

Abstract
Bladder cancer has been associated with chronic arsenic exposure. Monomethylarsonous acid [MMA(III)] is a metabolite of inorganic arsenic and has been shown to transform an immortalized urothelial cell line (UROtsa) at concentrations 20-fold less than arsenite. MMA(III) was used as a model arsenical to examine the mechanisms of arsenical-induced transformation of urothelium. A microarray analysis was performed to assess the transcriptional changes in UROtsa during the critical window of chronic 50 nM MMA(III) exposure that leads to transformation at three months of exposure. The analysis revealed only minor changes in gene expression at one and two months of exposure, contrasting with substantial changes observed at three months of exposure. The gene expression changes at three months were analyzed showing distinct alterations in biological processes and pathways such as a response to oxidative stress, enhanced cell proliferation, anti-apoptosis, MAPK signaling, as well as inflammation. Twelve genes selected as markers of these particular biological processes were used to validate the microarray and these genes showed a time-dependent changes at one and two months of exposure, with the most substantial changes occurring at three months of exposure. These results indicate that there is a strong association between the acquired phenotypic changes that occur with chronic MMA(III) exposure and the observed gene expression patterns that are indicative of a malignant transformation. Although the substantial changes that occur at three months of exposure may be a consequence of transformation, there are common occurrences of altered biological processes between the first two months of exposure and the third, which may be pivotal in driving transformation.
Keywords: Arsenic, Monomethylarsonous Acid, Bladder Cancer, UROtsa, Gene Expression
1. INTRODUCTION
Arsenic is a ubiquitous environmental metalloid. Risks for the development of skin, lung, and bladder cancers have been associated with chronic exposure to low concentrations of inorganic arsenic (Wu et al., 1989; Chen et al., 1992; NRC 2001; IARC 2004). A major source of exposure occurs through drinking of contaminated water. Although the EPA and World Health Organization (NRC, 2001) have recommended inorganic arsenic limits in drinking water not to exceed 10 ppb, there are still populations worldwide exposed to higher levels (Tapio and Grosche, 2006).
The exact biological events by which inorganic arsenic causes cancer have not been fully elucidated. Oxidative stress, increased cell proliferation, inhibited DNA repair, genotoxicity, and altered cellular signals have all been suggested as critical events in arsenic-induced carcinogenesis (Hei and Filipic, 2004; Platanias, 2009; Kitchin and Conolly, 2010). Additionally, long-term exposure to arsenic has been associated with diabetes, hypertension, cardiovascular ailments, and neuropathies (Tseng et al., 1996; Chen et al., 1996; Vahidnia et al., 2007; Druwe and Vaillancourt, 2010).
Inorganic arsenate [As(V)] and arsenite [As(III)] are the primary species of arsenic consumed by humans. The biotransformation of inorganic arsenic in humans progresses through biochemical reduction and methylation steps leading to several detectable metabolites (Vahter, 1994, 1999). The activities and toxicities of these various metabolites may partly account for the diverse physical manifestations seen with chronic arsenic exposure. Of notable interest are the methylated arsenicals in the trivalent state, namely, monomethylarsonous acid [MMA(III)] and dimethylarsonous acid [DMA(III)]. Both the trivalent forms MMA(III) and DMA(III) have been shown to be more cytotoxic than the methylated pentavalent forms and inorganic arsenic (Hughes, 2002; Styblo et al., 2002; Hirano et al., 2004). In particular, MMA(III) has been shown to be a potent generator of reactive oxygen species (ROS), to produce more DNA damage, and appears to be more clastogenic than inorganic arsenic (Mass et al., 2001; Kligerman et al., 2003; Kitchin and Ahmad, 2003). The genotoxic effects of MMA(III) have been attributed to the generation of ROS (Nesnow et al., 2002; Schoen et al., 2004). Since these various metabolites have been detected in the urine of individuals chronically exposed to inorganic arsenic, the bladder urothelium is clearly a site of exposure (Vahter, 1999; Valenzuela et al., 2005; Xie et al., 2006).
The ability of arsenicals to modify the expression of genes in keratinocytes and urothelial cells has been previously investigated (Hamadeh et al., 2002; Rea et al., 2003; Su et al., 2006; Baily et al., 2010). Additionally, Gentry et al. (2010) conducted a comprehensive literature search on the gene expression changes associated with arsenic exposure with primary cells and immortalized cell lines. This review of in vitro effects with inorganic arsenic exposure on immortalized cells revealed a consistent set of gene expression changes in stress response (increase in AP-1 and Trx), inflammation (i.e., S100A8), proliferation (Fos, Jun, CFER, CDH1, MAP3K11), cell cycle control, oxidative stress, and apoptosis (p53, NFkB). However, few studies have examined the effects of methylated arsenic species in vitro. Bailey et al. (2010) and Su et al. (2006) demonstrated distinct profiles of cellular responses to inorganic arsenic and the methylated metabolites. Bailey et al. (2010) reported that MMA(III) appeared to exhibit a greater carcinogenic potential, and involved more pro-inflammatory signals and the expression of growth factors compared to inorganic arsenic exposure at a comparable concentration in human keratinocytes. Su et al. (2006) provided insight on the perturbations caused by arsenicals in immortalized urothelial cells (SV-HUC-1). Key similarities and differences were observed between As(III), MMA(III), and DMA(III), and that some changes in gene expression could be explained by epigenetic alterations. Even with these bodies of work, the understanding and research on the relationship between chronic MMA(III) exposure and the development of bladder cancer is still limited.
UROtsa (immortalized human urothelial cells) are a suitable model to study long-term exposures to arsenicals (Eblin et al., 2008a). Although immortalized by a stable transfection of the SV40 large T-antigen, UROtsa retain normal morphological and phenotypic qualities of the urothelium (Petzoldt et al., 1995). UROtsa have been described as non-tumorigenic in immuno-compromised mice. UROtsa cells are not fully differentiated, and as such may be more sensitive to carcinogens as they lack the uroplakin-containing asymmetric unit membrane that serves as a major barrier between the urine and the bladder urothelium. With these limitations in mind, the cell line provides a potential model to investigate chronic exposure to environmental carcinogens and the development of bladder cancer.
Bredfeldt et al. (2006) first demonstrated that UROtsa chronically exposed to 50 nM MMA(III) for a period of 24 weeks underwent phenotypic changes consistent with malignant transformation, i.e., increased rate of proliferation, anchorage-independent growth, and, at 52 weeks of exposure, formation of tumors in immuno-compromised mice. However, Wnek et al. (2010) were able to observe these changes as early as 12 weeks of exposure, and this acquired phenotype was sustained even after withdrawal of MMA(III). Therefore, the documented biochemical effects from MMA(III) exposure in UROtsa, such as the induction of ROS, DNA damage, production of pro-inflammatory cytokines, activation of mitogenic signals, and the increase in proliferation rate, exemplifies the utility of the model in the study of arsenic carcinogenesis and the unique effects of MMA(III) on the biology of the urothelium (Eblin et al., 2008b; Wnek et al., 2009; Escudero-Lourdes et al., 2010). Unlike previous studies, the research herein associates transcriptional changes with the malignant transformation of a human urothelial cell line, allowing for a more comprehensive understanding of the biological changes associated with chronic MMA(III) exposure.
2. MATERIALS AND METHODS
2.1 Chemicals
Dulbecco’s Modified Eagle Medium (DMEM), fetal bovine serum (FBS), antibiotic-antimycotic, and 1X trypsin-ethylenediaminetetraacetic acid (EDTA) (0.25%) were acquired from Gibco Invitrogen/Molecular Probes Corporation (Carlsbad, CA). Diiodomethylarsine [MMA(III) iodide, CH3AsI2] was prepared by the Synthetic Chemistry Facility Core (Southwest Environmental Health Sciences Center, Tucson, AZ) using the technique of Millar et al. (1960). Dissolution of diiodomethylarsine in water yields monomethylarsonous acid [MMA(III)] and the concentration of MMA(III) is verified using high performance liquid chromatography – inductively coupled mass spectrometry (HPLC ICP-MS) in the analytical core of our NIEHS Superfund Research Program. MMA(III) solutions in distilled, de-ionized water were stable for approximately 4 months at 4 °C with no degradation observed when monitored using HPLC ICP-MS (Gong et al., 2001).
2.2 Cell Culture Procedures
UROtsa cells were a generous gift from Drs. Donald and Maryann Sens (University of North Dakota, ND). Cell culture conditions were derived from those previously described by Rossi et al. (2001) and Bredfeldt et al. (2004). UROtsa cells were cultured in a growth media of DMEM containing 5% (v/v) FBS and 1% (v/v) antibiotic–antimycotic. Cultured cells were incubated at 37 °C and 5% CO2. Confluent cells were removed from plates with trypsin–EDTA (0.25%) and subcultured at a ratio of 1:3. All UROtsa cells used in this study tested negative for the presence of mycoplasma contamination. Treated cultures of UROtsa were continuously grown in media supplemented with 50 nM MMA(III) and refreshed every two days. The use of 50 nM (or approximately 6.2 ppb) of MMA(III) in the assessment of effects from chronic arsenic exposure is a relevant physiological concentration, which falls within the range of MMA(III) detected in urine of individuals exposed to environmentally relevant concentrations of inorganic arsenic from the water supply (Aposhian et al., 2000; Le et al., 2000; Mandal et al., 2001, 2004).
2.3 Gene Expression Analysis
UROtsa (at passage 23) were grown until they were 80% confluent in 75 cm2 flasks with DMEM (as described above). Six conditions were maintained at the time of harvest: control (3 replicates), 24 hr (1 replicate), 1 month-exposed (3 replicates), 2 month-exposed (4 replicates), 3 month-exposed (4 replicates), and 4 month- exposed (1 replicate) cultures. Total RNA was prepared from each sample according to the RNeasy Mini Kit (Qiagen, Valencia, CA). RNA concentrations and quality were determined spectrophotometrically using A260 and A260/A280, respectively. All RNA samples were stored at −80°C until analysis. The isolated total RNA samples were used to produce labeled target, hybridized to Affymetrix GeneChip® Human Gene 1.0 ST Array, and read using the Agilent/Affymetrix 2500A scanner according to manufacturer’s protocols. Raw data (CEL files) were normalized and summarized according to Irizarry et al. (2003), as implemented in package aroma.affymetrics (ref: H. Bengtsson; K. Simpson; J. Bullard; K. Hansen, aroma.affymetrix: A generic framework in R for analyzing small to very large Affymetrix data sets in bounded memory, Tech Report). Differential expression was tested using package LIMMA in R programming environment (Smyth, 2005). All p values were adjusted according Benjamini and Hochberg’s method to control the false discovery rate using a global method. Adjustment of p-values was performed separately in each tested group (significance level used, p value=0.05). Batch correction was performed according to Johnson et al. (2007). It has been shown previously that using additional criteria such as fold-change in expression does not lead to a better gene list (Zhang and Cao, 2009). Therefore, no cut-off in the fold-change was applied to the analyses.
