

Abstract
Mammalian metabolism is highly circadian and major hormonal circuits involving nuclear hormone receptors (NRs) display interlinked diurnal cycling1,2. However, mechanisms that logically explain the coordination of NRs and the clock are poorly understood. Here we show that two circadian co-regulators, cryptochromes 1 (Cry1) and 2 (Cry2), interact with the glucocorticoid receptor (GR) in a ligand-dependent fashion and globally alter the transcriptional response to glucocorticoids in mouse embryonic fibroblasts (MEFs): Cry deficiency vastly decreases gene repression and approximately doubles the number of dexamethasone (Dex) induced genes suggesting that cryptochromes broadly oppose GR activation and promote repression. In mice, genetic loss of Cry1 and/or Cry2 resulted in glucose intolerance and constitutively high levels of circulating corticosterone, suggesting reduced suppression of the hypothalamic-pituitary-adrenal (HPA) axis coupled with increased glucocorticoid transactivation in the liver. Genomically, Cry1 and Cry2 associate with a glucocorticoid response element (GRE) in the phosphoenolpyruvate carboxykinase 1 (Pck1) promoter in a hormone-dependent manner, and Dex-induced transcription of pck1 was strikingly increased in Cry-deficient livers. These results reveal a specific mechanism through which cryptochromes couple the activity of clock and receptor target genes to complex genomic circuits underpinning normal metabolic homeostasis.
Glucocorticoids (cortisol in humans and corticosterone in rodents) are critical regulators of many aspects of mammalian physiology, including glucose homeostasis and immune function, and these stress hormones exhibit a robust diurnal rhythm in the circulation. Synthetic glucocorticoids are widely used as anti-inflammatory drugs, but cause undesirable side effects including hyperglycemia, insulin resistance and suppression of adrenal function. A paradox of nuclear receptor signalling is that potent agonists typically repress as many genes as they activate. The cloning of the GR3 revealed that the same gene product mediated both agonist induced activation and repression via dichotomous mechanisms that still remain poorly understood. Ligand stimulation results in translocation of GR from the cytoplasm to the nucleus where it binds to GREs and alters the transcription of hundreds to thousands of genes. Several cofactors have been identified that mediate transcriptional activation, including the steroid receptor coactivators (SRC1-3), the histone acetyltransferases CBP/p300 and the nuclear methylase coactivator-associated arginine methyltransferase 1 (CARM1)4. In regards to GR-dependent repression, combinations of direct and indirect mechanisms have been proposed. For example, GR may `tether' or bind to proinflammatory transcription factors, such as nuclear factor of kappa light polypeptide gene enhancer in B-cells (NFκB), and `trans-repress' gene expression by interfering with their activation mechanism5. In addition, glucocorticoids can directly repress metabolic targets, particularly in the HPA axis, in a way that does not seem to require NFκB or other inflammatory transcription factors. In this case, GR acts via direct binding to negative response elements that presumably promote recruitment of one or more hypothetical repressive co-factors.
Circadian clocks drive rhythms in physiology and behaviour that enable organisms to keep track of the time of day and to adjust their physiology to adapt to recurrent, and therefore predictable, changes in the external environment. Mammalian circadian clocks are based on a transcription and translation feedback loop in which a heterodimer of the transcription factors circadian locomotor output cycles kaput (CLOCK) and brain and muscle ARNT-like 1 (BMAL1) drives transcription from E-box elements, including that of their own repressors, the Period (Per1, Per2 and Per3) and Cryptochrome (Cry1 and Cry2) genes2. For many years, mammalian clocks were thought to reside solely in the hypothalamic suprachiasmatic nucleus (SCN), in which the clock is set by light signals via the retinohypothalamic tract, and from which secreted signals drive rhythms in locomotor activity. Subsequently, circadian clocks have been identified throughout mammalian peripheral organs and their timing was demonstrated to be determined by metabolic cues rather than in direct response to light6. Several molecular mechanisms have been described that contribute to metabolic resetting of clock time in peripheral organs7–9.
The existence of clocks in peripheral organs and the demonstration that their timing is set by metabolic cues led to the question of whether they participate in physiological regulation that enables organisms to adapt to changing metabolic needs over the course of the day. Tissue-specific ablation of clock function demonstrated that clocks in the liver10 and pancreas11,12 indeed contribute to glucose homeostasis at specific times of the day. Several studies have demonstrated that thousands of transcripts are subject to circadian regulation in myriad organs13,14, and that most of this regulation is lost upon tissue-specific clock disruption10,15, suggesting that rhythmic transcription of rate-limiting enzymes and transporters contributes to circadian physiological regulation14.
The time of peak expression for these oscillating transcripts is distributed across the day, suggesting that multiple mechanisms contribute to their diurnal regulation. In addition to cascades of rhythmically expressed transcription factors, including NRs, which could be driven by clock-dependent rhythmic expression, circadian repression of subsets of target genes by the negative arm of the clock may contribute to oscillation of specific genetic programs. The direct regulation of cryptochromes by metabolic signals7 makes them ideal potential cross-over regulators for circadian and metabolic gene expression programs. Because nuclear receptors are prototypic regulators of metabolism, we examined the possibility that Cry1 and Cry2 might contribute to diurnal aspects of their regulatory output1.
