, Weibezahn J, Bukau B. Roles of individual domains and conserved motifs of the AAA+ chaperone ClpB in oligomerization, ATP-hydrolysis and chaperone activity. J Biol Chem. 2003;278:15–24. doi: 10.1074/jbc.M209686200. [DOI] [PubMed] [Google Scholar]
- Schirmer EC, Queitsch C, Kowal AS, Parsell DA, Lindquist S. The ATPase Activity of Hsp104, Effects of Environmental Conditions and Mutations. J Biol Chem. 1998;273:15546–15552. doi: 10.1074/jbc.273.25.15546. [DOI] [PubMed] [Google Scholar]
- Schirmer EC, Ware DM, Queitsch C, Kowal AS, Lindquist SL. Subunit interactions influence the biochemical and biological properties of Hsp104. Proc Natl Acad Sci U S A. 2001;98:914–919. doi: 10.1073/pnas.031568098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett ME, Zolkiewski M. Site-Directed Mutagenesis of Conserved Charged Amino Acid Residues in ClpB from Escherichia coli(,) Biochemistry. 2002;41:11277–11283. doi: 10.1021/bi026161s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattendorf DA, Lindquist SL. Cooperative kinetics of both Hsp104 ATPase domains and interdomain communication revealed by AAA sensor-1 mutants. Embo J. 2002;21:12–21. doi: 10.1093/emboj/21.1.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlee S, Groemping Y, Herde P, Seidel R, Reinstein J. The chaperone function of ClpB from Thermus thermophilus depends on allosteric interactions of its two ATP-binding sites. J Mol Biol. 2001;306:889–899. doi: 10.1006/jmbi.2001.4455. [DOI] [PubMed] [Google Scholar]
- Li J, Sha B. Crystal Structure of the E. coli Hsp100 ClpB N-Terminal Domain. Structure (Camb) 2003;11:323–328. doi: 10.1016/S0969-2126(03)00030-3. [DOI] [PubMed] [Google Scholar]
- Liu Z, Tek V, Akoev V, Zolkiewski M. Conserved amino acid residues within the amino-terminal domain of ClpB are essential for the chaperone activity. J Mol Biol. 2002;321:111–120. doi: 10.1016/S0022-2836(02)00591-0. [DOI] [PubMed] [Google Scholar]
- Beinker P, Schlee S, Groemping Y, Seidel R, Reinstein J. The N Terminus of ClpB from Thermus thermophilus Is Not Essential for the Chaperone Activity. J Biol Chem. 2002;277:47160–47166. doi: 10.1074/jbc.M207853200. [DOI] [PubMed] [Google Scholar]
- Clarke AK, Eriksson MJ. The truncated form of the bacterial heat shock protein ClpB/HSP100 contributes to development of thermotolerance in the cyanobacterium Synechococcus sp. strain PCC 7942. J Bacteriol. 2000;182:7092–7096. doi: 10.1128/JB.182.24.7092-7096.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser CM, Gocayne JD, White O, Adams MD, Clayton RA, Fleischmann RD, Bult CJ, Kerlavage AR, Sutton G, Kelley JM, et al. The minimal gene complement of Mycoplasma genitalium. Science. 1995;270:397–403. doi: 10.1126/science.270.5235.397. [DOI] [PubMed] [Google Scholar]
- Himmelreich R, Hilbert H, Plagens H, Pirkl E, Li BC, Herrmann R. Complete sequence analysis of the genome of the bacterium Mycoplasma pneumoniae. Nucleic Acids Res. 1996;24:4420–4449. doi: 10.1093/nar/24.22.4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cashikar AG, Schirmer EC, Hattendorf DA, Glover JR, Ramakrishnan MS, Ware DM, Lindquist SL. Defining a pathway of communication from the C-terminal peptide binding domain to the N-terminal ATPase domain in a AAA protein. Mol Cell. 2002;9:751–760. doi: 10.1016/s1097-2765(02)00499-9. [DOI] [PubMed] [Google Scholar]
