

Abstract
Coenzyme Q (CoQ) is an essential component of the respiratory chain but also participates in other mitochondrial functions such as regulation of the transition pore and uncoupling proteins. Furthermore, this compound is a specific substrate for enzymes of the fatty acids β–oxidation pathway and pyrimidine nucleotide biosynthesis. Furthermore, CoQ is an antioxidant that acts in all cellular membranes and lipoproteins. A complex of at least ten nuclear (COQ) genes encoded proteins synthesizes CoQ but its regulation is unknown. Since 1989, a growing number of patients with multisystemic mitochondrial disorders and neuromuscular disorders showing deficiencies of CoQ have been identified. CoQ deficiency caused by muta-tion(s) in any of the COQ genes is designated primary deficiency. Other patients have displayed other genetic defects independent on the CoQ biosynthesis pathway, and are considered to have secondary deficiencies. This review updates the clinical and molecular aspects of both types of CoQ deficiencies and proposes new approaches to understanding their molecular bases.
Keywords: Mitochondria, Coenzyme Q deficiency, Neuromuscular diseases
8.1 Introduction
Coenzyme Q (CoQ) is an electron carrier in the mitochondrial respiratory chain (MRC) transferring electrons from complex I and II to complex III [13]. It also transfers protons to the mitochondrial inter-membrane space contributing to establish the membrane potential and then to the ATP biosynthesis. Furthermore, CoQ has a key role in the mitochondrial respiratory chain function through the stabilization of the complex III [53]. In addition, CoQ is an essential factor for uncoupling proteins activation [17] and in the opening of the permeability transition pore [19], and acts as antioxidant in the different cellular membranes and even in cholesterol transport lipoproteins [57]. The antioxidant function of CoQ directly prevents the progression of lipid peroxidation in membranes or by recycling other antioxidants such as vitamin E or ascorbate [26, 61]. CoQ of plasma membrane also regulates the ceramide-dependent apoptosis pathway by the non-competitive inhibition of the neutral sphingomyelinase activity [40]. Redox functions of CoQ are due to its ability to exchange two electrons in a redox cycle between the oxidized (ubiquinone) and the reduced (ubiquinol) forms. The redox reaction can be driven by one step of two electrons or by two steps of one electron through the semiquinone form (Fig. 8.1).
Fig. 8.1. Scheme of CoQ redox isoforms.
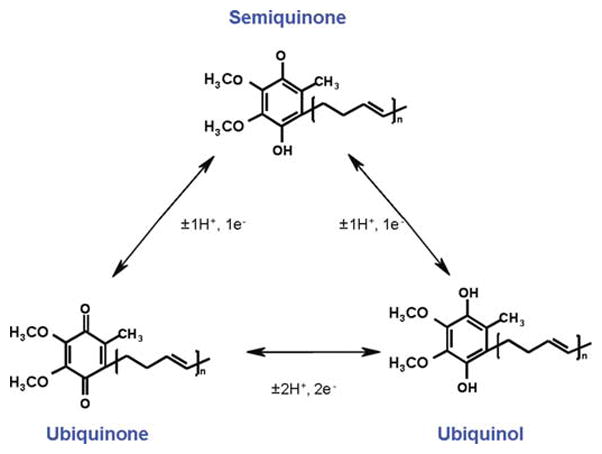
It includes the fully oxidized form ubiquinone, the fully reduced form ubiquinol, and the intermediate redox form semiubiquinone. The polyprenyl residue in carbon 2 contains isoprene units repeated several times (n). The number of isoprene residues n is specific in different organisms
CoQ is composed of a benzoquinone ring, synthesized from tyrosine through 4. hydroxybenzoate, and a polyprenyl side-chain, generated from acetyl-coA through the mevalonate pathway [57], which is also required for the synthesis of other compounds, such as cholesterol and dolichol-phosphate. The polyprenyl side-chain is built with different isoprenoid units, which is species specific. Human CoQ contains predominantly ten isoprenoid units and is therefore designated CoQ10.
8.2 CoQ Biosynthesis Pathway
The biosynthesis of CoQ is a very complex process (Fig. 8.2), which involves at least ten genes as shown in Table 8.1.
Fig. 8.2. A model for the biosynthesis pathway of CoQ in eukaryotic organisms.