We first explored whether Cry1 can interact with two NRs known to modulate clock function—the retinoid-related orphan receptors (ROR α/β/γ) and Reverb α/β. Unexpectedly, we find robust association between Cry1 and RORα and γ but not with RORβ or Reverb α/β though we did reproduce the reported association of PER2 with Reverb α16 (Fig. 1A–B). This is intriguing for two reasons: 1) RORα and RORγ but not RORβ can activate Bmal1 transcription17 and 2) Cry1 preferentially associates with the activation (ROR) and not the repressive (Reverb) arm of the pathway. Together these results suggest that the so-called “secondary loop” in which ROR's and Rev-Erb's modulate BMAL1 transcription and are thought to make clocks more robust and accurate involves cryptochromes.
Figure 1.

Cryptochromes interact with GR. (a–e, h) Immunoblots showing recovery of the indicated proteins from 293T cells expressing the indicated plasmids following IP with the indicated antibodies. In (e), cells were treated with vehicle (−) or 1 μM dexamethasone (+) for 16 hours. (f) Immunoblots showing recovery of endogenous GR and PER2 from MEFs stably expressing empty vector (−) or FLAG-Cry1 (+) following FLAG IP. (g) Luciferase activity in CV-1 cells transfected as indicated. Data represent the mean ± s.e.m. of triplicate samples. ***P < 0.001. Data represent typical results of 2–6 independent experiments.
We next initiated a broader survey of NR interactions and unexpectedly discovered a robust physical association of Cry1 with the glucocorticoid and androgen receptors (GR and AR, respectively) (Fig. 1A). Although Per2 can interact with some NRs (such as Reverb16), it does not appear to bind GR (Fig. 1C). Further analysis reveals that Cry2 also interacts with GR (Fig. 1D) and that the association is stimulated by glucocorticoids (Fig. 1E–F). Notably, Cry1 represses the ability of GR to drive expression of a luciferase reporter from a GRE-containing promoter (Fig. 1G). Finally, we used a series of deletion mutants of GR to demonstrate that Cry1 interacts with the C-terminus of GR (Fig. 1H), which is required for either activation or repression of transcription in response to ligand. Steroid hormones (glucocorticoids, testosterone, progesterone and aldosterone) are critical regulators of metabolic and reproductive physiology so the possibility that they could be directly regulated by cryptochromes suggests a new mechanism by which circadian clocks modulate physiological rhythms.
In order to determine whether cryptochromes modulate glucocorticoid-stimulated changes in transcription of endogenous genes in vivo, we analyzed global changes in gene expression following glucocorticoid treatment of primary fibroblasts derived from wild type and cry1−/−;cry2−/− double knockout (DKO) littermate mouse embryos (Fig. 2A). Circadian rhythms of transcription can be synchronized by dexamethasone treatment of fibroblasts18 and these changes are expected to be lost in Cry-deficient cells, such that a subset of both positive and negative transcriptional responses to dexamethasone will be absent. However, if cryptochromes are independently capable of repressing transcription via GR, we would expect to see a global positive shift in the transcriptional response to dexamethasone (e.g. an increase in gene activation and a decrease in repression). Indeed, we found that the transcriptional response to the synthetic glucocorticoid dexamethasone (Dex) was significantly more positive in cryptochrome-deficient cells: 205 more genes (191→396) were activated by Dex in Cry-deficient cells compared to the controls indicating that Cry limits induction of a larger network of GR responsive genes. Perhaps even more unexpectedly, of 657 Dex repressed genes only 64 were suppressed in DKO cells (Fig. 2B). We confirmed the observed expression patterns for several individual genes in an independently derived pair of control and cryptochrome-deficient MEFs (Fig. 2C). Because the interpretation of the results for individual transcripts is complicated by the potential role of circadian synchronization, we also examined the expression profile of the established GR transcriptional target serum/glucocorticoid regulated kinase 1 (sgk1) in response to dexamethasone stimulation and found that sgk1 mRNA is more robustly activated in cry1−/−;cry2−/− cells compared to control cells within two hours of exposure to glucocorticoids (Fig. 2D) and over a range of dosages (Fig. 2E). In general, the dynamic response to Dex is complex and transcript-dependent but more positive in the absence of cryptochromes (Fig. S2). Together these results lead to the surprising suggestion that Cry1 and Cry2, central regulators of the circadian clock, also directly oppose Dex-induced GR activation.
Figure 2.
