

Abstract
Objectives
This overview is intended to highlight connections between monomer structure and the development of highly crosslinked photopolymer networks including the conversion dependent properties of shrinkage, modulus and stress.
Methods
A review is provided that combines the polymer science and dental materials literature along with examples of relevant experimental results, which include measurements of reaction kinetics, photorheology as well as polymerization shrinkage and stress.
Results
While new monomers are continually under development for dental materials applications, mixtures of dimethacrylate monomers persist as the most common form of dental resins used on composite restorative materials. Monomer viscosity and reaction potential is derived from molecular structure and by employing real-time near-infrared spectroscopic techniques, the development of macromolecular networks is linked to the evolution of polymerization shrinkage (measured by linometer), modulus (measured by photorheometer), and stress (measured by tensometer). Relationships between the respective polymer properties are examined.
Significance
Through a better understanding of the polymer network formation and property development processes using conventional dimethacrylate monomer formulations, the rational design of improved materials is facilitated with the ultimate goal of achieving dental polymers that deliver enhanced clinical outcomes.
Keywords: Dental composite, polymer networks, photopolymerization, shrinkage, stress
Introduction
Polymer network formation is highly dependent on the structure of the monomer or comonomers used as well as the polymerization conditions employed. In direct dental restorative applications, the monomers are typically selected to yield densely crosslinked, glassy polymer networks that provide high modulus and strength, resistance to swelling and staining, and when used with inorganic fillers, composite wear rates that are comparable to that of sound enamel. Since direct placement restorations require that polymerization processes be conducted at near-ambient conditions, the use of pressure and heat, which can be applied in laboratory-processed indirect materials, leave photopolymerization as the preferred curing mode. Photopolymerization provides the distinct advantage of one-part materials that can be cured on demand with the reaction rate readily manipulated through the combination of the photoinitiator choice and concentration, as well as the irradiance and wavelength range introduced by the light source. The ability to spatially localize the irradiation and alter the photo flux over a very wide range offers both physical and chemical approaches to control polymer network development including the evolution of properties and the final properties achieved. Visible light activation over the range of 400 to 500 nm, primarily based on camphorquinone and tertiary amine photoinitiation [1–3], remains the convention used in dentistry to avoid the potential for biological damage to both patients and practitioners. The shorter UV wavelengths compared with visible light also limit the light penetration depth, particularly in highly scattering materials such as composites [4–5]. However, the lower energy levels of the photons associated with the longer visible light wavelengths also mean that direct photodecomposition of the initiator to produce active radicals is not typically energetically allowed. This means that two- and three-component photoinitiating systems are used with visible light polymerizations that are reliant on reductive or in some cases, oxidative transitions of the photosensitizer component to generate the initiating radicals [6]. Acyl and bisacyl phosphine oxide initiators can be utilized as single component visible light alpha-cleavage (or Norrish type I) initiators at wavelengths generally below 450 nm. While particular aspects of selected photoinitiators or irradiation conditions will be covered in the subsequent discussion here, the visible light photopolymerization process as applied to dental materials, which includes a variety of irradiation modes [7–10], has been well covered in recent reviews [11–12], and is not a primary topic in this report. This paper is intended to provide a general overview of dimethacrylate photopolymer network development and describe how monomer choice and curing conditions can affect the network structure and polymer properties. The aim is to integrate literature in the field of basic polymer science with relevant recent dental materials studies while augmenting this with a number of applicable examples from the author’s laboratory.
In free radically induced dimethacrylate photopolymerizations, which represent the large majority of all commercial resins used in dental composite restorative materials [13–15], there are several distinct stages to the polymerization process as the reaction progresses from a liquid pre-gel regime to a rubbery gelled phase and finally reaches a glassy state. Under ambient conditions, the limiting conversion due to vitrification of polymeric networks in dental resins based on commonly used dimethacrylate monomers can range from less than 50 % to more 80 % [16–18]. The initial liquid monomeric state of the resin is based on mixtures of comonomers selected to fulfill a variety of material property demands prior to polymerization as well as in the final polymer. Monomers are typically selected on the basis of their viscosity, refractive index, hydrophilic/hydrophobic character, reactivity, and potential contribution to the eventual crosslink density of the polymer network. Structure and included functionality (other than the reactive methacrylate groups) of the monomers determines these properties, which are largely but not exclusively, additive in nature when comonomer mixtures are formulated. In particular, polymerization reaction rates can be synergistically enhanced by appropriate combinations of monomers [19–20] and certain monomer structures contribute unusually high rates of polymerization [21].
Monomeric viscosity
The size and shape of a liquid monomer molecule as well as its potential for intermolecular interactions determines bulk viscous flow behavior. In liquids, the individual molecules are mutually attracted through van der Waal’s forces that range from very weak to relatively strong interactions. Short chain branching within the molecular structure can reduce viscosity for equivalent molecular weight monomers since branching enhances intermolecular interactions that don’t directly affect viscosity. Dipole-dipole interactions, of which hydrogen bonding is an important example, represent the strongest interactions between neutral molecules. The strength of the intramolecular interactions is important since these effectively make small molecules behave as larger structures. Unlike many photocured industrial coating materials, dental resins tend to make relatively little use of high molecular weight reactive oligomers, such as the one developed for Kalore (GC America). Small molecule monomers such as triethylene glycol dimethacrylate (TEGDMA) or hexanediol dimethacrylate (HDDMA), that also lack hydrogen bond donor functionality present very low viscosities. On the other hand, BisGMA {2,2-bis[p-(2′-hydroxy-3′-methacryloxypropoxy)phenylene]propane}, which is an interesting example of a monomer molecule designed specifically for use in dental materials (and now is found in a diverse range of high-performance, polymer-based material applications), exhibits dramatically high viscosities. The range of viscosity of BisGMA (approximately 700 to more than 1300 Pa.s) is based on small differences in the degree of oligomerization that occurs during the synthetic procedure. The extreme room temperature viscosity in BisGMA is derived from the relatively strong hydrogen bonding between hydroxyl groups, which is exclusively intermolecular in this monomer due to the rigid aromatic core structure [22]. The weaker, but still significant contribution of π-π aromatic ring stacking interactions in BisGMA is demonstrated by comparison of its viscosity with those of ethoxylated bisphenol A dimethacrylate (BisEMA; 3 Pa.s) and TEGDMA (0.05 Pa.s) [19]. Another perspective on this comes from the comparison of BisGMA with its essentially half-sized monovinyl analog prepared by the reaction of 4-tert-butylphenyl glycidyl ether with methacrylic acid. With a single hydroxyl group, the analog does not engage in the extended hydrogen bonding interactions and it has a viscosity several orders of magnitude lower than that of BisGMA [23].