. The tentative gene participating in the different steps is indicated. 5-demethoxy-CoQ (DMQ) is accumulated in yeast and C. elegans clk-1/coq-7 mutants. SAM=s-adenosyl-methionine; SAH=S-Adenosyl-homocysteine (modified from [43])
Table 8.1.
Homologue sequences of genes required for coenzyme Q biosynthesis in S. cerevisiae and H. sapiens. GeneBank(http://www.ncbi.nlm.nih.gov/Genbank) accession numbers for genes and proteins (in parenthesis) are listed
| Common name | Chromosome location | H. sapiens |
|---|---|---|
| coq-1(PDSS1) | 10p12.1 | NM_014317(NP_055132.1) |
| coq-1(PDSS2) | 6q21 | NM_020381.3(NP_065114.3) |
| coq-2(COQ2) | 4q21.3 | NM_015697(AAH20728.1) |
| coq-3(COQ3) | 6q16.3 | NM_017421(NP_059117) |
| coq-4(COQ5) | 9q34.13 | NM_016035(NP_057119.1) |
| coq-5(COQ5) | 12q24.31 | NM_032314(NP_115690.1) |
| coq-6(COQ6) | 14q24.1 | NM_182476, IsoA(NP_872282) |
| NM_182480, IsoB(NP_872286) | ||
| coq-7/clk-1(COQ7) | 16p13.11-p12.3 | NM_016138(NP_057222) |
| coq-8 (COQ8) (ABC1, ADCK3) | 1q42.13 | NM_020247(NP_064632.1) |
| coq-9(COQ9) | 16q13 | NM_020312.1(NP_064708.1) |
Most of the proteins of these genes have not been yet purified and, although the regulation of this biosynthesis pathway is largely unknown [56, 57], it is up-regulated under oxidative stress in rats [15]. The yeast coq-1 homologue encodes for a polyprenyl diphosphate synthase and is a branch-point enzyme of the mevalonate pathway [42].
Yeast coq-2 and human homologue, COQ2, encodes a polyprenyltransferase that catalyses the prenylation of the 4-hydroxybenzoic acid intermediate [6, 20]. The gene products of yeast and rat coq-3 homologues catalyze two O-methyltransferase steps in CoQ biosynthesis [44], essential for the final structure of quinone ring. Yeast coq-5 encodes for a C-methyltransferase although a direct demonstration of its kinetic function is not available [24], it is required for the stability of other coq-encoded proteins [7]. Protein encoded by yeast and nematode clk-1/coq-7 has been described as a membrane-bound hydroxylase of the benzoquinonic ring [25, 55], and it has been also demonstrated to participate in CoQ biosynthesis in mammal species such as mouse and human [59].
Yeast Coq-6 protein catalytic participation in CoQ biosynthesis is still unknown, but its sequence has a high homology with a large family of proteins that function as flavoprotein monoxygenases [22]. No catalytic function on CoQ biosynthesis has been shown for coq-4-encoded peptide although it has been demonstrated to be required for CoQ biosynthesis in eukaryotic species including humans [5, 9, 11]. It has been demonstrated that the gene coq-8/ABC1 participates in CoQ biosynthesis in S. cerevisiae and C. elegans [5, 16]. There is no current demonstration of the exact role of coq-8 in CoQ biosynthesis, but based on sequence similarity, coq-8 belongs to a protein kinase family harboring an ATP-binding cassette similar to those found in regulatory kinases [30]. C. elegans coq-8 knockout animals showed embryo development arrest that coincided with the triggering of expression of coq-8 in embryo, and these animals are the only coq knockout strain that maintain a basal CoQ biosynthesis level [4]. This gene encodes probably a regulatory protein of the CoQ biosynthesis complex through a phosphorylation mechanism.
The gene COQ10 encodes for a CoQ binding protein that is required for the activity of CoQ in respiration. COQ10 null mutants of both Saccharomyces cere-visiae and Schizosaccharomyces pombe have normal levels of CoQ but partially lack respiration [8, 14]. A human COQ10 ortholog is able to complement S. pombe null strain [14]. Although this gene is not required for CoQ biosynthesis, it must be considered in the study of mitochondrial disorders based on CoQ deficiency or dysfunction.