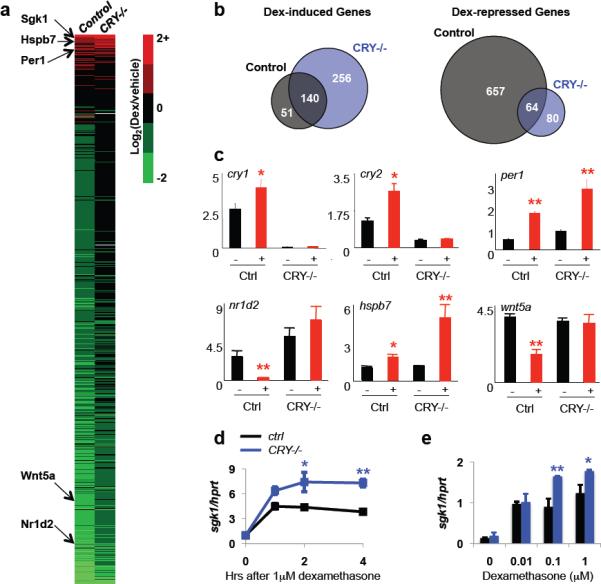
Cryptochromes modulate GR-dependent transcription. (a) Heat map: color denotes dexamethasone-induced change in expression of all transcripts significantly altered by genotype or glucocorticoid stimulation. Selected genes are indicated on the left. (b) Transcripts altered by dexamethasone treatment in control (gray circles) or DKO (blue circles) MEFs. (c) Expression of indicated transcripts in control or DKO MEFs following overnight treatment with vehicle (−) or dexamethasone (+). (d,e) Expression of sgk1 in MEFs following treatment with dexamethasone for 1–4 hours. In (c–e), data represent the mean ± s.e.m. of triplicate samples analyzed in triplicate. * P < 0.01, ** P < 0.001.
Some of the best-studied physiological roles of glucocorticoids are suppression of the immune system and activation of gluconeogenesis. We examined whether cryptochromes are required for transrepression of inflammatory genes in primary bone marrow macrophages from wildtype and DKO mice. Following lipopolysaccharide (LPS) stimulation with or without prior Dex treatment, the expression of tumor necrosis factor alpha (TNFα) and chemokine (C–C motif) ligand 4 (Ccl4) was indistinguishable in wildtype and DKO macrophages (Fig. S3). The basal and LPS-induced expression of monocyte chemoattractant protein 1 (Mcp1), interleukin 6 (IL-6), and inducible nitric oxide sythase (iNOS) was elevated in macrophages lacking Cry1 and Cry2 but these transcripts were equally suppressed by dexamethasone treatment regardless of the presence or absence of cryptochromes (Fig. S3). Thus, cryptochromes appear to regulate a distinct, albeit substantial, subset of GR regulated target genes, which does not include the NFκB inflammatory gene network.
Chronic treatment with gluocorticoids to suppress inflammation often results in hyperglycemia due to GR-induced expression of Pck1, a rate-limiting gluconeogenic enzyme in the liver. If cryptochrome repression of GR-mediated transcription were relevant in the liver, we would expect the ability of glucocorticoids to induce pck1 transcription to be lower when cryptochromes are present. Indeed, pck1 induction following a one-hour exposure to dexamethasone was dependent on the time of day and the induction of pck1 is inversely correlated with the amount of cryptochrome protein in the nucleus (Fig. 3A). Excluding the possibility that the observed effect of day-time involves altered expression or chromatin accessibility of GR, in an independent experiment, we observed a greater increase in pck1 expression in response to Dex at ZT4 (ZT, zeitgeber time, denotes hours after the lights are turned on) than at ZT16, even though the glucocorticoid-induced association of GR with the Pck1 promoter was relatively elevated at ZT16 (Fig. S4). Furthermore, using antibodies that we generated to detect endogenous Cry1 and Cry2 (Fig. S5), we found that cryptochromes interact with GR in the liver following ligand stimulation in vivo at night (ZT18) when glucocoticoids are less effective at inducing the expression of pck1 (Fig. 3B). (Cry1−/− and Cry2−/− samples were used in this experiment to ensure antibody specificity without abolishing circadian rhythmicity). Strikingly, dexamethasone stimulated pck1 transcription 12.8-fold at ZT16 in Cry-deficient livers, even though glucocorticoid-induced association of GR with the Pck1 promoter was reduced in the DKO livers (Fig. 3C). Finally, chromatin immunoprecipitation of Cry1 and Cry2 revealed that cryptochromes are associated with the Pck1 promoter GRE following dexamethasone treatment in vivo in mouse livers, while dexamethasone had no effect on the Bmal1-mediated association of Cry1 and Cry2 with the Dbp promoter (Fig. 3D). Together, these data demonstrate that cryptochromes directly repress GR induction of pck1 in the liver and thus may limit glucocorticoid-induced hyperglycemia.
Figure 3.

Cryptochromes interact with GR on chromatin to regulate pck1. (a) Top, pck1 in mouse livers. *, ** P < 0.05, 0.01 vs. saline; #, ## P < 0.05, 0.01 vs. ZT2. Bottom, Cry1 immunoblots. (b) Immunoblots of liver lysates or IPs. (c) Left, pck1 at ZT16. Right, recovery of Pck1 GRE. *, ** P < 0.05, 0.01 vs. saline; #, ## P < 0.05, 0.01 vs. wildtype. (d) Recovery of Pck1 GRE or Dbp promoter E-box at ZT16. Data represent the mean ± s.e.m. of three samples (a,c) or duplicate samples (d) analyzed in triplicate.
