

Abstract
A composite cytomegalovirus-immediate early gene enhancer/chicken β-actin promoter (CAG) was utilized to generate transgenic mice that overexpress human spermidine synthase (SpdS) in order to determine the impact of elevated spermidine synthase activity on murine development and physiology. CAG-SpdS mice were viable and fertile and tissue SpdS activity was increased up to 9-fold. This increased SpdS activity did not result in a dramatic elevation of spermidine or spermine levels but did lead to a 1.5 to 2-fold reduction in tissue spermine:spermidine ratio in heart, muscle and liver tissues with the highest levels of SpdS activity. This new mouse model enabled simultaneous overexpression of SpdS and other polyamine biosynthetic enzymes by combining transgenic animals. The combined overexpression of both SpdS and spermine synthase (SpmS) in CAG-SpdS/CAG-SpmS bitransgenic mice did not impair viability or lead to overt developmental abnormalities but instead normalized the elevated tissue spermine:spermidine ratios of CAG-SpmS mice. The CAG-SpdS mice were bred to MHC-AdoMetDC mice with a >100-fold increase in cardiac S-adenosylmethionine decarboxylase (AdoMetDC) activity to determine if elevated dcAdoMet would facilitate greater spermidine accumulation in mice with SpdS overexpression. CAG-SpdS/MHC-AdoMetDC bitransgenic animals were produced at the expected frequency and exhibited cardiac polyamine levels comparable to MHC-AdoMetDC littermates. Taken together these results indicate that SpdS levels are not rate limiting in vivo for polyamine biosynthesis and are unlikely to exert significant regulatory effects on cellular polyamine content and function.
Keywords: polyamine, aminopropyltransferase, transgenic mice, S-adenosylmethionine decarboxylase, spermidine, spermine
INTRODUCTION
The polyamines putrescine, spermidine and spermine are cationic molecules that are found in all eukaryotic cells and are clearly essential to growth and development (reviewed in (Pegg 2009a; Wallace et al. 2003)). Using arginine (Wu et al. 2009) and methionine (Pegg 2009b) precursors, mammalian polyamine biosynthesis is achieved by the combined function of two decarboxylases, ornithine decarboxylase (ODC, EC 4.1.1.17) and S-adenosylmethionine decarboxylase (AdoMetDC, EC 4.1.1.50), and two distinct aminopropyltransferases, spermidine synthase (SpdS, EC 2.5.1.16) and spermine synthase (SpmS, EC 2.5.1.22). ODC catalyzes the conversion of ornithine to putrescine, while AdoMetDC converts S-adenosylmethionine (AdoMet) to decarboxylated S-adenosylmethionine (dcAdoMet). SpdS transfers an aminopropyl group from dcAdoMet to putrescine to produce spermidine and SpmS transfers an aminopropyl group from a second dcAdoMet molecule to spermidine to produce spermine.
Cellular polyamine biosynthesis and catabolism is tightly regulated to maintain levels that permit critical biological processes such as DNA replication, gene transcription, mRNA translation, modulation of ion channels and protection from oxidative stress (Pegg 2009a; Wallace et al. 2003; Rider et al. 2007). An extensive body of literature supports the paradigm that polyamine biosynthetic rates are regulated through modulation of the levels of the decarboxylases, both of which are present at very low levels in the absence of an inducing stimulus (Pegg 2006, 2009b). SpdS and SpmS are expressed constitutively; therefore, the levels of the substrates dcAdoMet and putrescine, or dcAdoMet and spermidine, determine spermidine or spermine synthesis rates, respectively. A few studies suggest that the aminopropyltransferase levels may be inducible in certain physiological situations (see (Pegg 2009a; Ikeguchi et al. 2006; Forshell et al. 2010) and references therein). Transgenic overexpression of SpmS in mice had little effect on spermine levels as described below, but no in vivo studies have addressed the question of whether unregulated elevation of SpdS activity alters polyamine content.
Deregulated polyamine metabolism is linked to pathological processes and the pathway has been proposed as a promising target for the treatment and prevention of diverse diseases such as cancer and parasitic infection (Basuroy and Gerner 2006; Casero and Marton 2007; Heby et al. 2007; Shantz and Levin 2007). Generally it is difficult to assign a particular biological function to a specific polyamine due to their presence at millimolar concentrations in most cells, the inability to distinguish bound versus free polyamine concentrations and the fact that they are readily interconverted by polyamine metabolic enzymes (reviewed in (Pegg 2009a; Wallace et al. 2003)). However, the production of active eukaryotic translation initiation factor 5A (eIF-5A) requires a post-translational spermidine-dependent hypusine modification (Park et al. 2010), which is essential for growth in yeast (Chattopadhyay et al. 2002). In addition, recent studies have implicated elevated spermidine levels in the induction of autophagy and increased longevity (Eisenberg et al. 2009; Madeo et al. 2010).
The advent of methods to manipulate the mouse genome facilitated the production of numerous valuable mouse models with altered polyamine metabolism in order to dissect the critical enzymes and/or polyamines in specific physiological or pathological processes (Janne et al. 2004; Pegg et al. 2003; Pegg and Wang 2009; Janne et al. 2006). Both ODC (Pendeville et al. 2001) and AdoMetDC (Nishimura et al. 2002) gene knockouts in the mouse are lethal at a very early stage of embryonic development. Spermidine is essential for growth in lower eukaryotes (Chattopadhyay et al. 2002) and, although mouse embryonic stem cells with targeted SpdS deletion have been produced (http://www.komp.org/geneinfo.php?geneid=80254), the attempted generation and characterization of SpdS knockout mice has not been reported. However, the in vivo functions of spermine have been studied in several mouse models (Pegg and Wang 2009). Male Gyro (Gy) mice with a large deletion on the X chromosome that includes most of the SpmS gene are viable despite a nearly complete absence of tissue spermine, yet they exhibit profound abnormalities such as deafness, inner ear abnormalities, circling behavior, sterility and a greatly reduced lifespan (Wang et al. 2004; Wang et al. 2009). In humans, mutations in the SMS gene that abolish nearly all SpmS activity have been identified in the rare genetic disorder Snyder-Robinson syndrome, which is characterized by mild-to-moderate mental retardation, hypotonia, cerebellar circuitry dysfunction, facial asymmetry, thin habitus, osteoporosis and kyphoscoliosis (Cason et al. 2003; Becerra-Solano et al. 2009; de Alencastro et al. 2008).
A composite cytomegalovirus-immediate early gene enhancer/chicken β-actin promoter (CAG) enabled ubiquitous overexpression of SpmS in the mouse. CAG-SpmS animals exhibited up to 2,000-fold increases in SpmS activity but only a corresponding 2 to 4-fold increase in Spm:Spd ratio and no appreciable increase in total polyamine levels (Ikeguchi et al. 2004). SpdS activity was not affected by SpmS overexpression, but dcAdoMet content was decreased slightly (Pegg et al. 2011). The developmental abnormalities in the SpmS-deleted Gy mice were reverted completely by the CAG-SpmS transgene, with the exception of those that are linked to the hypophosphatemia caused by co-deletion of the Phex gene in Gy mice (Wang et al. 2004; Wang et al. 2009). It is important to note that, in addition to an absence of spermine, Gy mice also have dramatically elevated spermidine levels that are also normalized by the CAG-SpmS transgene.
