

Abstract
Histone deacetylases are key regulators of gene expression and have recently emerged as important therapeutic targets for cancer and a growing number of non-malignant diseases. Many widely studied inhibitors of HDACs such as SAHA are thought to have low selectivity within or between the human HDAC isoform classes. Using an isoform-selective assay, we have shown that a number of the known inhibitors have in fact a low activity against HDAC8. Based on the wealth of structural information available for human HDAC8, we use a combination of docking and molecular dynamics simulations to determine the structural origin of the experimental results. A close relationship is found between the activity and the high surface malleability of HDAC8. These results provide a rationale for the recently described “linkerless” HDAC8 selective inhibitors and design criteria for HDAC8 selective inhibitors.
Introduction
Histone deacetylases (HDACs) regulate gene expression through the deacetylation of histone tails and are promising targets in drug development for cancer therapy.1-14 They have been linked mechanistically to the pathogenesis of cancer and several other diseases.11, 15-23 Small-molecule inhibitors of HDACs have significant effects in preclinical models of cancer.24-27 The increased focus on HDAC inhibitors for cancer treatment stems from their ability to alter several cellular functions known to be important in cancer cells. The anticancer properties of these drugs may, for example, be due to the accumulation of acetylated histones that leads to the activation (and/or repression) of transcription of genes, and inhibition of tumor cell growth.26
Eukaryotic HDACs have been classified into four groups on the basis of a phylogenetic analysis.28 Class I enzymes comprise HDACs 1,2,3 and 8 (homologous to yeast Rpd3) and class II HDACs include 4–7, 9 and 10 (homologous to yeast Hda1), which are divided into two subclasses: IIa (HDACs 4, 5, 7, 9) with one catalytic domain and IIb (HDACs 6, 10) with two HDAC domains. HDAC11 is distinct from those in classes I and II; therefore, it has been placed in class IV, and class III refers to the unrelated, NAD-dependent sirtuin deacetylases. Class I and class II, as well as class IV HDACs are Zn-dependent hydrolases. The active site of the enzyme, containing the Zn ion occupies the bottom of a narrow channel, likely to accommodate the acetylated lysine side chain during hydrolysis.
A wide range of structures have been identified that are able to inhibit the activity of the different classes, several of which are in clinical trials.25, 26 Two HDAC inhibitors, SAHA and FK228, already received FDA approval under the names of vorinostat and istodax, respectively. Four types of HDAC inhibitors can be differentiated according to the chemical structure: simple aliphatic carboxylic acids such as phenylbutyrate and valproic acid; hydroxamic acids such as SAHA and PCI-34051; Benzamides such as MS275, and; cyclic peptides and depsipeptides such as apicidin and FK228, respectively. They all share a common pharmacophore pattern consisting of: (i) a metal binding domain which complexes Zn, (ii) a linker domain which mimics the substrate and occupies the enzymatic channel, (iii) a connecting unit, (iv) a surface domain, which makes contact with the rim. Although hydroxamic acids such as SAHA were widely considered to be non-selective inhibitors of class I and II enzymes, more recent work from some of us29 demonstrated that problems with the widely used assay require a re- assessment of this assumption. In particular, HDAC8 was found to have a much lower affinity to hydroxamic acids than previously reported.30 In comparison, MS275 is a class I selective inhibitor which blocks the activities of HDAC1,2 and less efficiently HDAC3,31 with no inhibition of HDAC8 or HDAC6. Following this approach, selective inhibitors of HDAC1 and HDAC2 have been developed from rational modifications of the benzamide moiety.32, 33 HDAC8 selectivity has been also recently achieved34, 35 and Class IIa selective inhibitors have been generated,36 marking the onset of the feasible dissection of the various activities of HDACs with chemical biology tool compounds. The rationalization of the structural origin of these experimentally observed selectivity is therefore an attractive starting point for the refinement of more potent isoform selective inhibitors, a widely accepted goal in the area of HDAC inhibitors.37, 38
On the basis of sequence homology, HDAC8 is considered to be a class I enzyme, although the phylogenetic analysis has shown it to lay near the boundary of the class I and class II enzymes.28 Its importance has been revealed by knockdown experiments of selective HDAC isoforms showing it as essential for cell survival. The three dimensional crystal structure of human HDAC8 was the first to be solved, and 14 human HDAC8 structures co-crystallized with different inhibitors are presently available (pdb codes 1T64, 1T67, 1T69, 1VKG, 2W22, 2V5W, 2V5X, 3EW8, 3EWF, 3EZF, 3EZT, 3F06, 3F07, 3FOR).7, 9, 39, 40 These structures have helped to understand how catalysis occurs within the HDAC family of enzymes, revealing unique features of HDAC8 as its conformational flexibility proximal to the binding site pocket, mediated by the L1 and L2 active site loops. However, the range of structural features observed for the same protein as a function of the co-crystallized inhibitor also poses a number of questions.
In the HDAC8 structures co-crystallized with different inhibitors, the active site topology shows large structural differences depending on which inhibitor is bound. These changes derive from the presence of two deep pockets one leading to the active site and a second one close to the active site and lined by Tyr306 and Phe152.7 The opening of the second pocket is mediated by a movement of loop L1. It is open in the structure 1T64, and closed but well defined in the bulk of the protein in 1W22, 2V5W and even in the SAHA-co-crystallized structure1T69. A large mobility is also observed for the loop comprising residues 85-105, which has no resolution in 1T67 and 1VKG.7 Figure 1 show lateral views of surface representations of 1W22, 1VKG, 1T64 and 1T69. All of them feature internal cavities that can be superimposed with the second pocket in 1T64, with Trp141 defining the bottom of the cavity.
Figure 1.
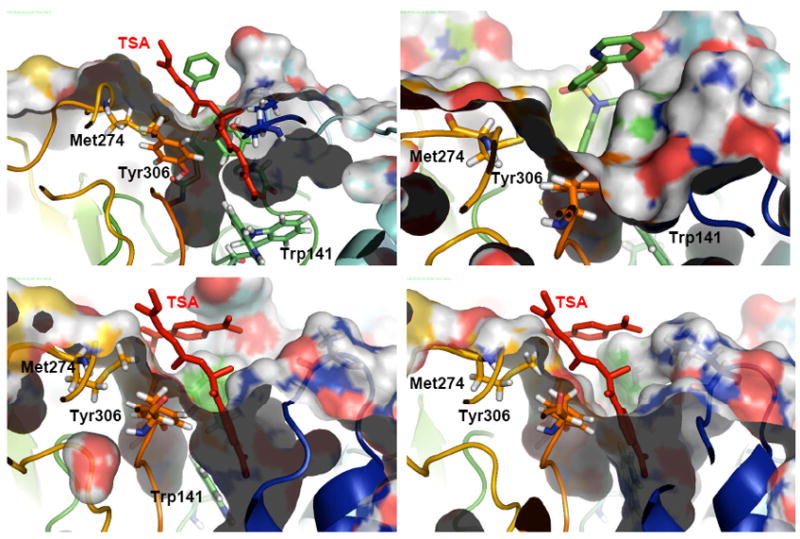
Lateral view of surface representations of 1T69 (top, left), 1W22 (top, right), 1T64 (bottom, left) and 1VKG (bottom, right). The position occupied by the second TSA molecule (shown in red) in the second pocket is highlighted for 1T69, 1T64, 1VKG.