To examine the role of cryptochromes in glucocorticoid-dependent physiology, we measured several GR-dependent parameters in wildtype and Cry-deficient mice. (Importantly, Cry-deficient mice were maintained in 12:12 light:dark conditions and were found to have normal behavioural patterns, Fig. S6). In addition to suppressing inflammation and increasing gluconeogenesis via pck1, a principal role of glucocorticoids is to promote negative feedback in all three organs of the (HPA) axis19. However, in DKO mice, we find that glucocorticoids, which normally show robust cycling rhythm, fail to be suppressed during the day as they are in wildtype mice (Fig. 4A). Perhaps not surprisingly, glucose homeostasis is severely disrupted in Cry-deficient mice, consistent with the observed regulation of pck1 transcription: thus, DKO mice exhibit elevated blood glucose in response to acute feeding after an overnight fast (Fig. 4B) and severely impaired glucose clearance in a glucose tolerance test (GTT) (Fig. 4C). Even mice lacking either Cry1 (cry1−/−) or Cry2 (cry2−/−) were significantly impaired in their ability to restore normal blood glucose following glucose injection (Fig. 4C). In contrast, Cry-deficient animals were normally responsive to insulin (Fig. 4D).
Figure 4.

Genetic loss of cryptochromes alters physiology. (a) Serum corticosterone. #P < 0.05 vs. ZT3-4. *P < 0.05, **P < 0.01 vs. wildtype. (b) Fasted and refed blood glucose **P < 0.01 vs. wildtype. (c) Glucose tolerance test. * P < 0.05, ** P < 0.01, ***P < 0.001 vs. wildtype. (d) Insulin tolerance test. (e) Corticosterone in sera collected at ZT10–11. (f) Fasted blood glucose. **P < 0.01 vs. saline-treated animals. ##P < 0.01 vs. wildtype. (g) Glucose tolerance tests. **P < 0.01 vs. saline-treated animals. In (a–d, f–g) data represent the mean ± s.e.m. for 6–8 animals per group.
To directly examine the role of cryptochromes in repression of the HPA axis and in glucocorticoid-induced hyperglycemia, we subjected wildtype and littermate DKO mice to 8 weeks of chronic glucocorticoid treatment. After long-term exposure to Dex, endogenous production of corticosterone was completely shut down in wildtype mice as expected, and was incompletely suppressed in Cry-deficient mice (Fig. 4E), suggesting that cryptochromes directly participate in GR-mediated repression of glucocorticoid synthesis via the HPA axis. Furthermore, circulating adrenocorticotropic hormone (ACTH) seems to be elevated in DKO mice (Fig. S7), suggesting that cryptochromes may regulate steroidogenesis upstream of pituitary secretion of ACTH. Chronic glucocorticoid treatment caused significant fasting hyperglycemia in both wildtype and DKO mice, but it was more severe in mice lacking cryptochromes (Fig. 4F). In addition, Cry-deficient mice exposed to chronic glucocorticoid treatment exhibited greater glucose intolerance compared to Cry-deficient mice treated with saline for the same time period while wildtype mice had the same ability to respond to a mild glucose challenge whether they were exposed to saline or dexamethasone over the 8 weeks of treatment (Fig. 4G). Together, these data suggest that cryptochrome repression of GR-mediated transcription is an important mechanism by which circadian clocks modulate metabolic physiology in vivo.
It has become increasingly clear that circadian clocks play an important role in optimizing the temporal coordination of tissue physiology with metabolic demands that fluctuate over the course of the day2. While oscillating transcriptional activation of Clock:Bmal1 target genes contributes to these physiological rhythms, some transcriptional targets, including pck1, undergo circadian oscillations independent of Clock and Bmal110. Some of these transcripts are likely to be regulated by rhythms of fasting and feeding and subsequent oscillations in insulin, glucagon and other hormones20 and may be modulated by circadian repressors in the Period and Cryptochrome families.
In this study, we describe an interaction between cryptochromes and the glucocorticoid receptor, through which Cry1 and Cry2 repress GR-dependent transcription, including the induction of pck1 in the liver (Fig. S1). We demonstrate that Cry1 and Cry2 interact with GR in mouse liver and are associated with a GRE in the Pck1 promoter in a glucocorticoid-dependent manner. The physiological relevance of these associations is apparent in the increased susceptibility of cryptochrome-deficient mice to glucocorticoid-induced hyperglycemia. Notably, cry1−/−, cry2−/− and cry1−/−;cry2−/− mice were also strikingly intolerant of a high dose of glucose even in the absence of supplemental glucocorticoid treatment, reflecting a combination of increased glucocorticoid production, increased glucocorticoid-induced pck1 transcription and other cryptochrome-dependent pathways, including both Clock/Bmal1-dependent transcriptional programs and recently reported Cry-dependent effects on cAMP responsive element binding protein (CREB)-mediated transcription21. Past studies have indicated a role for PER2 up-regulation in the hyperglycemia induced by glucocorticoids via GR binding a GRE in the Per2 promoter22; we have found a distinct regulatory axis, involving direct physical association between cryptochormes and GR, by which circadian clocks modulate the response to glucocorticoids.