Here we describe the generation and characterization of a transgenic mouse line that overexpresses human spermidine synthase from the CAG promoter in order to determine the impact of elevated spermidine synthase activity on murine development and physiology, and to enable simultaneous overexpression of SpdS and other polyamine biosynthetic enzymes by combining transgenic models. CAG-SpdS mice were viable and fertile and exhibited a reduced tissue Spm:Spd ratio that correlated with the level of SpdS activity. We also demonstrated that the combined overexpression of both aminopropyltransferases in CAG-SpdS/CAG-SpmS bitransgenic mice does not impair murine viability or lead to overt developmental abnormalities but instead normalizes the elevated tissue Spm:Spd ratios of CAG-SpmS mice. Finally, upon breeding of CAG-SpdS mice to MHC-AdoMetDC mice with a >100-fold increase in cardiac AdoMetDC activity (Nisenberg et al. 2006), CAG-SpdS/MHC-AdoMetDC bitransgenic animals were produced at the expected frequency and exhibit cardiac polyamine levels comparable to MHC-AdoMetDC littermates. Taken together these results indicate that SpdS levels are unlikely to exert significant regulatory effects on cellular polyamine content and function.
MATERIALS AND METHODS
Materials
All chemicals, unless noted, were purchased from Sigma Chemical Company (St. Louis, MO). Restriction enzymes were purchased from New England Biolabs (Beverly, MA). Oligonucleotides used as primers were synthesized and purified in the Macromolecular Core Facility of the Pennsylvania State University College of Medicine. [35S]-dcAdoMet was synthesized from L-[35S]methionine (PerkinElmer Life Sciences, Boston, MA) as described previously (Mackintosh and Pegg 2000). S-adenosyl-L-[carboxyl-14C]methionine (55 mCi/mmol) was obtained from American Radiolabeled Chemicals (St. Louis, MO).
Transgene construction and microinjection
The transgene for widespread expression of SpdS was constructed essentially as described previously for CAG-SpmS mice (Ikeguchi et al. 2004). Human SpdS cDNA was inserted in a vector containing a composite CMV-IE enhancer–β-actin (CAG) promoter to replace the enhanced green fluorescent protein coding sequence by EcoRI digestion of plasmid pCX-EGFP (Sawicki et al. 1998) (Fig. 1). Human SpdS cDNA was amplified by polymerase chain reaction (PCR) from plasmid pET28a-LIC-hSpdS (Wu et al. 2007) and was engineered to contain a nine amino acid amino-terminal hemagglutinin (HA) epitope coding sequence with sense primer 5′-GACGCATTAGGAATTCGCCACCATGGGATACCCCTAC GACGTCCCCGACTACGCCGAGCCCGGCCCCGACGG-3′ and antisense primer 5′-GACGCATTAGGAATTCTCATCAGCTCACATCATTCAGGG-3′ (EcoRI sites in italics and HA epitope coding sequence underlined). The PCR reaction was carried out in a 0.1 ml volume containing 2.5 units of Pfu polymerase (Stratagene, La Jolla, CA), 25 ng of template DNA and 25 pmol of each primer under the following conditions: initial denaturation for 2 min at 94°C, followed by 25 cycles of denaturation (94°C for 30 s), annealing (58°C for 30 s) and extension (72°C for 2 min), with a final extension at 72°C for 5 min. The entire SpdS cDNA insert was sequenced at the DNA Sequencing Core (Pennsylvania State University College of Medicine) to ensure the correct orientation and that no secondary mutations were introduced during the plasmid construction.
Figure 1. Transgene construct and PCR identification of transgenic mice.
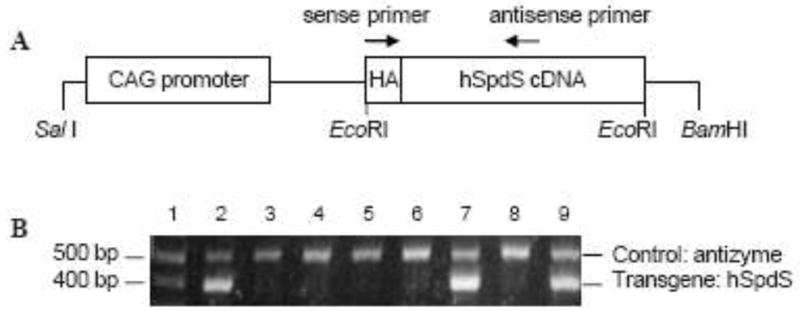
A, The human SpdS cDNA was placed under the control of composite cytomegalovirus-immediate early gene enhancer/chicken β-actin (CAG) promoter elements to generate the transgene expression construct. An HA epitope was added to the amino terminus of the hSpdS to enable detection of transgene-derived SpdS protein. Arrows indicate the positions of primers used for PCR identification of transgenic mice in B. B, A representative analytic gel of tail DNA extracts from eight potential transgenic mice (lanes 2 to 9) and a DNA standard (lane 1). Genotyping by PCR yielded a 397 bp product from the genomic DNA from transgenic animals (lanes 2, 7 and 9). Primers that amplify a 520 bp product from the mouse antizyme gene were included in all reactions as a positive control.
The plasmid pCAG-SpdS was digested with SalI and BamHI to remove prokaryotic vector sequences and liberate a 3.4 kb transgene fragment. The transgene was purified using the Perfectprep gel cleanup kit (Eppendorf, Hauppauge, NY) and Elutip-D minicolumns (Schleicher & Schuell, Keene, NH) and then precipitated and resuspended in microinjection buffer (10 mM Tris/HCl, pH 7.4, and 0.1 mM EDTA). The transgene was microinjected into fertilized C57BL/6 oocytes using standard techniques in the Transgenic Mouse Facility (Pennsylvania State University, University Park, PA) and transgenic founder animals were identified by PCR as described below.
Breeding and PCR identification of transgenic mice
All animal studies were reviewed and approved by the Institutional Animal Care and Use Committee of the Pennsylvania State University College of Medicine. Transgenic CAG-SpdS lines were established by breeding transgenic founders and their progeny to wild type C57BL/6J mice (The Jackson Laboratory, Bar Harbor, ME) to produce equal numbers of transgenic mice and wild type littermate controls. Genomic DNA was extracted from tail biopsies from potential transgenic mice and subjected to PCR analysis using the REDExtract-N-Amp Tissue PCR Kit (Sigma, St. Louis, MO) to detect the transgene DNA. PCR analysis utilized one of two independent primer sets. For the first, a sense primer (5′-TACGACGTCCCCGACTACG-3′) binds within the HA epitope coding region and the antisense primer (5′-TCGATCTCACACTGGACCAC-3′) binds within the hSpdS region (Figure 1A) to amplify a 397 bp product only in genomic DNA samples from CAG-SpdS mice. Alternatively, the transgene was detected with a sense primer (5′-TTCGGCTTCTGGCGTGTGAC-3′) that binds in the β-actin promoter region and an antisense primer (5′-CTTACTGCGGAAGACGAGG-3′) that binds in the human SpdS coding region to amplify a 330 bp product. A second primer pair that yields a 520 bp product from the mouse antizyme 1 gene (Oaz1) was also included in the reaction, which provides a positive control for successful PCR amplification for each genomic DNA sample (Feith et al. 2007).
