

Abstract
We previously reported the identification and structure-activity analysis of bithiazole-based correctors of defective cellular processing of the cystic fibrosis-causing CFTR mutant, ΔF508-CFTR. Here, we report the synthesis and uptake of a functional, fluorescently labeled bithiazole corrector. Following synthesis and functional analysis of four bithiazole-fluorophore conjugates, we found that 5, a bithazole-based BODIPY conjugate, had low micromolar potency for correction of defective δF508-CFTR cellular misprocessing, with comparable efficacy to benchmark corrector corr-4a. Intravenous administration of 5 to mice established its stability in extrahepatic tissues for tens of minutes. By fluorescence imaging of whole-body frozen slices, fluorescent corrector 5 was visualized strongly in gastrointestinal organs, with less in lung and liver. Our results provide proof-of-concept for mapping the biodistribution of a ΔF508-CFTR corrector by fluorophore labeling and fluorescence imaging of whole-body slices.
Introduction
The genetic disease cystic fibrosis (CF) is caused by mutations in the gene encoding the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR), a cAMP-regulated chloride channel expressed in epithelial cells in lung, pancreas, testis, intestine and other organs.1,2 The most common CF-causing CFTR mutation is deletion of phenylalanine at residue 508 (ΔF508), which produces a misfolded protein that is retained at the endoplasmic reticulum (ER) and rapidly degraded.3-6 A major focus in CF research is the identification and development of compounds, termed correctors, that normalize defective ΔF508-CFTR cellular processing to promote its plasma membrane targeting.7-10
By high-throughput screening, we previously reported the identification of bithiazole-based ΔF508-CFTR correctors such as corr-4a.11 Corr-4a improved ΔF508-CFTR trafficking, resulting in partial restoration of cell chloride permeability. Follow-on structure-activity relationship (SAR) studies conducted on 148 methylbithiazole analogs revealed that the substitution of the phenyl amide at position X (2; Figure 1) for a pivoyl amide improved potency,12 and that the addition of a dimethylamino-substituted phenyl moiety at position Y improved water solubility while retaining corrector activity.13
Figure 1.

(A) Bithiazole-based ΔF508-CFTR correctors. (B) 3-6: fluorescent dye analogs of corr-4a.
These findings have led us to further assess the utility of functional bithiazole derivatives at positions X and Y as ΔF508-CFTR correctors. With the intent to monitor the in vitro and in vivo efficacies of these compounds, we have incorporated photostable fluorescent dyes onto the bithiazole core (3-6; Figure 1). Herein, we report the synthesis and characterization of four fluorescently labeled bithiazole-based compounds, along with their corrector activity, as a proof-of-concept demonstration of the use of fluorescent CF correctors to probe uptake. While these derivatives are not development candidates, the work here demonstrates the possibility of synthesizing a functional fluorescent corrector whose uptake can be studied using imaging methodology.
Experimental Procedures
General Methods and Materials
All chemicals were purchased from commercial suppliers and used without further purification unless otherwise noted. Analytical thin layer chromatography was carried out on pre-coated glass plates (silica gel 60 F254, 250 µm thickness) and visualized with UV light at 245 nm. Flash chromatography was performed with 60 Å, 32-63 µm silica gel (Scientific Absorbents). Synthesized products were concentrated by rotary evaporation under reduced pressure. 1H NMR spectra were recorded at 400 or 600 MHz at ambient temperature with DMSO-d6, CDCl3, or CD3OD as solvent. 13C NMR spectra were recorded at 100 or 150 MHz at ambient temperature with DMSO-d6, CDCl3, or CD3OD as solvent. Chemical shifts are reported in parts per million relative to CHCl3 (1H, δ 7.26, 13C, 77.16), DMSO-d5 (1H, δ 2.50, 13C, 39.52) or CHD2OD (1H, δ 3.31, 13C, 49.00). Infrared spectra were recorded on an ATI-FTIR spectrometer (Mattson Genesis II). The specifications of the LC-MS are as follows: electrospray (+) ionization, mass range 150-1500 Da, 20 V cone voltage, and Xterra MS C18 column (2.1 mm × 50 mm × 3.5 µm). Dye precursors 4-(4,4-difluoro-1,3,5,7-tetramethyl-4-bora-3a-diaza-s-indacene-8-yl)-butyric acid (7)14 and 7-hydroxycoumarin-3-carboxylic acid (8)15,16 were synthesized and characterized according to literature methods. Bithiazole intermediates 1-(2-amino-4-methylthiazol-5-yl)ethanone HCl (9),17 1-(2-amino-4-methylthiazol-5-yl)-2-bromoethanone (10), 1-(2-chloro-5-(dimethylaminophenyl)thiourea (11), and N-(5-acytyl-4-methylthiazol-2-yl)pivalamide (13)18 were synthesized as previously described.