To detect differentially expressed genes, replicates for each time point were compared to the expression level of control. Additionally, for detection of differential expression in the first two months of MMA(III) exposure for only the Gene Ontology analysis, samples were pooled into groups to gain more sensitivity. First group included the control (t=0) and the 24 hr exposure. This was performed based on the observation that genes involved in response to MMA(III) exposure did not change substantially in 24 hr. The second group contained samples from 1 and 2 months of exposure. Again, this was performed based on the observation that there were not many genes that changed between first and second months of exposure. Application of this pooling follows the rationale that using groups with more replicates will increase the statistical power of the test.
The method of Signaling Pathway Impact Analysis (SPIA) according to Tarca et al. (2009) was applied to assess significant pathways associated with the condition of chronic arsenic exposure. The version of SPIA used in this analysis applies the Kyoto Encyclopedia of Genes and Genomes (KEGG) signaling pathway data (http://www.genome.jp/kegg/pathway.html). SPIA uses the information from a set of differentially expressed genes and their fold changes, as well as pathway topology in order to assess the significance of the pathways in the conditions under study. Gene Ontology terms were used for functional annotation of differentially express genes. Enrichment analysis of Gene Ontology terms was performed using method implemented as R package topGO by Alexa et al. (2010).
2.4 Quantitative Reverse Transcription PCR
Total RNA was isolated from all cells using the RNeasy Mini Kit (Qiagen, Valencia, CA). Gene expression was measured using quantitative real-time reverse transcription (QRT)-PCR. Total RNA was reverse transcribed to complementary DNA (cDNA) (Applied Biosystems, Foster City, CA). Converted cDNA was added to Universal PCR Master Mix (Applied Biosystems, Foster City, CA) and gene-specific Taqman Primers/Probes for each gene target (Applied Biosystems, Foster City, CA); and subjected to real-time PCR analysis using Roche Universal Probe technology (Roche) on an ABI 7500 Real-Time Detection System (Applied Biosystems, Foster City, CA). Results were calculated using the delta delta Ct method normalized to glyceraldehydes 3-phosphate dehydrogenase (GAPDH) expression for each sample according to manufacturer’s instructions.
3. RESULTS
3.1 Gene Array Analysis
To evaluate global changes in gene expression from long-term low-dose exposure to MMA(III), samples from 0, 24 hr, 1 month, 2 months, 3 months, and 4 months of exposure were analyzed. Since replicates were available only for control (t=0), 1, 2, and 3 months of exposure, the sample from the 4 months exposure time point were not used in statistical tests, but the 24 hr sample was included in the 1–2 month statistical analysis. Three months of exposure to 50 nM MMA(III) were required before a substantial number of genes was altered in their expression (Table 1). The number of significantly altered genes in the first and second months of exposure was small (52 genes and 88 genes, respectively), with predominantly down-regulation occurring. At the third month of exposure the number of differentially expressed genes was increased to 2779, with genes distributed more or less equally between up- and down-regulation.
TABLE 1.
Expression of genes in UROtsa cells exposed continuously to 50 nM MMA(III).
| Change in Expression | 1 MONTH | 2 MONTH | 3 MONTH |
|---|---|---|---|
| up-regulated after exposure | 12 | 23 | 1240 |
| down-regulated after exposure | 40 | 65 | 1539 |
| no change | 33200 | 33164 | 30473 |
3.2 Comparison of Gene Expression Changes Between Time Points
Fifteen differentially expressed genes were common between each month of exposure (Figure 1). This group of genes includes MRPL23, GBP5, IFIT2, PARP14, XRN1, CXCL10 and 11, CCRL1, and PDCD1LG2. Between 1 and 2 months of exposure 19 differentially expressed genes were found to be common; between 2 and 3 months, 58 genes, and between 1 and 3 months, 22 genes. A majority of these genes were down-regulated in the first two months. By the third month of exposure substantial alterations were seen in the number of genes changed, and the number of genes found up-regulated in comparison to the number of genes down-regulated were about the same.
FIGURE 1.
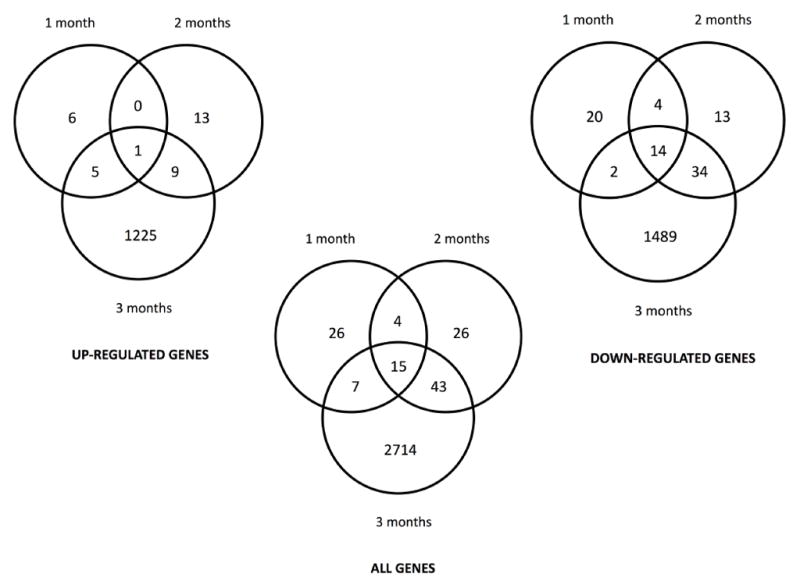
Venn diagrams illustrating common differentially expressed genes between first, second, and third month of continuous exposure to 50 nM MMA(III).
Line plots show only minor changes in gene expression at one and two months of exposure (Figure 2). The most substantial and significant changes occurred at three months of exposure, with multi-fold changes in expression for many genes.
FIGURE 2.
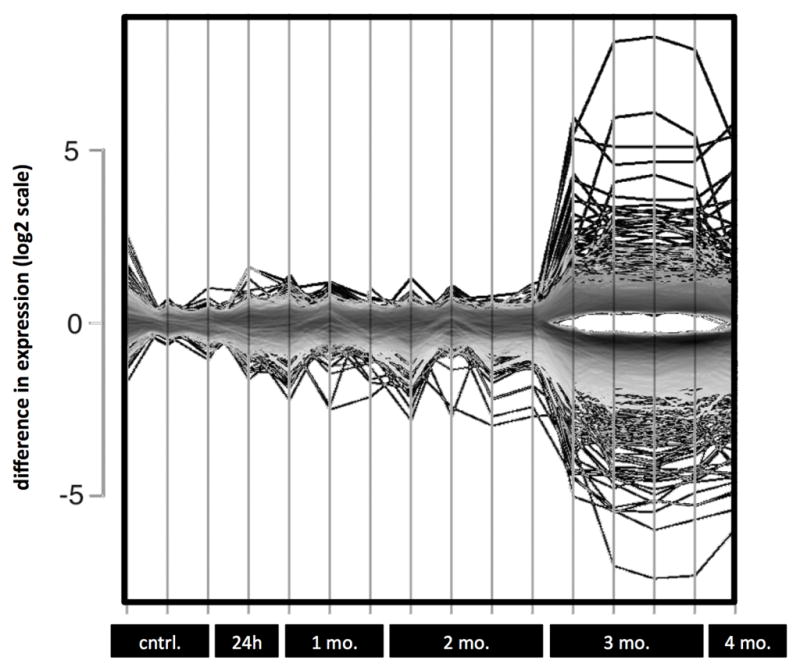
Line plot depicting gene expression changes in UROtsa with continuous exposure to 50 nM MMA(III). Each vertical line represents a replicate for the given time point listed along the X-axis. Y-axis is shown in log2 scale.
3.3 PCR Validation and Markers of Bladder Cancer
Twelve genes were selected as representatives of cell processes: proliferation, cell cycle changes, oxidative stress response, DNA repair, surface adhesion, and differentiation. Some of these genes have been found altered in human and mouse bladder cancers (see second paragraph in Discussion). To confirm the accuracy of the microarray these genes were subjected to quantitative PCR. Time-dependent changes in the expression of these genes were observed with the greatest changes occurring at 3 months of exposure (Figure 3 and 4). At the first and second months of exposure TMPRSS11A, CXCR4, CRYAB, CTNND2, PDGFRA, and ERCC2 displayed varying levels of expression, but by the third month of exposure these genes were significantly elevated with TMPRSS11A, PDGFRA, and CTNND2 showing 8-, 12-, and 32-fold increases over control, respectively. At the first and second months of exposure the expression of KRT7, TGFBI, DUSP1, DCLRE1C, IRF1, and ICAM1 were observed to be either slightly depressed, significantly depressed, or slightly elevated (i.e., KRT7 at 2 months of exposure). At the third month of exposure the expression of these genes were significantly depressed by as much as 80%. The observed trends with PCR validations of these genes were consistent with the observed changes in the microarray results.
FIGURE 3.

Confirmation of microarray results by QRT-PCR analysis of selected gene transcripts elevated in UROtsa exposed continuously to 50 nM MMA(III). Graph represents selected genes up-regulated at 3 months of exposure to MMA(III) as well as expression levels at 1 and 2 months of exposure. mRNA was normalized to UROtsa control and relative to GAPDH levels. Error bars indicate standard deviation of triplicates.
FIGURE 4.

Confirmation of microarray results by QRT-PCR analysis of selected gene transcripts suppressed in UROtsa exposed continuously to 50 nM MMA(III). Graph represents selected genes down-regulated at 3 months of exposure to MMA(III) as well as expression levels at 1 and 2 months of exposure. mRNA was normalized to UROtsa control and relative to GAPDH levels. Error bars indicate standard deviation of triplicates.
3.4 Pathways and Categories of Gene Expression Changes
3.4.1 Summary of Functional Categories and Differentially Expression Gene Transcripts
A summary of differentially expressed genes presented in functional categories is given in the following sections (Table 2 and 3):
TABLE 2.