The CAG-SpdS mice were crossed with other transgenic mice with altered polyamine metabolism including CAG-SpmS that overexpress spermine synthase in many tissues and MHC-AdoMetDC that overexpress AdoMetDC in the heart. To generate and characterize bitransgenic animals, heterozygous mice from two different transgenic lines were bred and offspring were genotyped for both transgenes by PCR. CAG-SpmS line 8, which had been backcrossed to the C57BL/6J inbred strain for >10 generations, were identified as described previously (Ikeguchi et al. 2004). The MHC-AdoMetDC transgene was maintained on a mixed B6D2 genetic background and transgenic animals were identified as described previously (Nisenberg et al. 2006). All experimental groups included both male and female mice and no sex-dependent differences were observed unless noted.
SpdS and SpmS enzymatic activity assays and SpdS western blotting
Mice were euthanized at the indicated age and tissues were harvested and flash frozen in liquid nitrogen. To determine aminopropyltransferase activity, each tissue sample was placed in ice-cold spermidine/spermine synthase harvesting buffer (50 mM sodium phosphate (pH 7.2), 0.3 mM EDTA, 10 mM 2-mercaptoethanol and 1× protease inhibitor cocktail (Calbiochem, LaJolla, CA)), homogenized on ice using a Polytron for 30 s with 15 s on/15 s off and centrifuged at 20000 ×g for 30 min at 4°C. Epidermis was separated from dermis as described (Feith et al. 2001) and then processed on ice by sonication for 30 s with 10 s on/10 s off. Tissues and extracts were stored at −80°C before use. SpdS and SpmS activity were assayed by measuring the production of [35S]methylthioadenosine from [35S]-dcAdoMet in the presence of the appropriate amine acceptor as reported previously (Wiest and Pegg 1998; Mackintosh and Pegg 2000). (50 μg) were fractionated by SDS-PAGE, transferred to PVDF membrane, and SpdS protein was detected by western blotting using a rabbit polyclonal antibody to human SpdS (anti-SRM, Sigma, St. Louis, MO) or to the HA epitope (sc-805, Santa Cruz Biotechnology, Santa Cruz, CA). Signals were visualized with a chemiluminescence detection system (Cell Signaling Technology, Beverly, MA).
HPLC analysis of polyamine and AdoMet/dcAdoMet content
For polyamine quantification, tissue samples were homogenized in 10% trichloroacetic acid (Fisher Scientific, Pittsburgh, PA) and analyzed by HPLC using an ion-pair reverse-phase separation method with post-column derivatization using o-phthaldialdehyde as described previously (Pegg et al. 1989) and normalized to tissue wet weight. For AdoMet and dcAdoMet quantification, tissue extracts were reacted with chloroacetaldehyde to convert AdoMet and dcAdoMet to fluorescent 1, N6-etheno derivatives, which were then separated and quantified by HPLC as described previously (Pegg et al. 2011) and normalized to tissue wet weight.
AdoMetDC activity assay
Tissue samples were placed in ice-cold AdoMetDC harvesting buffer (50 mM sodium phosphate (pH 6.8), 2.5 mM putrescine, 2.5 mM dithiothreitol, 0.1 mM EDTA and 1× protease inhibitor cocktail (Calbiochem, LaJolla, CA)), homogenized on ice using a Polytron for 30 s with 15 s on/15 s off and centrifuged at 20000 × g for 30 min at 4°C. AdoMetDC activity was assayed by measuring the production of [14C]-labeled CO2 as described previously (Nisenberg et al. 2006).
Statistical analysis
All comparisons between wild type controls and transgenic mice utilized the two-tailed, unpaired Student’s t test.
RESULTS
Generation and identification of CAG-SpdS transgenic mice
Our initial goal was to generate a transgenic mouse line with ubiquitous SpdS expression that is devoid of the normal regulatory elements that govern SpdS transcription and translation. To achieve this goal, we utilized the CAG promoter to drive expression of a human SpdS cDNA that was modified to add an HA epitope to the amino terminus to facilitate the detection of transgene-derived SpdS and differentiate this protein from the endogenous mouse SpdS protein (Fig. 1A). Genomic DNA screening (Fig. 1B) identified two transgenic founders (lines A and B) and both were found to transmit the transgene in a Mendelian fashion. Transgenic mice were produced and maintained on a C57BL/6J background and no obvious phenotypic abnormalities were observed in any of the founder mice or their progeny up to 1.5 years of age.
Aminopropyltransferase activity in CAG-SpdS Transgenic Mice
To evaluate transgene expression in CAG-SpdS mice, SpdS activity and protein levels were determined in several tissues from 7-week-old mice from the two founder lines. Transgenic mice from line B exhibited elevated SpdS activity relative to wild type controls in most tissues examined (Table 1). Heart and skeletal muscle showed the greatest fold increase (6.0 and 8.9, respectively) in SpdS activity, liver had a moderate 2.6-fold increase, epidermis and dermis had an approximately 1.5-fold increase, while brain and kidney showed no statistically significant increase in SpdS activity. The higher expression in heart and skeletal muscle is likely related to the β-actin promoter and a similar effect was seen previously in CAG-SpmS mice (Ikeguchi et al. 2004). Tissues of transgenic mice from line A failed to show any evidence of increased SpdS activity or alterations in polyamine content (data not shown). Subsequent studies utilized progeny of CAG-SpdS line B unless noted.
Table 1.
Aminopropyltransferase activity in control and CAG-SpdS mice
| Tissue | Spermidine synthase (pmol/mg/h)
|
Spermine synthase (pmol/mg/h)
|
||
|---|---|---|---|---|
| Control | CAG-SpdS | Control | CAG-SpdS | |
| Heart | 272 ± 23 | 1630 ± 211** | 20 | 19 |
| Muscle | 169 ± 61 | 1506 ± 428** | 119 | 114 |
| Liver | 1928 ± 452 | 4974 ± 1032** | 11 | 9 |
| Kidney | 731 ± 128 | 814 ± 95 | 57 | 61 |
| Brain | 5052 ± 1246 | 5050 ± 1213 | 102 | 98 |
| Epidermis | 1953 ± 374 | 2856 ± 479* | ||
| Dermis | 2908 ± 406 | 3977 ± 710* | ||
All measurements were done in CAG-SpdS mice from founder line B. Spermidine synthase activity (mean ± S.D., n=4) was measured in 7-week-old mice. Spermine synthase activity (average of 2 mice) was measured in mice at the same age.
p<0.05,
p<0.005 vs. wild type controls.
Tissue extracts were also subjected to Western blot analysis using anti-hSpdS and anti-HA antibodies (Fig. 2). Significant amounts of SpdS protein were detected in skeletal muscle, heart and liver from transgenic mice of line B. Transgene-derived SpdS was detected to a lesser extent in epidermis and dermis but not in brain and kidney of mice from line B (data not shown), which is in agreement with the SpdS activity data in Table 1. SpdS protein was below the limit of detection in all tissues from wild type and CAG-SpdS mice of line A. Given that SpdS activity is quite high in some wild type mouse tissues such as brain and liver (Table 1), this result indicates that the anti-hSpdS antibody does not efficiently recognize the mouse SpdS protein. Elevated SpdS activity did not result in a compensatory alteration of SpmS activity in any of the five tissues examined (Table 1). Similarly, dramatic elevation of SpmS activity failed to influence SpdS activity in CAG-SpmS mice (Ikeguchi et al. 2004).