In addition, we found that Cry1 and Cry2 participate in glucocorticoid-dependent suppression of the HPA axis and the production of endogenous glucocorticoids. The observed effects on corticosterone production are reminiscent of a recent study in which cry1−/−;cry2−/− mice were found to have constitutively elevated production of aldosterone due to adrenal de-repression of type VI 3-β hydroxyl-steroid dehydrogenase (hsd3b6)23. Combined with our observation of direct interactions between cryptochromes and multiple steroid hormone receptors (Fig. 1A and data not shown), these data suggest that cryptochromes may play a general role in feedback inhibition of steroid hormone biosynthesis, though the anatomic location of action may differ as previous studies did not observe a role of cryptochromes in corticosteroid biosynthetic gene expression in adrenal cortex while they do seem to modulate aldosterone-synthesizing pathways in the adrenal gland23.
These results suggest a specific mechanism through which cryptochromes couple the activity of clock and receptor target genes to complex genomic circuits underpinning normal metabolic homeostasis. While we found that cryptochromes participate in glucocorticoid regulation of gluconeogenesis and steroidogenesis, they are not required for trans-repression of inflammatory gene transcription. Thus, the undesirable metabolic side effects of glucocorticoids used to suppress inflammation may be alleviated by altering the timing of treatment or by combining them with agents that can stabilize Cry1 and/or Cry2 in the liver. As metabolic syndrome is a complex blend of chronic inflammation and poorly controlled glucose homeostasis, the ability of cryptochromes to integrate metabolism and circadian rhythms suggests that targeting the GR-Cry interface could be a new therapeutic strategy.
Methods Summary
The methods section provides detailed information about all experimental procedures including (1) the generation, culture, and transfection of cell lines (2) generation or sources of plasmids and antibodies, (3) luciferase assays, (4) preparation of samples for western blotting, immunoprecipitation and chromatin immunoprecipitation, (5) performance and analysis of microarray experiments, (6) quantitative PCR, (7) analysis of behaviour and metabolism in mice, (8) measurement of ACTH and corticosterone by immunoassay, (9) acute and chronic treatment of mice with synthetic glucocorticoids.
METHODS
Cells and Cell Culture
HEK 293T cells were purchased from the American Type Culture Collection (ATCC) and were grown in complete Dulbecco's Modified Eagle Medium (DMEM) (Invitrogen) supplemented with 10% dialyzed fetal bovine serum, penicillin and streptomycin in a 37°C incubator maintained at 5% CO2. Mouse embryonic fibroblasts (MEFs) stably expressing FLAG-Cry1 were previously described6. Primary wildtype and cry1−/−;cry2−/− mouse embryonic fibroblasts (MEFs) were generated from E15.5 embryos derived from mating cry1+/−;cry2+/− male mice with cry1+/−;cry2+/− female mice. A pregnant female was euthanized by CO2 inhalation, her uteri were dissected and rinsed in PBS and each embryo was extracted from the surrounding tissue. After removal of the heads and fetal livers, the remaining embryonic tissue was minced with razor blades, digested with trypsin and resuspended in DMEM containing 15% FBS to generate a homogeneous cell slurry, which was plated in 6-cm dishes. MEFs were expanded slowly and were used for experiments after 8 to 10 passages. Genomic DNA isolated from the fetal livers was used to genotype the resulting cell lines. Primary macrophages were isolated and cultured as described24.
Plasmids and Transfection
pcDNA3-2xFlag-mCRY1, pcDNA3-2xFlag-mCRY2, pcDNA3-Myc-Cry1 and pcDNA3-mPER2 were gifts from Dr. Charles Weitz; pcDNA3-Flag-Rev-Erb-a was a gift from Dr. Han Cho; pcDNA3.1-RORa-v5, pcDNA3.1-RORb-v5, pcDNA3.1-RORg-v5, pcDNA3-GR-v5, pcDNA3-AR-v5, pcDNA3-ERR1-v5, pcDNA3-ERR2-v5, pcDNA3-ERR3-v5 were gifts from Dr. Johan Jonker; hGRa, hGRaΔ262–404, hGRaΔ490–515 and hGRaΔ532–697 were previously described3. Transfections were carried out using FuGene HD (Roche).
Luciferase Assays
CV-1 cells were transfected with the indicated plasmids and treated with either vehicle (ethanol) or 1 μM dexamethasone 24 hours after transfection. After overnight treatment, cells were lysed in passive lysis buffer (Promega) and analyzed using Dual-Glo Luciferase Reporter System (Promega). Firefly luciferase signal was normalized to β-galactosidase. All luciferase assay data represents the mean ± s.e.m. of triplicate samples.