N2-(2-Chloro-5-(dimethylamino)phenyl)-4′-methyl-4,5′-bithiazole-2,2′-diamine (12)
Bromothiazole 10 (3.14 g, 11.56 mmol) and thiourea 11 (2.28 g, 9.92 mmol) were dissolved in ethanol (27 mL) and the mixture was heated to reflux for 2 h. After cooling to room temperature, the solid was collected by filtration and rinsed with cold ethanol to afford aminobithiazole 12 (3.68 g, 79%) as a pale pink solid. IR (neat) νmax 3388, 3210, 3035, 1628, 1609, 1586, 1544, 1514, 1474, 1450, 1406, 1068, 1048, 879, 870, 807 cm-1; 1H NMR (400 MHz, DMSO-d6) δ 2.40 (s, 3H), 3.08 (s, 6H), 7.00 (d, J = 8.8 Hz, 1H), 7.15 (s, 1H), 7.46 (d, J = 8.8 Hz, 1H), 8.24 (s, 1H), 9.47 (br s, 2H), 10.01 (s, 1H); 13C NMR (150 MHz, DMSO-d6) δ 17.05, 40.32, 101.12, 104.46, 107.57, 108.99, 114.41, 129.34, 137.41, 143.05, 143.88, 149.84, 162.79, 165.73; MS (ESI) m/z calcd. 365.05, found 366.04 [M + H]+.
General Procedure for Preparation of Fluorescent Bithazole Probes 3-6 via CDI-Mediated Amide Formation
Fluorophore 7 (or 8; 1.5 equiv) was dissolved in DMF (2.21 mL/mmol fluorophore) followed by the addition of carbonyldiimidazole (CDI; 1.5 equiv). The mixture was stirred for 30 min at room temperature, and aminobithiazole 12 (or 15; 1 equiv) was added in one portion. The reaction was warmed to 80 °C and stirred for 48 h. After cooling to room temperature, the mixture was poured into water to precipitate the product. The solid was collected by filtration, rinsed with water, and dried under vacuum to yield 3-6, each as a crude solid.
10-(4-(2-(2-Chloro-5-(dimethylamino)phenylamino)-4′-methyl-4,5′-bithiazol-2′-ylamino)-4-oxobutyl)-5,5-difluoro-1,3,7,9-tetramethyl-5H-dipyrrolo[1,2-c:1′,2′-f][1,3,2]diazaborinin-4-ium-5-uide (3)
According to the general procedure for amide formation, 7 (138 mg, 0.41 mmol), DMF (0.91 mL), CDI (66 mg, 0.41 mmol), and 12 (100 mg, 0.27 mmol) gave crude solid, which was purified by flash column chromatography (1.25:1 hexane/EtOAc) to produce 3 (36 mg, 19%) as a red solid; IR (neat) νmax 2916, 1686, 1600, 1581, 1535, 1513, 1431, 1017, 843 cm-1; 1H NMR (400 MHz, CDCl3) δ 2.04 (m, 2H), 2.35 (s, 6H), 2.49 (s, 6H), 2.54 (s, 3H), 2.58 (m, 2H), 3.01 (m, 2H), 3.05 (s, 6H), 6.01 (s, 2H), 6.35 (dd, J = 2.8, 8.9 Hz, 1H), 6.62 (s, 1H), 7.20 (d, J = 8.9 Hz, 1H), 7.62 (br s, 1H), 7.88 (d, J = 2.8 Hz); 13C NMR (100 MHz, CDCl3) δ 14.61, 16.49, 17.28, 26.97, 27.45, 36.06, 40.92, 102.68, 102.82, 107.32, 108.77, 122.02, 122.32, 129.37, 131.54, 136.82, 140.49, 142.40, 144.62, 150.44, 154.42, 155.90, 162.92, 169.28; MS (ESI) m/z calcd. 681.21, found 682.19 [M + H]+.
N-(2-(2-Chloro-5-(dimethylamino)phenylamino)-4′-methyl-4,5′-bithiazol-2′-yl)-7-hydroxy-2-oxo-2H-chromene-3-carboxamide (4)
According to the general procedure for amide formation, 8 (154 mg, 0.75 mmol), DMF (1.65 mL), CDI (121 mg, 0.75 mmol), and 12 (200 mg, 0.50 mmol) gave crude solid, which was purified by recrystalization in EtOH to yield 4 (108 mg, 20%) as an orange solid; IR (neat) νmax 3276, 2208, 2164,1695, 1679, 1529, 1509, 1373, 1264, 1195, 1134, 834 cm-1; 1H NMR (400 MHz, DMSO-d6) δ 3.02 (s, 6H), 6.44 (dd, J = 2.9, 8.9 Hz, 1H), 6.86 (d, J = 1.9, 1H), 6.94 (dd, J = 2.1, 8.6 Hz, 1H), 7.01 (s, 1H), 7.22 (d, J = 8.9 Hz, 1H), 7.91 (d, J = 8.7 Hz, 1H), 8.07 (d, J = 2.8 Hz, 1H), 8.96 (s, 1H), 9.63 (s, 1H), 11.33 (br s, 1H), 11.85 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 17.09, 40.54, 102.02, 103.96, 104.44, 108.94, 111.26, 111.84, 114.85, 121.90, 129.41, 132.63, 137.43, 141.87, 142.91, 149.33, 149.76, 153.72, 156.66, 159.71, 161.31, 163.24, 164.63; MS (ESI) m/z calcd. 553.06, found 554.11 [M + H]+.
N-(5-(2-Bromoacetyl)-4-methylthiazol-2-yl)pivalamide (14)
A solution of 13 (6.5 g, 27.1 mmol) in 30% HBr in AcOH (40 mL) was treated with PryH+Br3- (11.25 g, 35.17 mmol) and stirred at room temperature for 48 h. Water was added and the aqueous layer was extracted with CH2Cl2. The combined organic extracts were washed with saturated NaHCO3 and brine. The organic layer was dried over sodium sulfate, filtered, and evaporated under reduced pressure. The residue was purified by flash column chromatography (20-40% EtOAc/hexane) to yield 4.23 g (49%) of the title compound 14 as a white solid; IR (neat) νmax 3260, 2970, 2928, 1687, 1660, 1530, 1493, 1370, 1318, 1280, 1220, 1196, 1136, 975, 819 cm-1; 1H NMR (600 MHz, CDCl3) δ 1.28 (s, 9H), 2.52 (s, 3H), 4.17 (s, 2H); 13C NMR (150 MHz, CDCl3) δ 17.85, 26.82, 33.58, 39.37, 121.19, 157.27, 160.69, 177.27, 184.34; MS (ESI) m/z calcd. 318.00, found 318.97 [M + H]+.