The CoQ biosynthetic intermediate 3-hexaprenyl-4-hydroxybenzoic acid (HHB) is the metabolite only accumulated in yeast coq null mutants suggesting that interactions among Coq proteins lead to the formation of a CoQ biosynthetic complex [24, 56]. These results also would indicate a regulation point at the Coq2 enzyme reaction [57]. Furthermore, mutations in coq7/clk-1 induced the accumulation of 2-hexaprenyl-3-methyl-6-methoxy-1,4-benzoquinone (demethoxy-Q, DMQ) in yeast and nematodes indicating a second regulation point of CoQ biosynthesis pathway. This regulation is independent on Coq7p and modulated by either CoQ or Coq8p in yeast [43]. As a consequence, we would speculate that a mutation in any of the coq genes but also in those unknown that regulate the CoQ biosynthesis pathway would induce a CoQ deficiency as a primary event.
8.3 Primary CoQ Deficiencies
8.3.1 Clinical and Biochemical Diagnosis
In 1989, Ogasahara et al. reported the first two siblings with a syndrome characterized by myoglobinuria, brain involvement, ragged-red-fibers and lipid storage. The patients had severe CoQ deficiency in muscle causing an impairment of the mitochondrial respiratory chain (MRC). Since then, more than 30 patients with deficiencies of CoQ have been reported and with diverse clinical phenotypes with proven or postulated autosomal recessive inheritance [45]. In addition to the original encephalomyopathic form, with myoglobinuria, epilepsy, mental retardation, and ataxia [41], other phenotypes include: a severe infantile multisystemic syndrome with prominent central nervous system involvement, including initially nystagmus, followed by bilateral visual loss, due to retinitis pigmentosa and optic nerve atrophy, sensorineural deafness, progressive ataxia, and a nephrotic syndrome causing terminal renal failure [50] and an ataxic form presenting with cerebellar ataxia and atrophy, nystagmus, seizures, absence of tendon reflexes, and in some cases, mental retardation [3, 28, 39]. This phenotype includes patients with early- or late-onset presentation and diagnosis and is the most frequent one associated with CoQ deficiency. Finally, other forms are expanding the clinical phenotype, since CoQ deficiency has been related with cases of neonatal presentation and fatal evolution, presenting poor feeding, hypothermia, hypotonia, renal tubulopathy, cardiac involvement, seizures with generalized cerebral and cerebellar atrophy [49], or failure of liver and pancreas and Leigh syndrome [31]. It has even been reported in adult patients with Leigh syndrome plus growth retardation, infantilism, ataxia, deafness and altered neuroimaging [60], or an adult form was diagnosed in a 48-year-old man who was completely normal up to 39 years of age and then presented late-onset ataxia with cerebellar atrophy and hypergonadotrophic hypogonadism [23]. Thus, considering this variety of clinical phenotypes, in any patient suspected of having a mitochondrial disorder CoQ deficiency should be considered, and therefore the next step in the investigation is the biochemical analysis of CoQ content in tissues. In all patients reported hitherto, metabolic investigations for the diagnosis of MRC defects, metabolite analysis in body fluids showed controversial results: some cases revealed increased blood or CSF lactate levels [10], while other patients did not show such metabolic alterations [23].
Therefore, peripheral markers of MRC disorders appear to have limited value in the initial diagnosis and selection of patients with CoQ deficiency syndrome.
The hallmark of CoQ deficiency syndrome is a decreased CoQ concentration in muscle and/or fibroblasts, which is usually measured by HPLC with electrochemical detection procedures. Patients showed a variable degree of CoQ deficiency in muscle and/or fibroblasts (with normal plasma CoQ concentration) causing decreased activities of NADH:cytochrome c oxidoreductase (complex I + CoQ + III) and suc-cinate:cytochrome c oxidoreductase (complex II + CoQ + III) of the MRC. The degree of the deficiency is very broad, ranging from extremely low CoQ concentration in muscle [10, 50, 54] to milder deficiencies [3, 39], or even normal results in fibroblasts. Another biochemical sign commonly observed was a dramatic recovery of MRC enzyme activities in muscle and/or fibroblasts (complex I + III and II + III) after the addition of CoQ analogues in vitro [50]. Other very helpful laboratory tool for CoQ deficiency diagnosis is the quantification of CoQ biosynthesis in fibroblasts by means of the measurement of the incorporation of radiolabeled 4-hydroxy[U-14C] benzoic acid to the quinone ring of CoQ, or the incorporation of radiolabeled 3H-mevalonate to the polyisoprenoid side chain [36, 47]. After incubation of fibroblasts, the CoQ synthesized is separated by reverse phase HPLC and quantified by radiometric detection.