List of gene transcripts ELEVATED in UROtsa with 3 month continuous exposure to 50 nM MMA(III).
| Oxidative Stress or Similar Response | |
|
| |
| OXSR1 | regulates actin cytoskeleton in response to stress |
| SOD1 | catalyzes the dismutation of superoxide into oxygen |
| GSTM3 | detoxifies endogenous compounds |
| GSTA1, GSTA4, GSTZ1 | defense against toxic, carcinogenic and pharmacologically |
| HSPB8 | chaperone |
| MT1X | heavy metal binding and control of oxidative stress |
|
| |
| Apoptosis | |
|
| |
| DNASE1L3 | mediates breakdown of DNA during apoptosis |
| DAPL1 | apoptosis; early epithelial differentiation-associated |
| GPC3 | induce apoptosis in certain cell types; peptidyl-dipeptidase inhibitor activity |
| BNIP3 | protein heterodimerization activity; induce apoptosis, even in the presence of BCL2 |
|
| |
| Inflammatory Response | |
|
| |
| CXCR4 | chemokine receptor |
| CYP4F11 | aromatase activity; heme binding; metal ion binding; monooxygenase activity |
| S100A8 | involved in inflammatory response, cell cycle progression and differentiation |
|
| |
| Growth & Proliferation | |
|
| |
| EMP1 | cell growth; cell proliferation and differentiation; multicellular organismal development |
| IGFBP5 | alter the interaction of IGFs with their receptors |
| KGFLP1 | growth factor activity |
| NTS | stimulation of growth and proliferation |
| NAP1L2 | regulate neuronal cell proliferation |
| PIK3CB | involved in signaling pathways regulating cell growth |
| PDGFRA | cell proliferation, differentiation, and growth |
|
| |
| DNA Repair Elements | |
|
| |
| BCL11B | zinc ion binding; a role in response to DNA replication stress and damages |
| XPD (or ERCC2) | transcription-coupled nucleotide excision repair |
|
| |
| Surface Adhesion | |
|
| |
| CDH4, CDH16 | cell-to-cell adhesion |
| CTNND2 | cell-to-cell adhesion |
| CD302 | is a C-type lectin receptor involved in cell adhesion and migration |
| IGSF9 | proportion of cell adhesion molecules |
| LPHN1 | G-protein coupled receptor activity; cell adhesion and signal transduction |
| TMPRSS11A | regulates cell-cell and cell-matrix interactions |
|
| |
| Developmental/Differentiation | |
|
| |
| BMP3, BMP7, BMP8A | growth-inhibitory factors; angiogenesis |
| NELL2 | involved in growth and differentiation |
| SPRR3 | keratinocyte differentiation marker |
| WNT5A | regulation of cell fate & development |
|
| |
| Bladder Cancer and Metastasis Markers | |
|
| |
| FGF1, FGF2 | involved in angiogenesis |
| FGFR1, FGFR3 | FGF receptors |
| MMP11, MMP14 | degrades non-collagen matrix |
| TNC | promotes cell growth, cell migration, and angiogenesis |
TABLE 3.
List of gene transcripts SUPPRESSED in UROtsa with 3 month continuous exposure to 50 nM MMA(III).
| Oxidative Stress | |
|
| |
| CYP24A1 | drug metabolism and synthesis of cholesterol; oxidoreductase activity |
| CYP2J2 | aromatase activity; electron carrier activity; heme binding; metal ion binding |
| DUOX2 | ROS generator |
| DUSP1 | induced by stress and growth factors; negative regulation of cellular proliferation |
| FAM129A | response to stress |
| GPX8 | reduces peroxides |
| SQSTM1 | scaffolding/adaptor protein |
|
| |
| Apoptosis | |
|
| |
| IFI27 | impact cellular apoptosis |
| IFI6 | protein binding; role in the regulation of apoptosis |
| CARD11 | guanylate kinase activity; regulator of cell apoptosis and NF-kappaB activation |
| CEACAM1 | roles in differentiation and arrangement of tissue apoptosis |
| FAS | regulation of programmed cell death |
| GSDMC | a tumor suppressors gene; regulation of cell proliferation; regulation of apoptosis |
| GSDMB | regulation of apoptosis in epithelial cells |
| XAF1 | inhibitor of apoptosis; bind and inhibit caspases; zinc ion binding |
|
| |
| Inflammatory or Immune Response | |
|
| |
| CTSS | cysteine-type endopeptidase activity; peptidase activity |
| CXCL10 | inflammatory response |
| CCL2 | heparin binding; signal transducer activity; protein binding |
| FYB | protein binding; protein amino acid phosphorylation |
| GDF15 | cytokine activity; growth factor activity |
| GBP2 | GTPase activity; cytokines; antiviral effects and inhibit tumor cell proliferation |
| HCP5 | to control disease progression soon after infection |
| IFIT1 | cellular antiviral responses |
| IFIT2 | microtubule-associated protein |
| IFI44L | immune response |
| IL1A | copper ion binding; interleukin-1 receptor binding |
| IRF1 | regulates transcription of interferons; involved in the JAK-STAT signaling pathway |
| IRF7 | plays a role in the transcriptional activation of virus-inducible cellular genes |
| IL6 | pro-inflammatory and anti-inflammatory cytokine |
| IL7 | lymphocyte regulatory factor |
| IL8 | induction of chemotaxis |
| HLA | antigen presentation |
| NFKB1 | transcription factor involved in immune and inflammatory response |
| PTGS1, PTGS2 (or COX1 & 2) | biosynthesis of prostanoids |
| RSAD2 | iron-sulfur cluster binding; metal ion binding; antiviral defense |
| STAT1, STAT3, STAT6 | interferon signaling |
| SOCS3 | inhibits activity of JAK2 kinase |
| SP100 | interferon-stimulated antigen |
| TRIM22 | metal ion binding; transcription corepressor activity; ligase activity |
| TNFAIP3 | DNA binding; cysteine-type peptidase activity; protein binding; a zinc finger protein |
| TLR3 | pathogen recognition and activation of innate immunity |
|
| |
| Proliferation | |
|
| |
| HBEGF | growth factor activity |
| BMPR1A, BMPR2 | receptor for BMPs |
| GBP2 | GTPase activity; cytokines; antiviral effects and inhibit tumor cell proliferation; |
| PLAU | involved in degradation of the extracellular matrix; tumor cell migration/proliferation |
|
| |
| DNA Repair Elements | |
|
| |
| DDB2 | participates in nucleotide excision repair; mediates ubiquitylation of histones H3, H4 |
| DCLRE1C | involved in DNA repair |
| XRCC4 | involved in DNA repair |
|
| |
| Surface Adhesion | |
|
| |
| ALCAM | receptor binding; cell adhesion molecule that binds to CD6 |
| ASAM | a role in cell-cell adhesion; component of epithelial tight junctions |
| AMTN | protein binding; cell adhesion |
| AMIGO2 | protein binding; neuronal activity-dependent; cell-to-cell interaction |
| CDH6, CDH13 | cell-to-cell adhesion |
| ICAM1 | expressed on endothelial cells; binds to integrins |
| PVRL3 | cell adhesion molecule binding; protein binding; protein homodimerization activity |
| PCDH1 | involved in neural cell adhesion and neuronal development; |
| TGFBI | binds to type I, II, IV, VI collagens and fibronectin |
|
| |
| Cell Cycle Control | |
|
| |
| CDKN1A | regulator of cell cycle progression |
| DRAM1 | regulated as part of the p53 tumor suppressor pathway |
| TP53 | tumor suppressor |
| RB1 | tumor suppressor |
| SPARC | calcium ion binding; extracellular matrix binding; inhibits cell-cycle |
|
| |
| Bladder Cancer Markers | |
|
| |
| CFHR1 | marker for transitional cell carcinoma |
| DBC1 | inhibits cell proliferation |
| KRT7 | type II keratin specifically expressed in simple epithelia |
| LCN2 | innate immune response |
| ZFP36L2 | metal ion binding; nucleic acid binding; transcription factor activity |
DNA Repair
In the gene array, by the third month of exposure, two critical genes involved in DNA double-strand break repair, DCLRE1C and XRCC4, were found to be down-regulated (Table 3). P53 gene transcripts and target gene, p21 (a direct inhibitor of cell-cycle progression), were found to be suppressed by 3 months of exposure to MMA(III). Moreover, gene transcripts of proteins directly involved in apoptosis and/or cell-cycle progression were unchanged or slightly depressed (RRM2B, TP53INP1, and GADD45B).
Oxidative Stress
Oxidative stress is a significant category assessed by Gene Ontology analysis in chronically exposed cells. Wnek et al. (2009) observed elevated ROS and DNA damage in UROtsa at 12, 24, and 52 weeks of chronic exposure to MMA(III). The over-expression of genes associated with oxidative stress, such as SOD-1, glutathione peroxidase 1, metallothionein 1X, and oxidative stress response 1, is evident in the gene array data (Table 2). Additionally, several glutathione S-transferases were found to be elevated: GSTM3, GSTA1 and A4, GSTZ1, and MGST1. Only slight elevations were observed with GSTTP1 and GSTT1. Although arsenic has been reported to be an inducer of NRF2 and activated in mouse bladder tissue with arsenic exposure (Jiang et al., 2009), the target gene transcripts for NQO1 and HMOX1 had not been changed appreciably at 1, 2, and 3 months of exposure to MMA3; and TXNRD1, GCLC, PRDX1 were found only slightly elevated while GCLM was slightly depressed at 3 months of exposure. NRF2 activation has been observed in UROtsa with acute exposures to higher concentrations of As(III) and MMA(III) (Wang et al., 2007), but to our knowledge there is no precedent on the behavior of NRF2 and the expression of target genes with chronic low-dose MMA(III).
Apoptosis
SPIA analysis indicates apoptosis is an active biological pathway in the first two months of exposure, and by the third month anti-apoptotic mechanisms are in effect. Important activators of apoptosis were also found to be down-regulated, such as Fas, CASP8 and FADD-like apoptosis regulator, interleukin 1a, and suppressor of cytokine signaling 2 (Table 3). Other transcripts of reported anti-apoptotic genes were found to be elevated, such as crystallin alpha B (Arya et al., 2007), and T-box 3, an inhibitor of senescence (Brummelkamp et al., 2002) (Table 2). Apoptosis-inducing genes shown to be elevated in the third month of exposure included DNASE1L3, DAPL1, GPC3, and BNIP3. In promoting cell growth and survival, a slight over-expression of Akt1 was observed as well as over-expression of several HSP90 transcripts. HSP90 plays a role in Akt stabilization and preventing proteolytic removal (Liao and Hung, 2010).
Proliferation And Cell Growth
In the present study, PDGFRA, an activator of the MAPK pathway, was found to be over-expressed (Table 2). Additionally, the gene array data showed that DUSP1, DUSP6, and DUSP10 were down-regulated, possibly enhancing the activity of this pathway (Table 3). Furthermore, BMP7, 8A, and 3 were found to be elevated in the gene array at three months of exposure to MMA(III) and concomitant down-regulation was seen with BMPR1A and BMPR2. Other genes involved in promoting cell growth and proliferation were found to be elevated at the third month of exposure including EMP1, NTS, IGFBP5, NAP1L2, and PIK3CB. Several fibroblast growth factors (FGF1, FGF2) and receptors (FGFR1, FGFR3) were found to be elevated. Finally, cyclin D3 was found to be slightly depressed at three months of exposure, and cyclin D1 and D2, CDK4, cyclin E2, and TFDP1, genes that promote G1 to S phase transition, were not appreciably changed.