The differences in the experimentally observed structures present an interesting conundrum for the understanding and the design of HDAC8 selective inhibitors. Despite the wealth of information, it is hard to understand the origin of these differences and to guess which conformation is the relevant one. Several questions need to be answered to better understand HDAC8 : (i) How many pockets does the inhibitor see in the event of coordination, one pocket as in 1T67, two pockets as in 1T64, or one groove as in 1VKG? (ii) Are there low-energy pathways linking the different conformations of the protein? (iii) Does HDAC8 have one groove which becomes organized after inhibitor binding? (iv) Can the conformational flexibility be used to understand and design HDAC8- selective inhibitors? Using a detailed computational analysis of the interactions of several inhibitors (Chart 1) with different available HDAC8 crystal structures selected for their different surface topology, namely 1T67, 1T64, and 1VKG, we here address these questions. After discussing the results organized by overall surface topology of the protein, we show that molecular dynamics (MD) results provide detailed insights in the origin of the weak inhibition of HDAC8 by many inhibitors and provide structural principles for the design of HDAC8 selective inhibitors.
Chart 1.
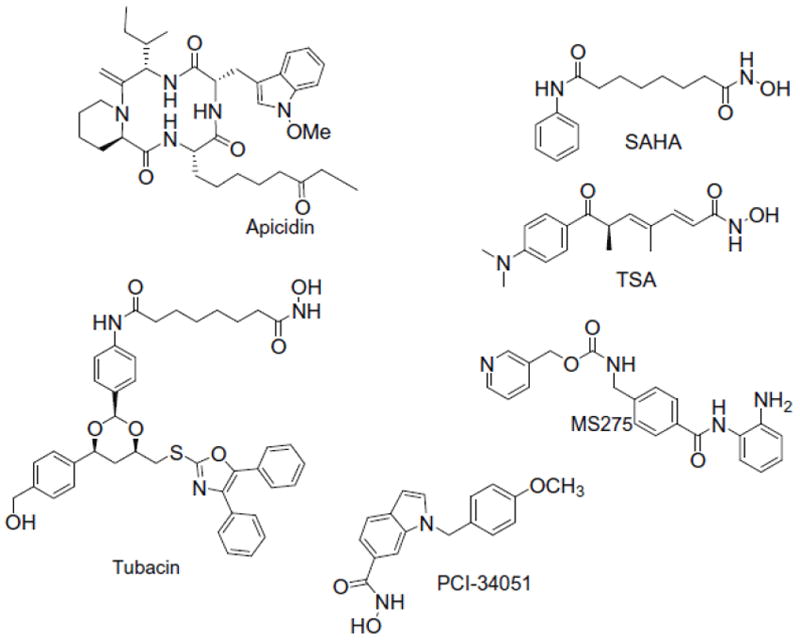
Structures of the selected HDAC inhibitors
Methodology
Molecular dynamics simulations
Initial coordinates for the protein atoms were taken from the crystallographic structures of human HDAC8 (pdb codes 1T67, 1T64). The ionizable residues were set to their normal ionization states at pH 7, except for the His residue that is hydrogen bonded to the anionic hydroxamate ligand, which has been modeled as positively charged. The protein atoms and water molecules resolved in the crystal structure were surrounded by a periodic box of TIP3P41 water molecules that extended 10 Å from the protein. Na+ counterions were placed by LEaP42 to neutralize the system.
The ff03 version43 of the all-atom AMBER force field was used, together with GAFF,44 to model the system. Atom-centered partial charges were derived by using the AMBER antechamber program (RESP methodology),45-47 after geometry optimization at the B3LYP/6-31G* level.48 Solvent molecules and counterions were initially relaxed by means of energy minimizations. The full system was then minimized to remove bad contacts in the initial geometry. All MD simulations were carried out using the pmemd version included in the AMBER10 suite of programs.49 The time step was chosen to be 2 fs, and SHAKE50 was used to constrain all bonds involving hydrogen atoms. A nonbonded cutoff of 10.0 Å was used, and the nonbonded pair list was updated every 25 steps. Langevin dynamics was used to control the temperature (300K) using a collision frequency of 1.0 ps-1, with isotropic position scaling to maintain the pressure (1 atm).51 Periodic boundary conditions were applied to simulate a continuous system. To include the contributions of long-range interactions, the Particle-Mesh-Ewald (PME)52, 53 method was used with a grid spacing of ~1 Å combined with a fourth-order B-spline interpolation to compute the potential and forces in between grid points. The trajectories were analyzed using the PTRAJ module of AMBER.
Docking
The crystal structures of human HDAC8 complexed with hydroxamic inhibitors (pdb codes 1T67, 1T64, 1VKG) were employed in the docking calculations, using Glide 4.5 XP.54 Solvent and ligands were removed and Macromodel atom types were assigned. Before the docking calculations, the ligands were submitted to a Macromodel conformational search55 saving 10 conformations for each one. The protein and the ligands were further prepared using the protein preparation and ligprep modules of the Schrodinger 2007 program package. The results of the docking were evaluated using GlideScore, saving the ten best poses for each run.
HDAC8 assays
All experimental assays were performed as outlined in Ref.29 except where noted.
Results and Discussion
Table 1 compares the results for the inhibition constants (Ki, nM) of several established, widely studied HDAC inhibitors, measured against different HDAC isoforms. It can be seen that contrary to the widely held belief that many of these inhibitors do not show isoform selectivity,30 inhibition of HDAC8 is in fact much diminished compared to other class I as well as class II HDACs. Only the “linkerless” inhibitors like PCI-34051 have developed as HDAC8 selective, showing a promising therapeutic modality for the treatment of leukemia and several autoimmune disorders.34, 56 However, the structural origin of these observations is not clear.
Table 1.
Inhibition of HDAC isoforms by selected HDAC inhibitors (IC50, nM)29
| Compound | HDAC1 | HDAC2 | HDAC3 | HDAC6 | HDAC8 |
|---|---|---|---|---|---|
| SAHA | 11 | 36 | 31 | 26 | 2000 |
| TSA | 0.15 | 1 | 0.6 | 13 | 300 |
| Tubacin | 240 | 260 | 630 | 13 | 5000 |
| MS-275 | 25 | 15 | 120 | 13000 | >40000 |
| Apicidin | 0.7 | 1 | 0.6 | 9600 | 300 |
| PCI-34051* | 4000 | >50000 | >50000 | 2900 | 10 |
From reference 33
The HDAC1-3 selectivity of benzamides such as MS275 has been rationalized by differences in the bottom of the active site channel of HDAC8 and HDAC1, the latter exhibiting a lateral cavity that is thought to be involved in product release.14 In HDAC8, a non-conserved residue (Trp141) was proposed to block the connection to any internal cavity, precluding the binding of bulky binders as those characterizing HDAC1 selective inhibition.3, 5 The results in Table 1 show that the weak inhibition of HDAC8 is much more widespread and cannot be explained using this rationalization. Not only is Trp141 more flexible than thought previously, as will be discussed in more detail later, it also does not explain the weak HDAC inhibition by small hydroxamates such as SAHA, nor the HDAC8 activity of PCI-34051. Using the small molecules as probes, a comparative analysis of the interactions of representative compounds of Table 1 (SAHA, Tubacin, MS275, TSA, apicidin, PCI-34051) with HDAC8 structures showing different topologies (such as observed in 1T67, 1T64, and 1VKG), offers an explanation for the different HDAC8 inhibition of this compounds and provides a rational basis for the design of HDAC8 selective inhibitors.