Figure 2. Detection of SpdS protein in tissue extracts from CAG-SpdS mice.

Tissues extracts of skeletal muscle, heart and liver from wild type (wt) control and transgenic mice from each founder line (A and B) were blotted for SpdS protein using anti-hSpdS antibody (top) or anti-HA antibody (bottom). Each lane contains 50 μg of cytosolic proteins.
Polyamine, AdoMet and dcAdoMet content in tissues from CAG-SpdS mice
The impact of SpdS overexpression on tissue polyamine levels and AdoMet/dcAdoMet content was measured in tissues of CAG-SpdS mice and controls at 7 weeks of age. Spermidine levels increased significantly and the Spm:Spd ratio decreased significantly in heart, skeletal muscle and liver (Table 2), which are the tissues with the greatest increase in SpdS activity (Table 1). The elevations in spermidine were relatively small given the fold increase in SpdS activity in those tissues. There was no significant alteration in putrescine or spermine content associated with SpdS overexpression, except for a modest 13.6% reduction in liver spermine levels.
Table 2.
Polyamine content in control and CAG-SpdS mice
| Tissue | Group | Polyamine content (pmol/mg tissue)
|
Spm:Spd ratio | ||
|---|---|---|---|---|---|
| Putrescine | Spermidine | Spermine | |||
| Heart | Control | 8.0 ± 0.4 | 183 ± 18 | 328 ± 23 | 1.8 ± 0.2 |
| CAG-SpdS | 6.3 ± 1.3 | 278 ± 39** | 345 ± 82 | 1.2 ± 0.2* | |
| Muscle | Control | 9.6 ± 3.0 | 113 ± 23 | 179 ± 17 | 1.6 ± 0.4 |
| CAG-SpdS | 8.4 ± 2.7 | 222 ± 69* | 161 ± 26 | 0.8 ± 0.4* | |
| Liver | Control | 16 ± 4 | 693 ± 86 | 693 ± 64 | 1.0 ± 0.1 |
| CAG-SpdS | 19 ± 5 | 832 ± 31* | 599 ± 21* | 0.7 ± 0.0** | |
| Kidney | Control | 64 ± 17 | 383 ± 49 | 644 ± 39 | 1.7 ± 0.1 |
| CAG-SpdS | 51 ± 26 | 410 ± 7 | 631 ± 33 | 1.5 ± 0.1 | |
| Brain | Control | 9.6 ± 1.4 | 317 ± 33 | 268 ± 18 | 0.9 ± 0.1 |
| CAG-SpdS | 9.8 ± 2.3 | 257 ± 58 | 306 ± 15* | 1.2 ± 0.3* | |
Polyamine content (mean ± S.D.) was determined in 7-week-old mice (n=4 per genotype).
p<0.05,
p<0.005 vs. wild type controls.
The synthesis of spermidine from putrescine is not only dependent on SpdS activity but also on the levels of the aminopropyl donor, dcAdoMet (Pegg 2009a). Therefore, AdoMet and dcAdoMet levels were determined in heart and skeletal muscle (Table 3) since these tissues exhibited the greatest increase in SpdS activity. Heart AdoMet and dcAdoMet levels were not altered by SpdS overexpression even though increased SpdS activity is expected to consume dcAdoMet for spermidine synthesis. The stable dcAdoMet pool may be maintained by a compensatory change in AdoMetDC activity, which was increased nearly 2-fold in the heart of CAG-SpdS mice. AdoMet, dcAdoMet and AdoMetDC measurements in muscle extracts revealed clear differences between male and female mice regardless of genotype, and there was a nominal decrease in dcAdoMet content associated with SpdS overexpression in this tissue.
Table 3.
AdoMet and dcAdoMet content and AdoMetDC activity in control and CAG-SpdS mice
| Tissue | Group | Sex | AdoMet (pmol/mg tissue) | dcAdoMet (pmol/mg tissue) | AdoMetDC activity (pmol CO2/min/mg protein) |
|---|---|---|---|---|---|
| Heart | Control | M and F | 36.5 ± 6.2 | 0.24 ± 0.04 | 3.4 ± 0.3 |
| CAG-SpdS | M and F | 33.2 ± 2.9 | 0.26 ± 0.09 | 6.1 ± 0.3** | |
| Muscle | Control | F | 14.2, 17.2 | 0.23, 0.23 | 37, 37 |
| M | 10.9, 9.0 | 1.53, 1.50 | 191, 161 | ||
| CAG-SpdS | F | 17.7, 25.3 | 0.14, 0.13 | 34, 31 | |
| M | 11.5, 12.1 | 0.78, 0.25 | 450, 205 |
AdoMet and dcAdoMet content and AdoMetDC enzymatic activity were determined in 7-week-old mice (n=4 per genotype). Heart values represent mean ± S.D. of 2 male and 2 female mice. Muscle values differed between male and female mice so individual values are presented for each of 2 male and 2 female mice.
p<0.005 vs. wild type controls.
Simultaneous overexpression of SpdS and SpmS in bitransgenic mice
CAG-SpmS mice with ubiquitous overexpression of SpmS using the same CAG promoter have been reported previously (Ikeguchi et al. 2004). In those mice, up to 2000-fold increases in SpmS activity led to only small changes in spermine and spermidine levels and SpdS activity was unaltered. We bred CAG-SpdS and CAG-SpmS mice to determine whether simultaneous overexpression of both aminopropyltransferases would have a more dramatic effect on tissue polyamine profiles. CAG-SpdS/CAG-SpmS mice were generated at the expected frequency (9 of 34 offspring, 26.5%) and did not exhibit any obvious phenotypic abnormalities. In heart and liver from 5-week-old bitransgenic mice we demonstrated that both aminopropyltransferase activities were elevated to levels that are comparable to values from single transgenic animals (Tables 1 and 4) and to those observed in previous studies of CAG-SpmS mice (Ikeguchi et al. 2004). Next, polyamine content was measured in heart, skeletal muscle, liver, kidney and brain (Table 5). In agreement with previous results (Table 2 and (Ikeguchi et al. 2004)), CAG-SpdS mice exhibited increased spermidine levels and decreased Spm:Spd ratios while CAG-SpmS mice exhibited increased spermine levels, decreased spermidine levels and increased Spm:Spd ratios. In bitransgenic mice, spermidine and spermine levels as well as Spm:Spd ratios were intermediate between the values obtained in individual single transgenic mice in all tissues except brain, which lacks transgene-derived SpdS activity (Table 1). Putrescine levels did not appear to be depleted in response to SpdS or SpmS overexpression. These results clearly show that overexpression of SpdS, SpmS or both aminopropyltransferases is not sufficient to yield considerable increases in the higher polyamines spermidine and spermine.
Table 4.