Inflammation
Several key pro-inflammatory cytokines and chemokine, such as IL-6, IL-8, TNF, and COX have been reported to elevate with arsenic exposure (Lin and Karin, 2007). Although previously shown that the protein level of several of these cytokines were elevated in UROtsa exposed chronically to MMA(III) for 3 months (Escudero-Lourdes, 2010), the transcripts for these and other cytokines were found to be reduced in the present study (IL1A, IL1B, IL7, IL6, FAS, and LIFR) (Table 3). Additionally, a number of other inflammatory-related gene transcripts (IRF7, CD86, TLR3) was found to be down-regulated at three months of exposure. Of the cytokine receptors, CXCL1, CXCL2, CXCL10, and CXCL11 were found down-regulated; and CCL2 and CCL20 were also down-regulated. The JAK-STAT signaling pathway partially mediates the regulation of cytokines and growth factors in development (Li, 2008). Suppression of the JAK-STAT pathway was evident from the gene array. STAT1, STAT2, STAT3, STAT4, STAT5A gene transcripts were found depressed at three months of exposure. Additionally, JAK2, SOCS2, SOCS3, and IL6 gene transcripts were down-regulated. Eblin et al. (2007) showed that UROtsa exposed chronically to MMA(III) for 12, 24, and 52 weeks had persistent elevated protein levels of COX-2 protein. In the present study, COX-2 gene transcripts were found to be depressed at 3 months, as well as phospholipase A2. Although COX-1 gene transcripts were found to be only slightly elevated at 1 and 2 months of exposure, at 3 months there was a substantial decrease.
ECM-Receptor Interactions
ECM-receptor interactions have also been significantly altered. Tenascin gene transcripts were found up-regulated. Laminin, collagen type IV, CD36, and integrin beta 8 and alpha 5 gene transcripts were found to be depressed.
3.4.2 Summary of Signaling Pathway Impact Analysis (SPIA)-Based Analysis
The most relevant KEGG diseases and biochemical systems perturbed at one and two months of MMA(III) were immune system and related diseases, infectious diseases, and cell growth and death. Of notable interest are the inhibition of chemokine signaling, inhibition of cytosolic DNA-sensing pathway, inhibition of cytokine-cytokine receptor interactions, and activation of apoptosis.
The most relevant diseases and biochemical systems which were perturbed at three months of MMA(III) exposure, were found to be immune system and related diseases, metabolic disease, cardiovascular disease, MAPK signaling, and other signaling components. Of notable interest are the activation of the MAPK signaling pathway, alterations in the cytosolic DNA-sensing pathway, alterations in cytokine-cytokine receptor interaction, changes in antigen processing and presentation, alterations in toll-like receptor signaling pathway, and the inhibition of ECM-receptor interactions.
3.4.3 Summary of Gene Ontology Analysis
The most significant Gene Ontology categories altered in the third month of exposure included immune and inflammatory responses, antigen processing and presentation, regulation of cell proliferation, response to oxidative stress and other stressors (i.e., wound healing), multi-organism processes, response to stimulus (i.e., chemotaxis, response to virus and drug and cytokines), intracellular protein kinase cascade (i.e., JAK-STAT cascade), and anti-apoptosis (Table 4).
TABLE 4.
Most significant biological processes, molecular functions, and cellular components altered in 1–2 months and 3 months of continuous exposure to 50 nM MMA(III). The grayed listings are those identified as common terms between the 1–2 month exposure interval and the 3 month. Lists have been ordered with the most significant terms at the top. The top 30 terms in each category are listed. For a more complete presentation of this table with numerical data, please see supplementary material.
| GENE ONTOLOGY TERMS ALTERED IN 1–2 MONTHS | |
|---|---|
| BIOLOGICAL PROCESSES | |
| GO.ID | Term |
| GO:0006878 | cellular copper ion homeostasis |
| GO:0042130 | negative regulation of T cell proliferat... |
| GO:0006955 | immune response |
| GO:0006954 | inflammatory response |
| GO:0002376 | immune system process |
| GO:0006213 | pyrimidine nucleoside metabolic process |
| GO:0032088 | negative regulation of NF-kappaB transcr... |
| GO:0045087 | innate immune response |
| GO:0006952 | defense response |
| GO:0001525 | angiogenesis |
| GO:0042157 | lipoprotein metabolic process |
| GO:0032103 | positive regulation of response to exter... |
| GO:0048646 | anatomical structure formation involved ... |
| GO:0048514 | blood vessel morphogenesis |
| GO:0048002 | antigen processing and presentation of p... |
| GO:0051707 | response to other organism |
| GO:0007156 | homophilic cell adhesion |
| GO:0006418 | tRNA aminoacylation for protein translat... |
| GO:0043038 | amino acid activation |
| GO:0043039 | tRNA aminoacylation |
| GO:0050729 | positive regulation of inflammatory resp... |
| GO:0051591 | response to cAMP |
| GO:0048872 | homeostasis of number of cells |
| GO:0001568 | blood vessel development |
| GO:0016579 | protein deubiquitination |
| GO:0001701 | in utero embryonic development |
| GO:0001944 | vasculature development |
| GO:0070646 | protein modification by small protein re... |
| GO:0043433 | negative regulation of transcription fac... |
| GO:0090048 | negative regulation of transcription reg... |
| MOLECULAR FUNCTIONS | |
|
| |
| GO.ID | Term |
| GO:0008252 | nucleotidase activity |
| GO:0016787 | hydrolase activity |
| GO:0017111 | nucleoside-triphosphatase activity |
| GO:0003723 | RNA binding |
| GO:0000166 | nucleotide binding |
| GO:0003924 | GTPase activity |
| GO:0032553 | ribonucleotide binding |
| GO:0032555 | purine ribonucleotide binding |
| GO:0016788 | hydrolase activity, acting on ester bond... |
| GO:0004843 | ubiquitin-specific protease activity |
| GO:0017076 | purine nucleotide binding |
| GO:0019783 | small conjugating protein-specific prote... |
| GO:0019825 | oxygen binding |
| GO:0008009 | chemokine activity |
| GO:0004812 | aminoacyl-tRNA ligase activity |
| GO:0016875 | ligase activity, forming carbon-oxygen b... |
| GO:0016876 | ligase activity, forming aminoacyl-tRNA ... |
| GO:0016779 | nucleotidyltransferase activity |
| GO:0042379 | chemokine receptor binding |
| GO:0070011 | peptidase activity, acting on L-amino ac... |
| GO:0001664 | G-protein-coupled receptor binding |
| CELLULAR COMPONENTS | |
|
| |
| GO.ID | Term |
| GO:0005796 | Golgi lumen |
|
| |
| GENE ONTOLOGY CATEGORIES ALTERED AT 3 MONTHS | |
|
| |
| BIOLOGICAL PROCESSES | |
|
| |
| GO.ID | Term |
| GO:0006955 | immune response |
| GO:0002474 | antigen processing and presentation of p... |
| GO:0009615 | response to virus |
| GO:0044419 | interspecies interaction between organis... |
| GO:0034097 | response to cytokine stimulus |
| GO:0009408 | response to heat |
| GO:0042060 | wound healing |
| GO:0019885 | antigen processing and presentation of e... |
| GO:0051384 | response to glucocorticoid stimulus |
| GO:0033280 | response to vitamin D |
| GO:0032355 | response to estradiol stimulus |
| GO:0008284 | positive regulation of cell proliferatio... |
| GO:0006954 | inflammatory response |
| GO:0045086 | positive regulation of interleukin-2 bio... |
| GO:0002504 | antigen processing and presentation of p... |
| GO:0032570 | response to progesterone stimulus |
| GO:0032496 | response to lipopolysaccharide |
| GO:0006935 | chemotaxis |
| GO:0043129 | surfactant homeostasis |
| GO:0030335 | positive regulation of cell migration |
| GO:0045087 | innate immune response |
| GO:0006916 | anti-apoptosis |
| GO:0006979 | response to oxidative stress |
| GO:0051240 | positive regulation of multicellular org... |
| GO:0008015 | blood circulation |
| GO:0019882 | antigen processing and presentation |
| GO:0007259 | JAK-STAT cascade |
| GO:0019221 | cytokine-mediated signaling pathway |
| GO:0042493 | response to drug |
| GO:0050729 | positive regulation of inflammatory resp... |
| MOLECULAR FUNCTIONS | |
|
| |
| GO.ID | Term |
| GO:0032393 | MHC class I receptor activity |
| GO:0015197 | peptide transporter activity |
| GO:0004298 | threonine-type endopeptidase activity |
| GO:0008083 | growth factor activity |
| GO:0004719 | protein-L-isoaspartate (D-aspartate) O-m... |
| GO:0042301 | phosphate binding |
| GO:0008252 | nucleotidase activity |
| GO:0016628 | oxidoreductase activity, acting on the C... |
| GO:0016836 | hydro-lyase activity |
| GO:0005041 | low-density lipoprotein receptor activit... |
| GO:0008009 | chemokine activity |
| GO:0004033 | aldo-keto reductase activity |
| GO:0032395 | MHC class II receptor activity |
| GO:0008237 | metallopeptidase activity |
| GO:0005178 | integrin binding |
| GO:0046982 | protein heterodimerization activity |
| GO:0005149 | interleukin-1 receptor binding |
| GO:0043548 | phosphoinositide 3-kinase binding |
| GO:0005509 | calcium ion binding |
| GO:0005161 | platelet-derived growth factor receptor ... |
| GO:0008191 | metalloendopeptidase inhibitor activity |
| GO:0008238 | exopeptidase activity |
| GO:0005111 | type 2 fibroblast growth factor receptor... |
| GO:0016813 | hydrolase activity, acting on carbon-nit... |
| GO:0016918 | retinal binding |
| GO:0017017 | MAP kinase tyrosine/serine/threonine pho... |
| GO:0019841 | retinol binding |
| GO:0008235 | metalloexopeptidase activity |
| GO:0005125 | cytokine activity |
| GO:0005520 | insulin-like growth factor binding |
| CELLULAR COMPONENTS | |
|
| |
| GO.ID | Term |
| GO:0042612 | MHC class I protein complex |
| GO:0042824 | MHC class I peptide loading complex |
| GO:0005615 | extracellular space |
| GO:0042613 | MHC class II protein complex |
| GO:0005887 | integral to plasma membrane |
| GO:0005839 | proteasome core complex |
| GO:0005576 | extracellular region |
| GO:0009986 | cell surface |
| GO:0031093 | platelet alpha granule lumen |
| GO:0005578 | proteinaceous extracellular matrix |
| GO:0005604 | basement membrane |
| GO:0005764 | lysosome |
| GO:0042598 | vesicular fraction |
| GO:0005741 | mitochondrial outer membrane |
| GO:0005626 | insoluble fraction |
| GO:0005783 | endoplasmic reticulum |
| GO:0005792 | microsome |
| GO:0005737 | cytoplasm |
| GO:0005788 | endoplasmic reticulum lumen |
| GO:0045121 | membrane raft |
| GO:0031901 | early endosome membrane |
| GO:0010008 | endosome membrane |
| GO:0044440 | endosomal part |
| GO:0005911 | cell-cell junction |
| GO:0043005 | neuron projection |
| GO:0005789 | endoplasmic reticulum membrane |
| GO:0005624 | membrane fraction |
| GO:0009897 | external side of plasma membrane |
| GO:0044297 | cell body |
| GO:0043025 | neuronal cell body |
GO.ID = Gene Ontology identifier.