N-(2-Amino-4′-methyl-4,5′-bithiazol-2′-yl)pivalamide (15)
To a solution of 14 (2.38 g, 7.46 mmol) in EtOH (15 mL) was added thiourea (568 mg, 7.46 mmol). The mixture was heated to reflux and stirred for 12 h. After cooling to room temperature, hexane was added to fully precipitate the product and the solid was collected by filtration and washed with cold ethanol to obtain 607 mg (27%) of 15 as a cream colored solid; IR (neat) νmax 3198, 3180, 2967, 2932, 1675, 1621, 1597, 1515, 1484, 1353, 1304, 1141, 994, 898 cm-1; 1H NMR (600 MHz, CD3OD) δ 1.31 (s, 9H), 2.41 (s, 3H), 4.91 (br s, 2H), 6.88 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 16.36, 27.20, 40.22, 106.73, 113.08, 131.93, 149.20, 159.55, 172.11, 179.12; MS (ESI) m/z calcd. 296.08, found 297.08 [M + H]+.
5,5-Difluoro-1,3,7,9-tetramethyl-10-(4-(4′-methyl-2′-pivalamido-4,5′-bithiazol-2-ylamino)-4-oxobutyl)-5H-dipyrrolo[1,2-c:1′,2′-f][1,3,2]diazaborinin-4-ium-5-uide (5)
According to the general procedure for amide formation, 7 (140 mg, 0.42 mmol), DMF (0.93 mL), CDI (68 mg, 0.42 mmol), and 15 (83 mg, 0.28 mmol) gave crude solid, which was purified by flash column chromatography (1.25:1 hexane/EtOAc) to produce 5 (94 mg, 55%) as a neon orange solid; mp 266 °C dec; IR (neat) νmax 3189, 2960, 1655, 1547, 1509, 2923, 1706, 1546, 1506, 1406, 1197, 1061, 983, 834 cm-1; 1H NMR (400 MHz, CDCl3) δ 1.30 (s, 9H), 2.01 (m, 2H), 2.36 (s, 6H), 2.50 (s, 6H), 2.51 (s, 3H), 2.53 (m, 2H), 2.97 (m, 2H), 6.03 (s, 2H), 6.91 (s, 1H), 8.91 (br s, 1H), 10.05 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 14.59, 16.49, 16.94, 26.94, 27.26, 27.40, 35.82, 39.24, 108.53, 121.09, 122.02, 131.59, 140.58, 142.19, 143.35, 144.80, 154.39, 155.96, 157.67, 170.02, 176.26; HRMS: Anal. C29H35BF2N6O2S2 m/z calcd. 612.2433, found 613.2405 [M + H]+.
7-Hydroxy-N-(4′-methyl-2′-pivalamido-4,5′-bithiazol-2-yl)-2-oxo-2H-chromene-3-carboxamide (6)
According to the general procedure for amide formation, 8 (209 mg, 1.01 mmol), DMF (2.25 mL), CDI (164 mg, 1.01 mmol), and 15 (200 mg, 0.68 mmol) gave crude solid, which was purified by recrystalization from DMF to yield 6 (56 mg, 17%) as a canary yellow solid; IR (neat) νmax 3376, 2967, 2959, 1698, 1616, 1521, 1505, 1266, 1192, 1129 cm-1; 1H NMR (400 MHz, DMSO-d6) δ 1.25 (s, 9H), 6.86 (d, J = 1.8 Hz, 1H), 6.93 (dd, J = 2.1, 8.6 Hz, 1H), 7.29 (s, 1H), 7.88 (d, J = 8.7, 1H), 8.94 (s, 1H), 11.32 (br s, 1H), 11.78 (s, 1H), 11.99 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 16.86, 26.61, 35.75, 102.06, 108.65, 111.21, 111.88, 114.89, 119.51, 132.64, 142.30, 143.20, 149.40, 156.12, 156.42, 156.70, 160.25, 161.17, 164.72, 176.56; MS (ESI) m/z calcd. 484.09, found 485.03 [M + H]+.
Short-circuit Current Measurements
Fisher rat thyroid (FRT) epithelial cells stably expressing human ΔF508-CFTR were generated as described previously.19 The FRT cells were cultured on Snapwell porous support for 7–9 days. Test compounds were incubated with FRT cells for 18-24 h at 37 °C prior to measurements. Standard short-circuit current measurement procedures were followed as previously described.20 The basolateral solution contained 130 mM NaCl, 2.7 mM KCl, 1.5 mM KH2PO4, 1 mM CaCl2, 0.5 mM MgCl2, 10 mM glucose, and 10 mM Na-HEPES (pH 7.3). In the apical bathing solution, 65 mM NaCl was replaced by Na gluconate, and CaCl2 was increased to 2 mM. Solutions were bubbled with air and maintained at 37 °C. The basolateral membrane was permeabilized with 250 μg/ml amphotericin B. Hemichambers were connected to a DVC-1000 voltage clamp (World Precision Instruments Inc.) via Ag/AgCl electrodes and 1 M KCl agar bridges for recording of short-circuit current.