Aminopropyltransferase activity in control, CAG-SpdS, CAG-SpmS and CAG-SpdS/CAG-SpmS mice
| Tissue | Group | Spermidine synthase (pmol/mg/h) | Spermine synthase (pmol/mg/h) |
|---|---|---|---|
| Heart | Control | 574 ± 69 | 59 ± 2 |
| CAG-SpdS | 2908 ± 433** | 51 ± 8 | |
| CAG-SpmS | 432 ± 41* | 16030 ± 801** | |
| CAG-SpdS/CAG-SpmS | 2064 ± 179** | 15517 ± 357** | |
|
| |||
| Liver | Control | 4330 ± 312 | 11 ± 1 |
| CAG-SpdS | 11952 ± 861** | 7 ± 2 | |
| CAG-SpmS | 3742 ± 501 | 9240 ± 940** | |
| CAG-SpdS/CAG-SpmS | 9069 ± 1676* | 9575 ± 1125** | |
Spermidine and spermine synthase activity (mean ± S.D.) was assayed in tissues from 5-week-old mice (n=4 per genotype).
p<0.05,
p<0.005 vs. wild type controls.
Table 5.
Polyamine content in control, CAG-SpdS, CAG-SpmS and CAG-SpdS/CAG-SpmS mice
| Tissue | Group | Polyamine content (pmol/mg tissue)
|
Spm:Spd ratio | ||
|---|---|---|---|---|---|
| Putrescine | Spermidine | Spermine | |||
| Heart | Control | 32 ± 5 | 219 ± 15 | 383 ± 14 | 1.8 ± 0.1 |
| CAG-SpdS | 25 ± 1* | 322 ± 28** | 330 ± 24** | 1.0 ± 0.1** | |
| CAG-SpmS | 46 ± 9* | 128 ± 18** | 477 ± 28** | 3.8 ± 0.6** | |
| CAG-SpdS/CAG-SpmS | 44 ± 6* | 165 ± 12** | 430 ± 24** | 2.6 ± 0.2** | |
|
| |||||
| Muscle | Control | 36 ± 5 | 172 ± 54 | 286 ± 89 | 1.8 ± 0.7 |
| CAG-SpdS | 24 ± 3** | 323 ± 15** | 212 ± 29 | 0.7 ± 0.1* | |
| CAG-SpmS | 47 ± 9* | 75 ± 24** | 309 ± 33 | 4.4 ± 1.4** | |
| CAG-SpdS/CAG-SpmS | 36 ± 11 | 151 ± 50 | 278 ± 68 | 2.0 ± 0.6 | |
|
| |||||
| Liver | Control | 32 ± 6 | 879 ± 61 | 814 ± 62 | 0.9 ± 0.1 |
| CAG-SpdS | 26 ± 1 | 1009 ± 142* | 486 ± 71** | 0.5 ± 0.1** | |
| CAG-SpmS | 58 ± 18* | 527 ± 112** | 1252 ± 140** | 2.4 ± 0.4** | |
| CAG-SpdS/CAG-SpmS | 58 ± 22* | 539 ± 51** | 1040 ± 79** | 1.9 ± 0.1** | |
|
| |||||
| Kidney | Control | 41 ± 5 | 397 ± 40 | 821 ± 30 | 2.1 ± 0.2 |
| CAG-SpdS | 38 ± 9 | 474 ± 35* | 773 ± 40 | 1.6 ± 0.1* | |
| CAG-SpmS | 67 ± 13** | 232 ± 34** | 964 ± 61** | 4.2 ± 1.0** | |
| CAG-SpdS/CAG-SpmS | 61 ± 13** | 250 ± 22** | 920 ± 81* | 3.7 ± 0.5** | |
|
| |||||
| Brain | Control | 21 ± 3 | 270 ± 71 | 322 ± 29 | 1.3 ± 0.3 |
| CAG-SpdS | 25 ± 4 | 275 ± 51 | 333 ± 22 | 1.2 ± 0.3 | |
| CAG-SpmS | 29 ± 3** | 208 ± 22 | 405 ± 58* | 2.0 ± 0.1** | |
| CAG-SpdS/CAG-SpmS | 35 ± 7** | 206 ± 22* | 398 ± 94 | 1.9 ± 0.4** | |
Polyamine content (mean ± S.D.) was measured in tissues from 5-week-old control (n=6), CAG-SpdS (n=4), CAG-SpmS (n=5) and CAG-SpdS/CAG-SpmS (n=8) mice.
p<0.05,
p<0.005 vs. wild type controls.
Combined overexpression of SpdS and AdoMetDC in the mouse heart
In order to test whether insufficient dcAdoMet content was limiting the ability of SpdS to mediate a greater increase in spermidine and spermine levels, we bred CAG-SpdS mice with MHC-AdoMetDC mice in which AdoMetDC expression is driven by the cardiac-specific αMHC promoter (Nisenberg et al. 2006). These mice exhibit a >100-fold increase in cardiac AdoMetDC activity and a >400-fold increase in cardiac dcAdoMet content (Pegg et al. 2011). Bitransgenic CAG-SpdS/MHC-AdoMetDC animals were generated at the expected frequency (7 of 30 offspring, 23.3%). Therefore, overexpression of both AdoMetDC and SpdS in the heart is compatible with the production of viable bitransgenic mice, whereas crosses of MHC-AdoMetDC and CAG-SpmS mice did not yield any viable bitransgenic offspring at 3 weeks of age (Ikeguchi et al. 2004). CAG-SpdS/MHC-AdoMetDC mice exhibited cardiac polyamine levels that were nearly identical to MHC-AdoMetDC littermates (Table 6). This result demonstrates that factors other than SpdS and AdoMetDC activity control cardiac spermidine and spermine accumulation.
Table 6.
Cardiac polyamine content in control, CAG-SpdS, MHC-AdoMetDC and CAG-SpdS/MHC-AdoMetDC mice
| Group | Polyamine content (pmol/mg tissue)
|
Spm:Spd ratio | ||
|---|---|---|---|---|
| Putrescine | Spermidine | Spermine | ||
| Control | 15 ± 4 | 187 ± 14 | 332 ± 47 | 1.8 ± 0.2 |
| CAG-SpdS | 10 ± 2* | 217 ± 22* | 287 ± 36 | 1.3 ± 0.0** |
| MHC-AdoMetDC | 9 ± 3* | 109 ± 19** | 354 ± 7 | 3.3 ± 0.6* |
| CAG-SpdS/MHC-AdoMetDC | 9 ± 1* | 104 ± 9** | 355 ± 24 | 3.4 ± 0.4** |
Polyamine content (mean ± S.D.) was measured in tissues from 5-week-old control (n=6), CAG-SpsS (n=6), MHC-AdoMetDC (n=4) and CAG-SpdS/MHC-AdoMetDC (n=6) mice.
p<0.05,
p<0.005 vs. wild type controls.