Hydrogen bonding interactions associated with urethane functionality, such as is found in urethane dimethacrylate (UDMA; 1,6-bis(methacryloxy-2-ethoxycarbonylamino)-2,4,4-trimethylhexane) are significantly weaker than those based on hydroxyl groups as shown by the conversion of BisGMA to a series of urethane-containing dimethacrylate monomers with greatly reduced viscosities (Fig. 1) [24]. That prior study also highlighted another interesting attribute of BisGMA. In spite of the much lower degree of conversion achieved during the homopolymerization of BisGMA relative to the lower viscosity (higher molecular mobility) urethane derivatives, the mechanical strength was greatest for the BisGMA polymer as a result of the strong hydrogen bonding interactions that serve as non-covalent physical crosslinks in the network. The hydrogen bond strength of BisGMA-containing resins was also found to be unaffected by polymerization whereas the interactions involving urethane groups in UDMA-based polymers were modestly reduced compared to the monomeric state [22]. Hydrogen bonding is transient in nature with cycling rates comparable to small molecule diffusion rates and well in excess of molecular diffusion processes in polymeric systems. Therefore, hydrogen bonding can serve a significant role in the reinforcement of polymeric network structures, especially in processes involving high strain rates. The practical approach of using reactive diluent comonomers to adjust a formulated resin viscosity to a specific target is readily predictable, although the viscosity of the mixture varies in logarithmic fashion with the composition rather than as a simple arithmetic mean of the individual constituent viscosities (Fig. 2) [19]. A similar logarithmic dependence is found between temperature and viscosity of a resin [22]. The choice of monomers also alters the handling properties of a resultant composite paste. Obviously, the size, shape and composition of the filler or mixed fillers used, as well as the surface treatment applied to the fillers will also affect the paste rheology, but differences in monomer composition in terms of viscosity and functionality are factors in composite paste cohesion and how the paste adheres or releases from various surfaces at different shear rates. Studies have recently begun to focus on how subtle compositional variations significantly influence composite handling properties [25–28].
Fig. 1.
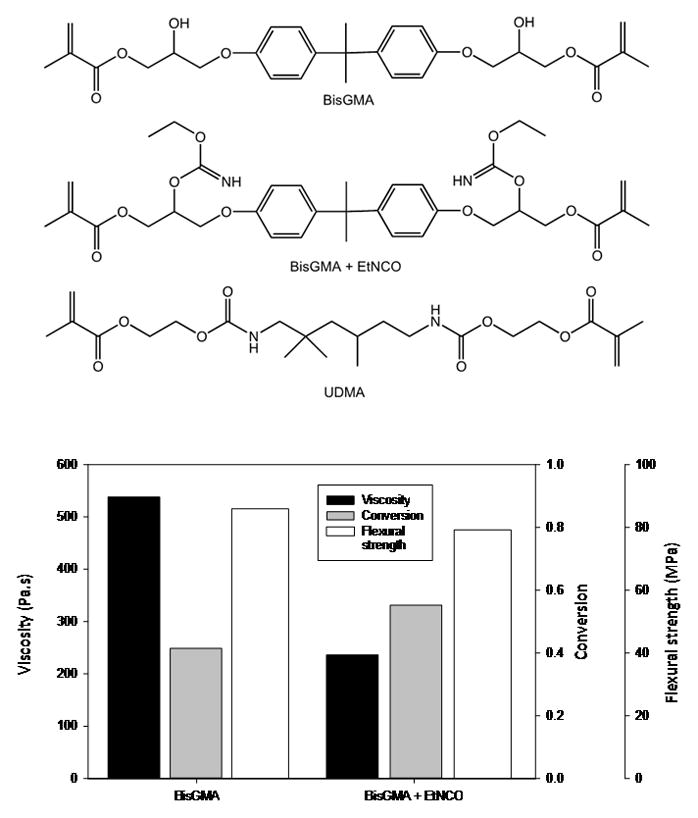
Structures of BisGMA, urethane analog of BisGMA prepared by the urethane-forming reaction between BisGMA and ethyl isocyanate, and UDMA. The plot provides the monomer viscosity at 25 °C (for comparison: UDMA viscosity = 10 Pa.s) as well as the degree of conversion and flexural strength for the photocured homopolymers [adapted from reference 24].
Fig. 2.
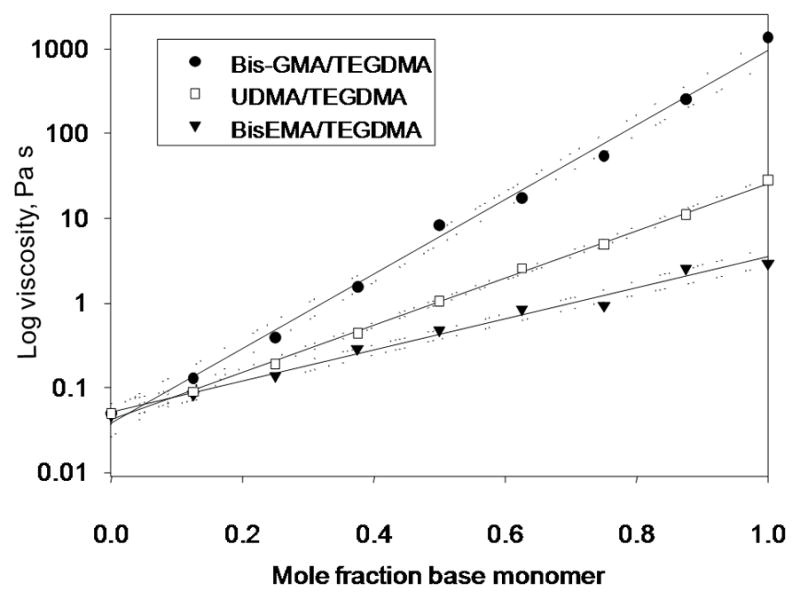
Viscosity (log scale) of BisGMA, UDMA or BisEMA as base monomers combined with TEGDMA as a reactive diluent comonomer; measured with a parallel plate rheometer at 30 °C and 1 Hz [adapted from reference 19].
Network development
Prior to polymerization, monomers typically exist as homogeneous liquid solutions. While the glass transition temperature (Tg) of the final polymer network that is ultimately formed as a consequence of the crosslinking polymerization reaction is a critical parameter, it can be informative to look at how the Tg and modulus development evolves during the polymerization. These details then can help direct the design of new materials and process controls that will potentially deliver materials with improved properties and clinical performance. As already mentioned, individual monomers exhibit widely varied viscosities, and in a related manner, they also differ considerably in their monomeric Tg’s. Some monomers will undergo crystallization when cooled below room temperature; however, the majority of dimethacrylate monomers commonly utilized in dental resins become amorphous glassy structures upon cooling in bulk. TEGDMA, which has a low viscosity, also has a relatively low monomeric Tg: −80 °C; as determined by differential scanning calorimetry [19]. In line with the progressively higher viscosities of BisEMA, UDMA and BisGMA, their monomeric Tg’s were likewise established at −42, −38 and −10 °C, respectively. For comonomer mixtures, the monomeric Tg is linearly additive since most monomer combinations yield homogeneous amorphous glasses upon cooling based on the principle that like dissolves like. However, some combinations that represent greater structural divergence between the monomers, such as BisGMA and long-chain aliphatic structures [29], can undergo phase separation as the temperature is reduced. These sorts of marginally thermodynamic compatible monomer formulations have potential application as resins in which polymerization-induced phase separation occurs to intentionally create heterogeneous polymer networks that display reduced degrees of polymerization shrinkage and stress development [13, 30].