8.3.2 Molecular Studies of CoQ Biosynthesis
At present, 10 genes have directly been associated in CoQ biosynthesis in humans.
All of the genes participate in the terminal part of CoQ biosynthesis, including decaprenyl diphosphate synthesis, the incorporation of this polyprenyl side chain to the benzoquinone ring, and both the condensation and modification of the quinone moiety to its final structure as CoQ (Fig. 8.1). Among these genes, at present primary CoQ deficiency has been linked to 4 of them (PDSS1, PDSS2, COQ2 and COQ8, also known as CABC1 or ADCK3). We will briefly review these different molecular defects:
8.3.2.1 Mutations in COQ2 Gene (MIM #609825)
Mutations in COQ2 gene were the first genetic defects in the CoQ biosynthesis pathway in humans [47]. COQ2 encodes for an enzyme called parahydroxybenzoate polyprenyl transferase, which catalyses the conjugation of the benzoquinone ring with the decaprenyl side chain. This paper describes a homozygous missense mutation (A→G transition in position 890) that changes a highly conserved tyrosine to cysteine at amino acid 297 in Coq2 peptide in 2 siblings presenting a severe CoQ deficiency (18% of mean control value in fibroblasts). This pathogenic mutation was associated with the infantile form of CoQ deficiency characterized by encephalopathy and nephropathy (one of the well-recognized clinical phenotypes of CoQ deficiency syndromes) [52]. Biochemically, both patients showed clear CoQ deficiency both in muscle and fibroblasts (more severe in the latter), affecting MRC enzymes dependent on CoQ (complexes I + III and II + III), and also the incorporation of both radiolabeled para-hydroxybenzoate and decaprenyl-PP into CoQ. Interestingly, further pathogenic mechanisms besides bioenergetics problems were demonstrated in these patients. Cultured fibroblasts from the patients revealed decreased ATP synthesis, increased production of reactive oxygen species and oxidative stress [48]. The decrease of CoQ biosynthesis impaired de novo pyrimidine nucleotide synthesis since ubiquinone is an essential cofactor for dihydro-orotate dehydrogenase, an enzyme located in the inner mitochondrial membrane [33]. Later, Mollet et al. [35] reported a new patient presenting pathogenic mutations in COQ2 gene associated with CoQ deficiency (24% of mean control value in fibroblasts) In this case, the clinical phenotype was more severe than the previous description since the patient presented a fatal infantile multiorgan disease. The mutation was a homozygous single base pair frameshift deletion resulting in a premature stop codon (c.1198delT, N401fsX415). Biochemical data and pathogenic mechanism investigations consistently demonstrated that these mutations were the cause of the oxidative phosphorylation defect in this new case.
8.3.2.2 Mutations in PDSS1 and PDSS2 Subunits of Prenyldiphosphate Synthase
Prenyldiphosphate synthase, one of the rate-limiting enzymes in CoQ biosynthesis, is an enzyme that catalyses the elongation of geranyl-geranyl diphophate to decaprenyl diphosphate prior to conjugation of this polyprenyl side chain to the benzoquinone ring. It is therefore an essential enzyme for CoQ biosynthesis in humans, which encodes the human ortholog of the yeast (Saccharomyces cere-visiae) COQ1 gene. Prenyldiphosphate synthase in humans is a heterotetramer composed by 2 copies of both subunit 1 and subunit 2 molecules encoded by different genes. Mutations in both subunits have been demonstrated as pathogenic in several patients.