DISCUSSION
The CAG enhancer/promoter element was chosen to drive overexpression of a human SpdS cDNA in the mouse without the endogenous regulatory controls that govern SpdS transcription and translation. Although this promoter was chosen to enable ubiquitous overexpression, the greatest increase in activity in CAG-SpdS line B mice was detected in heart and muscle as seen previously with CAG-SpmS mice (Ikeguchi et al. 2004). It is unclear whether the lesser activity in other tissues of SpdS mice is due to the lack of muscle-specific transcription factors or positional effects related to transgene integration site. The increase in SpdS activity in CAG-SpdS tissues (Table 1) was modest relative to the 2000-fold increase in SpmS activity that was achieved in some tissues of CAG-SpmS mice. However, there is a much higher constitutive level of SpdS activity compared to SpmS activity in most tissues and it is readily apparent from both the enzymatic assays and the western blots that a significant amount of additional SpdS enzyme was produced. The increased SpdS activity led to a 1.5 to 2-fold reduction in the Spm:Spd ratio (Table 2) as compared to the 2 to 4-fold increase in Spm:Spd ratio in tissues of CAG-SpmS mice. There was little or no compensatory change detected in either SpdS or SpmS in response to transgenic overexpression of the other synthase (Tables 1 and 4 and (Ikeguchi et al. 2004)); therefore, we conclude that the activity of these enzymes is not responsive to cellular Spm:Spd ratios in vivo.
The decarboxylation of AdoMet irreversibly commits it to polyamine biosynthesis rather than cellular methylation reactions and dcAdoMet levels are typically less than 5% of AdoMet. dcAdoMet levels are elevated in mice or humans lacking SpmS activity and widespread overexpression of SpmS slightly depletes tissue dcAdoMet, thus there is an inverse relationship between dcAdoMet content and SpmS activity (Pegg et al. 2011). However, there was no reduction in cardiac dcAdoMet levels in CAG-SpdS mice, possibly due to a concurrent increase in AdoMetDC activity (Table 3). Conversely, cardiac dcAdoMet levels were reduced in CAG-SpmS mice that also exhibited decreased cardiac AdoMetDC activity (Pegg et al. 2011). These opposing results may be related to the differing cellular Spm:Spd ratios in CAG-SpdS and CAG-SpmS mice and the fact that spermine is a stronger repressor of AdoMetDC than spermidine (Pegg 2009b). Interestingly, measurements of muscle AdoMet, dcAdoMet and AdoMetDC showed clear differences between male and female mice regardless of genotype. In both sexes, CAG-SpdS mice exhibit decreased dcAdoMet content that correlates with a lack of AdoMetDC induction in samples from female animals and modest AdoMetDC induction in males. The greatly elevated AdoMetDC activity in males relative to females requires further investigation and may be related to continued increases in muscle mass in young adult males (age 7 weeks old) but not females.
Substantial increases in SpdS or SpmS activity altered the Spm:Spd ratio in transgenic tissues and this was normalized somewhat by the combined overexpression of both aminopropyltransferases (Table 5). Overexpression of SpdS, SpmS or both enzymes failed to yield a dramatic elevation in spermidine and spermine content, which indicates that either low capacity or compensatory alterations in ODC and AdoMetDC limit polyamine synthesis. CAG-SpdS and MHC-AdoMetDC mice were bred to determine if elevated dcAdoMet would enable a greater cardiac spermidine accumulation but polyamine levels in bitransgenic animals did not support this hypothesis. In fact, the data indicate that increased dcAdoMet availability in CAG-SpdS mice facilitates spermine accumulation rather than spermidine (Table 6), which reinforces the concept that SpmS and its access to dcAdoMet are limiting for cardiac spermine accumulation (Ikeguchi et al. 2004; Pegg et al. 2011). Previous studies demonstrated that the combined overexpression of SpmS and AdoMetDC in the heart is lethal because this limiting step is removed and presumably putrescine and spermidine are rapidly converted to spermine (Ikeguchi et al. 2004). Conversely, our results suggest that endogenous cardiac SpmS levels are compatible with normal development even in the face of seemingly unlimited dcAdoMet and spermidine availability in CAG-SpdS/MHC-AdoMetDC mice. The cardiac overexpression of both ODC and AdoMetDC is also lethal (Nisenberg et al. 2006), again demonstrating that the decarboxylases provide the regulatory steps that limit toxic polyamine accumulation. We did not attempt to generate tri-transgenic CAG-SpdS/MHC-AdoMetDC/MHC-ODC mice. Studies of cardiac physiology in these animals, and the combined overexpression of all four polyamine biosynthetic enzymes, will require more advanced models with conditional transgene expression.
Our results in CAG-SpdS mice and a previous study of mice with extra copies of the complete human SpdS coding region plus flanking regulatory sequences (Kauppinen et al. 1993) show little change in polyamine content. Transgenic mice that overexpress both ODC and AdoMetDC also fail to exhibit dramatic polyamine accumulation (Heljasvaara et al. 1997). In fact, numerous studies with transgenic animals that overexpress one or even two polyamine biosynthetic enzymes have been unable to yield massive polyamine accumulation. This may be related to toxic effects, and combined SpmS/AdoMetDC (Ikeguchi et al. 2004) or ODC/AdoMetDC (Nisenberg et al. 2006) overexpression in the heart results in embryonic lethality. There is a dramatic accumulation of putrescine and to a lesser extent spermidine upon massive ODC overexpression (Shantz et al. 2001; O’Brien et al. 1997). Compensatory mechanisms such as polyamine catabolism or excretion may actively limit spermidine and spermine accumulation when the decarboxylase and aminopropyltransferase enzymes are present in excess. However, spermine oxidase and spermidine/spermine-N1-acetyltransferase were not altered in CAG-SpmS tissues (Ikeguchi et al. 2004) and were not evaluated in SpdS mice. Alternatively, there may be cellular mechanisms to compartmentalize or sequester polyamine metabolic enzymes or the polyamines themselves. Putrescine depletion was minimal or undetected in tissues with SpdS or SpdS/SpmS or SpdS/AdoMetDC overexpression (Tables 2, 5 and 6), which may indicate that some fraction of cellular putrescine is in a bound or otherwise sequestered state that is inaccessible to SpdS. Future combination of the CAG-SpdS and MHC-ODC mice will address whether putrescine availability limits SpdS-mediated polyamine accumulation.
We studied two founder animals identified out of 53 pups that resulted from microinjection of fertilized oocytes with the transgene, one of which failed to exhibit any increase in SpdS activity. The failure to detect any evidence of increased SpdS in CAG-SpdS line A mice is likely due to the integration of transgene DNA in a transcriptionally silent region of the genome or a loss of structural integrity upon integration, but these possibilities were not pursued. The CAG-SpdS line B mice reported here are viable and phenotypically normal, which may argue against a causative role for high spermidine levels in the abnormalities of Gy mice or Snyder-Robinson syndrome patients. However, it remains a formal possibility that additional founders with ubiquitous and extreme SpdS expression were not generated due to adverse consequences from very high levels of SpdS activity. The increased SpdS activity in CAG-SpdS mice was not uniform in all tissues and was not sufficient to cause elevations in spermidine of the same magnitude as observed in Gy mice (up to 5-fold (Wang et al. 2004)), but there was robust SpdS expression in the heart of CAG-SpdS animals and the sudden death in Gy mice is likely due to arrhythmias related to polyamine modulation of cardiac ion channels (Pegg and Wang 2009). Future studies to evaluate the consequences of SpdS overexpression in the Gy background may provide additional evidence of a role for increased spermidine in the Gy phenotype. CAG-SpdS/Gy mice expressing the human SpdS would also address the provocative question of whether spermine is detected in cells from Snyder-Robinson syndrome patients, but not Gy mice, because the human SpdS enzyme displays less substrate specificity than the mouse enzyme (Ikeguchi et al. 2006).