- Kim YI, Levchenko I, Fraczkowska K, Woodruff RV, Sauer RT, Baker TA. Molecular determinants of complex formation between Clp/Hsp100 ATPases and the ClpP peptidase. Nat Struct Biol. 2001;8:230–233. doi: 10.1038/84967. [DOI] [PubMed] [Google Scholar]
- Glover JR, Lindquist S. Hsp104, Hsp70, and Hsp40: A novel chaperone system that rescues previously aggregated proteins. Cell. 1998;94:73–82. doi: 10.1016/s0092-8674(00)81223-4. [DOI] [PubMed] [Google Scholar]
- Glover JR, Tkach JM. Crowbars and ratchets: hsp100 chaperones as tools in reversing protein aggregation. Biochem Cell Biol. 2001;79:557–568. doi: 10.1139/bcb-79-5-557. [DOI] [PubMed] [Google Scholar]
- Wang J, Song JJ, Franklin MC, Kamtekar S, Im YJ, Rho SH, Seong IS, Lee CS, Chung CH, Eom SH. Crystal structures of the HslVU peptidase-ATPase complex reveal an ATP-dependent proteolysis mechanism. Structure (Camb) 2001;9:177–184. doi: 10.1016/S0969-2126(01)00570-6. [DOI] [PubMed] [Google Scholar]
- Song HK, Hartmann C, Ramachandran R, Bochtler M, Behrendt R, Moroder L, Huber R. Mutational studies on HslU and its docking mode with HslV. Proc Natl Acad Sci U S A. 2000;97:14103–14108. doi: 10.1073/pnas.250491797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada-Inagawa T, Okuno T, Karata K, Yamanaka K, Ogura T. Conserved pore residues in the AAA protease, FtsH, are important for proteolysis and its coupling to ATP hydrolysis. J Biol Chem. 2003. [DOI] [PubMed]
- Goloubinoff P, Mogk A, Peres Ben Zvi A, Tomoyasu T, Bukau B. Sequential mechanism of solubilization and refolding of stable protein aggregates by a bichaperone network. Proc Natl Acad Sci USA. 1999;96:13732–13737. doi: 10.1073/pnas.96.24.13732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motohashi K, Watanabe Y, Yohda M, Yoshida M. Heat-inactivated proteins are rescued by the DnaK.J-GrpE set and ClpB chaperones. Proc Natl Acad Sci USA. 1999;96:7184–7189. doi: 10.1073/pnas.96.13.7184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolkiewski M. ClpB cooperates with DnaK, DnaJ, and GrpE in suppressing protein aggregation. A novel multi-chaperone system from Escherichia coli. J Biol Chem. 1999;274:28083–28086. doi: 10.1074/jbc.274.40.28083. [DOI] [PubMed] [Google Scholar]
- Diamant S, Ben-Zvi AP, Bukau B, Goloubinoff P. Size-dependent disaggregation of stable protein aggregates by the DnaK chaperone machinery. J Biol Chem. 2000;275:21107–21113. doi: 10.1074/jbc.M001293200. [DOI] [PubMed] [Google Scholar]
- Krzewska J, Langer T, Liberek K. Mitochondrial Hsp78, a member of the Clp/Hsp100 family in Saccharomyces cerevisiae, cooperates with Hsp70 in protein refolding. FEBS Lett. 2001;489:92–96. doi: 10.1016/S0014-5793(00)02423-6. [DOI] [PubMed] [Google Scholar]
- Dougan DA, Reid BG, Horwich AL, Bukau B. ClpS, a substrate modulator of the ClpAP machine. Mol Cell. 2002;9:673–683. doi: 10.1016/s1097-2765(02)00485-9. [DOI] [PubMed] [Google Scholar]
- Zeth K, Ravelli RB, Paal K, Cusack S, Bukau B, Dougan DA. Structural analysis of the adaptor protein ClpS in complex with the N-terminal domain of ClpA. Nat Struct Biol. 2002;9:906–911. doi: 10.1038/nsb869. [DOI] [PubMed] [Google Scholar]