In 2006, Lopez et al. [32] reported a single case with a severe infantile phenotype characterized by Leigh syndrome (a severe encephalopathy) with nephrotic syndrome associated with severe CoQ deficiency (13% of mean control value in fibrob-lasts) and MRC function impairment. Direct sequencing of eight genes involved in CoQ biosynthesis revealed the presence of 2 non-synonymous nucleotide changes in the PDSS2 gene; a heterozygous C→T transition at nucleotide 964, changing amino acid 322 from glutamine to a stop codon, and a heterozygous C→T transition at nucleotide 1145 changing amino acid 382 from serine to leucine in the seventh conserved domain of the enzyme. These changes (MIM #610564) were not found in DNA from 210 chromosomes from controls (analyzed by RFLP). Pathogenic effect of this mutation was further confirmed by a clear reduction of the incorporation of radiolabeled substrate 14C-PHB to CoQ in fibroblasts. Interestingly, the incorporation of other substrate to CoQ, 3H-decaprenyl-PP, was normal, indicating that the biosynthetic pathway after decaprenyl diphosphate synthase was intact.
In 2007, Mollet et al. reported a family (2 siblings) presenting a multisystem disease with early-onset deafness, encephaloneuropathy, obesity, livedo reticularis and valvulopathy associated with a profound CoQ deficiency (3.3% of the mean control values in fibroblasts), and thus, expanding the clinical phenotype of these syndromes. A genome-wide search for homozygosity identified several candidate chromosome regions, including 10p12.2-p12.1, which corresponds to the PDSS1 gene.
Direct sequencing of this gene revealed a homozygous T→G transversion at nucleotide 977, resulting in the change of a highly conserved aspartic acid into a glutamic acid (D308E). This mutation (MIM #607429) probably affects some of the catalytic properties of the PDSS1 encoded enzyme. Pathogenic effect of this mutation was further confirmed by MRC analysis and CoQ biosynthesis in either in muscle or fibroblasts, which was clearly impaired, and by functional complementation studies in yeasts.
8.3.2.3 Mutations in COQ8 Gene (MIM #606980)
COQ8 gene, also called ADCK3 or CABC1, function is still unknown in CoQ biosynthesis pathway, although it has been proposed that could act as a protein kinase, since several kinase motifs have been found in its amino acid sequence [30], and it has been demonstrated to have regulatory functions in C. elegans [4]. In 2008, 2 groups reported independently and simultaneously mutations in this gene associated with CoQ deficiency and an ataxic form with seizures.
In one report [34], 3 unrelated families (4 patients) were reported presenting pathogenic mutations in different parts of the COQ8 gene. Pathogenicity was consistently demonstrated by analysis of the amino acid residues conservation, by the absence of these mutations in 460 chromosomes in healthy controls, and by the study of functional complementation studies in yeasts. Furthermore, MRC function was impaired as well. CoQ values in muscle were clearly reduced in 3 patients, but this defect was not demonstrated in fibroblasts, because CoQ content was normal in 2 cases. These findings support the necessity of studying both muscle and fibroblast samples for CoQ deficiency syndrome diagnosis. Clinically, all these patients showed ataxia and cerebellar atrophy, with different degrees of affectation of other neurological functions (seizures) or extra-neurological signs, such as exercise intolerance.
In the second report, Lagier-Tourenne et al. [27] after a SNP-based genome widescan in a consanguineous family, found linkage of the ataxia to chromosome 1q41 that led to COQ8 gene sequencing analysis. After this initial analysis, 7 additional patients presenting with childhood-onset ataxia andcerebellar atrophy were found to harbor several types of mutations in both alleles of COQ8 gene. Pathogenicity of these mutations was demonstrated by different ways, including molecular studies of COQ8 gene, complementation analysis in yeasts, and MRC function analysis. Direct quantification of CoQ in fibroblasts revealed that also one case did not present a CoQ deficiency when compared with controls, while the other 3 patients studied presented a moderate deficiency. In the index case however, a more severe reduction in CoQ content in muscle biopsy was demonstrated. The authors proposed for this new disorder the name ARCA2 (autosomal-recessive cerebellar ataxia 2).
8.4 Secondary CoQ Deficiency in Mitochondrial Disorders
The first studies concerning CoQ deficiency and mitochondrial encephalomy-opathies were performed in Kearns-Sayre syndrome patients and other conditions caused by mutations in mitochondrial DNA (mtDNA) [18, 62]. Other nuclear and mitochondrial DNA defects has been described to be associated with CoQ defi-ciency, but no mechanism were described to be causing this deficiency. Some patients showing non-Friedreich ataxia showed a CoQ deficiency but it was not demonstrated any mutation in COQ gene sequences, indicating that either other COQ genes are unknown or it is a secondary defect [2]. In fact, some of the ataxic patients with CoQ deficiency harbor mutations in aprataxin that encodes for a DNA repair factor [29, 46].