One pocket - 1T67
HDAC8 co-crystallized with MS-344 (pdb code 1T67)7 features a narrow pocket that is well defined and terminated at the bottom by Trp141. The position of this residue, which is not conserved but replaced by a leucine in HDAC1, impairs the coordination of bulky metal binding groups like the benzamide in MS-275. Figure 2 left shows the best docking pose that demonstrates the effect of this steric interference in a rigid protein. The ligand is coordinated by the pyridine end rather than the benzamide moiety, i.e. in an upside down orientation relative to the one found for HDAC1. In Figure 2 right, the pose obtained from the docking in HDAC1 is overlapped with HDAC8, showing how Trp141 impedes MS275 coordination. It is, however, clear that Trp141 is flexible as is for example observed in the related X-ray structure 1T69, where Trp141 is shifted away from the limiting region between the two pockets, opening a cavity similar to the one observed in HDAC1. The “out” conformation of Trp141 has been also found in the MS-344 co-crystallized 3EW8 and 3F06 structures, where the change in the conformation was thought as related to the 101L and 101N mutations.40 In both cases the lateral cavity is filled by a solvent molecule.
Figure 2.
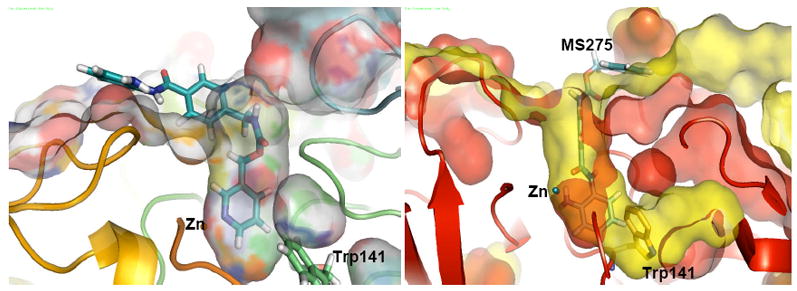
left: Surface representation of a lateral view of the Glide-XP docking of MS275 in 1T67. right: Superposition of surface representations of the active site channel of HDAC8 (red surface) and HDAC1 (yellow surface) with position of Trp141 is highlighted for HDAC8. MS275 is shown docked in the active site of human HDAC1 homology model.
Hydroxamates are very potent bidentate zinc chelators. The hydroxamate moiety is small enough to fit in the narrow pocket of HDAC8, which is supported by the literature57 and in the co-crystallized structures (pdb codes 1T64, 1T67, 1T69).5, 7, 9, 58 In these structures, the coordination with the zinc ion is reinforced by hydrogen-bond interactions with His142, His143 and Tyr306 as well as lipophilic contacts with residues Phe152 and Phe205.59 The same interactions are observed in the structures derived from the Glide-XP docking (Figure 3), and remain stable during long MD runs, increasing our confidence in the computations. These findings indicate that the approximately 102 difference in inhibition between HDAC1 and HDAC8 by SAHA or TSA must have a different reason.
Figure 3.
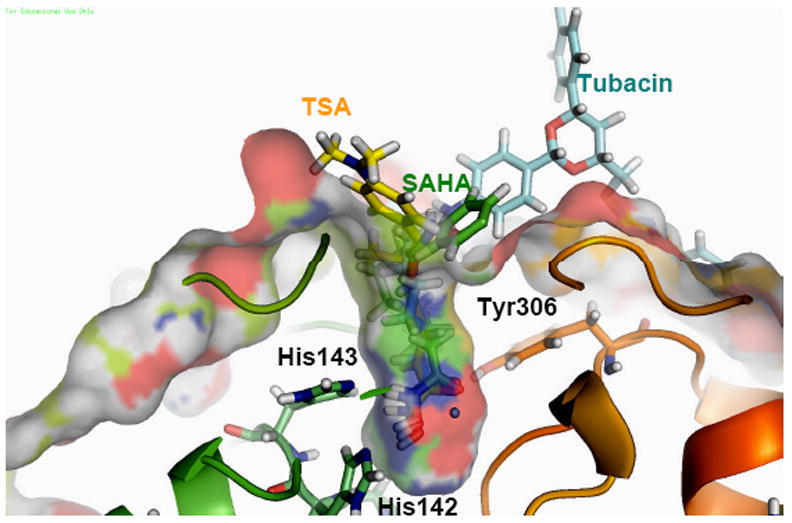
Docked poses of SAHA (green), TSA (yellow) and Tubacin (blue) in the active site of human HDAC8 (1T67)
Based on the argument above, it is unlikely that the origin of the experimentally observed selectivities can be found in the metal binding unit. We therefore hypothesized that, in analogy to the finding in the case of the HDAC6 selective inhibitor tubacin,37 selectivity might be determined by the size and shape of the cap group, prompting us to study both small and large cap inhibitors of HDAC8. For a detailed investigation of small and large-cap hydroxamate ligands, we studied the HDAC8-SAHA and HDAC8-tubacin complexes using 30ns and 20ns MD simulations, respectively (rms deviations are represented in Figure S1, Supporting Information). The results of the simulations of HDAC6 have already been discussed in detail37 and only the most relevant features for isoform selectivity are discussed here. During the 30ns MD simulations, the phenyl ring in the cap of SAHA adopts two preferred orientations, shown in yellow and green in Fig. 4 left, which are determined by hydrophobic interactions with Phe207, Phe208, and Tyr306, Phe152 respectively. More importantly, the second orientation opens a channel in the surface leaving Tyr306 solvent exposed (orange shadow in Figure 4 right), with the phenyl end of SAHA parallel to Phe152 and in a T-shape orientation relative to Tyr306. Non-bonded interactions of the SAHA phenyl cap group with Met274 and Lys21 further stabilize this binding mode. These orientations provide the structural rationalization of the results of a recent binding ensemble profiling study using photoaffinity labeling (BEProFL).60
Figure 4.