- Wawrzynów A, Wojtkowiak D, Marszalek J, Banecki B, Jonsen M, Graves B, Georgopoulos C, Zylicz M. The ClpX heat-shock protein of Escherichia coli, the ATP-dependent substrate specificity component of the ClpP-ClpX protease, is a novel molecular chaperone. EMBO J. 1995;14:1867–1877. doi: 10.1002/j.1460-2075.1995.tb07179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roggiani M, Hahn J, Dubnau D. Suppression of early competence mutations in Bacillus subtilis by mec mutations. J Bacteriol. 1990;172:4056–4063. doi: 10.1128/jb.172.7.4056-4063.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turgay K, Hamoen LW, Venema G, Dubnau D. Biochemical characterization of a molecular switch involving the heat shock protein ClpC, which controls the activity of ComK, the competence transcription factor of Bacillus subtilis. Genes Dev. 1997;11:119–128. doi: 10.1101/gad.11.1.119. [DOI] [PubMed] [Google Scholar]
- Turgay K, Hahn J, Burghoorn J, Dubnau D. Competence in Bacillus subtilis is controlled by regulated proteolysis of a transcription factor. EMBO J. 1998;17:6730–6738. doi: 10.1093/emboj/17.22.6730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger E, Volker U, Hecker M. Stress induction of clpC in Bacillus subtilis and its involvement in stress tolerance. J Bacteriol. 1994;176:3360–3367. doi: 10.1128/jb.176.11.3360-3367.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Msadek T, Kunst F, Rapoport G. MecB of Bacillus subtilis, a member of the ClpC ATPase family, is a pleiotropic regulator controlling competence gene expression and growth at high temperature. Proc Natl Acad Sci U S A. 1994;91:5788–5792. doi: 10.1073/pnas.91.13.5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger E, Witt E, Ohlmeier S, Hanschke R, Hecker M. The clp proteases of Bacillus subtilis are directly involved in degradation of misfolded proteins. J Bacteriol. 2000;182:3259–3265. doi: 10.1128/JB.182.11.3259-3265.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurgen B, Hanschke R, Sarvas M, Hecker M, Schweder T. Proteome and transcriptome based analysis of Bacillus subtilis cells overproducing an insoluble heterologous protein. Appl Microbiol Biotechnol. 2001;55:326–332. doi: 10.1007/s002530000531. [DOI] [PubMed] [Google Scholar]
- Schlothauer T, Mogk A, Dougan DA, Bukau B, Turgay K. MecA, an adaptor protein necessary for ClpC chaperone activity. Proc Natl Acad Sci U S A. 2003;100:2306–2311. doi: 10.1073/pnas.0535717100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persuh M, Turgay K, Mandic-Mulec I, Dubnau D. The N and the C-terminal domains of MecA recognize different partners in the competence molecular switch. Mol Microbiol. 1999;33:886–894. doi: 10.1046/j.1365-2958.1999.01544.x. [DOI] [PubMed] [Google Scholar]
- Taylor JP, Hardy J, Fischbeck KH. Toxic proteins in neurodegenerative disease. Science. 2002;296:1991–1995. doi: 10.1126/science.1067122. [DOI] [PubMed] [Google Scholar]
- Perutz Max F., Johnson Tony, Suzuki Masashi, Finch John T. Glutamine repeats as polar zippers: Their possible role in inherited neurodegenerative diseases. Proc Natl Acad Sci USA. 1994;91:5355–5358. doi: 10.1073/pnas.91.12.5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krobitsch S, Lindquist S. Aggregation of huntingtin in yeast varies with the length of the polyglutamine expansion and the expression of chaperone proteins. Proc Natl Acad Sci U S A. 2000;97:1589–1594. doi: 10.1073/pnas.97.4.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunkes A, Mandel JL. A cellular model that recapitulates major pathogenic steps of Huntington's disease. Hum Mol Genet. 1998;7:1355–1361. doi: 10.1093/hmg/7.9.1355. [DOI] [PubMed] [Google Scholar]
- Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- Warrick JM, Paulson HL, Gray-Board GL, Bui QT, Fischbeck KH, Pittman RN, Bonini NM. Expanded polyglutamine protein forms nuclear inclusions and causes neural degeneration in Drosophila. Cell. 1998;93:939–949. doi: 10.1016/s0092-8674(00)81200-3. [DOI] [PubMed] [Google Scholar]
- Satyal SH, Schmidt E, Kitagawa K, Sondheimer N, Lindquist S, Kramer JM, Morimoto RI. Polyglutamine aggregates alter protein folding homeostasis in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2000;97:5750–5755. doi: 10.1073/pnas.100107297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A, Lucas JJ, Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell. 2000;101:57–66. doi: 10.1016/S0092-8674(00)80623-6. [DOI] [PubMed] [Google Scholar]
- Serio TR, Lindquist SL. Protein-only inheritance in yeast: something to get [PSI+]-ched about. Trends Cell Biol. 2000;10:98–105. doi: 10.1016/S0962-8924(99)01711-0. [DOI] [PubMed] [Google Scholar]
- Chernoff Yury O., Lindquist Susan L., Ono Bun-ichiro, Inge-Vechtomov Sergei G., Liebman Susan W. Role of the Chaperone Protein Hsp104 in Propagation of the Yeast Prion-like Factor [psi+] Science. 1995;268:880–884. doi: 10.1126/science.7754373. [DOI] [PubMed] [Google Scholar]
- Kushnirov VV, Ter-Avanesyan MD. Structure and Replication of Yeast Prions. Cell. 1998;94:13–16. doi: 10.1016/s0092-8674(00)81216-7. [DOI] [PubMed] [Google Scholar]
- Sakahira H, Breuer P, Hayer-Hartl MK, Hartl FU. Molecular chaperones as modulators of polyglutamine protein aggregation and toxicity. Proc Natl Acad Sci U S A. 2002;99 Suppl 4:16412–16418. doi: 10.1073/pnas.182426899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodman PG. p97, a protein coping with multiple identities. J Cell Sci. 2003;116:4283–4290. doi: 10.1242/jcs.00817. [DOI] [PubMed] [Google Scholar]
- Hirabayashi M, Inoue K, Tanaka K, Nakadate K, Ohsawa Y, Kamei Y, Popiel AH, Sinohara A, Iwamatsu A, Kimura Y, Uchiyama Y, Hori S, Kakizuka A. VCP/p97 in abnormal protein aggregates, cytoplasmic vacuoles, and cell death, phenotypes relevant to neurodegeneration. Cell Death Differ. 2001;8:977–984. doi: 10.1038/sj.cdd.4400907. [DOI] [PubMed] [Google Scholar]
- Higashiyama H, Hirose F, Yamaguchi M, Inoue YH, Fujikake N, Matsukage A, Kakizuka A. Identification of ter94, Drosophila VCP, as a modulator of polyglutamine-induced neurodegeneration. Cell Death Differ. 2002;9:264–273. doi: 10.1038/sj.cdd.4400955. [DOI] [PubMed] [Google Scholar]
- Breakefield XO, Kamm C, Hanson PI. TorsinA: movement at many levels. Neuron. 2001;31:9–12. doi: 10.1016/s0896-6273(01)00350-6. [DOI] [PubMed] [Google Scholar]
- Liu Z, Zolkiewska A, Zolkiewski M. Characterization of human torsinA and its dystonia-associated mutant form. Biochem J. 2003;374:117–122. doi: 10.1042/BJ20030258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kustedjo K, Bracey MH, Cravatt BF. Torsin A and its torsion dystonia-associated mutant forms are lumenal glycoproteins that exhibit distinct subcellular localizations. J Biol Chem. 2000;275:27933–27939. doi: 10.1074/jbc.M910025199. [DOI] [PubMed] [Google Scholar]
- Hewett J, Ziefer P, Bergeron D, Naismith T, Boston H, Slater D, Wilbur J, Schuback D, Kamm C, Smith N, Camp S, Ozelius LJ, Ramesh V, Hanson PI, Breakefield XO. TorsinA in PC12 cells: localization in the endoplasmic reticulum and response to stress. J Neurosci Res. 2003;72:158–168. doi: 10.1002/jnr.10567. [DOI] [PubMed] [Google Scholar]