Isolated myopathy has been also associated to CoQ deficiency but it is also a secondary event because it has been described a pathogenic mutation in the ETFDH gene [21]. This gene encodes an electron transport flavoprotein dehydrogenase that depends on CoQ as substrate and it is an essential step of β–oxidation of fatty acids. It has been also recently demonstrated a case of a child that presented a secondary CoQ deficiency associated to a depletion of mtDNA [37]. Finally, secondary CoQ deficiency has been reported in a patient with cardiofaciocutaneous syndrome due to a BRAF gene mutation [1].
In recent years, decreased plasma and tissue concentrations of CoQ have been associated with several diseases, such as cardiomyopathies, degenerative muscle and central nervous system diseases, cancer, phenylketonuria, and an impaired CoQ reduced status in plasma, free radical damage and lipid peroxidation in vascular diseases [58]. Thus, a significant impairment of the distribution of CoQ between plasma and blood cells has been demonstrated in fibromyalgia [12]. The mechanisms that may explain these deficiencies in this heterogeneous group of diseases is far to be understood but it could be explained by either a decreased endogenous CoQ biosynthesis or an increased catabolism, although the involvement of dietary CoQ supply can not be discarded.
The existence of the secondary CoQ deficiency indicates that CoQ would be a marker of mitochondria homeostasis and also that CoQ biosynthesis complex must have a connection to other complexes such as MRC or pathways inside mito-chondria. The highly conserved biosynthesis pathway and structure of CoQ could guarantee its function as central sensor of mitochondria homeostasis and would coordinate the equilibrium between bioenergetics and nucleotides available for cellular physiology.
8.5 Therapy of CoQ Deficient Patients
Primary CoQ deficiency is unique among mitochondrial disease (and among genetic diseases in general) because an effective therapy is available, at least for some for patients. It has been known since the first report by Ogasahara et al. [41] that CoQ deficient patients benefit from oral CoQ10 supplementation. Subsequent reports have clearly shown that high-dose oral CoQ supplementation can stop the progression of the encephalopathy in patients with the multisystemic form of the disease [50, 52]. Moreover in 2008 two reports have shown that oral CoQ supplementation is effective also on renal manifestations, both in a patient with COQ2 mutations [38], and in PDSS2 knockout mice [51]. However, it is clear from these reports that therapy must be instituted as early as possible to be effective, because only limited recovery is seen after development of significant tissue damage.
CoQ10 appears to be less effective in patients with the ataxic form of the disease caused by ADCK3 mutations. No effect was noted in the patients reported by Mollet et al. [34], while there were only mild improvements in the only patient treated by Lagier-Tourenne et al. [27]. It is not clear if the lack of significant clinical response is due to the fact that treatment was instituted too late in the course of the disease to be effective, or because ADCK3 has also other biological functions unrelated to CoQ biosynthesis. The optimal dosage and formulation of this supplement are also still debated. Doses ranging from 5 mg/kg/day [50] to 30 mg/kg/day [38] have been employed without significant side effects in patients, while a dose of 200 mg/kg/day was shown to be effective in mice [51]. To date, however, there are no studies that correlate CoQ10 dosage with plasma or tissue levels in patients, and there is no practical way to monitor the efficacy of therapy. Moreover, CoQ10 is available in different formulations, and there are no data regarding their bioavailability in CoQ10-deficient patients of individual formulations.
8.6 Concluding Remarks
The importance of CoQ in human metabolism was increased in the last years after the description of patients with neuromuscular diseases associated to a deficiency of this compound. The primary deficiency implies not only the role of CoQ in bioener-getics but also the different functions that would have the COQ encoded proteins in mitochondria. The secondary deficiency opens a new view of mitochondria home-ostasis through the coordination of the different pathways acting in the organelle. This coordination would involve mainly the MRC and CoQ biosynthesis complexes through either direct interactions or intermediate regulatory proteins.
Acknowledgments
This work has been partially supported by the European Union contract LSHB-CT-2004-005151 and the NIH grant 1R01HD057543-01.