When a monomer or resin is photocured, the viscosity, modulus and glass transition temperature all increase as the proportion of free monomer is steadily reduced with respect to the growing polymer phase, which is composed of polymer backbone chains, crosslinks and pendant reactive groups in ratios that also vary dependent upon the degree of conversion. A demonstration of the progression in glass transition temperature profiles as a function of conversion is shown in Fig. 3. Dynamic mechanical analysis (DMA) was used to obtain the tan δ plots of a series of partially cured BisGMA/TEGDMA polymers that are static representations of various extents of network formation. A photo-iniferter (XDT) was used in place of a conventional free radical initiator since the iniferter produces a thermally stable, dormant chain-end group after the photopolymerization is discontinued [31–32]. Therefore, unlike free radically cured polymer networks that contain persistent trapped radicals [33–35], there is no thermally induced post-cure in the iniferter-based photopolymers when probed by the DMA technique. This gives a clear example of how the Tg, taken as the maximum of the tan δ peak, and polymer heterogeneity, taken as the breadth of the tan δ peak, evolve during the polymerization. For a polymer-based direct restorative material to be clinically successful, its operational Tg must exceed not only the imposed cure temperature but also the temperature excursions encountered in a moisture-rich oral environment that includes periodic high stress conditions. It is evident that the polymer Tg advances significantly beyond the cure temperature primarily as a result of the polymer network heterogeneity [36]. Dimethacrylate network heterogeneity is well established [37–40] with initial formation of independent microgel domains advancing ahead of the surrounding matrix structure that subsequently fills in with progressively denser network structure. This creates microscopic localized gradient regions of higher and lower crosslink density that vary in proportion throughout the polymerization process such that there are glassy regions at relatively low levels of conversion as well as rubbery regions even at the limiting conversion when the Tg is well above the curing temperature.
Fig. 3.
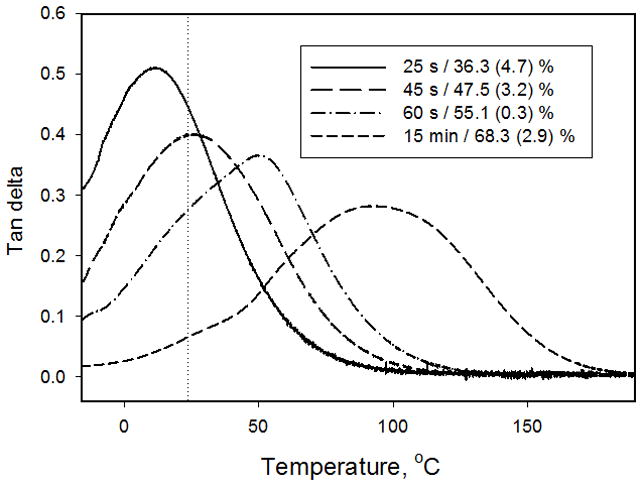
Dynamic mechanic analyzer characterization of tan δ for BisGMA/TEGDMA (7:3 mass ratio) resin photocured with 320–500 nm light at 70 mW/cm2 using p-xylylenebis(N,N-diethyl dithiocarbamate) (XDT) at 0.1 wt% as a photo-iniferter. The stable degree of conversion (with standard deviation) obtained by interrupting the photopolymerization at intermediate stages. Dotted line indicates the ambient cure temperature.
Gelation can be determined by a variety of techniques with photorheology being a particularly useful experimental technique to apply to measure not only gelation, but also the full range of viscoelastic property development that occurs during polymerization. Several groups have implemented related versions of photorheometers that have been modified to permit simultaneous real-time conversion monitoring [41–43]. This coupling of analytical techniques allows the gel point conversion to be accurately assigned as long as the issue of oxygen inhibition, which creates substantial edge effect problems in the rotational shear methodology of a parallel plate rheometry, is addressed. The photorheology indicates that the gel point is achieved at approximately 5 % conversion in the homopolymerization of BisEMA [43] under the reported conditions (Fig. 4). The choice of monomer(s) and curing condition both have the potential to affect the gel point to modest degrees. A dimethacrylate monomer with a connecting group structure that is relatively compact and presents significant flexibility is more likely to engage in cyclization reactions than is a monomer that conveys a more rigid and extended linkage. Cyclization reactions entail a propagating chain radical reacting with a pendant reactive group either already attached to that same chain (primary cycle) or attached to another chain that has previously been connected to the first by a crosslink (secondary cycle). These cyclization reactions produce ineffective crosslinks and so their occurrence delays the gel point conversion compared with monomers that more efficiently form effective crosslinks. As an example, TEGDMA would be expected to have a higher gel point conversion than BisGMA since the former incorporates approximately three-fold more cyclization into the polymer structure [44]. The number of reactive functional groups on a monomer also influences the gel point. In selecting a comonomer as a reactive diluent, the inclusion of a higher functionality monomer, such as trimethyloylpropane trimethacrylate rather than TEGDMA, would tend to promote an earlier gel point conversion due to the greater statistical likelihood of crosslink formation in the former. Use of a monomethacrylate does not avoid the potential for crosslink formation since chain transfer events involving both the monomeric and polymeric states can introduce crosslink formation, although at much lower concentrations than in dimethacrylate systems. A recent study using radical trapping agents provides evidence for radical production on multi-ethylene glycol segments of monovinyls [45] and similar behavior involving the crosslinking segments of monomers such as TEGDMA and BisEMA may be likely as well.
Fig. 4.
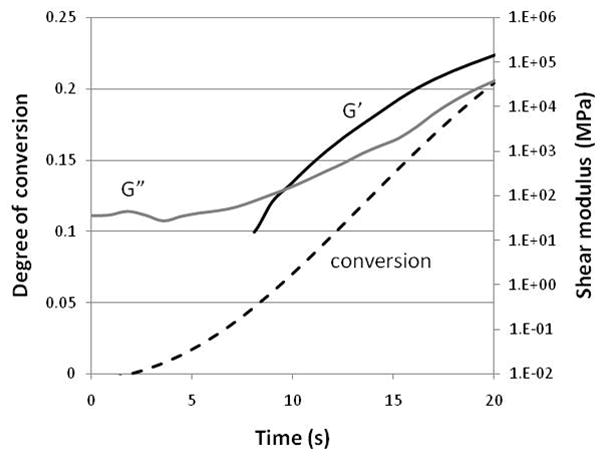
Nitrogen-purged photorheometer coupled with near-infrared spectroscopic analysis conveyed by fiber optics to simultaneously measure the storage (G′) and loss (G″) modulus development profiles along with degree of conversion. The polymerization involves BisEMA containing 0.1 wt% of 2,2-dimethoxy-2-phenylacetophenone with UV irradiation at 320–390 nm and 0.3 mW/cm2. The point of cross-over for the G′ and G″ profiles is a generally used representation of the gel point.
Photopolymerization conditions and initiator concentration control the initiation rate and the initial active radical concentration. The concentration of accessible radicals affects the chain length of propagating radicals at the point of termination. During the early stage of polymerization, short propagating chains display much higher termination rates due to disparities in terms of diffusion rates [46]. Higher initiation rates and propagating radical concentrations lead to shorter chain lengths and therefore, higher conversion values at the gel point since shorter chains, with fewer pendant reactive groups present statistically reduced likelihood for gelation compared with longer chains. This means that high curing light intensities can be expected to delay the conversion at which gelation occurs. Conversely, while a photopolymerization initiated with a low light intensity will extend the time to gelation relative to a fast polymerization, it will actually produce gelation at an earlier stage of conversion. As polymerization continues beyond the gel point, there is eventually a transition from chain-length dependent termination to reaction diffusion control of the termination process, which means that as the viscosity of the reacting medium increases with advancing conversion, propagating chain radicals rely on spatial extension based on reaction to encounter other radical species and terminate [47]. Monomers and resins that present higher initial viscosities before polymerization tend to enter this transition to reaction diffusion controlled kinetics at an earlier stage of conversion and also reach a lower limiting conversion [20]. Due to the heterogeneous nature of methacrylate networks, trapped radicals are present from the early stages of conversion. Inaccessible radicals isolated in densely crosslinked regions contribute a unimolecular termination process to the polymerization mechanism that perturbs the classical reaction kinetics based on bi-radical termination events [35].