Preparation of Protein Extracts, Immunoprecipitation, and Immunoblotting
Cell and liver extracts were prepared in lysis buffer containing 1% Triton X-100 (cell extracts) or 1% NP-40 (liver extracts) as previously described25. Immunoprecipitation antibodies were M2-agarose (Sigma A2220), PER21A (Alpha Diagnostic International), anti-GR M-20 (Santa Cruz Biotechnology sc-1004), and polyclonal antibodies raised in guinea pigs against the C-termini of Cry1 (amino acids 583–606) or Cry2 (amino acids 563–592) crosslinked to KLH and affinity purified against the immunogenic peptides crosslinked to a column (AminoLink Immobilization Kit,Thermo Scientific 44890). Antibodies used for Western Blotting were anti-v5, anti-myc and anti-Flag polyclonal antibodies (Sigma), PER21A, GR135 antiserum3, and anti-Cry1-CT and anti-Cry2-CT described above.
Microarrays
Total RNA was extracted using Trizol reagent (Invitrogen) and purity of the RNA was assessed by Agilent 2100 Bioanalyzer. 500 ng of RNA was reverse transcribed into cRNA and biotin-UTP labeled using the Illumina TotalPrep RNA Amplification Kit (Ambion). cRNA was quantified using an Agilent Bioanalyzer 2100 and hybridized to the Illumina mouseRefseq-8v2 Expression BeadChip using standard protocols (Illumina). Image data was converted into unnormalized Sample Probe Profiles using the Illumina BeadStudio software and analyzed on the VAMPIRE microarray analysis framework. Stable variance models were constructed for each of the experimental conditions (n=2). Differentially expressed probes were identified using the unpaired VAMPIRE significance test with a 2-sided, Bonferroni-corrected threshold of αBonf = 0.05. The VAMPIRE statistical test is a Bayesian statistical method that computes a model-based estimate of noise at each level of gene expression. This estimate was then used to assess the significance of apparent differences in gene expression between 2 experimental conditions. Lists of altered genes generated by VAMPIRE were mapped to pathways using the VAMPIRE tool GOby to determine whether any KEGG categories were overrepresented using a Bonferroni error threshold of αBonf = 0.05. Heat map was constructed with cubic spline-normalized values using the CIMminer program at http://discover.nci.nih.gov/, a development of the Genomics and Bioinformatics Group, Laboratory of Molecular Pharmacology (LMP), Center for Cancer Research (CCR) National Cancer Institute (NCI).
Gene Expression
RNA was extracted from livers or cultured cells with Trizol or using the Qiagen RNeasy purification system. cDNA was prepared using the SuperscriptII reverse transcriptase (Invitrogen) and analyzed for gene expression using quantitative real-time PCR with SYBR green (Invitrogen SybrGreenER or Biorad iQ SybrGreen supermix) chemistry. Primer sequences are available upon request. All quantitative PCR data represent the mean ± s.e.m. for three samples of each condition, analyzed in triplicate.
Chromatin Immunoprecipitation
Freshly dissected mouse livers were rinsed in ice-cold PBS, homogenized in 167ng/ml DSG in PBS using a Dounce homogenizer, and incubated at room temperature for 30 minutes. The homogenates were centrifuged at 3000 × g for 5 minutes and the pellets were resuspended in 1% formaldehyde in PBS and incubated at room temperature for 10 minutes. Crosslinking was stopped by addition of glycine to a final concentration of 100 mM and samples were placed on ice. Following cross-linking, nuclei were purified using a sucrose gradient by the method of Lavery and Schibler26. Purified nuclei were washed three times in ice-cold PBS and resuspended in buffer containing 150 mM NaCl, 5 mM EDTA, 50 mM Tris pH 7.5, 0.5% NP-40 and 1% Triton X-100 and chromatin immunoprecipitation was continued as described by Nelson, Denisenko and Bomzstyk27 using a modified shearing buffer (1% SDS, 10 mM EDTA, 50 mM Tris) and 3 micrograms of immunoprecipitating antibody per reaction. All chromatin immunoprecipitation data represent the mean ± s.e.m. for duplicate samples each analyzed in triplicate.
Mice
Cry1−/−;Cry2−/− mice were from Dr. Aziz Sancar28. Indirect calorimetry studies were conducted in a Comprehensive Lab Animal Monitoring System (eight-chamber system, Columbus Instruments, Columbus, OH). All animal care and treatments were in accordance with the Salk Institute guidelines for the care and use of animals.
Corticosterone and ACTH Measurements
Corticosterone and ACTH were measured in mouse serum collected by tail bleed using the Corticosterone double antibody 125I RIA Kit (MP Biomedicals 7120103) or ACTH 125I RIA Kit (Diasorin catalog #24130).
Fasted and Refed Blood Glucose Measurements
Mice were placed in clean cages (without food) and fasted from ZT11 to ZT2. For refeeding, food was returned to the cages for 2 hours from ZT2 to ZT4. Glucose was measured in blood collected from the tail vein using a OneTouch Basic glucometer.