Cellular Uptake
FRT cells19 were cultured on coverglasses and mounted in a perfusion chamber maintained at 37 ºC. Cells were incubated with serum-free culture medium containing 10 µM 5 in culture medium. Cells were imaged over 45 min using a laser-scanning confocal microscope (Nikon C1, 100× oil objective, NA 1.49) using appropriate filter sets. Cells expressing endoplasmic reticulum-targeted green fluorescent protein (GFP) were also imaged.
In Vivo Metabolic Stability
All experiments involving live animals were performed according to procedures approved by the Institution of Animal Care and Usage Committee (IACUC) at UCSF. Compound 5 (250 μg/100 μL; 10% DMSO, 10% Tween 80, 40% propylene glycol, and 20% H2O; sterilized through a 20 µm filter prior to injection) or saline was injected intraorbitally in adult CD1 mice. Mice were sacrificed by anesthetic overdose and cervical dislocation at 10 or 30 min. Following perfusion with PBS, organs were harvested and homogenized with PBS (1 mL/g of tissue). An equal volume of cold CH2Cl2 was added to extract 5 from the tissue/aqueous layer. Homogenates were centrifuged for 15 min at 4000 rpm and 4 °C. The organic layer was evaporated with nitrogen to 100 μL and the residue was analyzed by TLC. TLC was carried out on pre-coated glass plates (silica gel 60 F254, 250 µm thickness), using a solvent system of 1:1 ethyl acetate/hexane, and fluorescent images were acquired on a Kodak 4000mm image station (Eastman Kodak Company, Rochester, NY). TLC plates were imaged with excitation and emission bandpass filters of 465 nm and 535 nm respectively with f-stop at 2.8 and an exposure time of 2 min.
In a separate experiment, 5 was administered by tail vein injection (250 μg/100 μL; 10% DMSO, 10% Tween 80, 40% propylene glycol, and 20% H2O; sterilized through a 20 µm filter prior to injection) in adult mice. Mice were sacrificed by anesthetic overdose and cervical dislocation at 10 or 30 min. Dye-containing extracts (obtained as outlined in the preceding paragraph) were dissolved in eluent (30 μL DMSO and 120 μL 3:1 CH3CN/H2O with 0.5% formic acid). Reverse-phase HPLC was carried out using a C18 column (Supelco, 2.1 mm × 100 mm × 5 μm) connected to a solvent delivery system (Waters model 2690, Milford, MA). Elution conditions were: 1 min at 95% CH3CN/5% H2O, followed by a linear gradient from 95% CH3CN/5% H2O to 0% CH3CN/100% H2O over 9 min; 3 min at 0% CH3CN/100% H2O; a linear gradient from 0% CH3CN/100% H2O to 95% CH3CN/5% H2O over 3 min; and a linear gradient from 95% CH3CN/5% H2O to 100% CH3CN/0% H2O over 2 min (0.2 mL/min flow rate). Mass spectra were acquired to confirm compound identity using a mass spectrometer (Alliance HT 2790 + ZQ) using positive ion detection, scanning from 150 to 850 Da.
Whole-body Slice Imaging
Optical imaging studies were performed on a reflectance optical scanner (IVIS-50, Caliper Life Sciences, Hopkinton, MA), and data acquisition and processing were performed using the Living Image Software v3.2. Compound 5 was formulated in 10% DMSO, 10% Tween-80, 40% propylene glycol, 20% ethanol, and 20% water (2 nmole, 150 µL). The formulated solution was sterilized through a 20 µm filter prior to injection. Male nu/nu mice (Charles Rivers, MA) that had been on a non-fluorescent diet more than 10 days were injected intravenously via tail vein with 150 µL of 5 (2 nmol) or saline. Mice were euthanized 10 min post-injection and immediately frozen and stored at -80 °C. Whole body slices at 40-µm thickness were cut using a cryotome (Hacker-Bright Sledge Microtome, Winnsboro, SC). Coronal slices through the midsection were imaged using excitation and emission bandpass filters of 468 ± 22 nm and 545 ± 30 nm, respectively. Images were collected using the same field of view with binning set at 4, f-stop at 2, and exposure time 20 s. Total photon efficiency is a measure of fluorescent photons from the specimen normalized against the excitation intensity; the resulting efficiency is expressed in units of cm2.
Results
Chemistry
Synthesis of the BODIPY dye containing a free carboxyl group (7) was achieved via a one-pot condensation between two equivalents of 2,4-dimethylpyrrol and glutaric anhydride followed by the coordination of BF3·OEt2 (Scheme 1).14,21 The hydroxy-substituted coumarin-3-carboxylic acid 8 was prepared by Knoevenagel condensation of 2,4-dihydroxybenzaldehyde with Meldrum's acid directly releasing a free carboxyl group in one pot.15 The percent yield for this one-pot transformation was 76%.
Scheme 1.

Synthesis of fluorescent dyesa. a Reagents: (a) i. BF3·OEt2, CH2Cl2, reflux, ii. BF3·OEt2, Et3N; (b) piperidinium acetate, EtOH, reflux.