Returning to the dynamic rheological data, a composite plot constructed from multiple experiments conducted under conditions of overlapping strain, provides modulus development as a function of conversion (Fig. 5). The initial rise in the storage modulus as gelation progresses is evident on the log axis while the equivalent linear data more clearly demonstrates the very dramatic rise in modulus that corresponds with vitrification. The rubbery modulus regime is located between these two features, which constitutes the bulk of the polymerization process. Rubbery modulus is directly dependent on the effective crosslink density and as known from studies focused on the evolution of gel fraction [48], the gel fraction develops rapidly with respect to conversion in the low to moderate conversion range but much more slowly towards the end of the polymerization process. This indicates that crosslink density develops slowly at low conversion, with an expected gradual increase in the rubbery modulus, whereas the majority of late stage conversion is directed towards crosslink formation through reaction of pendant groups. This is obviously important for dental materials in terms of residual free monomer content at the end of polymerization.
Fig. 5.
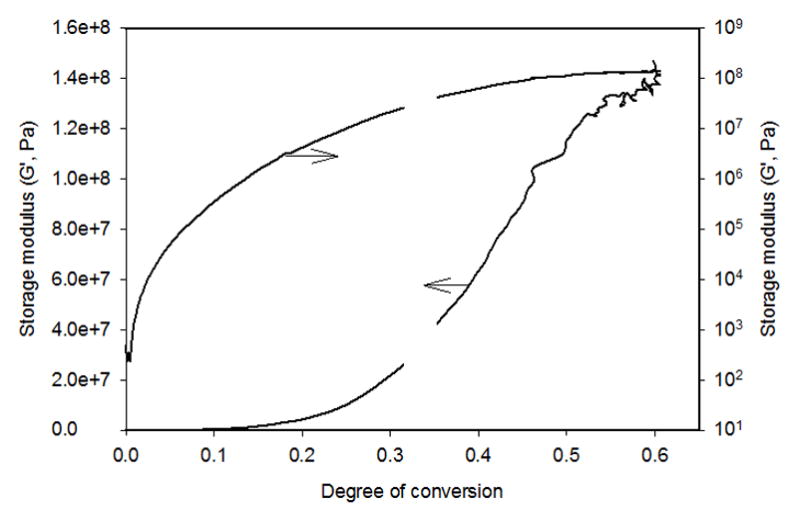
A photorheomometer-based analysis of the copolymerization of BisEMA/BisGMA 1:1 (mol ratio) with 10 mol% dodecanethiol and 2,2-dimethoxy-2-phenylacetophenone (0.1 wt%). Irradiated with 365 nm UV light at 300 μW/cm2 for 10 min. Identical data plotted on either linear (left) or log (right) scales.
Unlike the gel point, which is a well defined point in conversion, vitrification is dependent on the reaction conditions since it is determined by mobility restrictions that are affected by factors such as temperature and free volume. This means a faster polymerization reaction with a greater exotherm will delay vitrification to a later stage of conversion compared with a slower polymerization. The heat released by the exothermic reaction, as well as radiant heat produced by the curing light provides the energy to maintain greater mobility within the forming network. The residual heat causes reduced density, which also coincides with greater mobility. The coefficient of thermal expansion decreases during polymerization [49], probably related mainly to the free monomer content, which may trap additional free volume in the polymer after the temperature excursion subsides. Monomers with higher reactive group concentrations, which can be calculated readily based on monomer molecular weight, degree of functionality and density, contributes to the exothermic potential. Beyond that, the sample geometry and substrate materials, the presence of fillers, and the imposed reaction rate will determine the extent of the internal temperature rise [50]. As demonstrated in Fig. 3, dimethacrylate networks are created with substantial heterogeneity, which means very wide variations in segmental chain mobility and relaxation times. Therefore, the transition from the rubbery to the glassy state occurs over a range of conversion. Due to the process of microgel formation and coalescence in dimethacrylate networks, bulk vitrification does not occur until the crosslink density of the regions between the microgels reaches the threshold of significant mobility restriction. This allows for a sharper onset of vitrification than would be anticipated simply based on the breadth of the tan δ as vitrification is approached. With the transition to the bulk glassy state, the polymerization reaction rate slows several orders of magnitude but network density, and thus the polymer properties, such as modulus and shrinkage stress that are dependent on crosslink density, continue to develop.
Polymerization shrinkage
The incorporation of free monomer into a polymer backbone structure severely limits both its mobility and rate of motion, which leads to increased polymer densities relative to the corresponding monomer. The monomeric densities of BisGMA and TEGDMA are known to be additive [51] but this should not be assumed for all comonomer mixtures since the free volume of mixing can be positive or negative depending on the monomer combination used. Since polymerization shrinkage is based on the initial monomeric reactive group concentration and the degree of conversion attained, monomers with higher molar mass and lower degrees of functionality will produce lower absolute shrinkage results. The practical issue that polymer mechanical properties are determined in large part by the network crosslink density is a primary reason why very large monomer structures and extended reactive oligomers are not widely used in dental composite materials where clinically noncompromisable properties such as mechanical strength and hardness are quite challenging to meet. As already discussed in relation to BisGMA, non-covalent interactions can be employed to provide some degree of reinforcement that allows larger monomer structures to be applied [52]. Polymerization shrinkage can be normalized across a wide range of monomer structures by calculating the shrinkage per mole of methacrylate functional group that has reacted. Several studies have examined a variety of mono- and di-methacrylate structures to determine their molar shrinkage coefficients [53–55]. The results show that there are structurally dependent differences but that the range of values is relatively small and can reasonable be approximated by a simple average. Crosslink formation does not appear to impart any additional free volume into networks that is not seen in materials based on linear polymers; however, a recent study of network development in BisGMA/TEGDMA resins that included polymerization shrinkage measurements along with free volume assessment by positron annihilation lifetime spectroscopic analysis demonstrated that the more flexible TEGDMA crosslinker introduces greater free volume into the polymer networks compared with BisGMA [56].
The temperature at which the polymerization is conducted can significantly affect the polymerization rate and final conversion as well as the amount of shrinkage produced. Polymerization shrinkage testing of commercial composite materials under simulated oral conditions (35 °C and 92 % relative humidity) consistently yielded higher shrinkage than obtained in ambient laboratory conditions [57]. The higher shrinkage follows from the higher values of conversion that are expected with delayed vitrification when higher cure temperatures are used [58]. Several studies have examined the relationship between composite preheating and polymerization shrinkage strain [59–61]. Dynamic post-gel polymerization shrinkage data obtained using a linometer can be indexed with fiber optic near-IR spectroscopic conversion simultaneously on the same specimen (Fig. 6). The resulting data shows an initially low rate of shrinkage with respect to conversion as the reaction exotherm develops. This phase is followed by a relatively uniform progression in shrinkage with a lower rate with respect to conversion associated with the higher irradiance condition. With the exotherm subsidence, there is an increase in the shrinkage rate with the higher irradiance case displaying the greater delay and a higher rate of shrinkage relative to conversion. The experimental process concludes with a region, which accounts for the large majority of the actual photopolymerization time, where shrinkage is significantly suppressed relative to conversion. This feature is not present in lower Tg polymers that display much more rapid volume relaxation kinetics. This phenomenon of trapped excess free volume in glassy photopolymer networks has been described previously [62–63] and is consistent with the more recent observation of an essentially conversion-independent densification or physical-aging process that leads to increasing modulus and Tg values for dental polymers [64]. This type of behavior is expected if volumetric relaxation of the glassy polymer network occurs on a significantly longer timescale than that associated with end-stage conversion kinetics. The most notable difference based on the irradiance level used is not that the higher reaction rate produced a higher final degree of conversion, but that the volumetric shrinkage does not show a corresponding increase with the more advanced extent of reaction. This type of irradiance dependent higher level of conversion without higher shrinkage has been noted previously [65]. Potential subtle variations in network structure and density that are based on reaction rate and physical aging rather than exclusively on conversion may contribute to some of the uncertainty that exists with regard to how photocuring protocols affect crosslink density, Tg and solvent-based softening [66–69]. As already mentioned, it should also be recognized that it only requires small variations in conversion in the vitrified state produce significant network structural and property differences, which means the accuracy and precision of degree of conversion measurements can be critical. This is also a reason that conversion measurements and polymer property testing should be conducted either simultaneously or, if done independently, the respective specimens should be prepared to be as identical as possible at the time of testing.