References
- 1.Aeby A, Sznajer Y, Cave H, Rebuffat E, Van Coster R, Rigal O, Van Bogaert PJ. Inherit Metab Dis. 2007;30:827. doi: 10.1007/s10545-007-0612-0. [DOI] [PubMed] [Google Scholar]
- 2.Artuch R, Brea-Calvo G, Briones P, Aracil A, Galván M, Espinós C, Corral J, Volponi V, Ribes A, Andreu AL, Palau F, Sánchez-Alcázar JA, Navas P, Pineda M. J Neurol Sci. 2006;246:153. doi: 10.1016/j.jns.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 3.Artuch R, Briones P, Aracil A, Ribes A, Pineda J, Galván M, Sánchez-Alcázar JA, Brea G, Navas P, Pineda M. J Inherit Metab Dis. 2004;27 (Suppl 1):118. [Google Scholar]
- 4.Asencio C, Rodríguez-Aguilera JC, Johnson TE, Cabello J, Schnabel R, Navas P. Mech Ageing Dev. 2009;130:145. doi: 10.1016/j.mad.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 5.Asencio C, Rodriguez-Aguilera JC, Ruiz-Ferrer M, Vela J, Navas P. FASEB J. 2003;17:1135. doi: 10.1096/fj.02-1022fje. [DOI] [PubMed] [Google Scholar]
- 6.Ashby MN, Kutsunai SY, Ackerman S, Tzagoloff A, Edwards PA. J Biol Chem. 1992;267:4128. [PubMed] [Google Scholar]
- 7.Baba SW, Belogrudov GI, Lee JC, Lee PT, Strahan J, Shepherd JN, Clarke CF. J Biol Chem. 2004;279:10052. doi: 10.1074/jbc.M313712200. [DOI] [PubMed] [Google Scholar]
- 8.Barros MH, Johnson A, Gin P, Marbois BN, Clarke CF, Tzagoloff A. J Biol Chem. 2005;280:42627. doi: 10.1074/jbc.M510768200. [DOI] [PubMed] [Google Scholar]
- 9.Belogrudov GI, Lee PT, Jonassen T, Hsu AY, Gin P, Clarke CF. Arch Biochim Biophys. 2001;392:48. doi: 10.1006/abbi.2001.2448. [DOI] [PubMed] [Google Scholar]
- 10.Boitier E, Degoul F, Desguerre I, Charpentier C, Francois D, Ponsot G, Diry M, Rustin P, Marsac C, Desguerre I. J Neurol Sci. 1998;156:41. doi: 10.1016/s0022-510x(98)00006-9. [DOI] [PubMed] [Google Scholar]
- 11.Casarin A, Jimenez-Ortega JC, Trevisson E, Pertegato V, Doimo M, Ferrero-Gomez ML, Abbadi S, Artuch R, Quinzii C, Hirano M, Basso G, Santos-Ocaña C, Navas P, Salviati L. Biochem Biophys Res Commun. 2008;372:35. doi: 10.1016/j.bbrc.2008.04.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cordero MD, Moreno-Fernández AM, de Miguel M, Bonal P, Campa F, Jiménez-Jiménez LM, Ruiz-Losada A, Sánchez-Domínguez B, Sánchez-Alcázar JA, Salviati L, Navas P. Clin Biochem. 2009;42:732. doi: 10.1016/j.clinbiochem.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 13.Crane FL, Hatefi Y, Lester RL, Widmer C. Biochim Biophys Acta. 1957;25:220. doi: 10.1016/0006-3002(57)90457-2. [DOI] [PubMed] [Google Scholar]
- 14.Cui T-Z, Kawamukai M. FEBS J. 2008 doi: 10.111/j.1742-4658.2008.06821.x. [DOI] [Google Scholar]
- 15.De Cabo R, Burgess JR, Navas P. J Bioenerg Biomembr. 2006;38:309. doi: 10.1007/s10863-006-9050-1. [DOI] [PubMed] [Google Scholar]
- 16.Do TQ, Hsu AY, Jonassen T, Lee PT, Clarke CF. J Biol Chem. 2001;276:18161. doi: 10.1074/jbc.M100952200. [DOI] [PubMed] [Google Scholar]