Next, two amino bithiazole intermediates (12 and 15) were synthesized (Scheme 2); each intermediate was designed to allow for late stage attachment of the fluorescent dye. Aminobithiazole building block 12 was synthesized by α-bromination of aminothiazole 9 in 86% yield followed by condensation with 1-(2-chloro-5-(dimethylaminophenyl)thiourea 11 in 79% yield. CDI-mediated condensation of 12 with fluorescent acids 7 and 8 provided probes 3 and 4 in modest yields (3, 19%; and 4, 20%, respectively). To synthesize the aminobithiazole precursor 15, aminothiazole 9 was first treated with CDI-activated pivalic acid to give 13 (83% yield) followed by α-bromination with 30% HBr in AcOH to produce 14 (49% yield). Bromination of 13 proved to be more difficult than bromination of 9; therefore, a stronger acidic medium (HBr vs. HOAc) was required. Subsequent condensation with thiourea afforded aminobithiazole intermediate 15 (27% yield). Acids 7 and 8 were then activated with CDI and subsequently treated with 15 to obtain fluorescent compounds 5 and 6 in modest yields (5, 55%; and 6, 17%, respectively).
Scheme 2.

Synthetic route to fluorescent bithiazole derivativesa. a Reagents: (a) Br2, AcOH; (b) 1-(2-Chloro-5-dimethylaminophenyl)thiourea (11), EtOH, reflux; (c) 7, CDI, DMF, 80 °C; (d) 8, CDI, DMF, 80 °C (e) pivalic acid, CDI, DMF, 80 °C; (f) PyrH+Br3-, 33% HBr in AcOH; (g) NH2C(S)NH2, EtOH, reflux [See Figure 1 for 3-6 structures].
Corrector Activity
We followed established standard short-circuit current measurement procedures20 to determine the corrector activity of each fluorescently labeled bithiazole. Apical membrane chloride current was measured in δF508-CFTR expressing FRT cells after basolateral membrane permeabilization and in the presence of a transepithelial chloride gradient. Cells were incubated for 18-24 h with 10 µM test compound prior to short-circuit current assay. DMSO vehicle was used as a control. Figure 2A shows minimum apical membrane current with DMSO alone, with a small increase following addition of the cAMP-elevating agent forskolin (20 µM) and the potentiator genistein (50 µM). The increased apical membrane current was inhibited upon addition of the CFTR inhibitor CFTRinh-172. While conjugates 3 and 4 (Figure 2A) had little corrector activity at 10 µM, conjugate 6 had moderate activity, and 5 had efficacy comparable to that of 5 µM corr-4a. Based on its corrector activity, we chose 5 for further studies.
Figure 2.
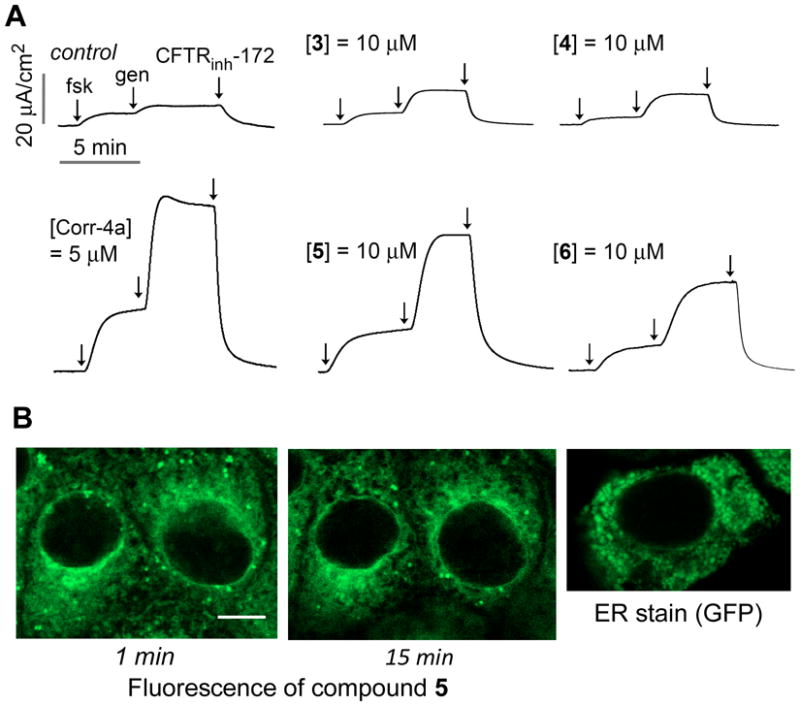
δF508-CFTR corrector activity and cellular uptake of 5. (A) Short-circuit measurements showing apical membrane chloride current after incubation for 24 h at 37 °C with 10 µM of indicated fluorescent bithiazoles. Negative control was incubation with DMSO vehicle alone and positive control was incubation with corr-4a (5 µM). Forskolin (20 µM), genistein (50 µM) and CFTRinh-172 (10 µM) were added as indicated. (B) Confocal micrographs of live FRT cells after incubation with 5 for 1 or 15 min; for comparison, ER staining shown of FRT cells expressing an ER-targeted GFP. Scale bar, 5 µm.
Intracellular Uptake
To assess cellular uptake and localization of conjugate 5, FRT cells were imaged by confocal microscopy at different times after exposure to 10 µM 5. Figure 2B shows rapid uptake of 5 within 1 min, which remained stable for >45 min. Fluorescence was observed in an intracellular compartment consistent with a characteristic membrane pattern of the endoplasmic reticulum, which is similar to that in FRT cells expressing ER-targeting GFP (Figure 2B). While not conclusive, the high lipophilicity of 5 would account for its intracellular staining pattern, as endoplasmic reticulum membranes constitute the vast majority of cell membranes.