Fig. 6.
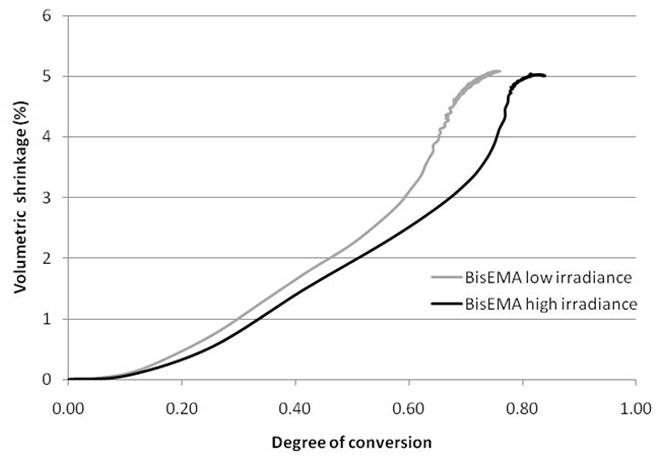
Dynamic volumetric shrinkage of BisEMA containing 0.1wt% 2,2-dimethoxy-2-phenylacetophenone photopolymerized with 365 nm light at either an irradiance of either 20 or 190mW/cm2 for 10 min; specimen geometry: 1.25 × 6 mm.
Polymerization shrinkage stress
Stress is produced by the polymerization of a resin or composite sample under conditions of constraint, whether based on a surface adherent film as may be the case with a sealant coating, an adhesive layer as with cementation of an indirect veneer or prosthetic, or a multi-walled cavity being filled as with a direct composite restoration. Just as modulus is observed as a stress-strain response, polymerization stress development is largely dependent on the evolving modulus of a polymer and its potential for shrinkage that is frustrated by attachment to a bonded interface. Free shrinkage from an open or non-bonded surface, compliance of the bonded substrate or damage zones and defects generated in the polymer or at/near the interface all constitute stress relief mechanisms. Polymerization shrinkage stress in dental composites is recognized as a significant material limitation that substantially complicates the placement of restorations. The challenge prompted by stress development requires careful observance to technique sensitive adhesive bonding protocols as well as incremental layering techniques and even when the prescribed regimens are followed, the reliability of the dentin bond remains suspect [70–71]. While improvements are still needed in the materials and techniques used in adhesive bonding to dentin, approaches that limit the extent of the challenge posed by the restorative to the bonded interface continue to drive much of the development work in resins in dental composites [43, 72–74].
A substantial number of studies have been focused on measurements of dental composite stress levels in light of polymerization shrinkage and modulus considerations of the materials [75–79]. To develop a more comprehensive understanding of the stress evolution process, it is beneficial to simultaneously couple dynamic stress measurements with the real-time near-IR conversion monitoring technique [65, 80]. As with the assessment of the conversion dependence of shrinkage and modulus, indexing stress with conversion rather than simply relating property development to time, not only allows different materials and polymerization protocols to be rationally compared, but it also provides a means to directly examine the key interrelationships of shrinkage, modulus and stress. In Fig. 7, the stress development profile shows an initial gradual rise in the rubbery state followed by a rapid increase at the onset of vitrification. The series of partial cure experiments demonstrates a relatively smooth transition towards higher stress as conversion increases. This plot of final stress values is reminiscent of a prior study in which Tg was evaluated for a series of BisGMA/TEGDMA polymers prepared by widely varied curing protocols [81]. A significant difference is that the increase in stress with conversion noted here occurs significantly more steeply than the increase in polymer Tg with conversion. This means that stress is highly sensitive to small variations in the end-stage conversion. Also, the dynamic conversion-stress profiles shown here, as well as the stress profiles with respect to time that are reported in the literature, represent the stress state during and immediately following the photopolymerization process. As discussed in the previous section, a low level of shrinkage may continue, driven by either limited post-cure conversion or physical densification, which might result in an appreciable additional increment in stress. Since there is minimal stress buildup during the initial post-gel stage of polymerization, methods that target this phase seem of questionable value. Rather, it may be most profitable to focus efforts on understanding how polymer heterogeneity or other aspects of polymer structure can be used to specifically affect the vitrification stage of polymerization where stress development is concentrated. As just one example of this, the introduction of small amounts of a chain transfer agent into a dimethacrylate photopolymerization was found to not only delay gelation but also delayed the vitrification process with the result that significantly higher limiting conversion and modulus were achieved while stress was simultaneously and dramatically reduced [43].
Fig. 7.
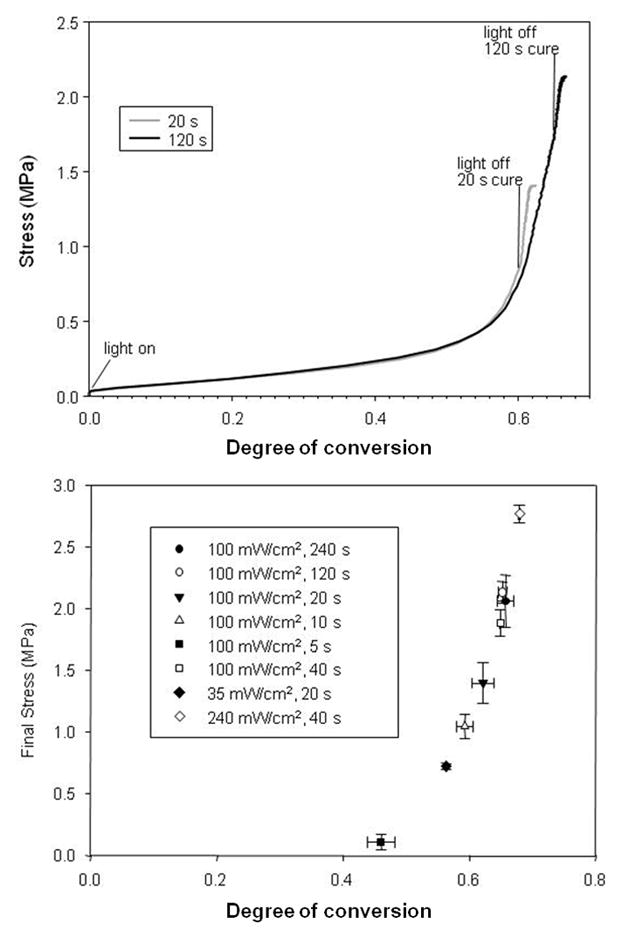
Stress development profiles for BisGMA/TEGDMA (7:3 mass ratio) resin photopolymerized with 0.1 wt% 2,2-dimethoxy2-phenylacetophenone at 320–390 nm for up to 4 min. Different exposure times and irradiances were used to obtain dynamic stress (left) and final stress levels (right) related by conversion.