Glucose Tolerance Test (GTT)
Mice were placed in clean cages (without food) at ZT10–11 one day prior to the experiment and were injected with glucose (1 or 2 mg/g bodyweight; 20% glucose in 0.9% NaCl). Blood glucose levels were measured before the injection of glucose and at 15, 30, 60, 120 and 180 min following injection.
Insulin Tolerance Test (ITT)
Mice were placed in clean cages (without food) at ZT4 and were injected intraperitoneally 2 h later with 0.5 – 1.0 Unit per kg bodyweight of Novolin-R in 0.9% NaCl. Blood glucose was measured using a One Touch Basic glucometer (Lifescan) before injection of insulin and at 15, 30, 45, 60, and 90 min following insulin injection.
Acute Dexamethasone Treatment
For experiments measuring the in vivo transcriptional response to dexamethasone (Figure 3), mice were treated with 1 mg dexamethasone per kilogram bodyweight by intraperitoneal injection one hour before dissection. All data represent the mean ± s.e.m. for three animals treated with saline (−) or dexamethasone (+), with each sample analyzed in triplicate.
Chronic Dexamethasone Treatment
Mice were treated with 1 mg dexamethasone per g bodyweight by intraperitoneal injection of an equimolar amount of dexamethasone 21-phosphate sodium salt (Sigma D1159) dissolved in sterile saline every other day at ZT11–12 for eight weeks. Mice in the control group were injected with sterile saline at the same time.
Statistics
Luciferase assay data were analyzed using two-tailed t-test. Q-PCR data were analyzed by ANOVA (Figure 3A) or two-tailed t-test. Glucose and insulin tolerance tests data were analyzed using repeated measures ANOVA for effects of genotype × time or treatment × time. Corticosterone and glucose measurements were analyzed by two-tailed t-test.
Supplementary Material
Acknowledgements
The authors thank Samantha Kaufman for assistance with glucose tolerance tests, Jacqueline Alvarez for RNA sample preparation, Henry Juguilon for luciferase assays, Joan Vaughan for corticosterone and ACTH measurements, antigenic peptide design and peptide-KLH coupling reactions, Han Cho for sharing unpublished plasmids and Reuben Shaw for comments on the manuscript. This work was supported by NIH grants DK057978 and DK062434 (to R.M.E.) and DK090188 (to K.A.L.), by support from the Glenn Foundation for Aging Research (to R.M.E. and K.A.L.), from the Helmsley Trust (to R.M.E.), and by a Merck fellowship from the Life Sciences Research Foundation (to K.A.L.).
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Contributions: K.L. and R.E. conceived the project and designed the research. K.L., S.P., G.B., and N.U. performed the experiments. J.J. provided critical reagents. K.L. and R.Y. analyzed the data. K.L. and R.E. wrote the paper. All authors edited the manuscript.
Author Information: Microarray data have been deposited to GEO with record #GSE24469. Reprints and permissions information is available at www.nature.com/reprints. The authors declare no competing financial interests.
References
- 1.Yang X, et al. Nuclear receptor expression links the circadian clock to metabolism. Cell. 2006;126(4):801–810. doi: 10.1016/j.cell.2006.06.050. [DOI] [PubMed] [Google Scholar]
- 2.Green CB, Takahashi JS, Bass J. The meter of metabolism. Cell. 2008;134(5):728–742. doi: 10.1016/j.cell.2008.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weinberger C, et al. Identification of human glucocorticoid receptor complementary DNA clones by epitope selection. Science. 1985;228(4700):740–742. doi: 10.1126/science.2581314. [DOI] [PubMed] [Google Scholar]
- 4.Lonard DM, O'Malley BW. The expanding cosmos of nuclear receptor coactivators. Cell. 2006;125(3):411–414. doi: 10.1016/j.cell.2006.04.021. [DOI] [PubMed] [Google Scholar]
- 5.De Bosscher K, Vanden Berghe W, Haegeman G. Cross-talk between nuclear receptors and nuclear factor kappaB. Oncogene. 2006;25(51):6868–6886. doi: 10.1038/sj.onc.1209935. [DOI] [PubMed] [Google Scholar]
- 6.Damiola F, et al. Restricted feeding uncouples circadian oscillators in peripheral tissues from the central pacemaker in the suprachiasmatic nucleus. Genes Dev. 2000;14(23):2950–2961. doi: 10.1101/gad.183500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lamia KA, et al. AMPK regulates the circadian clock by cryptochrome phosphorylation and degradation. Science. 2009;326(5951):437–440. doi: 10.1126/science.1172156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Asher G, et al. SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell. 2008;134(2):317–328. doi: 10.1016/j.cell.2008.06.050. [DOI] [PubMed] [Google Scholar]