The most significant molecular functions altered in the third month of MMA(III) exposure included growth factor activity, MHC class I receptor activity, oxidoreductase activity, and phosphate binding. Cellular components altered by the third month of exposure include those integral to the plasma membrane, extracellular space, and MHC class I and II protein complexes. This contrasts with the 1-to-2 month exposure results that indicated alterations in transcriptional activity (i.e., negative regulation of NF-kappaB transcription factor activity), regulation of protein stability, vasculature development (i.e., angiogenesis), protein modification, homeostasis of cell number, and immune and inflammatory responses. Molecular functions altered in first two months of exposure included nucleotide binding, hydrolase activity, G-protein-coupled receptor binding, chemokine activity and receptor binding, ligase activity, and peptidase activity (Table 4).
4. DISCUSSION
The present work supports earlier findings that chronic 12-week exposure to 50 nM MMA(III) leads to substantial biological and functional changes in UROtsa. The work by Wnek and colleagues (2010) suggested that the observed phenotypic changes should coincide with perturbations in the transcriptional activity of the cells. Hyper-proliferation, morphological changes, acquisition of anchorage-independent growth in semi-solid media, and the formation of tumors in immuno-compromised mice all coincide with alterations in pathways associated with cancer progression (from Gene Ontology and SPIA-based analysis). Implicated in this process are the activation of the MAPK signaling, perturbations in ECM-receptor interactions, enhancement of anti-apoptotic processes, and the activity of growth factors. Additionally, the time-dependent analysis of gene expression revealed that at three months of exposure other changes are consistent with an induction of a stress response to ROS, and alterations to the expression of genes involved in DNA repair and the inflammatory response. These concomitant events (increased cellular proliferation, unregulated cell cycle control, oxidative stress response) may be particularly important as indicators of carcinogenic processes and coincide strongly with the transcriptional changes between 2 and 3 months of exposure. When presented with the observed time-dependent phenotypic changes reported from previous studies, the line plots visually relate the profound transcriptional changes that occur in this window of time (Figure 5).
FIGURE 5.

Comparison of observed phenotypic changes with the gene expression profile (from Figure 2) that occurs in UROtsa with continuous exposure to 50 nM MMA(III). The phenotypic changes listed in the curved arrows are reported by Wnek et al. (2010).
With PCR analysis, specific markers of carcinogenesis showed time-dependent changes in gene expression, with the most prominent changes occurring at three months of exposure. With the corresponding enhancement in proliferation, the MAPK signaling pathway appears to be active through the down-regulation of phosphatases (i.e., DUSP1, DUSP6) that attenuate the pathway, and by the up-regulation of growth factors and corresponding receptors (i.e., PDGFRA, FGFR3). The role of DUSPs in cancer is not well understood. The variations in the expression of these proteins have been observed in various cancers and cell lines (Patterson et al., 2009; Bermudez et al., 2010; Moncho-Amor et al., 2010). In bladder cancer, DUSP1 over-expression has been observed in early stages of the cancer, decreasing with higher grade and metastases. The decline in gene expression of these proteins possibly enhances MAPK signaling in UROtsa. KRT7 was selected for PCR analysis since previous studies on UROtsa exposed to 50 nM MMA(III) found significant gene promoter methylation by 24 weeks of exposure (Jensen et al., 2009), and a decrease in gene expression with 12 weeks of exposure followed by withdrawal of MMA(III) for 12 weeks (Wnek et al., 2010). Hypermethylation and silencing have also been proposed as a mechanism regulating TGFBI expression in lung and prostate cancers (Shah et al., 2008). Since TGFBI possesses a tumor suppressing function, its down-regulation may be a contributing factor in the transformation process in conjunction with the suppression of IRF1 gene transcripts. IRF1 has been proposed to act as a tumor suppressor, and shown to be reduced in hematopoietic, gastric, and breast cancer (Ogasawara et al., 1996; Nozawa et al., 1998; Cavalli et al., 2010). In contrast, elevation in TMPRSS11A gene transcripts at 3 months, which is involved in cell cycle regulation and inhibition of tumor progression (Yueying et al., 2008; Li et al., 2011), does not appear to have a sufficient impact on the control of cellular proliferation. CTNND2 has been implicated in promoting anchorage-independent growth (Zeng et al., 2009), which is a phenotypic change observed in UROtsa just beyond three months of exposure. The over-expression of this gene is consistent with expression patterns found in bladder cancer specimens (Zheng et al., 2004). Alterations in the expression of DCLRE1C, CRYAB, ERCC2, and CXCR4, all of which have been reported to be modified in human cancers, are consistent with decreased capacity for DNA repair, oxidative stress, anti-apoptosis, and enhanced proliferation (Arrigo et al., 2007; Malats, 2008; Morio and Kim, 2008; Sun et al., 2010).
It has been proposed that arsenic promotes carcinogenesis through the modification of cellular signal transduction and biological functions (Kitchin, 2001). The use of SPIA-based and Gene Ontology provides insight into the alterations of specific pathways by mapping genes against the KEGG database of signaling pathways and Gene Ontology terms. The analyses revealed that several critical pathways and biological functions were altered by chronic exposure to MMA(III) which support the observed phenotypic changes. Earliest perturbed pathways are associated with the inflammatory response, and these perturbations continued into the third month of exposure. At the third month of exposure the MAPK signaling pathway was activated, which up-regulates cell proliferation and this was reflected in the enhanced growth seen in UROtsa with chronic exposure to MMA(III). Disruption in ECM-receptor interactions due to the altered expression of integrins, collagen, laminin, and glycoprotein gene transcripts lend some support to the colony-forming potential of the cells. An unexpected result was the suppression of many important pro-inflammatory mediators. The presence of these mediators has been considered a pre-disposing factor in the malignant transformation of cells (Balkwill and Mantovani, 2001; Balkwill et al., 2003; Dobrovolskaia and Kozlov, 2005; Kundu and Surh, 2008; Escudero-Lourdes et al., 2010). The implications of these suppressed gene transcripts are not clear and will require further analysis. Another intriguing outcome of these analyses was the attenuation of HLA class I and II (MHC class I and II) antigens, FAS, and CCL5 (a chemotactic cytokine that recruits lymphocytes to a site of inflammation), which is suggestive of an aggressive and invasive phenotype. Carcinomas deficient in HLA class I antigens are strongly associated with tumor grade and probability of recurrence in patients with bladder cancer (Du and Wang, 2011). This may be the first observation where low-level chronic-exposure of a methylated metabolite of arsenic suppresses the gene expression of components of the JAK-STAT pathway, and the pro-inflammatory response in a human urothelial cell line.
Bredfeldt et al. (2006) first demonstrated the malignant transformation of UROtsa from chronic 53-week exposure to MMA(III). The criteria used to assess transformation were an increase in the cell proliferation rate, colony formation in soft agar, and tumor formation in nude mice. This was the same criteria used by Sens et al. (2004) to assess transformation of UROtsa from chronic exposure to inorganic arsenic over 52 weeks. However, if we examine the original transformation model developed by Bredfeldt et al. (2006), it is apparent that UROtsa can form tumors in nude mice at a high passage number (>100), despite previously being characterized as non-tumorigenic, and raises concern about the stability of UROtsa in long-term culture. This observation suggests that MMA(III) may be accelerating spontaneous transformation of UROtsa. This is a possible scenario given that the SV40 T-antigen oncoprotein disrupts the activity of p53, an important mediator of the cellular response to DNA damage, cellular senescence, and apoptosis (Pipas and Levine, 2001). However, UROtsa in long-term culture did not form colonies in soft agar, and it was proposed that the formation of tumors might have been an artifact of the nude mice system. Additionally, it should be noted that there is no association between germline mutations in p53 and the risk of developing bladder cancer, excluding environmental factors such as exposure to carcinogens (Birch et al., 2001). Therefore, to address the observed inconsistency with UROtsa in long-term culture, the cells used in the present study were started at a much lower passage number. The control cells remained stable over the three-month period, showing no sign of anchorage-independent growth or tumorigenicity.
The changes observed between the first two months of exposure and the third month may very well encompass the earliest modifications that promote malignant transformation characteristic to MMA(III) exposure. This was exemplified by the fact that altered biological processes that were identified in the first two months of exposure were also present in the third. However, the overall changes occurring at the third month may be viewed as a consequence of a transformed population, and not necessarily the continued effects of MMA(III) exposure. Tokar et al. (2010) have reported that the arsenic-induced malignant transformation of a human prostate cell line was a result of an over-accumulation of cancer stem cells. Such stem-like cancer cells have been considered as a sub-population in urothelial carcinomas (Brandt et al., 2009). Support for a stem-like population in the UROtsa cell line can be inferred from the gradual acquisition of anchorage-independent growth with MMA(III) exposure. Wnek et al. (2010) first reported the ability of UROtsa, chronically exposed to MMA(III), to form colonies in soft agar by three months. The fraction of cells that displayed this phenotype was small compared to the fraction of cells that acquired the phenotype at 4 and 5 months of exposure. Additionally, when 3-month exposed cells were allowed to continue growing in the absence of MMA(III), the number of cells that form colonies increased in a time-dependent manner and the changes were very substantial the longer the cells were grown.
Few studies have investigated the effects of MMA(III) in vivo. A majority of studies focused on the pentavalent methylated metabolites. One of the earliest indications of the toxic effects of MMA(III) on bladder tissue was through the preliminary findings of Shen et al. (2006). They showed that the effects of methylated metabolites on F344 female rats resulted in pathological changes of the bladder which were attributed to MMA(III) following DMA(V) treatments in the diet. Other studies have also provided some evidence of the carcinogenicity of MMA(III) in rodent models (Delker et al., 2009; Krishnamohan et al., 2006). In an attempt to elicit some type of toxicological response, researchers had employed very high concentrations of MMA(III) in these studies, far beyond the relevant levels of exposures in human populations. Additionally, humans are generally exposed to inorganic arsenic from water sources, and the methylated species are formed from hepatic biotransformation. Furthermore, arsenic has been shown to have significantly different toxicokinetic properties in rodents compared to humans (Aposhian, 1997).