Conclusions
It is clear that dimethacrylate polymers will continue to be used in dentistry for the foreseeable future although new material innovations that rely on hybrid or completely different polymerization mechanisms will also continue to be introduced. Photopolymerization serves as a very well established and versatile technique to effectively control the polymerization reaction process with great specificity and efficiency. Even though the majority of dental resin systems in use today are very closely related to the materials introduced by Ray Bowen at the inception of the modern age of dental composites, there remain significant opportunities to achieve meaningful improvements in the properties, performance and ease of use of these polymeric dental materials. As better understanding emerges concerning the complex material structure/property relationships and process parameters that control the formation and structure of the polymer networks, further advances are certain to follow.
Acknowledgments
Several of the experimental studies reported here were conducted with the financial support of NIH/NIDCR grant 5R01DE014227. In many of these studies, Esstech (Essington, PA) generously provided the monomers that were used.
Footnotes
The contributions of Carmem Pfeifer, Parag Shah, Steven Lewis and Jeff Garcia presented here are greatly appreciated.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Asmusen S, Arenas G, Cook WD, Vallo C. Photobleaching of camphorquinone during polymerization of dimethacrylate-based resins. Dent Mater. 2009;25:1603–11. doi: 10.1016/j.dental.2009.08.010. [DOI] [PubMed] [Google Scholar]
- 2.Cook WD. Photopolymerization kinetics of dimethacrylates using the camphorquinone amine initiator system. Polymer. 1992;33:600–9. [Google Scholar]
- 3.Pyszka I, Kucybala Z, Paczkowski J. Reinvestigation of the mechanism of the free radical polymerization photoinitiation process by camphorquinone-coinitiator systems: New results. Macromol Chem Phys. 2004;205:2371–5. [Google Scholar]
- 4.Griffith ML, Halloran JW. Scattering of ultraviolet radiation in turbid suspensions. J Appl Phys. 1997;81:2538–46. [Google Scholar]
- 5.Tomeckova V, Halloran JW. Predictive models for the photopolymerization of ceramic suspensions. J Eur Ceram Soc. 2010;30:2833–40. [Google Scholar]
- 6.Kim D, Stansbury JW. Kinetic pathway investigations of three-component photoinitiator systems for visible-light activated free radical polymerizations. J Polymr Sci, Polym Chem. 2009;47:887–98. doi: 10.1002/pola.23252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Costa SXS, Galvao MR, Jacomassi DP, Bernardi MIB, Hernandes AC, Rastelli AND, et al. Continuous and gradual photo-activation methods: influence on degree of conversion and crosslink density of composite resins. J Therm Anal Calorim. 2011;103:219–27. [Google Scholar]
- 8.Cunha LG, Alonso RCB, Pfeifer CSC, Correr-Sobrinho L, Ferracane JL, Sinhoreti MAC. Contraction stress and physical properties development of a resin-based composite irradiated using modulated curing methods a two C-factor levels. Dent Mater. 2008;24:392–8. doi: 10.1016/j.dental.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 9.De Santis R, Gloria A, Prisco D, Amendola E, Puppulin L, Pezzotti G, et al. Fast curing of restorative materials through the soft light energy release. Dent Mater. 2010;26:891–900. doi: 10.1016/j.dental.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 10.Ilie N, Jelen E, Hickel R. Is the soft-start polymerisation concept still relevant for modern curing units? Clin Oral Invest. 2011;15:21–9. doi: 10.1007/s00784-009-0354-5. [DOI] [PubMed] [Google Scholar]
- 11.Ikemura K, Endo T. A review of the development of radical photopolymerization initiators used for designing light-curing dental adhesives and resin composites. Dent Mater J. 2010;29(5):481–501. doi: 10.4012/dmj.2009-137. [DOI] [PubMed] [Google Scholar]
- 12.Rueggeberg FA. State-of-the-art: Dental photocuring-A review. Dent Mater. 2011;27:39–52. doi: 10.1016/j.dental.2010.10.021. [DOI] [PubMed] [Google Scholar]
- 13.Cramer NB, Stansbury JW, Bowman CN. Recent advances and developments in composite dental restorative materials. J Dent Res. 2011;90:402–16. doi: 10.1177/0022034510381263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferracane JL. Resin composite-State of the art. Dent Mater. 2011;27:29–38. doi: 10.1016/j.dental.2010.10.020. [DOI] [PubMed] [Google Scholar]
- 15.Moszner N, Salz U. Recent developments of new components for dental adhesives and composites. Macromol Mater Eng. 2007;292:245–71. [Google Scholar]
- 16.Goncalves F, Azevedo CLN, Ferracane JL, Braga RR. BisGMA/TEGDMA ratio and filler content effects on shrinkage stress. Dent Mater. 2011;27:520–6. doi: 10.1016/j.dental.2011.01.007. [DOI] [PubMed] [Google Scholar]
- 17.Goncalves F, Kawano Y, Pfeifer C, Stansbury JW, Braga RR. Influence of BisGMA, TEGDMA, and BisEMA contents on viscosity, conversion, and flexural strength of experimental resins and composites. Eur J Oral Sci. 2009;117:442–6. doi: 10.1111/j.1600-0722.2009.00636.x. [DOI] [PubMed] [Google Scholar]
- 18.Pfeifer CS, Silva LR, Kawano Y, Braga RR. Bis-GMA co-polymerizations: Influence on conversion, flexural properties, fracture toughness and susceptibility to ethanol degradation of experimental composites. Dent Mater. 2009;25:1136–41. doi: 10.1016/j.dental.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 19.Dickens SH, Stansbury JW, Choi KM, Floyd CJE. Photopolymerization kinetics of methacrylate dental resins. Macromolecules. 2003;36:6043–53. [Google Scholar]
- 20.Sideridou ID, Achilias DS, Kostidou NC. Copolymerization kinetics of dental dimethacrylate resins initiated by a benzoyl peroxide/amine redox system. J Appl Polym Sci. 2008;109:515–24. [Google Scholar]
- 21.Kilambi H, Cramer NB, Schneidewind LH, Shah P, Stansbury JW, Bowman CN. Evaluation of highly reactive mono-methacrylates as reactive diluents for BisGMA-based dental composites. Dent Mater. 2009;25:33–8. doi: 10.1016/j.dental.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trujillo-Lemon M, Jones MS, Stansbury JW. Hydrogen bonding interactions in methacrylate monomers and polymers. J Biomed Mater Res. 2007;83A:734–46. doi: 10.1002/jbm.a.31448. [DOI] [PubMed] [Google Scholar]
- 23.Kim LU, Kim JW, Kim CK. Effects of molecular structure of the resins on the volumetric shrinkage and the mechanical strength of dental restorative composites. Biomacromolecules. 2006;7:2680–7. doi: 10.1021/bm060453h. [DOI] [PubMed] [Google Scholar]
- 24.Khatri CA, Stansbury JW, Schultheisz CR, Antonucci JM. Synthesis, characterization and evaluation of urethane derivatives of Bis-GMA. Dent Mater. 2003;19:584–8. doi: 10.1016/s0109-5641(02)00108-2. [DOI] [PubMed] [Google Scholar]