Metabolic Stability
To determine a suitable time point for whole-body slice imaging of the organ distribution of conjugate 5, the metabolic stability of 5 was first assessed by fluorescence imaging of TLC plates (see Supporting Information p. S9). By 10 min, 5 was metabolized in liver, but remained largely intact in lung and kidney. By 30 min, 5 was not detected in lung or kidney, and little non-metabolized compound was seen in liver. Extracts from the lung, liver, kidney, brain, and heart obtained at 10 and 30 min were also analyzed by LC-MS. Rapid uptake and clearance of 5 was found in the liver, lung, kidney and heart, with no compound seen in brain. Figure 3 shows LC-MS analysis of lung extract at 10 and 30 min after intravenous injection of 5. While we have not analyzed the chemical basis of conjugate instability, which would be a major undertaking beyond the scope of the study focused here on a single fluorescent corrector, the detection of intact 5 supports the follow-on imaging studies described below. Chromatograms of liver, kidney, brain and heart are provided in the Supporting Information (p. S9-S12).
Figure 3.
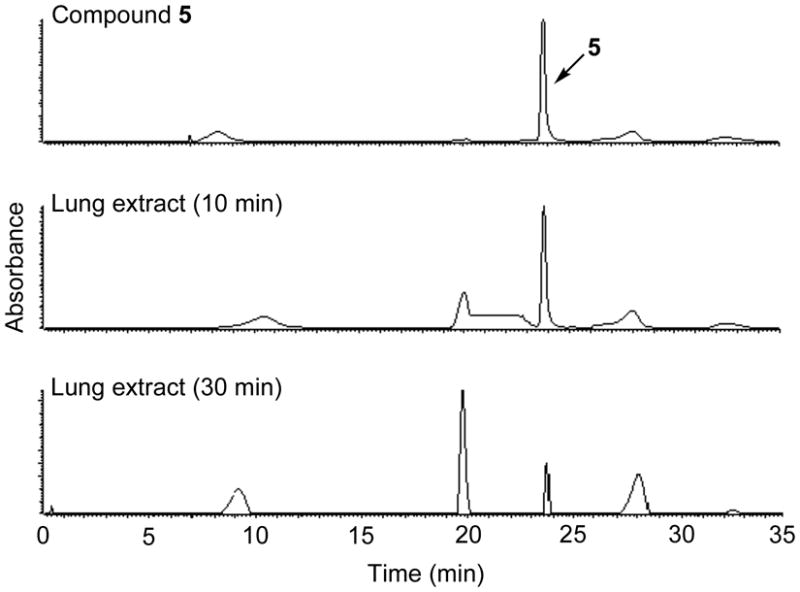
Qualitative LC-MS data for identification of 5 in lung extract. HPLC chromatograms (absorbance, 494 nm) of 5 (A), and lung extract at 10 min (B) and 30 min (C) post injection.
Uptake Measured in Whole-body Slices
Fluorescence imaging of 5 was done in whole-body slices obtained at 10 min after intravenous administration, in which the above data showed that 5 was largely intact in extrahepatic organs. Coronal slices at the mid section of mice were imaged. Figure 4 shows accumulation of 5 mainly in gastrointestinal organs, including stomach, small and large intestine and liver, with lesser fluorescence in lung. Little background signal was seen in slices from control mice.
Figure 4.
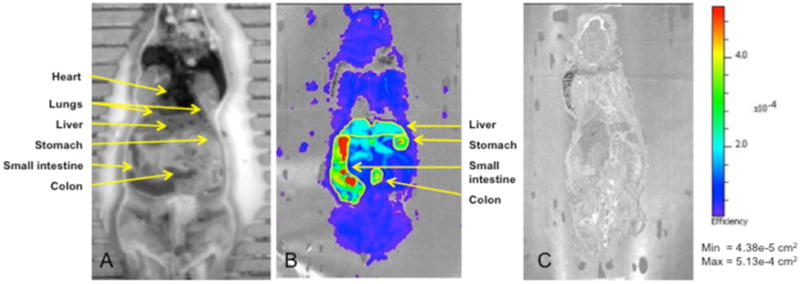
Fluorescence imaging of the coronal slices of mice at 10 min after injection of 2 nmole of 5 or saline. Shown are white light image (A) and fluorescence images of the coronal slice from mice injected with 5 (B) or saline (C).
Discussion
In a recent study, we synthesized a focused library of fifty-four thiazole-tethered pyrazoles that allowed for incorporation of property-modulating functionality (ester, acid, and amide) on C5 of the pyrazole ring (see Figure 5).18 These pyrazolylthiazoles retained δF508-CFTR corrector activity (< 1 µM) and improved the experimentally determined logP (1.2 log units lower than corr-4a). That work also established that the corrector binding site of δF508-CFTR affords a fair degree of plasticity with regard to the aniline-moiety binding pocket; compare 16 to corr-4a (Figure 5). Building on that insight, we speculated that perhaps the δF508-CFTR corrector binding site could accommodate the fluorescent dye moieties shown in analogs 5 and 6 where the dye is attached at the 2-amino position (see Figure 1). SAR insight regarding the plasticity of the presumed amide-binding pocket of δF508-CFTR, while less extensive,7 suggested that dye attachment via the 2′-amino as in analogs 3 and 4 might also retain corrector activity (see Figure 1). We did not attempt to generate conjugates with near infrared dyes (NIR dyes; for example, a bithaizole modified with a Cy7-based dye22) because, while they would offer the advantages of long-wavelength fluorescence and non-invasive imaging, their large size would likely interfere with corrector activity. Positron Emission Tomography (PET; another non-invasive technique) using an 18F-labeled CF corrector has the potential advantage of only very modestly modifying the small molecule. However, because of the higher costs and complexity, this technique was judged as inappropriate for our initial proof-of-concept work [note: we have synthesized an 19F-labeled analog of corr-4a (4-fluorobenzamide replacing benzamide; refer to Figure 1) that has corrector efficacy comparable to 5 µM corr-4a]. On the basis of this analysis, we synthesized fluorescent bithiazole analogs 3-6.