ROS and subsequent DNA damage have been associated with the transformation of the UROtsa cell line (Eblin et al., 2006, 2007). Although an elevation in DNA damage has been detected in UROtsa between one and three months of exposure to MMA(III) and attributed to elevation in ROS (Wnek et al., 2009, 2010, 2011), oxidative stress did not appear as a significant category in the Gene Ontology analysis of the first two months of exposure. It is certain that an increasing trend was observed with endogenous ROS, but only a significant change was measured at three months of exposure. This suggests ROS may not entirely be playing a pivotal role in the transformation process. It is possible that the biochemical alterations acquired at the third month of exposure become particularly important in significantly elevating ROS levels, but in the present study, changes with inflammatory-related genes and altered metabolic processes characterized the first two months of exposure. Although oxidative stress has been considered a mode of action in arsenic-induced carcinogenesis with a number of in vitro studies, this has not been well established in vivo (Clewell et al., 2011). 8-Hydroxy-2′-deoxyguanosine (8-OHdG) has long been used as an oxidative stress marker. Several studies have associated arsenic exposure and increased urinary 8-OHdG levels in rodents exposed to arsenicals (Nishikawa et al., 2002; Yamanaka et al., 2001). Studies on human populations and oxidative stress from arsenic exposure have yielded mixed results. Increased urinary 8-OHdG has been measured in humans living in arsenic-affected areas of Asia (Fujino et al., 2005; Kubota et al., 2006), yet Burgess et al. (2007) did not find an association between urinary 8-OHdG and arsenic when examining populations in Arizona and Sonora, Mexico exposed to arsenic in drinking water below 40 micrograms/L. Therefore, the role of oxidative stress in arsenic-induced bladder cancer in human populations is not entirely clear.
In conclusion, the previously observed phenotypic changes in UROtsa following 3 months exposure to 50 nM MMA(III) [hyper-proliferation, increases in DNA damage, ROS production, and colony formation in semi-solid media] coincides with the substantial gene expression changes presented here. These changes include the expression of genes involved in an oxidative stress response, decreases in specific DNA repair genes, up-regulation of genes involved in proliferation, and the suppression of inflammatory components. These events support the pleiotropic response of urothelial cells to chronic MMA(III) exposure, and indicate that the most critical window of time in the transformation of UROtsa from continuous exposure to MMA(III) occurs between two and three months of exposure.
Supplementary Material
HIGHLIGHTS.
Chronic exposure to 50 nM monomethylarsonous acid in UROtsa was investigated.
At three months of exposure substantial changes were observed in gene expression.
Notable changes occurred in mitogenic signaling, stress, immune and inflammatory responses.
Gene expression changes correlate with phenotypic changes from previous studies.
Acknowledgments
This study was supported by the Superfund Basic Research Program Grant (NIH grant ES04940) from National Institute of Environmental Health Sciences, and the Trainee in Toxicology and Toxicogenomics (NIEHS grant ES007091). The microarray data was generated by the Genomics Core at the Arizona Cancer Center (NIH grant CA23074) and supported the Southwest Environmental Health Sciences Center (NIEHS grant ES06694). The microarray analysis was supported by the Academy of Sciences of the Czech Republic (grant AVOZ50510513).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexa A, Rahnenfuher J. topGO: topGO: enrichment analysis for gene ontology. R package version 2.4.0 2010 [Google Scholar]
- Aposhian HV. Enzymatic methylation of arsenic species and other new approaches to arsenic toxicity. Annu Rev Pharmacol Toxicol. 1997;37:397–419. doi: 10.1146/annurev.pharmtox.37.1.397. [DOI] [PubMed] [Google Scholar]
- Aposhian HV, Gurzau ES, Le XC, Gurzau A, Healy SM, Lu X, Ma M, Yip L, Zakharyan RA, Maiorino RM, Dart RC, Tircus MG, Gonzalez-Ramirez D, Morgan DL, Avram D, Aposhian MM. Occurrence of monomethylarsonous acid in urine of humans exposed to inorganic arsenic. Chem Res Toxicol. 2000;13(8):693–7. doi: 10.1021/tx000114o. [DOI] [PubMed] [Google Scholar]
- Arrigo AP, Simon S, Gibert B, Kretz-Remy C, Nivon M, Czekalla A, Guillet D, Moulin M, Diaz-Latoud C, Vicart P. Hsp27 (HspB1) and alphaB-crystallin (HspB5) as therapeutic targets. FEBS Lett. 2007;581:3665–74. doi: 10.1016/j.febslet.2007.04.033. [DOI] [PubMed] [Google Scholar]
- Arya R, Mallik M, Lakhotia SC. Heat shock genes - integrating cell survival and death. J Biosci. 2007;32:595–610. doi: 10.1007/s12038-007-0059-3. [DOI] [PubMed] [Google Scholar]
- Bailey KA, Hester SD, Knapp GW, Owen RD, Thai SF. Gene expression of normal human epidermal keratinocytes modulated by trivalent arsenicals. Mol Carcinog. 2010;49:981–98. doi: 10.1002/mc.20677. [DOI] [PubMed] [Google Scholar]
- Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;17:539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- Balkwill F. Chemokine biology in cancer. Semin Immunol. 2003;15(1):49–55. doi: 10.1016/s1044-5323(02)00127-6. [DOI] [PubMed] [Google Scholar]
- Bermudez O, Pagès G, Gimond C. The dual-specificity MAP kinase phosphatases: critical roles in development and cancer. Am J Physiol Cell Physiol. 2010;299:C189–C202. doi: 10.1152/ajpcell.00347.2009. [DOI] [PubMed] [Google Scholar]
- Birch JM, Alston RD, McNally RJ, Evans DG, Kelsey AM, Harris M, Eden OB, Varley JM. Relative frequency and morphology of cancers in carriers of germline TP53 mutations. Oncogene. 2001;20:4621–8. doi: 10.1038/sj.onc.1204621. [DOI] [PubMed] [Google Scholar]
- Brandt WD, Matsui W, Rosenberg JE, He X, Ling S, Schaeffer EM, Berman DM. Urothelial carcinoma: stem cells on the edge. Cancer Metastasis Rev. 2009;28:291–304. doi: 10.1007/s10555-009-9187-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredfeldt TG, Kopplin MJ, Gandolfi AJ. Effects of arsenite on UROtsa cells: low-level arsenite causes accumulation of ubiquitinated proteins that is enhanced by reduction in cellular glutathione levels. Toxicol Appl Pharmacol. 2004;198(3):412–8. doi: 10.1016/j.taap.2003.10.013. [DOI] [PubMed] [Google Scholar]
- Bredfeldt TG, Jagadish B, Eblin KE, Mash EA, Gandolfi AJ. Monomethylarsonous acid induced transformation of human bladder cells. Toxicol Appl Pharmacol. 2006;216:69–79. doi: 10.1016/j.taap.2006.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brummelkamp TR, Kortlever RM, Lingbeek M, Trettel F, MacDonald ME, van Lohuizen M, Bernards R. TBX-3, the gene mutated in Ulnar-Mammary Syndrome, is a negative regulator of p19ARF and inhibits senescence. J Biol Chem. 2002;277:6567–72. doi: 10.1074/jbc.M110492200. [DOI] [PubMed] [Google Scholar]
- Burgess JL, Meza MM, Josyula AB, Poplin GS, Kopplin MJ, McClellen HE, Stürup S, Lantz RC. Environmental Arsenic Exposure and Urinary 8-OHdG in Arizona and Sonora. Clin Toxicol (Phila) 2007;45:490–8. doi: 10.1080/15563650701354119. [DOI] [PubMed] [Google Scholar]
- Cavalli LR, Riggins RB, Wang A, Clarke R, Haddad BR. Frequent loss of heterozygosity at the interferon regulatory factor-1 gene locus in breast cancer. Breast Cancer Res Treat. 2010;121:227–31. doi: 10.1007/s10549-009-0509-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CJ, Chen CW, Wu MM, Kuo TL. Cancer potential in liver, lung, bladder and kidney due to ingested inorganic arsenic in drinking water. Br J Cancer. 1992;66:888–92. doi: 10.1038/bjc.1992.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CJ, Chiou HY, Chiang MH, Lin LJ, Tai TY. Dose-response relationship between ischemic heart disease mortality and long-term arsenic exposure. Arterioscler Thromb Vasc Biol. 1996;16:504–10. doi: 10.1161/01.atv.16.4.504. [DOI] [PubMed] [Google Scholar]
- Christian BJ, Loretz LJ, Oberley TD, Reznikoff CA. Characterization of human uroepithelial cells immortalized in vitro by simian virus 40. Cancer Res. 1987;47:6066–73. [PubMed] [Google Scholar]
- Clewell HJ, Thomas RS, Kenyon EM, Hughes MF, Adair BM, Gentry PR, Yager JW. Concentration- and Time-dependent Genomic Changes in the Mouse Urinary Bladder Following Exposure to Arsenate in Drinking Water for up to 12 Weeks. Toxicol Sci. 2011;123:421–32. doi: 10.1093/toxsci/kfr199. [DOI] [PubMed] [Google Scholar]
- Delker DA, Geter DR, Roop BC, Ward WO, Ahlborn GJ, Allen JW, Nelson GM, Ouyang M, Wels W, Chen Y, O’Brien T, Kitchin KT. Oncogene expression profiles in K6/ODC mouse skin and papillomas following a chronic exposure to monomethylarsonous acid. J Biochem Mol Toxicol. 2009;23(6):406–18. doi: 10.1002/jbt.20304. [DOI] [PubMed] [Google Scholar]
- Dobrovolskaia MA, Kozlov SV. Inflammation and cancer: when NF-kB amalgamates the perilous partnership. Curr Cancer Drug Targets. 2005;5:325–344. doi: 10.2174/1568009054629645. [DOI] [PubMed] [Google Scholar]
- Druwe IL, Vaillancourt RR. Influence of arsenate and arsenite on signal transduction pathways: an update. Arch Toxicol. 2010;84:585–96. doi: 10.1007/s00204-010-0554-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du C, Wang Y. The immunoregulatory mechanisms of carcinoma for its survival and development. J Exp Clin Cancer Res. 2011;30:12. doi: 10.1186/1756-9966-30-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eblin KE, Bowen ME, Cromey DW, Bredfeldt TG, Mash EA, Lau SS, Gandolfi AJ. Arsenite and monomethylarsonous acid generate oxidative stress response in human bladder cell culture. Toxicol Appl Pharmacol. 2006;217(1):7–14. doi: 10.1016/j.taap.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Eblin KE, Bredfeldt TG, Buffington S, Gandolfi AJ. Mitogenic signal transduction caused by monomethylarsonous acid in human bladder cells: role in arsenic-induced carcinogenesis. Toxicol Sci. 2007;95:321–30. doi: 10.1093/toxsci/kfl160. [DOI] [PubMed] [Google Scholar]