- 9.Asher G, et al. Poly(ADP-Ribose) Polymerase 1 Participates in the Phase Entrainment of Circadian Clocks to Feeding. Cell. 142(6):943–953. doi: 10.1016/j.cell.2010.08.016. [DOI] [PubMed] [Google Scholar]
- 10.Lamia KA, Storch KF, Weitz CJ. Physiological significance of a peripheral tissue circadian clock. Proc Natl Acad Sci U S A. 2008;105(39):15172–15177. doi: 10.1073/pnas.0806717105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marcheva B, et al. Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature. 2010;466(7306):627–631. doi: 10.1038/nature09253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sadacca LA, Lamia KA, deLemos AS, Blum B, Weitz CJ. An intrinsic circadian clock of the pancreas is required for normal insulin release and glucose homeostasis in mice. Diabetologia. 2010;54(1):120–124. doi: 10.1007/s00125-010-1920-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Storch KF, et al. Extensive and divergent circadian gene expression in liver and heart. Nature. 2002;417(6884):78–83. doi: 10.1038/nature744. [DOI] [PubMed] [Google Scholar]
- 14.Panda S, et al. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell. 2002;109(3):307–320. doi: 10.1016/s0092-8674(02)00722-5. [DOI] [PubMed] [Google Scholar]
- 15.Kornmann B, Schaad O, Bujard H, Takahashi JS, Schibler U. System-driven and oscillator-dependent circadian transcription in mice with a conditionally active liver clock. PLoS Biol. 2007;5(2):e34. doi: 10.1371/journal.pbio.0050034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schmutz I, Ripperger JA, Baeriswyl-Aebischer S, Albrecht U. The mammalian clock component PERIOD2 coordinates circadian output by interaction with nuclear receptors. Genes Dev. 24(4):345–357. doi: 10.1101/gad.564110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sato TK, et al. A functional genomics strategy reveals Rora as a component of the mammalian circadian clock. Neuron. 2004;43(4):527–537. doi: 10.1016/j.neuron.2004.07.018. [DOI] [PubMed] [Google Scholar]
- 18.Balsalobre A, et al. Resetting of circadian time in peripheral tissues by glucocorticoid signaling. Science. 2000;289:2344–2347. doi: 10.1126/science.289.5488.2344. [DOI] [PubMed] [Google Scholar]
- 19.Reichardt HM, et al. DNA binding of the glucocorticoid receptor is not essential for survival. Cell. 1998;93(4):531–541. doi: 10.1016/s0092-8674(00)81183-6. [DOI] [PubMed] [Google Scholar]
- 20.Vollmers C, et al. Time of feeding and the intrinsic circadian clock drive rhythms in hepatic gene expression. Proc Natl Acad Sci U S A. 2009;106(50):21453–21458. doi: 10.1073/pnas.0909591106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang EE, et al. Cryptochrome mediates circadian regulation of cAMP signaling and hepatic gluconeogenesis. Nat Med. 2010;16(10):1152–1156. doi: 10.1038/nm.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.So AY-L, Bernal TU, Pillsbury ML, Yamamoto KR, Feldman BJ. Glucocorticoid regulation of the circadian clock modulates glucose homeostasis. Proc Natl Acad Sci U S A. 2009;106(41):17582–17587. doi: 10.1073/pnas.0909733106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Doi M, et al. Salt-sensitive hypertension in circadian clock-deficient Cry-null mice involves dysregulated adrenal Hsd3b6. Nat Med. 16(1):67–74. doi: 10.1038/nm.2061. [DOI] [PubMed] [Google Scholar]
- 24.Barish GD, et al. A Nuclear Receptor Atlas: macrophage activation. Mol Endocrinol. 2005;19(10):2466–2477. doi: 10.1210/me.2004-0529. [DOI] [PubMed] [Google Scholar]
- 25.Lamia KA, et al. Increased insulin sensitivity and reduced adiposity in phosphatidylinositol 5-phosphate 4-kinase beta−/− mice. Mol Cell Biol. 2004;24(11):5080–5087. doi: 10.1128/MCB.24.11.5080-5087.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lavery DJ, Schibler U. Circadian transcription of the cholesterol 7 alpha hydroxylase gene may involve the liver-enriched bZIP protein DBP. Genes Dev. 1993;7(10):1871–1884. doi: 10.1101/gad.7.10.1871. [DOI] [PubMed] [Google Scholar]
- 27.Nelson JD, Denisenko O, Bomsztyk K. Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat Protoc. 2006;1(1):179–185. doi: 10.1038/nprot.2006.27. [DOI] [PubMed] [Google Scholar]
- 28.Thresher RJ, et al. Role of mouse cryptochrome blue-light photoreceptor in circadian photoresponses. Science. 1998;282(5393):1490–1494. doi: 10.1126/science.282.5393.1490. [DOI] [PubMed] [Google Scholar]
- 29.Stratmann M, Stadler F, Tamanini F, van der Horst GT, Ripperger JA. Flexible phase adjustment of circadian albumin D site-binding protein (DBP) gene expression by CRYPTOCHROME1. Genes Dev. 24(12):1317–1328. doi: 10.1101/gad.578810. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.