Figure 5.

Presumed aniline- and amide-binding pockets of δF508-CFTR.
BODIPY and hydroxycoumarin fluorophores were selected for conjugation with bithiazole cores 12 and 15 (see Scheme 2) because each dye is readily generated in one synthetic operation (see Scheme 1), insensitive to changes in pH, affords bright fluorescence and photostability, and has modest molecular size. Also, the preparation of each final substrate (3-6) closely parallels already developed synthetic protocols.21
Of the four bithiazole-fluorophore conjugates 3-6, BODIPY 5 was the most efficacious corrector (see Figure 2A), having comparable corrector activity to that of corr-4a. We therefore selected BODIPY 5 for in vivo and imaging studies. The activity of 5 further supports the notion that the “analine-binding pocket” of δF508-CFTR affords exploitable plasticity.
Small molecular weight therapeutics are desirable in part due to their ability to permeate the cell membrane. An initial concern with the conjugation of a fluorescent dye to the core of corr-4a was the possibility of reduced cell plasma membrane permeability. However, as seen from the rapid appearance of intracellular fluorescence in Figure 2B, conjugate 5 is highly permeable across cell membranes. The rapid uptake and probable ER distribution of 5 is likely a consequence of its lipophilicity at position Y (see Figure 1).
Metabolic stability studies showed that in the lung, the primary organ of interest in cystic fibrosis, the main fluorescent species at 10 min after intravenous administration was the original compound, allowing unambiguous interpretation of whole-body slice fluorescence images. Unfortunately, however, conjugate 5 is rapidly metabolized by the liver, precluding analysis of its biodistribution at longer times by the methods used here. Interestingly, despite the highly lipophilic nature of the BODIPY moiety at position Y, 5 does not significantly cross the blood-brain barrier at 10 min.
Optical imaging is a sensitive tool to monitor the in vivo distribution of exogenous fluorescent agents in animals.23 Here, the incorporation of BODIPY on the bithiazole core has allowed detection of fluorescence at 510 nm. Because of the limited tissue penetration of light at 510 nm, images were obtained of whole-body coronal slices at 10 min following intravenous administration of 5. Of relevance to cystic fibrosis, 5 was largely targeted to gastrointestinal organs, where CFTR is expressed, and to lesser extent in lung.
Conclusion
In conclusion, our data define the early uptake in mice of a functional, fluorescently labeled bithiazole δF508-CFTR corrector. The BODIPY fluorophore did not appear to interfere with the corrector activity of conjugate 5, but is likely to affect its biodistribution, as lipophilic compounds often accumulate in adipose tissues such as the stomach and intestine. Although the rapid hepatic metabolism and clearance of 5 restricted our analysis of biodistribution to early times after administration, fluorescence imaging of coronal slices has provided information about the uptake of 5, and hence proof-of-concept for the strategy of fluorescent drug labeling and analysis of biodistribution by whole-body slice imaging.
Supplementary Material
Acknowledgments
The authors thank the Tara K. Telford Fund for Cystic Fibrosis Research at the University of California, Davis, the National Institutes of Health (Grants DK72517 and GM089153), and the National Science Foundation [CHE-0910870; grants CHE-0910870, CHE-0443516, CHE-0449845, and CHE-9808183 supporting NMR spectrometers].