- Eblin KE, Bredfeldt TG, Gandolfi AJ. Immortalized human urothelial cells as a model of arsenic-induced bladder cancer. Toxicology. 2008a;248:67–76. doi: 10.1016/j.tox.2008.03.020. [DOI] [PubMed] [Google Scholar]
- Eblin KE, Hau AM, Jensen TJ, Futscher BW, Gandolfi AJ. The role of reactive oxygen species in arsenite and monomethylarsonous acid-induced signal transduction in human bladder cells: acute studies. Toxicology. 2008b;250:47–54. doi: 10.1016/j.tox.2008.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escudero-Lourdes C, Medeiros MK, Cárdenas-González MC, Wnek SM, Gandolfi JA. Low level exposure to monomethyl arsonous acid-induced the over-production of inflammation-related cytokines and the activation of cell signals associated with tumor progression in a urothelial cell model. Toxicol Appl Pharmacol. 2010;244:162–73. doi: 10.1016/j.taap.2009.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujino Y, Guo X, Liu J, Matthews IP, Shirane K, Wu K, Kasai H, Miyatake M, Tanabe K, Kusuda T, Yoshimura T Japan Inner Mongolia Arsenic Pollution Study Group. Chronic arsenic exposure and urinary 8-hydroxy-2′-deoxyguanosine in an arsenic-affected area in Inner Mongolia, China. J Expo Anal Environ Epidemiol. 2005;15:147–52. doi: 10.1038/sj.jea.7500381. [DOI] [PubMed] [Google Scholar]
- Gentry PR, McDonald TB, Sullivan DE, Shipp AM, Yager JW, Clewell HJ. Analysis of genomic dose-response information on arsenic to inform key events in a mode of action for carcinogenicity. Environ Mol Mutagen. 2010;51:1–14. doi: 10.1002/em.20505. [DOI] [PubMed] [Google Scholar]
- Gong Z, Lu X, Cullen WR, Le C. Unstable Trivalent Arsenic Metabolites, Monomethylarsonous Acid and Dimethylarsinous Acid. J Anal At Spectrom. 2001;16:1409–1413. [Google Scholar]
- Hamadeh Hk, Trouba KJ, Amin RP, Afshari CA, Germolec D. Coordination of altered DNA repair and damage pathways in arsenite-exposed keratinocytes. Toxicological Sci. 2002;69:306–316. doi: 10.1093/toxsci/69.2.306. [DOI] [PubMed] [Google Scholar]
- Hei TK, Filipic M. Role of oxidative damage in the genotoxicity of arsenic. Free Radic Biol Med. 2004;37:574–81. doi: 10.1016/j.freeradbiomed.2004.02.003. [DOI] [PubMed] [Google Scholar]
- Hirano S, Kobayashi Y, Cui X, Kanno S, Hayakawa T, Shraim A. The accumulation and toxicity of methylated arsenicals in endothelial cells: important roles of thiol compounds. Toxicol Appl Pharmacol. 2004;198:458–67. doi: 10.1016/j.taap.2003.10.023. [DOI] [PubMed] [Google Scholar]
- Hughes MF. Arsenic toxicity and potential mechanisms of action. Toxicol Lett. 2002;133:1–16. doi: 10.1016/s0378-4274(02)00084-x. [DOI] [PubMed] [Google Scholar]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Some drinking-water disinfectants and contaminants, including arsenic. IARC Monogr Eval Carcinog Risks Hum. 2004;84:1–477. [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen TJ, Novak P, Wnek SM, Gandolfi AJ, Futscher BW. Arsenicals produce stable progressive changes in DNA methylation patterns that are linked to malignant transformation of immortalized urothelial cells. Toxicol Appl Pharmacol. 2009;241:221–9. doi: 10.1016/j.taap.2009.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T, Huang Z, Chan JY, Zhang DD. Nrf2 protects against As(III)-induced damage in mouse liver and bladder. Toxicol Appl Pharmacol. 2009;240:8–14. doi: 10.1016/j.taap.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson WE, Rabinovic A, Li C. Adjusting batch effects in microarray expression data using Empirical Bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- Kitchin KT. Recent advances in arsenic carcinogenesis: modes of action, animal model systems, and methylated arsenic metabolites. Toxicol Appl Pharmacol. 2001;172:249–61. doi: 10.1006/taap.2001.9157. [DOI] [PubMed] [Google Scholar]
- Kitchin KT, Ahmad S. Oxidative stress as a possible mode of action for arsenic carcinogenesis. Toxicol Lett. 2003;137:3–13. doi: 10.1016/s0378-4274(02)00376-4. [DOI] [PubMed] [Google Scholar]
- Kitchin KT, Wallace K. The role of protein binding of trivalent arsenicals in arsenic carcinogenesis and toxicity. J Inorg Biochem. 2008;102:532–9. doi: 10.1016/j.jinorgbio.2007.10.021. [DOI] [PubMed] [Google Scholar]
- Kitchin KT, Conolly R. Arsenic-induced carcinogenesis--oxidative stress as a possible mode of action and future research needs for more biologically based risk assessment. Chem Res Toxicol. 2010;23:327–35. doi: 10.1021/tx900343d. [DOI] [PubMed] [Google Scholar]
- Kligerman AD, Doerr CL, Tennant AH, Harrington-Brock K, Allen JW, Winkfield E, Poorman-Allen P, Kundu B, Funasaka K, Roop BC, Mass MJ, DeMarini DM. Methylated trivalent arsenicals as candidate ultimate genotoxic forms of arsenic: Induction of chromosomal mutations but not gene mutations. Environ Mol Mutagen. 2003;42:192–205. doi: 10.1002/em.10192. [DOI] [PubMed] [Google Scholar]
- Krishnamohan M, Qi L, Lam PK, Moore MR, Ng JC. Urinary arsenic and porphyrin profile in C57BL/6J mice chronically exposed to monomethylarsonous acid (MMAIII) for two years. Toxicol Appl Pharmacol. 2007;224:89–97. doi: 10.1016/j.taap.2007.04.020. [DOI] [PubMed] [Google Scholar]
- Kubota R, Kunito T, Agusa T, Fujihara J, Monirith I, Iwata H, Subramanian A, Tana TS, Tanabe S. Urinary 8-hydroxy-2′-deoxyguanosine in inhabitants chronically exposed to arsenic in groundwater in Cambodia. J Environ Monit. 2006;8:293–9. doi: 10.1039/b513652k. [DOI] [PubMed] [Google Scholar]
- Kundu JK, Surh YJ. Inflammation: Gearing the journey to cancer. Mutation Res. 2008;659:15–30. doi: 10.1016/j.mrrev.2008.03.002. [DOI] [PubMed] [Google Scholar]
- Le XC, Lu X, Ma M, Cullen WR, Aposhian HV, Zheng B. Speciation of key arsenic metabolic intermediates in human urine. Anal Chem. 2000;72:5172–7. doi: 10.1021/ac000527u. [DOI] [PubMed] [Google Scholar]
- Liao Y, Hung MC. Physiological regulation of Akt activity and stability. Am J Transl Res. 2010;2:19–42. [PMC free article] [PubMed] [Google Scholar]
- Li LW, Li YY, Li XY, Zhang CP, Zhou Y, Lu SH. A novel tumor suppressor gene ECRG4 interacts directly with TMPRSS11A (ECRG1) to inhibit cancer cell growth in esophageal carcinoma. BMC Cancer. 2011;11:52. doi: 10.1186/1471-2407-11-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WX. Canonical and non-canonical JAK-STAT signaling. Trends Cell Biol. 2008;18:545–51. doi: 10.1016/j.tcb.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin WW, Karin M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest. 2007;117:1175–1183. doi: 10.1172/JCI31537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malats N. Genetic epidemiology of bladder cancer: scaling up in the identification of low-penetrance genetic markers of bladder cancer risk and progression. Scand J Urol Nephrol Suppl. 2008;218:131–40. doi: 10.1080/03008880802285172. [DOI] [PubMed] [Google Scholar]
- Mandal BK, Ogra Y, Suzuki KT. Identification of dimethylarsinous and monomethylarsonous acids in human urine of the arsenic-affected areas in West Bengal, India. Chem Res Toxicol. 2001;14(4):371–8. doi: 10.1021/tx000246h. [DOI] [PubMed] [Google Scholar]
- Mandal BK, Ogra Y, Anzai K, Suzuki KT. Speciation of arsenic in biological samples. Toxicol Appl Pharmacol. 2004;198(3):307–18. doi: 10.1016/j.taap.2003.10.030. [DOI] [PubMed] [Google Scholar]
- Mass MJ, Tennant A, Roop BC, Cullen WR, Styblo M, Thomas DJ, Kligerman AD. Methylated trivalent arsenic species are genotoxic. Chem Res Toxicol. 2001;14:355–361. doi: 10.1021/tx000251l. [DOI] [PubMed] [Google Scholar]
- Millar IT, Heany H, Heinehey DM, Fernelius WC. Methyliiodoarsine. Inorg Synth. 1960;6:113–115. [Google Scholar]
- Moncho-Amor V, de Cáceres II, Bandres E, Martínez-Poveda B, Orgaz JL, Sánchez-Pérez I, Zazo S, Rovira A, Albanell J, Jiménez B, Rojo F, Belda-Iniesta C, García-Foncillas J, Perona R. DUSP1/MKP1 promotes angiogenesis, invasion and metastasis in non-small-cell lung cancer. Oncogene. 2011;30:668–78. doi: 10.1038/onc.2010.449. [DOI] [PubMed] [Google Scholar]
- Morio T, Kim H. Ku, Artemis, and ataxia-telangiectasia-mutated: signalling networks in DNA damage. Int J Biochem Cell Biol. 2008;40:598–603. doi: 10.1016/j.biocel.2007.12.007. [DOI] [PubMed] [Google Scholar]
- Nahas SA, Gatti RA. DNA double strand break repair defects, primary immunodeficiency disorders, and ‘radiosensitivity’. Curr Opin Allergy Clin Immunol. 2009;9:510–6. doi: 10.1097/ACI.0b013e328332be17. [DOI] [PubMed] [Google Scholar]
- Nesnow S, Roop BC, Lambert G, Kadiiska M, Mason RP, Cullen WR, Mass MJ. DNA damage induced by methylated trivalent arsenicals is mediated by reactive oxygen species. Chem Res Toxicol. 2002;15:1627–34. doi: 10.1021/tx025598y. [DOI] [PubMed] [Google Scholar]
- Nishikawa T, Wanibuchi H, Ogawa M, Kinoshita A, Morimura K, Hiroi T, Funae Y, Kishida H, Nakae D, Fukushima S. Promoting effects of monomethylarsonic acid, dimethylarsinic acid and trimethylarsine oxide on induction of rat liver preneoplastic glutathione S-transferase placental form positive foci: a possible reactive oxygen species mechanism. Int J Cancer. 2002;100:136–9. doi: 10.1002/ijc.10471. [DOI] [PubMed] [Google Scholar]