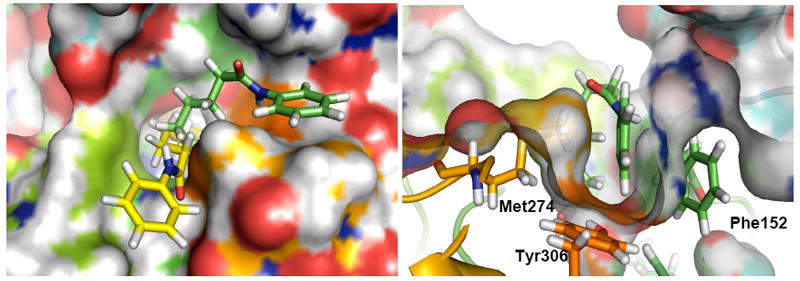
left: Top view of a surface representation of the most favorable orientations of the cap of SAHA in 1T67 resulting from interactions of the phenyl terminus with Phe205 (yellow) and Tyr306 (green). Right: surface representation of the surface channel opened to allow the interaction of SAHA with Tyr306.
The preferred coordination of tubacin to the structure 1T67 shows the alkyl linker accommodated in a similar channel as the one for SAHA shown in Fig.4 right, lined by Tyr306, Phe152 and Met274 (see Fig. S2, Supporting Information). In addition, the large cap region of tubacin can interact with different areas of the protein surface, and even open a second cavity in the course of the MD that accommodates part of tubacin’s diphenyl oxazole moiety (Fig.5) and perfectly overlaps with the one filled by TSA in 1T64 (shown for comparison purposes in white in Fig 5).
Figure 5.
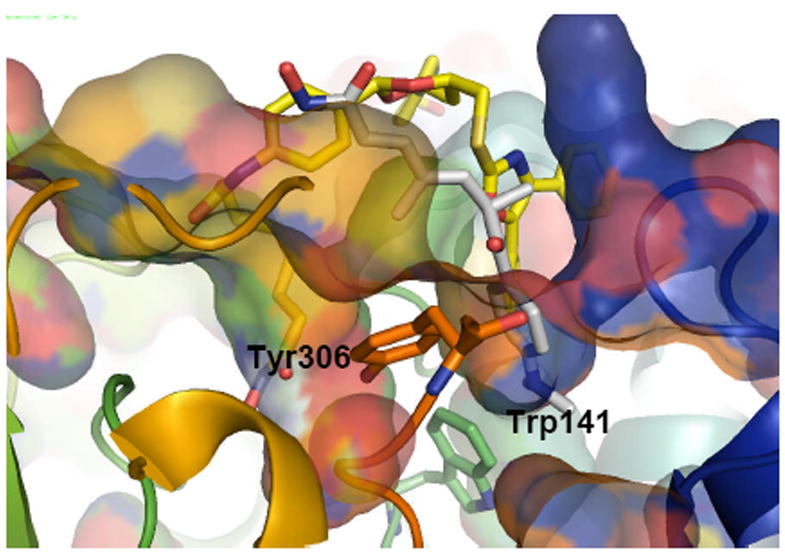
Top and side view of the coordination attained by tubacin in 1T67 starting from a configuration of the ligand bent towards the surface. Tubacin is shown in yellow. For comparison, TSA is shown in white in the position that it occupies in the second pocket of 1T64.
Cyclic tetrapeptides such as FK-228 and apicidin were experimentally found to show the same pattern of lower efficacy in HDAC8 than in HDAC1 inhibition, as shown in Table 1, but are still among the most potent HDAC8 inhibitors available. In the context of the hypothesis presented here, this is particularly interesting because the apicidin relies, in the absence of a strong metal binding group, predominately on surface interactions for inhibition. We thus focused on apicidin as representative of this class of HDAC inhibitors, analyzing its binding characteristics over 25 ns MD simulations. The molecule features a tetrapeptide macrocycle bound to a linear (S)-2-amino-8-oxodecanyl side chain that perfectly fits in the narrow pocket of HDAC8. The best pose from the docking in the 1T67 crystal structure positions the carbonyl oxygen at 2.37 A of the Zn atom, with the hydroxyl group of Tyr306 hydrogen bonded it. During the MD the protein evolves to better fit to the bonded ligand, leading to the structure shown in Figure 6. The N-methoxyindole moiety of the macrocycle is positioned above Tyr306 and the indole group is lined by Met 274 and Lys21. This orientation is showing the cap region of the ligand lying in a surface channel similar to the one shown for SAHA in Fig.4. (see Figure S3, Supporting Information). In summary, the interactions previously described for tubacin, SAHA and apicidin with the HDAC8 surface all show a well defined preferred orientation of the ligand, where the cap sits in a channel delimited by Tyr306 in the bottom. A superposition of the different ligands with the 1T67 crystallographic structure is shown in the Supporting Information (Figure S4).
Figure 6.
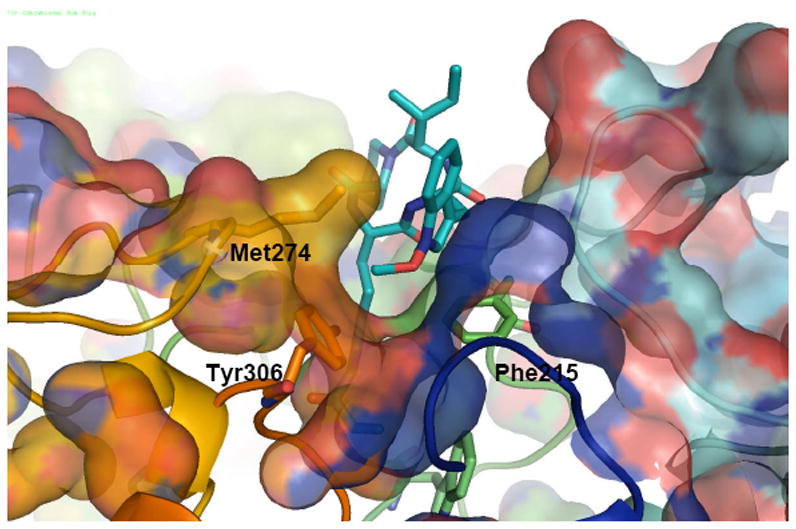
Snapshot after 20 ns of MD simulation of apicidin - HDAC8 complex
Two pockets-1T64
The crystal structure of HDAC8 co-crystallized with TSA (pdb code 1T64) shown a fundamentally different topology where both surface pockets are filled (with one ligand each. Interestingly, this leads to a structure similar to the one described for the bent conformation of tubacin shown in Figure 5, suggesting pathways between that conformations that are low enough in energy to be sampled by MD simulations on the 20-30 ns timescale.
TSA is a hydroxamate-type inhibitor with an unsaturated linker and the only one that has been experimentally observed as binding to the second surface cavity. Having the necessary computational tools at hand, we analyzed the probability of similar inhibitors of blocking both channels, provided the second one is open in the native protein or the ligand induces its opening.
We focused on the binding of TSA, for which experimental geometries are available, and analyzed the possible coordination patterns by means of 20ns MD simulations. The structure with two cavities filled, as shown in the X-ray data, is stable over 20ns MD simulations (see Figs S5, S6 Supporting Information). The ligand in the main pocket is bent over the second one and stabilized by π staking interactions of the cap with Tyr100. The separation between both pockets is delimited by Tyr306, Trp141 and Phe152.