- 25.Ertl K, Graf A, Watts D, Schedle A. Stickiness of dental resin composite materials to steel, dentin and bonded dentin. Dent Mater. 2010;26:59–66. doi: 10.1016/j.dental.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 26.Kaleem M, Satterthwaite JD, Watts DC. Effect of filler particle size and morphology on force/work parameters for stickiness of unset resin-composites. Dent Mater. 2009;25:1585–92. doi: 10.1016/j.dental.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 27.Kaleem M, Satterthwaite JD, Watts DC. A method for assessing force/work parameters for stickiness of unset resin-composites. Dent Mater. 2011;27:805–10. doi: 10.1016/j.dental.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 28.Lee IB, Min SH, Kim SY, Ferracane J. Slumping tendency and rheological properties of flowable composites. Dent Mater. 2010;26:443–8. doi: 10.1016/j.dental.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 29.Trujillo-Lemon M, Ge JH, Lu H, Tanaka J, Stansbury JW. Dimethacrylate derivatives of dimer acid. J Polym Sci, Polym Chem. 2006;44:3921–9. [Google Scholar]
- 30.Lu H, Trujillo-Lemon M, Ge J, Stansbury JW. Dental resins based on dimer acid dimethacrylates: A route to high conversion with low polymerization shrinkage. Compend Cont Ed Dent. 2010;31(SI2):1–4. [PubMed] [Google Scholar]
- 31.Lalevee J, Blanchard N, El-Roz M, Allonas X, Fouassier JP. New photoiniferters: Respective role of the initiating and persistent radicals. Macromolecules. 2008;41:2347–52. [Google Scholar]
- 32.Lovell LG, Elliott BJ, Brown JR, Bowman CN. The effect of wavelength on the polymerization of multi(meth)acrylates with disulfide/benzilketal combinations. Polymer. 2001;42:421–9. [Google Scholar]
- 33.Leprince JG, Lamblin G, Devaux J, Dewaele M, Mestdagh M, Palin WM, et al. Irradiation modes’ impact on radical entrapment in photoactive resins. J Dent Res. 2010;89:1494–8. doi: 10.1177/0022034510384624. [DOI] [PubMed] [Google Scholar]
- 34.Pavlinec J, Moszner N. Dark reactions of free radicals crosslinked polymer networks trapped in densely after photopolymerization. J Appl Polym Sci. 2003;89:579–88. [Google Scholar]
- 35.Zhang Y, Kranbuehl DE, Sautereau H, Seytre G, Dupuy J. Study of UV cure kinetics resulting from a changing concentration of mobile and trapped radicals. Macromolecules. 2008;41:708–15. [Google Scholar]
- 36.Ye S, Cramer NB, Bowman CN. Relationship between glass transition temperature and polymerization temperature for cross-linked photopolymers. Macromolecules. 2011;44:490–4. [Google Scholar]
- 37.Barszczewska-Rybarek IM. Structure-property relationships in dimethacrylate networks based on Bis-GMA, UDMA and TEGDMA. Dent Mater. 2009;25:1082–9. doi: 10.1016/j.dental.2009.01.106. [DOI] [PubMed] [Google Scholar]
- 38.Guo Z, Browne E, Compton J, Sautereau H, Kranbuehl D. Dielectric, dynamic mechanical and DSC evidence for a spatial and dynamic heterogeneity in a single phase polymer system. J Non-Cryst Solids. 2006;352:5025–8. [Google Scholar]
- 39.Krzeminski M, Molinari M, Troyon M, Coqueret X. Characterization by atomic force microscopy of the nanoheterogeneities produced by the radiation-induced cross-linking polymerization of aromatic diacrylates. Macromolecules. 2010;43:8121–7. [Google Scholar]
- 40.Rey L, Galy J, Sautereau H, Simon GP, Cook WD. PALS free volume and mechanical properties in dimethacrylate-based thermosets. Polym Int. 2004;53:557–68. [Google Scholar]
- 41.Alig I, Steeman PAM, Lellinger D, Dias AA, Wienke D. Polymerization and network formation of UV curable materials monitored by hyphenated real-time ultrasound reflectometry and near-infrared spectroscopy (RT US/NIRS) Prog Org Coat. 2006;55:88–96. [Google Scholar]
- 42.Botella A, Dupuy J, Roche AA, Sautereau H, Verney V. Photo-rheometry/NIR spectrometry: An in situ technique for monitoring conversion and viscoelastic properties during photopolymerization. Macromol Rapid Commun. 2004;25:1155–8. [Google Scholar]
- 43.Pfeifer CS, Wilson ND, Shelton ZR, Stansbury JW. Delayed gelation through chain-transfer reactions: Mechanism for stress reduction in methacrylate networks. Polymer. 2011;52:3295–303. doi: 10.1016/j.polymer.2011.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Elliott JE, Lovell LG, Bowman CN. Primary cyclization in the polymerization of bis-GMA and TEGDMA: a modeling approach to understanding the cure of dental resins. Dent Mater. 2001;17:221–9. doi: 10.1016/s0109-5641(00)00075-0. [DOI] [PubMed] [Google Scholar]
- 45.Tasdelen MA, Moszner N, Yagci Y. The use of poly(ethylene oxide) as hydrogen donor in type II photoinitiated free radical polymerization. Polym Bull. 2009;63:173–83. [Google Scholar]
- 46.Achilias DS, Verros GD. Modeling of diffusion-controlled reactions in free radical solution and bulk polymerization: Model validation by DSC experiments. J Appl Polym Sci. 2010;116:1842–56. [Google Scholar]
- 47.Bowman CN, Kloxin CJ. Toward an enhanced understanding and implementation of photopolymerization reactions. AIChE J. 2008;54:2775–95. [Google Scholar]
- 48.Stansbury JW, Dickens SH. Network formation and compositional drift during photo-initiated copolymerization of dimethacrylate monomers. Polymer. 2001;42:6363–9. [Google Scholar]
- 49.Mucci V, Arenas G, Duchowicz R, Cook WD, Vallo C. Influence of thermal expansion on shrinkage during photopolymerization of dental resins based on bis-GMA/TEGDMA. Dent Mater. 2009;25:103–14. doi: 10.1016/j.dental.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 50.Howard B, Wilson ND, Newman SM, Pfeifer CS, Stansbury JW. Relationships between conversion, temperature and optical properties during composite photopolymerization. Acta Biomaterialia. 2010;6:2053–9. doi: 10.1016/j.actbio.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Venhoven BAM, Degee AJ, Davidson CL. Polymerization contraction and conversion of light-curing BisGMA-based methacrylate resins. Biomaterials. 1993;14:871–5. doi: 10.1016/0142-9612(93)90010-y. [DOI] [PubMed] [Google Scholar]
- 52.Ge JH, Trujillo M, Stansbury J. Synthesis and photopolymerization of low shrinkage methacrylate monomers containing bulky substituent groups. Dent Mater. 2005;21:1163–9. doi: 10.1016/j.dental.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 53.Arney JS, Sanders L, Hardesty R. The impact of molecular structure and cross-link formation on the shrinkage of acrylic monomers. J Imaging Sci Tech. 1994;38:262–8. [Google Scholar]
- 54.Dewaele M, Truffier-Boutry D, Devaux J, Leloup G. Volume contraction in photocured dental resins: The shrinkage-conversion relationship revisited. Dent Mater. 2006;22:359–65. doi: 10.1016/j.dental.2005.03.014. [DOI] [PubMed] [Google Scholar]