- NRC (National Research Council) Arsenic in Drinking Water (2001 update) National Academy Press; Washington, DC: 2001. [Google Scholar]
- Nozawa H, Oda E, Ueda S, Tamura G, Maesawa C, Muto T, Taniguchi T, Tanaka N. Functionally inactivating point mutation in the tumor-suppressor IRF-1 gene identified in human gastric cancer. Int J Cancer. 1998;77:522–527. doi: 10.1002/(sici)1097-0215(19980812)77:4<522::aid-ijc8>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Ogasawara S, Tamura G, Maesawa C, Suzuki Y, Ishida K, Satoh N, Uesugi N, Saito K, Satodate R. Common deleted region on the long arm of chromosome 5 in esophageal carcinoma. Gastroenterology. 1996;110:52–57. doi: 10.1053/gast.1996.v110.pm8536888. [DOI] [PubMed] [Google Scholar]
- Patterson KI, Brummer T, Philippa M, O’Brien PM, Daly RJ. Dual-specificity phosphatases: critical regulators with diverse cellular targets. Biochem J. 2009;418:475–489. doi: 10.1042/bj20082234. [DOI] [PubMed] [Google Scholar]
- Petzoldt JL, Leigh IM, Duffy PG, Sexton C, Masters JR. Immortalisation of human urothelial cells. Urol Res. 1995;23:377–380. doi: 10.1007/BF00698738. [DOI] [PubMed] [Google Scholar]
- Pipas JM, Levine AJ. Role of T antigen interactions with p53 in tumorigenesis. Semin Cancer Biol. 2001;11:23–30. doi: 10.1006/scbi.2000.0343. [DOI] [PubMed] [Google Scholar]
- Platanias LC. Biological responses to arsenic compounds. J Biol Chem. 2009;284:18583–7. doi: 10.1074/jbc.R900003200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea MA, Gregg JP, Qin Q, Philips MA, Rice RH. Global alterations of gene expression in human keratinocytes by inorganic arsenic. Carcinogenesis. 2003;24(4):747–756. doi: 10.1093/carcin/bgg010. [DOI] [PubMed] [Google Scholar]
- Rossi MR, Masters JR, Park S, Todd JH, Garrett SH, Sens MA, Somji S, Nath J, Sens DA. The immortalized UROtsa cell line as a potential cell culture model of human urothelium. Environ Health Perspect. 2001;109:801–808. doi: 10.1289/ehp.01109801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoen A, Beck B, Sharma R, Dubé E. Arsenic toxicity at low doses: epidemiological and mode of action considerations. Toxicol Appl Pharmacol. 2004;198:253–67. doi: 10.1016/j.taap.2003.10.011. [DOI] [PubMed] [Google Scholar]
- Schuliga M, Chouchane S, Snow ET. Upregulation of glutathione-related genes and enzyme activities in cultured human cells by sublethal concentrations of inorganic arsenic. Toxicol Sci. 2002;70:183–92. doi: 10.1093/toxsci/70.2.183. [DOI] [PubMed] [Google Scholar]
- Sens DA, Park S, Gurel V, Sens MA, Garrett SH, Somji S. Inorganic cadmium- and arsenite-induced malignant transformation of human bladder urothelial cells. Toxicol Sci. 2004;79:56–63. doi: 10.1093/toxsci/kfh086. [DOI] [PubMed] [Google Scholar]
- Shah JN, Shao G, Hei TK, Zhao Y. Methylation screening of the TGFBI promoter in human lung and prostate cancer by methylation-specific PCR. BMC Cancer. 2008;8:284. doi: 10.1186/1471-2407-8-284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Wanibuchi H, Waalkes MP, Salim EI, Kinoshita A, Yoshida K, Endo G, Fukushima S. A comparative study of the sub-chronic toxic effects of three organic arsenical compounds on the urothelium in F344 rats; gender-based differences in response. Toxicol Appl Pharmacol. 2006;210:171–80. doi: 10.1016/j.taap.2005.04.018. [DOI] [PubMed] [Google Scholar]
- Smyth GK. Limma: linear models for microarray data. In: Gentleman R, Carey V, Dudoit S, Irizarry R, Huber W, editors. Bioinformatics and Computational Biology Solutions Using R and Bioconductor. Springer; New York: 2005. pp. 397–420. [Google Scholar]
- Stýblo M, Drobná Z, Jaspers I, Lin S, Thomas DJ. The role of biomethylation in toxicity and carcinogenicity of arsenic: a research update. Environ Health Perspect. 2002;110:767–71. doi: 10.1289/ehp.110-1241242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su PF, Hu YJ, Ho IC, Cheng YM, Lee TC. Distinct gene expression profiles in immortalized human urothelial cells exposed to inorganic arsenite and its methylated trivalent metabolites. Environ Health Perspect. 2006;114:394–403. doi: 10.1289/ehp.8174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Cheng G, Hao M, Zheng J, Zhou X, Zhang J, Taichman RS, Pienta KJ, Wang J. CXCL12/CXCR4/CXCR7 chemokine axis and cancer progression. Cancer Metastasis Rev. 2010;29:709–22. doi: 10.1007/s10555-010-9256-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapio S, Grosche B. Arsenic in the aetiology of cancer. Mutat Res. 2006;612:215–46. doi: 10.1016/j.mrrev.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Tarca AL, Draghici S, Khatri P, Hassan SS, Mittal P, Kim JS, Kim CJ, Kusanovic JP, Romero R. A novel signaling pathway impact analysis. Bioinformatics. 2009;25:75–82. doi: 10.1093/bioinformatics/btn577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokar EJ, Qu W, Liu J, Liu W, Webber MM, Phang JM, Waalkes MP. Arsenic-specific stem cell selection during malignant transformation. J Natl Cancer Inst. 2010;102:638–49. doi: 10.1093/jnci/djq093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng CH, Chong CK, Chen CJ, Tai TY. Dose-response relationship between peripheral vascular disease and ingested inorganic arsenic among residents in blackfoot disease endemic villages in Taiwan. Atherosclerosis. 1996;120:125–33. doi: 10.1016/0021-9150(95)05693-9. [DOI] [PubMed] [Google Scholar]
- Vahidnia A, van der Voet GB, de Wolff FA. Arsenic neurotoxicity--a review. Hum Exp Toxicol. 2007;26:823–32. doi: 10.1177/0960327107084539. [DOI] [PubMed] [Google Scholar]
- Vahter M. Species differences in the metabolism of arsenic compounds. Appl Organomet Chem. 1994;8:175–182. [Google Scholar]
- Vahter M. Methylation of inorganic arsenic in different mammalian species and population groups. Sci Prog. 1999;82:69–88. doi: 10.1177/003685049908200104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzuela OL, Borja-Aburto VH, Garcia-Vargas GG, Cruz-Gonzalez MB, Garcia-Montalvo EA, Calderon-Aranda ES, Del Razo LM. Urinary trivalent methylated arsenic species in a population chronically exposed to inorganic arsenic. Environ Health Perspect. 2005;113:250–4. doi: 10.1289/ehp.7519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XJ, Sun Z, Chen W, Eblin KE, Gandolfi JA, Zhang DD. Nrf2 protects human bladder urothelial cells from arsenite and monomethylarsonous acid toxicity. Toxicol Appl Pharmacol. 2007;225(2):206–13. doi: 10.1016/j.taap.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wnek SM, Medeiros MK, Eblin KE, Gandolfi AJ. Persistence of DNA damage following exposure of human bladder cells to chronic monomethylarsonous acid. Toxicol Appl Pharmacol. 2009;241:202–9. doi: 10.1016/j.taap.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wnek SM, Jensen TJ, Severson PL, Futscher BW, Gandolfi AJ. Monomethylarsonous acid produces irreversible events resulting in malignant transformation of a human bladder cell line following 12 weeks of low-level exposure. Toxicol Sci. 2010;116:44–57. doi: 10.1093/toxsci/kfq106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wnek SM, Kuhlman CL, Camarillo JM, Medeiros MK, Liu KJ, Lau SS, Gandolfi AJ. Interdependent genotoxic mechanisms of monomethylarsonous acid: Role of ROS-induced DNA damage and poly(ADP-ribose) polymerase-1 inhibition in the malignant transformation of urothelial cells. Toxicol Appl Pharmacol. 2011 Sep 10; doi: 10.1016/j.taap.2011.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MM, Kuo TL, Hwang YH, Chen CJ. Dose-response relation between arsenic concentration in well water and mortality from cancers and vascular diseases. Am J Epidemiol. 1989;130:1123–1132. doi: 10.1093/oxfordjournals.aje.a115439. [DOI] [PubMed] [Google Scholar]
- Xie R, Johnson W, Spayd S, Hall GS, Buckley B. Arsenic speciation analysis of human urine using ion exchange chromatography coupled to inductively coupled plasma mass spectrometry. Anal Chim Acta. 2006;578:186–94. doi: 10.1016/j.aca.2006.06.076. [DOI] [PubMed] [Google Scholar]
- Yamanaka K, Takabayashi F, Mizoi M, An Y, Hasegawa A, Okada S. Oral exposure of dimethylarsinic acid, a main metabolite of inorganic arsenics, in mice leads to an increase in 8-Oxo-2′-deoxyguanosine level, specifically in the target organs for arsenic carcinogenesis. Biochem Biophys Res Commun. 2001;287:66–70. doi: 10.1006/bbrc.2001.5551. [DOI] [PubMed] [Google Scholar]
- Yueying W, Jianbo W, Hailin L, Huaijing T, Liping G, Shih-Hsin L. ECRG1, a novel esophageal gene, cloned and identified from human esophagus and its inhibition effect on tumors. Carcinogenesis. 2008;29:157–60. doi: 10.1093/carcin/bgm203. [DOI] [PubMed] [Google Scholar]
- Zhang S, Cao J. A close examination of double filtering with fold change and T test in microarray analysis. BMC Bioinformatics. 2009;10:402. doi: 10.1186/1471-2105-10-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Abdallah A, Lu JP, Wang T, Chen YH, Terrian DM, Kim K, Lu Q. Delta-catenin promotes prostate cancer cell growth and progression by altering cell cycle and survival gene profiles. Mol Cancer. 2009;8:19. doi: 10.1186/1476-4598-8-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng M, Simon R, Mirlacher M, Maurer R, Gasser T, Forster T, Diener PA, Mihatsch MJ, Sauter G, Schrami P. TRIO amplication and abundant mRNA expression is associated with invasive tumor growth and rapid tumor cell proliferation in urinary bladder cancer. Amer J of Pathology. 2004;165:63–63. doi: 10.1016/S0002-9440(10)63275-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.