The effect of the binding of TSA to either of the two sites can be investigated by pair-wise deletion. Deletion of the TSA ligand bound to the secondary cavity does not lead to a closing of the pocket. Conversely, it is filled by the phenyl ring of TSA’s cap that lies between Tyr306 and Phe152, lined by Trp141 in the bottom (Figure 7). The binding resembles the one that results from the docking, but with the surface further relaxed to accommodate the phenyl ring of TSA cap. This relaxation pushes away Phe152 from Tyr306 through the tilting of the latter, which was originally oriented towards the hydroxamate binder. In this binding conformation the ligand goes deep into the second pocket and may compete with the X-ray found in 1T64 to justify the photolabelling of residues Asn136, Trp137 and Ser139 detected by BEProFL.60
Figure 7.
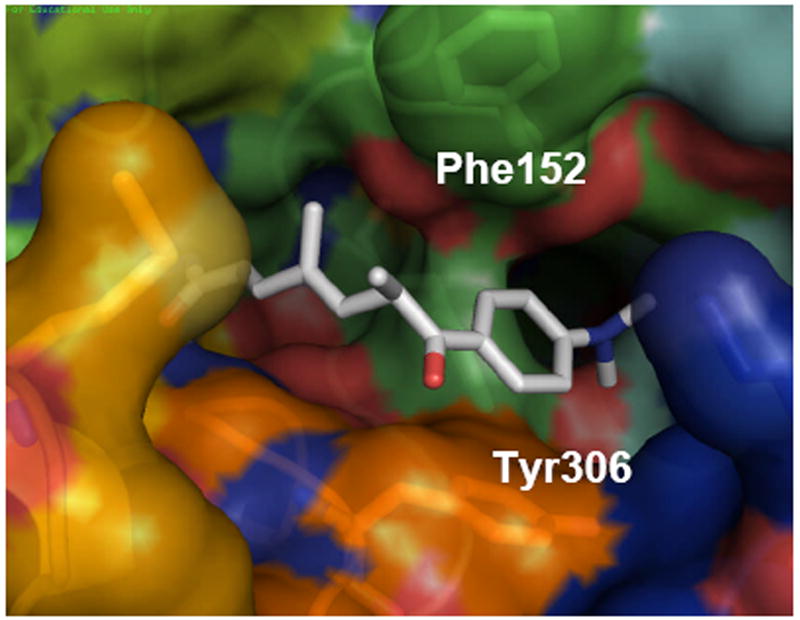
Top view of 20 ns MD snapshot of the HDAC8-TSA structure with no ligand in the secondary cavity.
Deletion of the ligand in the main cavity leads to an even more interesting result. During a simulation time of 20ns, the protein structure alternates between two and one pocket topologies shown in Figure 8. In this case, the protein conformation with two distinct pockets (shown in red in Fig. 8) leads to a topology with one large groove similar to the one observed in the co-crystal structure of HDAC8 with CRAA discussed below. The merging of the pockets proceeds by tilting of Phe152 which, together with Tyr306 forms the wall that separates the active site pocket from the second one (the evolution of the Phe152-Tyr306 distance over the 20ns MD is shown in Figure S7 in the Supporting Information). Whereas the tilting of Phe152 and Tyr306 relative to each other determines the shape of the pockets in the solvent exposed region as shown in Figure 8, the shape at the bottom of the channel is formed by Trp141 where bifurcated and merged cavities alternate during the course of the MD simulations. The fact that opening and closing of these two states are observed during the 20 ns simulation time indicates that these two conformations are in rapid equilibrium and suggest that different inhibitors can bind to the conformation most complementary to their shape.
Figure 8.
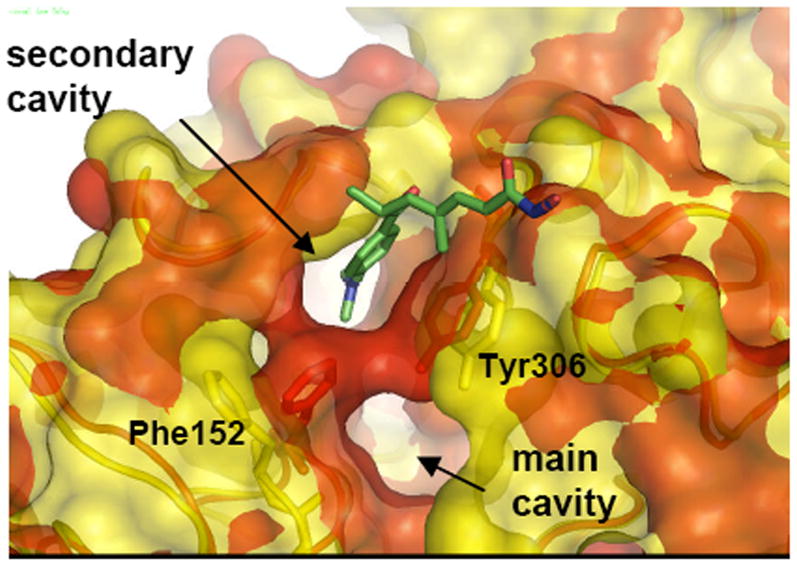
Surface representation of a superposition of two snapshots at 10 ns (red) and 20 ns (yellow) of 1T64 with main pocket TSA ligand deleted.
One groove – 1VKG
The structure of HDAC8 co-crystallized with CRAA (pdb code 1VKG) has been described as having a deep binding groove immediately adjacent to the acetyl-lysine binding site. As previously discussed, this “superpocket” can be created by the movement of Phe152, which can flip and lie parallel to Tyr306 at about 7A distance, defining together with the latter the lips of the wide cavity. Phe152 plays therefore a major role in shutting the gate to build the narrow channel (experimentally seen in 1T67, 1T69, 1W22. 2V5W) occupied by the four methylene groups of the acetylated lysine. The resulting channel displays a parallel arrangement of the aromatic side chains of Phe152 and Phe208, which has been given some importance in the stabilization of the linker portion of inhibitors. In this conformation, Phe152 also defines the wall between two cavities in 1T64. The coordination of a single TSA ligand in 1T64 triggers a tilting of Tyr306 when bending to fill the secondary cavity, having a similar effect as the tilting of Phe152, as both residues lie parallel defining the walls of a larger cavity. Nevertheless, both pockets are kept in the latter, well separated by Trp 141.