- McLean PJ, Kawamata H, Shariff S, Hewett J, Sharma N, Ueda K, Breakefield XO, Hyman BT. TorsinA and heat shock proteins act as molecular chaperones: suppression of alpha-synuclein aggregation. J Neurochem. 2002;83:846–854. doi: 10.1046/j.1471-4159.2002.01190.x. [DOI] [PubMed] [Google Scholar]
- Sharma N, Hewett J, Ozelius LJ, Ramesh V, McLean PJ, Breakefield XO, Hyman BT. A close association of torsinA and alpha-synuclein in Lewy bodies: a fluorescence resonance energy transfer study. Am J Pathol. 2001;159:339–344. doi: 10.1016/s0002-9440(10)61700-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell GA, Cao S, Sexton EG, Gelwix CC, Bevel JP, Caldwell KA. Suppression of polyglutamine-induced protein aggregation in Caenorhabditis elegans by torsin proteins. Hum Mol Genet. 2003;12:307–319. doi: 10.1093/hmg/ddg027. [DOI] [PubMed] [Google Scholar]
- Cummings CJ, Mancini MA, Antalffy B, DeFranco DB, Orr HT, Zoghbi HY. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nature Genetics. 1998;19:148–154. doi: 10.1038/502. [DOI] [PubMed] [Google Scholar]
- Waelter S, Boeddrich A, Lurz R, Scherzinger E, Lueder G, Lehrach H, Wanker EE. Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol Biol Cell. 2001;12:1393–1407. doi: 10.1091/mbc.12.5.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert U, Anton LC, Gibbs J, Norbury CC, Yewdell JW, Bennink JR. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature. 2000;404:770–774. doi: 10.1038/35004754. [DOI] [PubMed] [Google Scholar]
- Thomas JG, Ayling A, Baneyx F. Molecular chaperones, folding catalysts, and the recovery of active recombinant proteins from E. coli. To fold or to refold. Appl Biochem Biotechnol. 1997;66:197–238. doi: 10.1007/BF02785589. [DOI] [PubMed] [Google Scholar]
- Tomoyasu T, Mogk A, Langen H, Goloubinoff P, Bukau B. Genetic dissection of the roles of chaperones and proteases in protein folding and degradation in the Escherichia coli cytosol. Mol Microbiol. 2001;40:397–413. doi: 10.1046/j.1365-2958.2001.02383.x. [DOI] [PubMed] [Google Scholar]
- Carrio MM, Villaverde A. Protein aggregation as bacterial inclusion bodies is reversible. FEBS Lett. 2001;489:29–33. doi: 10.1016/S0014-5793(01)02073-7. [DOI] [PubMed] [Google Scholar]
- Levchenko I, Seidel M, Sauer RT, Baker TA. A specificity-enhancing factor for the ClpXP degradation machine. Science. 2000;289:2354–2356. doi: 10.1126/science.289.5488.2354. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Gottesman S, Hoskins JR, Maurizi MR, Wickner S. The RssB response regulator directly targets sigma(S) for degradation by ClpXP. Genes Dev. 2001;15:627–637. doi: 10.1101/gad.864401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez M, Rasulova F, Maurizi MR, Woodgate R. Subunit-specific degradation of the UmuD/D' heterodimer by the ClpXP protease: the role of trans recognition in UmuD' stability. Embo J. 2000;19:5251–5258. doi: 10.1093/emboj/19.19.5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher SB, Sauer RT, Baker TA. Distinct peptide signals in the UmuD and UmuD' subunits of UmuD/D' mediate tethering and substrate processing by the ClpXP protease. Proc Natl Acad Sci U S A. 2003;100:13219–13224. doi: 10.1073/pnas.2235804100. [DOI] [PMC free article] [PubMed] [Google Scholar]