- 55.Patel MP, Braden M, Davy KWM. Polymerization shrinkage of methacrylate esters. Biomaterials. 1987;8:53–6. doi: 10.1016/0142-9612(87)90030-5. [DOI] [PubMed] [Google Scholar]
- 56.Pfeifer CS, Shelton ZR, Braga RR, Windmoller D, Machado JC, Stansbury JW. Characterization of dimethacrylate polymeric networks: A study of the crosslinked structure formed by monomers used in dental composites. Eur Polym J. 2011;47:162–70. doi: 10.1016/j.eurpolymj.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tiba A, Charlton DG, Vandewalle KS, Cohen ME. Volumetric polymerization shrinkage of resin composites under simulated intraoral temperature and humidity conditions. Oper Dent. 2005;30:696–701. [PubMed] [Google Scholar]
- 58.Trujillo M, Newman SA, Stansbury JW. Use of near-IR to monitor the influence of external heating on dental composite photopolymerization. Dent Mater. 2004;20:766–77. doi: 10.1016/j.dental.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 59.El-Korashy DI. Post-gel shrinkage strain and degree of conversion of preheated resin composite cured using different regimens. Oper Dent. 2010;35:172–9. doi: 10.2341/09-072-L. [DOI] [PubMed] [Google Scholar]
- 60.Lohbauer U, Zinelis S, Rahiotis C, Petschelt A, Eliades G. The effect of resin composite preheating on monomer conversion and polymerization shrinkage. Dent Mater. 2009;25:514–9. doi: 10.1016/j.dental.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 61.Tantbirojn DTD, Chongvisal S, Augustson DG, Versluis A. Hardness and postgel shrinkage of preheated composites. Quintessence Int. 2011;42(3):e51–e9. [PubMed] [Google Scholar]
- 62.Anseth KS, Kline LM, Walker TA, Anderson KJ, Bowman CN. Reaction-kinetics and volume relaxation during polymerizations of multiethylene glycol dimethacrylates. Macromolecules. 1995;28:2491–9. [Google Scholar]
- 63.Kloosterboer JG, Lijten GFCM, Boots HMJ. Network formation by chain crosslinking photopolymerization and some applications in electronics. Makromol Chem, Macromol Symp. 1989;24:223–30. [Google Scholar]
- 64.Truffier-Boutry D, Demoustier-Champagne S, Devaux J, Biebuyck JJ, Mestdagh M, Larbanois P, et al. A physico-chemical explanation of the post-polymerization shrinkage in dental resins. Dent Mater. 2006;22:405–12. doi: 10.1016/j.dental.2005.04.030. [DOI] [PubMed] [Google Scholar]
- 65.Stansbury JW, Trujillo-Lemon M, Lu H, Ding XZ, Lin Y, Ge JH. Conversion-dependent shrinkage stress and strain in dental resins and composites. Dent Mater. 2005;21:56–67. doi: 10.1016/j.dental.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 66.Benetti AR, Asmussen E, Munksgaard EC, Dewaele M, Peutzfeldt A, Leloup G, et al. Softening and elution of monomers in ethanol. Dent Mater. 2009;25:1007–13. doi: 10.1016/j.dental.2009.01.104. [DOI] [PubMed] [Google Scholar]
- 67.Benetti AR, Peutzfeldt A, Asmussen E, Pallesen U, Franco EB. Influence of curing rate on softening in ethanol, degree of conversion, and wear of resin composite. Am J Dent. 2011;24:115–8. [PubMed] [Google Scholar]
- 68.da Silva EM, Poskus LT, Guimaraes JGA, Barcellos ADL, Fellows CE. Influence of light polymerization modes on degree of conversion and crosslink density of dental composites. J Mater Sci, Mater Med. 2008;19:1027–32. doi: 10.1007/s10856-007-3220-5. [DOI] [PubMed] [Google Scholar]
- 69.Dewaele M, Asmussen E, Peutzfeldt A, Munksgaard EC, Benetti AR, Finne G, et al. Influence of curing protocol on selected properties of light-curing polymers: Degree of conversion, volume contraction, elastic modulus, and glass transition temperature. Dent Mater. 2009;25:1576–84. doi: 10.1016/j.dental.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 70.Hilton TJ. Can modern restorative procedures and materials reliably seal cavities? In vitro investigations. Part 1 Am J Dent. 2002;15:198–210. [PubMed] [Google Scholar]
- 71.Perdigao J, Duarte S, Gomes G. Direct resin-based composite restorations - Clinical challenges. J Adhes Sci Tech. 2009;23:1201–14. [Google Scholar]
- 72.Cramer NB, Couch CL, Schreck KM, Carioscia JA, Boulden JE, Stansbury JW, et al. Investigation of thiol-ene and thiol-ene-methacrylate based resins as dental restorative materials. Dent Mater. 2010;26:21–8. doi: 10.1016/j.dental.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Moraes RR, Garcia JW, Barros MD, Lewis SH, Pfeifer CS, Liu JC, et al. Control of polymerization shrinkage and stress in nanogel-modified monomer and composite materials. Dent Mater. 2011;27:509–19. doi: 10.1016/j.dental.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Park HY, Kloxin CJ, Scott TF, Bowman CN. Covalent adaptable networks as dental restorative resins: Stress relaxation by addition fragmentation chain transfer in allyl sulfide-containing resins. Dent Mater. 2010;26:1010–6. doi: 10.1016/j.dental.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Boaro LCC, Goncalves F, Guimaraes TC, Ferracane JL, Versluis A, Braga RR. Polymerization stress, shrinkage and elastic modulus of current low-shrinkage restorative composites. Dent Mater. 2010;26:1144–50. doi: 10.1016/j.dental.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 76.Charton C, Falk V, Marchal P, Pla F, Colon P. Influence of Tg, viscosity and chemical structure of monomers on shrinkage stress in light-cured dimethacrylate-based dental resins. Dent Mater. 2007;23:1447–59. doi: 10.1016/j.dental.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 77.Goncalves F, Kawano Y, Braga RR. Contraction stress related to composite inorganic content. Dent Mater. 2010;26:704–9. doi: 10.1016/j.dental.2010.03.015. [DOI] [PubMed] [Google Scholar]
- 78.Min SH, Ferracane J, Lee IB. Effect of shrinkage strain, modulus, and instrument compliance on polymerization shrinkage stress of light-cured composites during the initial curing stage. Dent Mater. 2010;26:1024–33. doi: 10.1016/j.dental.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 79.Tantbirojn D, Pfeifer CS, Braga RR, Versluis A. Do low-shrink composites reduce polymerization shrinkage effects? J Dent Res. 2011;90:596–601. doi: 10.1177/0022034510396217. [DOI] [PubMed] [Google Scholar]
- 80.Lu H, Stansbury JW, Dickens SH, Eichmiller FC, Bowman CN. Probing the origins and control of shrinkage stress in dental resin composites. II. Novel method of simultaneous measurement of polymerization shrinkage stress and conversion. J Biomed Mater Res, Appl Biomater. 2004;71B:206–13. doi: 10.1002/jbm.b.30088. [DOI] [PubMed] [Google Scholar]
- 81.Lovell L, Lu H, Elliott J, Stansbury J, Bowman C. The effect of cure rate on the mechanical properties dental resins. Dent Mater. 2001;17:504–11. doi: 10.1016/s0109-5641(01)00010-0. [DOI] [PubMed] [Google Scholar]