The malleability of the HDAC8 surface and the possibility of opening a lateral subpocket has been recently targeted to design HDAC8 selective inhibitors.34, 35 The most promising scaffold seems to be associated with bulky linkers, which can fit into the HDAC8 groove. A highly selective HDAC8 inhibitor has been reported (PCI-34051), 34, 35 which shows more than 200-fold selectivity over other HDAC isoforms, and is 10 times more active than apicidin in HDAC8. The HDAC8-PCI structure stabilized after 30 ns MD starting from the two-pocket 1T64 shows a similar tilting of Tyr306 as described for TSA. The phenyl ring of PCI’s cap fits into the channel thus generated, and is further stabilized by non-bonded interactions with Met274. The aromatic linker fills the main pocket lined by Phe152 and Phe208, almost parallel to each another and in T-shape interactions with the PCI’s linker. (Figure 9). This binding mode is similar to the one described for TSA. However, TSA can fit one molecule in each cavity to gain further stabilization, as demonstrated by the 1T64 X-ray structure, whereas PCI has a unique binding motif that simultaneously bind to both cavities.
Figure 9.
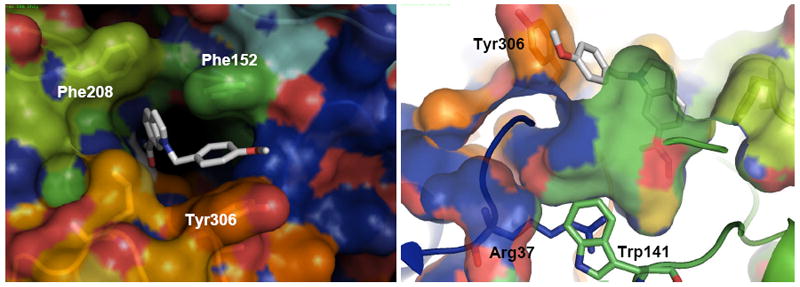
Top (a) and lateral (b) views of a 30 nd MD snapshot of the HDAC8-PCI structures. PCI-34051 is shown as white sticks.
Effects of surface malleability on substrate binding and inhibition in HDAC8
From both the topological differences observed in the available HDAC8 crystallographic structures, the photoaffinity labeling60 and from the results of the MD simulations, a remarkable malleability of the protein surface in HDAC8 is observed. Based on the MD simulations of homology models of other isoforms, there does not appear to be a counterpart in the other HDAC isoforms.10, 37 The simulations point to a dynamic equilibrium between structures with one or two pockets and with a wide-open groove in the protein surface that easily interconvert by movement of Tyr306 and Phe152 as shown in Fig. 8. The fact that most of the crystallographic structures with coordinated hydroxamates feature a single narrow pocket through which the alkyl linker directs the binder for Zn coordination suggests that a great deal of stabilization is attained through a ligand induced reorganization of the protein. Moreover, inspection of the internal protein topology of the different deposited structures shows a distinctive characteristic shape right in the position occupied by the second cavity in 1T64, shown in Figure 1.
Both the docking and the molecular dynamics simulations agree in describing interactions where the ligand and the protein accommodate to each other. In this way, the docking poses show an inhibitor filling both pockets when available, whereas the simulations show the opening of a lateral cavity. The quantification of this effect (Table 2) is should be treated with some caution because of the dependence of the docking score on the HDAC8 surface topology, which is treated as a rigid body. However, the scores clearly reflect the trend of increasing activity when maximizing surface interactions.
Table 2.
Glide XP Scores for selected inhibitors
| 1T67 | 1T64- | |
|---|---|---|
| Apicidin | -8.2 | -9.67 |
| Saha | -7.3 | -9.5 |
| Tubacin | -8.6 | -15.5 |
| TSA | -8.92 | -9.02 |
| PCI-34051 | - | -11.7* |
| MS-275 | -8.87** | -8.87** |
docked in 1VKG,
upside down (see Figure 2)
The results of these calculations strongly suggest answers to the questions posed in the introduction: (i) The surface topology that the inhibitor interacts with in the event of coordination can be described as an ensemble of structures in dynamic equilibrium, featuring one, two, or a single wide cavity. (ii) Different conformations are observed in the time frame of the molecular dynamics simulations, indicating that the protein can evolve among them at a low energy cost. (iii) Whereas the same key residues are involved in the stabilization of different ligands co-crystallized with HDAC8, their different structural characteristics lead to different surface topologies, originated in different orientations of these residues (Phe152, Tyr306), which become organized after inhibitor binding. As a result of this, binding to one of these rapidly interconverting protein conformations will be determined by the details of not only the metal binding and the cap groups, but also the previously underappreciated linker region59 that can interact either with the opened or closed wall formed by Phe152 and Tyr306. (iv) The design of a selective inhibitor has to take advantage of the differences between HDAC8 and the other isoforms, which is hampered by the lack of co-crystallized structures for the other class I HDACs. Nevertheless, some differences can be inferred from the MD analysis of the human HDAC1 homolog-PCI system. The evaluation of 30 ns HDAC1-PCI MD simulations shows that the active site channel is fairly stable in the timeframe of the dynamics, and no groove opens in the surface. Tyr306 does not tilt but rather delimitates the mouth of the pocket from below and the internal lateral channel already described for HDAC1/2/3 and HDLP from above. 1, 10, 14 (Figure 10). It is interesting to notice a Arg37-Tyr306 cation-π interaction61, 62 that is stable over the 30 ns simulation, and can help to keep the orientation of the Tyr residue and thus prevents an opening of the groove. Arg37 is conserved in HDAC8, but in this case the interaction can alternate between Tyr306 and Trp141 (Figure 9), releasing the orientation of Tyr306 and leading to the observed change of the cavity shape.
Figure 10.
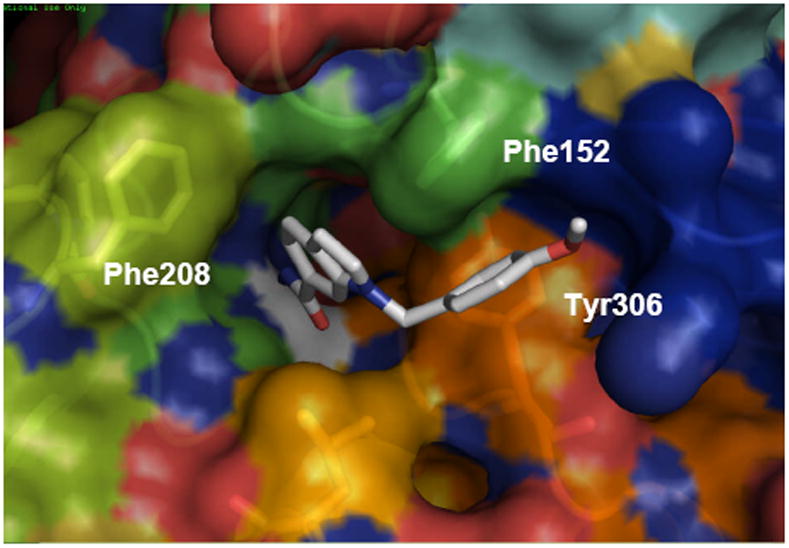
Top view of a 30 nd MD snapshot of the HDAC1-PCI structures (residue numbering as in HDAC8). PCI-34051 is shown as white sticks.