Footnotes
Supporting Information Available: 1H and 13C NMR spectra of all new compounds, fluorescent TLC image, and LC-MS chromatograms of organ homogenates. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Pilewski JM, Frizzell RA. Role of CFTR in airway disease. Physiol Rev. 1999;79:S215–S255. doi: 10.1152/physrev.1999.79.1.S215. [DOI] [PubMed] [Google Scholar]
- 2.Sheppard DN, Welsh MJ. Structure and function of the CFTR chloride channel. Physiol Rev. 1999;79:S23–S45. doi: 10.1152/physrev.1999.79.1.S23. [DOI] [PubMed] [Google Scholar]
- 3.Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, Welsh MJ. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature. 1992;358:761–764. doi: 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]
- 4.Lukacs GL, Mohamed A, Kartner N, Chang AB, Riordan JR, Grinstein S. Conformational maturation of CFTR but not its mutant counterpart (ΔF508) occurs in the endoplasmic reticulum and requires ATP. EMBO J. 1994;13:6076–6086. doi: 10.1002/j.1460-2075.1994.tb06954.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kopito RR. Biosynthesis and degredation of CFTR. Physiol Rev. 1999;79:S167–S173. doi: 10.1152/physrev.1999.79.1.S167. [DOI] [PubMed] [Google Scholar]
- 6.Du K, Sharma M, Lukacs GL. The ΔF508 cystic fibrosis impairs domain-domain interactions and arrests post-translational folding of CFTR. Nat Struct Mol Biol. 2005;12:17–25. doi: 10.1038/nsmb882. [DOI] [PubMed] [Google Scholar]
- 7.Carlile GW, Robert R, Zhang D, Teske KA, Luo Y, Hanrahan JW, Thomas DY. Correctors of protein trafficking defects identified by a novel high-throughput screening assay. ChemBioChem. 2007;8:1012–1020. doi: 10.1002/cbic.200700027. [DOI] [PubMed] [Google Scholar]
- 8.Becq F. On the discovery and development of CFTR chloride channel activators. Cur Pharm Des. 2006;12:471–484. doi: 10.2174/138161206775474459. [DOI] [PubMed] [Google Scholar]
- 9.Van Goor F, Straley KS, Cao D, Gonzalez J, Hadida S, Hazelwood A, Joubran J, Knapp T, Makings LR, Miller M, Neuberger T, Olson E, Panchenko V, Rader J, Singh A, Stack JH, Tung R, Gootenhuis PDJ, Negulescu P. Rescue of deltaF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am J Physiol. 2006;290:L1117–L1130. doi: 10.1152/ajplung.00169.2005. [DOI] [PubMed] [Google Scholar]
- 10.Yang H, Shelat AA, Guy RK, Gopinath VS, Ma T, Du K, Lukacs GL, Taddei A, Folli C, Pedemonte N, Galietta LJV, Verkman AS. Nanomolar affinity small molecule correctors of defective ΔF508-CFTR chloride channel gating. J Biol Chem. 2003;278:35079–35085. doi: 10.1074/jbc.M303098200. [DOI] [PubMed] [Google Scholar]
- 11.Pedemonte N, Lukacs GL, Du K, Caci E, Zegarra-Moran O, Galietta LJV, Verkman AS. Small molecule correctors of defective ΔF508-CFTR cellular processing identified by high-throughput screening. J Clin Invest. 2005;115:2564–2571. doi: 10.1172/JCI24898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoo CL, Yu GJ, Yang B, Robins LI, Verkman AS, Kurth MJ. 4′-Methyl-4,5′-bithiazole-based correctors of defective ΔF508-CFTR cellular processing. Bioorg Med Chem Lett. 2008;18:2610–2614. doi: 10.1016/j.bmcl.2008.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu GJ. Ph D dissertation. University of California, Davis; 2009. Golden Bridge to Cystic Fibrosis: Heterocycles; pp. 93–116. Chapter 3. [Google Scholar]
- 14.Li Z, Mintzer E, Bittman R. First synthesis of free cholesterol-BODIPY conjugates. J Org Chem. 2006;71:1718–1721. doi: 10.1021/jo052029x. [DOI] [PubMed] [Google Scholar]
- 15.Song A, Wang X, Lam KS. A convenient synthesis of coumarin-3-carboxylic acids via Knoevenagel condensation of Meldrum's acid with ortho-hydroxyaryl aldehydes or ketones. Tetrahedron Lett. 2003;44:1755–1758. [Google Scholar]
- 16.Mizukami S, Watanabe S, Hori Y, Kikuchi K. Covalent protein labeling based on noncatalytic β-lactamase and a designed FRET substrate. J Am Chem Soc. 2008;131:5016–5017. doi: 10.1021/ja8082285. [DOI] [PubMed] [Google Scholar]
- 17.Hantzsch A. Thiazoles from thiamides. Justus Liebigs Ann Chem. 1889;250:257–273. [Google Scholar]
- 18.Ye L, Knapp JM, Sangwung P, Fettinger JC, Verkman AS, Kurth MJ. Pyrazolylthiazole as ΔF508-cystic fibrosis transmembrane conductance regulator correctors with improved hydrophilicity compared to bithiazoles. J Med Chem. 2010;53:3772–3781. doi: 10.1021/jm100235h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang H, Shelat AA, Guy RK, Gopinath VS, Ma T, Du K, Lukacs GL, Taddei A, Folli C, Pedemonte N, Galietta LJ, Verkman AS. Nanomolar affinity small molecule correctors of defective Delta F508-CFTR chloride channel gating. J Biol Chem. 2003;278:35079–35085. doi: 10.1074/jbc.M303098200. [DOI] [PubMed] [Google Scholar]
- 20.Sheppard DN, Carson MR, Ostedgaard LS, Denning GM, Welsh MJ. Expression of cystic fibrosis transmembrane conductance regulator in a model epithelium. Am J Physiol. 1994;266:L405–13. doi: 10.1152/ajplung.1994.266.4.L405. [DOI] [PubMed] [Google Scholar]
- 21.Wang D, Fan J, Gao X, Wang B, Sun S, Peng X. Carboxyl BODIPY dyes from bicarboxylic anhydrides: one-pot preparation, spectral properties, photostability, and biolabeling. J Org Chem. 2009;74:7675–7683. doi: 10.1021/jo901149y. [DOI] [PubMed] [Google Scholar]
- 22.Oushiki D, Kojima H, Terai T, Arita M, Hanaoka K, Urano Y, Nagano T. Development and Application of a Near-Infrared Fluorescence Probe for Oxidative Stress Based on Differential Reactivity of Linked Cyanine Dyes. J Am Chem Soc. 2010;132:2795–2801. doi: 10.1021/ja910090v. [DOI] [PubMed] [Google Scholar]
- 23.Mahmood U, Weissleder R. Near-infrared optical imaging of proteases in cancer. Mol Cancer Ther. 2003;2:489–496. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.