The CAG-SpdS animals provide additional insight into the regulatory mechanisms that govern polyamine metabolism and are a valuable resource for future studies to explore the role of elevated spermidine levels in physiological or pathological processes. SpdS is a cMyc target gene and elevated SpdS is necessary for Myc-induced B-cell lymphoma (Forshell et al. 2010). CAG-SpdS mice may be useful to determine the effect of elevated SpdS activity and reduced Spm:Spd ratios on tumor development and progression in this model as well as other mouse models of cancer such as skin chemical carcinogenesis. The combination of CAG-SpdS animals with the numerous transgenic and knockout mice with altered polyamine metabolism (Pegg et al. 2003; Janne et al. 2004; Janne et al. 2006) will facilitate the manipulation of polyamine content and ratios to study the role of polyamines in cancer, ion channels function and processes where previous studies propose a role for spermidine or SpdS activity.
Acknowledgments
The authors wish to thank Tom Salada of the Penn State University Transgenic Mouse Facility for microinjection of the CAG-SpdS construct, the Penn State University College of Medicine Macromolecular Synthesis and DNA Sequencing Cores, and the technicians of the Penn State University College of Medicine Department of Comparative Medicine for expert animal care. We thank Dr. J. A. Sawicki for providing the pCX-EGFP plasmid and Dr. M. Okabe for permission to use this construct in our experiments. Supported by National Institutes of Health grants CA-018138 (DJF) and GM-26290 (AEP).
ABBREVIATIONS
- AdoMet
S-adenosylmethionine
- AdoMetDC
S-adenosylmethionine decarboxylase
- AZ
antizyme
- CAG
composite cytomegalovirus immediate early gene enhancer-chicken β-actin promoter
- dcAdoMet
decarboxylated S-adenosylmethionine
- DFMO
α-difluoromethylornithine
- Gy
Gyro
- HA
hemagglutinin
- MHC
α-myosin heavy chain
- ODC
ornithine decarboxylase
- PCR
polymerase chain reaction
- SpdS
spermidine synthase
- SpmS
spermine synthase
Footnotes
The authors declare that they have no conflict of interest.
References
- Basuroy UK, Gerner EW. Emerging concepts in targeting the polyamine metabolic pathway in epithelial cancer chemoprevention and chemotherapy. J Biochem. 2006;139 (1):27–33. doi: 10.1093/jb/mvj022. [DOI] [PubMed] [Google Scholar]
- Becerra-Solano LE, Butler J, Castaneda-Cisneros G, McCloskey DE, Wang X, Pegg AE, Schwartz CE, Sanchez-Corona J, Garcia-Ortiz JE. A missense mutation, p.V132G, in the X-linked spermine synthase gene (SMS) causes Snyder-Robinson syndrome. Am J Med Genet A. 2009;149A (3):328–335. doi: 10.1002/ajmg.a.32641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casero RA, Jr, Marton LJ. Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nat Rev Drug Discov. 2007;6 (5):373–390. doi: 10.1038/nrd2243. [DOI] [PubMed] [Google Scholar]
- Cason AL, Ikeguchi Y, Skinner C, Wood TC, Holden KR, Lubs HA, Martinez F, Simensen RJ, Stevenson RE, Pegg AE, Schwartz CE. X-linked spermine synthase gene (SMS) defect: the first polyamine deficiency syndrome. Eur J Hum Genet. 2003;11 (12):937–944. doi: 10.1038/sj.ejhg.5201072. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay MK, Tabor CW, Tabor H. Absolute requirement of spermidine for growth and cell cycle progression of fission yeast (Schizosaccharomyces pombe) Proc Natl Acad Sci U S A. 2002;99 (16):10330–10334. doi: 10.1073/pnas.162362899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Alencastro G, McCloskey DE, Kliemann SE, Maranduba CM, Pegg AE, Wang X, Bertola DR, Schwartz CE, Passos-Bueno MR, Sertie AL. New SMS mutation leads to a striking reduction in spermine synthase protein function and a severe form of Snyder-Robinson X-linked recessive mental retardation syndrome. J Med Genet. 2008;45 (8):539–543. doi: 10.1136/jmg.2007.056713. [DOI] [PubMed] [Google Scholar]
- Eisenberg T, Knauer H, Schauer A, Buttner S, Ruckenstuhl C, Carmona-Gutierrez D, Ring J, Schroeder S, Magnes C, Antonacci L, Fussi H, Deszcz L, Hartl R, Schraml E, Criollo A, Megalou E, Weiskopf D, Laun P, Heeren G, Breitenbach M, Grubeck-Loebenstein B, Herker E, Fahrenkrog B, Frohlich KU, Sinner F, Tavernarakis N, Minois N, Kroemer G, Madeo F. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol. 2009;11 (11):1305–1314. doi: 10.1038/ncb1975. [DOI] [PubMed] [Google Scholar]
- Feith DJ, Shantz LM, Pegg AE. Targeted antizyme expression in the skin of transgenic mice reduces tumor promoter induction of ornithine decarboxylase and decreases sensitivity to chemical carcinogenesis. Cancer Res. 2001;61 (16):6073–6081. [PubMed] [Google Scholar]
- Feith DJ, Shantz LM, Shoop PL, Keefer KA, Prakashagowda C, Pegg AE. Mouse skin chemical carcinogenesis is inhibited by antizyme in promotion-sensitive and promotion-resistant genetic backgrounds. Mol Carcinog. 2007;46 (6):453–465. doi: 10.1002/mc.20294. [DOI] [PubMed] [Google Scholar]
- Forshell TP, Rimpi S, Nilsson JA. Chemoprevention of B-cell lymphomas by inhibition of the Myc target spermidine synthase. Cancer Prev Res (Phila) 2010;3 (2):140–147. doi: 10.1158/1940-6207.CAPR-09-0166. [DOI] [PubMed] [Google Scholar]
- Heby O, Persson L, Rentala M. Targeting the polyamine biosynthetic enzymes: a promising approach to therapy of African sleeping sickness, Chagas’ disease, and leishmaniasis. Amino Acids. 2007;33 (2):359–366. doi: 10.1007/s00726-007-0537-9. [DOI] [PubMed] [Google Scholar]
- Heljasvaara R, Veress I, Halmekyto M, Alhonen L, Janne J, Laajala P, Pajunen A. Transgenic mice overexpressing ornithine and S-adenosylmethionine decarboxylases maintain a physiological polyamine homoeostasis in their tissues. Biochem J. 1997;323 (Pt 2):457–462. doi: 10.1042/bj3230457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeguchi Y, Bewley MC, Pegg AE. Aminopropyltransferases: function, structure and genetics. J Biochem. 2006;139 (1):1–9. doi: 10.1093/jb/mvj019. [DOI] [PubMed] [Google Scholar]
- Ikeguchi Y, Wang X, McCloskey DE, Coleman CS, Nelson P, Hu G, Shantz LM, Pegg AE. Characterization of transgenic mice with widespread overexpression of spermine synthase. Biochem J. 2004;381 (Pt 3):701–707. doi: 10.1042/BJ20040419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janne J, Alhonen L, Pietila M, Keinanen TA. Genetic approaches to the cellular functions of polyamines in mammals. Eur J Biochem. 2004;271 (5):877–894. doi: 10.1111/j.1432-1033.2004.04009.x. [DOI] [PubMed] [Google Scholar]