HDAC8 inhibition is strongly regulated by the surface malleability. The topology of the unbound state very likely offers a wide pocket that accommodates to the ligand after its coordination to Zn. In analogy to the inhibition by PCI-34051, an HDAC8 selective inhibitor has to be able to trap this state out of the ensemble of configurations in equilibrium that are displayed in the MD simulations. Binding to the narrow, single cavity state, as in the case of small hydroxamates such as SAHA, leads to weaker inhibition of HDAC8 because the malleability of the protein will remove several of the hydrophobic contacts in the linker region that contribute to binding.59 The structure of the HDAC8 selective inhibitors recently reported agrees with this model, as they display lateral bulky substituents capable of simultaneously blocking the active channel and the lateral superpocket. Specifically, the short linker regions in HDAC8 selective inhibitors such as CRA19156 or PCI34051 will bind selectively to the open-groove conformation that is unique to HDAC8, positioning the metal binding moiety in the active site and an aromatic group in the position of the secondary pocket, connected by a short alkyl or aryl linker.
Supplementary Material
Acknowledgments
We thank the Walther Cancer Institute (G. E. and O.W.), the Multiple Myeloma Research Foundation (J.E.B. and E.G) and the National Cancer Institute Initiative for Chemical Genetics (R.M.) for support of this research. The computations were performed on Kraken (a Cray XT5) at the National Institute for Computational Sciences (http://www.nics.tennessee.edu/) and in Queen Bee at the Louisiana Optical Network Initiative. Generous allocation of computing resources by the Center for Research Computing at the University of Notre Dame is also acknowledged.
Footnotes
Supporting Information Available. Rmsd plots and additional snapshots of the MD simulations (pdf format) are available free of charge via the Internet. Pymol sessions including the overlapped structures of the snapshots discussed in the article and additional structures of the HDAC-inhibitor complexes are available from the authors upon request.
References
- 1.Finin MS, Donigian JR, Cohen A, Richon V, Rifkind R, Marks P, Breslow R, Pavletich N. Nature. 1999;401:188. doi: 10.1038/43710. [DOI] [PubMed] [Google Scholar]
- 2.Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:4389. doi: 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hildmann C, Wegener D, Riester D, Hempel R, Schober A, Merana J, Giurato L, Guccione S, Nielsen TK, Ficner R, Schwienhorst A. Journal of Biotechnology. 2006;124:258. doi: 10.1016/j.jbiotec.2006.01.030. [DOI] [PubMed] [Google Scholar]
- 4.Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. Cell. 2003;115:727. doi: 10.1016/s0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- 5.Nielsen TK, Christian H, Dickmanns A, Schwienhorst A, Ficner R. J Mol Biol. 2005;354:107. doi: 10.1016/j.jmb.2005.09.065. [DOI] [PubMed] [Google Scholar]
- 6.Park JH, Jung Y, Kim T, Kim S, Jong H-S, Lee JW, Kim D-K, Lee J-S, Kim NK, Kim T-Y, Bang Y-J. Clin Cancer Res. 2004;10:5271. doi: 10.1158/1078-0432.CCR-03-0709. [DOI] [PubMed] [Google Scholar]
- 7.Somoza JR, Skene RJ, Katz BA, Moi C, Ho JD, Jennings AJ, Luong C, Arvai A, Buggy JJ, Chi E, Tang J, Sang BC, Verner E, Wynands R, Leahy EM, Dougan D, Snell G, Navre M, Knuth MW, Swanson RV, McRee D, Tari LW. Structure. 2004;12:1325. doi: 10.1016/j.str.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 8.Vannini A, Volpari C, Filocamo G, Casavola E, Brunetti M, Renzoni D, Chakravarty P, Paolini C, De Francesco R, Gallinari P, Steinkuhler C, Di Marco S. Proc Nat Acad Sci. 2004;101:15064. doi: 10.1073/pnas.0404603101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vannini A, Volpari C, Gallinari P, Jones P, Mattu M, Carfi A. EMBO. 2007;8:879. doi: 10.1038/sj.embor.7401047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang D-F, Helquist P, Wiech N, Wiest O. J Med Chem. 2005;48:6936. doi: 10.1021/jm0505011. [DOI] [PubMed] [Google Scholar]
- 11.Yang X-J, Gregoire S. Mol Cell Biol. 2005;25:2873. doi: 10.1128/MCB.25.8.2873-2884.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller TA, Witter D, Belvedere S. J Med Chem. 2003;46:5097. doi: 10.1021/jm0303094. [DOI] [PubMed] [Google Scholar]
- 13.Paris D, Porcelloni M, Binaschi M, Fattori D. J Med Chem. 2008;51:1505. doi: 10.1021/jm7011408. [DOI] [PubMed] [Google Scholar]
- 14.Wang D-F, Wiest O, Helquist P, Lan-Hargest H-Y, Wiech N. J Med Chem. 2004;47:3409. doi: 10.1021/jm0498497. [DOI] [PubMed] [Google Scholar]
- 15.Baylin SB, Ohm JE. Nature Rev Cancer. 2006;6:107. doi: 10.1038/nrc1799. [DOI] [PubMed] [Google Scholar]
- 16.Bhalla KN. J Clin Oncol. 2005;23:3971. doi: 10.1200/JCO.2005.16.600. [DOI] [PubMed] [Google Scholar]
- 17.Bolden J, Peart M, Johnstone R. Nature. 2006;5:769. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 18.Lindemann RK, Gabrielli B, Johnstone RW. Cell Cycle. 2004;3:779. [PubMed] [Google Scholar]
- 19.Lund AH, van Lohuizen M. Genes Dev. 2004;18:2315. doi: 10.1101/gad.1232504. [DOI] [PubMed] [Google Scholar]
- 20.Marks P, Jiang X. Cell Cycle. 2005;4:549. doi: 10.4161/cc.4.4.1564. [DOI] [PubMed] [Google Scholar]
- 21.Rosato RR, Grant S. Cancer Biol Ther. 2003;2:30. doi: 10.4161/cbt.190. [DOI] [PubMed] [Google Scholar]
- 22.Villar-Garea A, Esteller M. Int J Cancer. 2004;112:171. doi: 10.1002/ijc.20372. [DOI] [PubMed] [Google Scholar]
- 23.Mai A, Massa S, Rotili D, Cerbara I, Valente S, Pezzi R, Simeoni S, Ragno R. Medicinal Research Rev. 2005;25:261. doi: 10.1002/med.20024. [DOI] [PubMed] [Google Scholar]