- Janne J, Alhonen L, Pietila M, Keinanen TA, Uimari A, Hyvonen MT, Pirinen E, Jarvinen A. Genetic manipulation of polyamine catabolism in rodents. J Biochem. 2006;139 (2):155–160. doi: 10.1093/jb/mvj035. [DOI] [PubMed] [Google Scholar]
- Kauppinen L, Myohanen S, Halmekyto M, Alhonen L, Janne J. Transgenic mice over-expressing the human spermidine synthase gene. Biochem J. 1993;293 (Pt 2):513–516. doi: 10.1042/bj2930513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackintosh CA, Pegg AE. Effect of spermine synthase deficiency on polyamine biosynthesis and content in mice and embryonic fibroblasts, and the sensitivity of fibroblasts to 1,3-bis-(2-chloroethyl)-N-nitrosourea. Biochem J. 2000;351(Pt 2):439–447. [PMC free article] [PubMed] [Google Scholar]
- Madeo F, Tavernarakis N, Kroemer G. Can autophagy promote longevity? Nat Cell Biol. 2010;12 (9):842–846. doi: 10.1038/ncb0910-842. [DOI] [PubMed] [Google Scholar]
- Nisenberg O, Pegg AE, Welsh PA, Keefer K, Shantz LM. Overproduction of cardiac S-adenosylmethionine decarboxylase in transgenic mice. Biochem J. 2006;393 (Pt 1):295–302. doi: 10.1042/BJ20051196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura K, Nakatsu F, Kashiwagi K, Ohno H, Saito T, Igarashi K. Essential role of S-adenosylmethionine decarboxylase in mouse embryonic development. Genes Cells. 2002;7 (1):41–47. doi: 10.1046/j.1356-9597.2001.00494.x. [DOI] [PubMed] [Google Scholar]
- O’Brien TG, Megosh LC, Gilliard G, Soler AP. Ornithine decarboxylase overexpression is a sufficient condition for tumor promotion in mouse skin. Cancer Res. 1997;57 (13):2630–2637. [PubMed] [Google Scholar]
- Park MH, Nishimura K, Zanelli CF, Valentini SR. Functional significance of eIF5A and its hypusine modification in eukaryotes. Amino Acids. 2010;38 (2):491–500. doi: 10.1007/s00726-009-0408-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegg AE. Regulation of ornithine decarboxylase. J Biol Chem. 2006;281 (21):14529–14532. doi: 10.1074/jbc.R500031200. [DOI] [PubMed] [Google Scholar]
- Pegg AE. Mammalian polyamine metabolism and function. IUBMB Life. 2009a;61 (9):880–894. doi: 10.1002/iub.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegg AE. S-Adenosylmethionine decarboxylase. Essays Biochem. 2009b;46:25–45. doi: 10.1042/bse0460003. [DOI] [PubMed] [Google Scholar]
- Pegg AE, Feith DJ, Fong LY, Coleman CS, O’Brien TG, Shantz LM. Transgenic mouse models for studies of the role of polyamines in normal, hypertrophic and neoplastic growth. Biochem Soc Trans. 2003;31 (2):356–360. doi: 10.1042/bst0310356. [DOI] [PubMed] [Google Scholar]
- Pegg AE, Wang X. Mouse models to investigate the function of spermine. Commun Integr Biol. 2009;2 (3):271–274. doi: 10.4161/cib.2.3.8225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegg AE, Wang X, Schwartz CE, McCloskey DE. Spermine synthase activity affects the content of decarboxylated S-adenosylmethionine. Biochem J. 2011;433 (1):139–144. doi: 10.1042/BJ20101228. [DOI] [PubMed] [Google Scholar]
- Pegg AE, Wechter R, Poulin R, Woster PM, Coward JK. Effect of S-adenosyl-1,12-diamino-3-thio-9-azadodecane, a multisubstrate adduct inhibitor of spermine synthase, on polyamine metabolism in mammalian cells. Biochemistry. 1989;28 (21):8446–8453. doi: 10.1021/bi00447a026. [DOI] [PubMed] [Google Scholar]
- Pendeville H, Carpino N, Marine JC, Takahashi Y, Muller M, Martial JA, Cleveland JL. The ornithine decarboxylase gene is essential for cell survival during early murine development. Mol Cell Biol. 2001;21 (19):6549–6558. doi: 10.1128/MCB.21.19.6549-6558.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rider JE, Hacker A, Mackintosh CA, Pegg AE, Woster PM, Casero RA., Jr Spermine and spermidine mediate protection against oxidative damage caused by hydrogen peroxide. Amino Acids. 2007;33 (2):231–240. doi: 10.1007/s00726-007-0513-4. [DOI] [PubMed] [Google Scholar]
- Sawicki JA, Morris RJ, Monks B, Sakai K, Miyazaki J. A composite CMV-IE enhancer/beta-actin promoter is ubiquitously expressed in mouse cutaneous epithelium. Exp Cell Res. 1998;244 (1):367–369. doi: 10.1006/excr.1998.4175. [DOI] [PubMed] [Google Scholar]
- Shantz LM, Feith DJ, Pegg AE. Targeted overexpression of ornithine decarboxylase enhances beta-adrenergic agonist-induced cardiac hypertrophy. Biochem J. 2001;358 (Pt 1):25–32. doi: 10.1042/0264-6021:3580025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shantz LM, Levin VA. Regulation of ornithine decarboxylase during oncogenic transformation: mechanisms and therapeutic potential. Amino Acids. 2007;33 (2):213–223. doi: 10.1007/s00726-007-0531-2. [DOI] [PubMed] [Google Scholar]
- Wallace HM, Fraser AV, Hughes A. A perspective of polyamine metabolism. Biochem J. 2003;376 (Pt 1):1–14. doi: 10.1042/BJ20031327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Ikeguchi Y, McCloskey DE, Nelson P, Pegg AE. Spermine synthesis is required for normal viability, growth, and fertility in the mouse. J Biol Chem. 2004;279 (49):51370–51375. doi: 10.1074/jbc.M410471200. [DOI] [PubMed] [Google Scholar]
- Wang X, Levic S, Gratton MA, Doyle KJ, Yamoah EN, Pegg AE. Spermine synthase deficiency leads to deafness and a profound sensitivity to alpha-difluoromethylornithine. J Biol Chem. 2009;284 (2):930–937. doi: 10.1074/jbc.M807758200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiest L, Pegg AE. Assay of spermidine and spermine synthases. Methods Mol Biol. 1998;79:51–57. doi: 10.1385/0-89603-448-8:51. [DOI] [PubMed] [Google Scholar]
- Wu G, Bazer FW, Davis TA, Kim SW, Li P, Marc Rhoads J, Carey Satterfield M, Smith SB, Spencer TE, Yin Y. Arginine metabolism and nutrition in growth, health and disease. Amino Acids. 2009;37 (1):153–168. doi: 10.1007/s00726-008-0210-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Min J, Ikeguchi Y, Zeng H, Dong A, Loppnau P, Pegg AE, Plotnikov AN. Structure and mechanism of spermidine synthases. Biochemistry. 2007;46 (28):8331–8339. doi: 10.1021/bi602498k. [DOI] [PubMed] [Google Scholar]