- 24.Furumai R, Komatsu Y, Nishino N, Khochbin N, Yoshida M, Horinouchi S. Proc Nat Acad Sci. 2001;98:87. doi: 10.1073/pnas.011405598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Minucci S, Pelicci PG. Nat Rev Cancer. 2006;6:38. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 26.Moradei O, Maroun CR, Paquin I, Vaisburg A. Curr Med Chem-Anticancer Agents. 2005;5:529. doi: 10.2174/1568011054866946. [DOI] [PubMed] [Google Scholar]
- 27.Pandolfi PP. Cancer Chemother Pharmacol. 2001;48:S17. doi: 10.1007/s002800100322. [DOI] [PubMed] [Google Scholar]
- 28.Gregoretti L, Lee Y, Goodson HV. J Mol Biol. 2004:17. doi: 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 29.Bradner JE, L, W N, Grachan ML, Greenberg EF, Haggarty SJ, Warnow T, Mazitschek R. Nat Chem Biol. 2010;6:238. doi: 10.1038/nchembio.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kapustin GV, Fejer G, Gronlund JL, McCafferty DG, Seto E, Etzkorn FA. Org Lett. 2003;5:3053. doi: 10.1021/ol035056n. [DOI] [PubMed] [Google Scholar]
- 31.Inoue S, Mai A, D M, Cohen G. Cancer Res. 2006;66:6785. doi: 10.1158/0008-5472.CAN-05-4563. [DOI] [PubMed] [Google Scholar]
- 32.Methot J, Chakravarty P, Chenard M, Close J, Cruz J, Dahlberg W, Fleming J, Hamblett C, Hamill J, Harrington P, Harsch A, Heidebrecht R, Hughes B, Jung J, Kenific C, Kral A, Meinke P, Middleton R, Ozerova N, Sloman D, Stanton M, Szewczak A, Tyagarajan S, Witter D, Secrist P, Miller TA. Bioorg Med Chem Letters. 2008;18:973. doi: 10.1016/j.bmcl.2007.12.031. [DOI] [PubMed] [Google Scholar]
- 33.Witter D, Belvedere S, Chen L, Secrist J, Mosley R, Miller TA. Bioorg Med Chem Letters. 2007;17:4562. doi: 10.1016/j.bmcl.2007.05.091. [DOI] [PubMed] [Google Scholar]
- 34.Balasubramanian S, Ramos J, Luo W, Sirisawad M, Verner E, Buggy JJ. Leukemia. 2008;2008:1. doi: 10.1038/leu.2008.9. [DOI] [PubMed] [Google Scholar]
- 35.KrennHrubec K, Marshall B, Hedglin M, Verdin E, Ulrich SM. Bioorg Med Chem Letters. 2007;17:2874. doi: 10.1016/j.bmcl.2007.02.064. [DOI] [PubMed] [Google Scholar]
- 36.Mai A, Massa S, Pezzi R, Simeoni S, Rotili D, Nebbioso A, Scognamiglio A, Altucci L, Loidl P, Brosch G. J Med Chem. 2005;48:3344. doi: 10.1021/jm049002a. [DOI] [PubMed] [Google Scholar]
- 37.Estiu G, Greenberg E, Harrison CB, Kwiatkowski NP, Mazitschek R, Bradner JE, Wiest O. J Med Chem. 2008;51:2898. doi: 10.1021/jm7015254. [DOI] [PubMed] [Google Scholar]
- 38.Weerasinghe S, Estiu G, Wiest O, Pflum MKH. J Med Chem. 2008;51:5543. doi: 10.1021/jm800081j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vannini A, Volpari C, Filocamo G, Caroli Casavola E, Brunetti M, Renzoni D, Chakrabarti P, Paolini C, De Francesco R, Gallinari P, Steinkuhler C, Di Marco S. Proc Nat Acad Sci. 2004;101:15064. doi: 10.1073/pnas.0404603101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dowling DP, Gantt SL, Gattia SG, Fierke CA, Christianson DW. Biochemistry. 2008;47:13554. doi: 10.1021/bi801610c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jorgensen WLCJ, Madura J, Impey RW, Klein ML. J Chem Phys. 1983;79:926. [Google Scholar]
- 42.Schafmeister C, Ross WS, Romanovski V. University of California; San Francisco: 1995. [Google Scholar]
- 43.Cieplak P, Caldwell J, Kollman P. J Comput Chem. 2001;22:1048. [Google Scholar]
- 44.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Journal of Computational Chemistry. 2004;25:1157. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 45.Bayly CA, Cieplak P, Cornell WD, Kollman PA. J Phys Chem. 1993;97:10269. [Google Scholar]
- 46.Fox T, Kollman PA. J Phys Chem B. 1998;102:8070. [Google Scholar]
- 47.Pearlman DA, Case DA, Caldwell JW, Ross WS, Cheatham TE, Debolt S, Ferguson D, Seibel G, Kollman P. Comput Phys Commun. 1995;91:1. [Google Scholar]
- 48.Becke AD, Yarkony DR. In Modern Electronic Structure Theory Part II. Singapore: World Scientific; 1995. [Google Scholar]
- 49.Case DA, Darden TA, Cheatham ITE, Simmerling CL, Wang J, Duke RE, Luo R, Crowley M, Walker RC, Zhang W, Merz KMJ, Wang B, Hayik S, Roitberg A, Seabra G, Kolossvary I, Wong KF, Paesani F, Vanicek J, Wu X, Brozell S, Steinbrecher H, Gohlke H, Yang L, Tan C, Mongan J, Hornak V, Cui G, Mathews DH, Seetin MG, Sagui C, Babin V, Kollman PA. University of California; San Francisco, CA, USA: 2008. [Google Scholar]
- 50.Ryckaert JP, Ciccotti G, Berendsen HJC. J Comput Phys. 1977;23:327. [Google Scholar]
- 51.Pastor RW, Brooks BR, Szabo A. Mol Phys. 1988;65:1409. [Google Scholar]
- 52.Petersen HG. J Chem Phys. 1995;103:3668. [Google Scholar]
- 53.Essman V, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. J Chem Phys. 1995;103:8577. [Google Scholar]
- 54.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. J Med Chem. 2006;49:6177. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 55.Macromodel. Schödinger Inc.; Portland OR: [Google Scholar]
- 56.KrennHrubec K, Marshall BL, Hedglin M, Verdin E, Ulrich SM. Bioorg Med Chem Letters. 2007;17:2874. doi: 10.1016/j.bmcl.2007.02.064. [DOI] [PubMed] [Google Scholar]
- 57.Ortore G, Di Colo F, Martinelli A. J Chem Inf Model. 2009;49:2774. doi: 10.1021/ci900288e. [DOI] [PubMed] [Google Scholar]
- 58.Fininin MS, Donigian JR, Cohen A, Richon V, Rifkind R, Marks P, Breslow R, Pavletich N. Nature. 1999;401:188. doi: 10.1038/43710. [DOI] [PubMed] [Google Scholar]
- 59.Weerasinghe SVW, Estiu G, Wiest O, Pflum MK. J Med Chem. 2008;45:4429. doi: 10.1021/jm800081j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.He B, Velaparthi S, Pieffet G, Pennington C, Mahesh A, Holzie D, Brunsteiner M, von Breemen R, Blond S, Petukhov P. J Med Chem. 2009;52:7003. doi: 10.1021/jm9005077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gallivan JP, Dougherty D. Proc Nat Acad Sci. 1999;96:9459. doi: 10.1073/pnas.96.17.9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tatko C, Waters M. Protein Science. 2003;12:2443. doi: 10.1110/ps.03284003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.