- 17.Echtay KS, Bienengraeber M, Klingenberg M. Biochemistry. 2001;40:5243. doi: 10.1021/bi002130q. [DOI] [PubMed] [Google Scholar]
- 18.Fischer JC, Ruitenbeek W, Gabreels FJ, Janssen AJ, Renier WO, Sengers RC, Stadhouders AM, ter Laak HJ, Trijbels JM, Veerkamp JH. Eur J Pediatr. 1986;144:441. doi: 10.1007/BF00441735. [DOI] [PubMed] [Google Scholar]
- 19.Fontaine E, Ichas F, Bernardi P. J Biol Chem. 1998;273:25734. doi: 10.1074/jbc.273.40.25734. [DOI] [PubMed] [Google Scholar]
- 20.Forsgren M, Attersand A, Lake S, Grunler J, Swiezewska E, Dallner G, Climent I. Biochem J. 2004;382:519. doi: 10.1042/BJ20040261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gempel K, Topaloglu H, Talim B, Schneiderat P, Schoser BG, Hans VH, Palmafy B, Kale G, Tokatli A, Quinzii C, Hirano M, Naini A, DiMauro S, Prokisch H, Lochmuller H, Horvath R. Brain. 2007;130:2037. doi: 10.1093/brain/awm054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gin P, Hsu AY, Rothman SC, Jonassen T, Lee PT, Tzagoloff A, Clarke CF. J Biol Chem. 2003;278:25308. doi: 10.1074/jbc.M303234200. [DOI] [PubMed] [Google Scholar]
- 23.Gironi M, Lamperti C, Nemni R, Moggio M, Comi G, Guerini FR, Ferrante P, Canal N, Naini A, Bresolin N, DiMauro S. Neurology. 2004;62:818. doi: 10.1212/01.wnl.0000113719.67643.b7. [DOI] [PubMed] [Google Scholar]
- 24.Hsu AY, Do TQ, Lee PT, Clarke CF. Biochim Biophys Acta. 2000;1484:287. doi: 10.1016/s1388-1981(00)00019-6. [DOI] [PubMed] [Google Scholar]
- 25.Jonassen T, Proft M, Randez-Gil F, Schultz JR, Marbois BN, Entian KD, Clarke CF. J Biol Chem. 1998;273:3351. doi: 10.1074/jbc.273.6.3351. [DOI] [PubMed] [Google Scholar]
- 26.Kagan VE, Arroyo A, Tyurin VA, Tyurina YY, Villalba JM, Navas P. FEBS Lett. 1998;428:43. doi: 10.1016/s0014-5793(98)00482-7. [DOI] [PubMed] [Google Scholar]
- 27.Lagier-Tourenne C, Tazir M, López LC, Quinzii CM, Assoum M, Drouot N, Busso C, Makri S, Ali-Pacha L, Benhassine T, Anheim M, Lynch DR, Thibault C, Plewniak F, Bianchetti L, Tranchant C, Poch O, DiMauro S, Mandel JL, Barros MH, Hirano M, Koenig M. Am J Hum Genet. 2008;82:661. doi: 10.1016/j.ajhg.2007.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lamperti C, Naini A, Hirano M, De Vivo DC, Bertini E, Servidei S, Valeriani M, Lynch D, Banwell B, Berg M, Dubrovsky T, Chiriboga C, Angelini C, Pegoraro E, DiMauro S. Neurology. 2003;60:1206. doi: 10.1212/01.wnl.0000055089.39373.fc. [DOI] [PubMed] [Google Scholar]
- 29.Le Ber I, Dubourg O, Benoist JF, Jardel C, Mochel F, Koenig M, Brice M, Lombes A, Durr A. Neurology. 2007;68:295. doi: 10.1212/01.wnl.0000252366.10731.43. [DOI] [PubMed] [Google Scholar]
- 30.Leonard CJ, Aravind L, Koonin EV. Genome Res. 1998;8:1038. doi: 10.1101/gr.8.10.1038. [DOI] [PubMed] [Google Scholar]
- 31.Leshinsky-Silver E, Levine A, Nissenkorn A, Barash V, Perach M, Buzhaker E, Shahmurov M, Polak-Charcon S, Lev D, Lerman-Sagie T. Mol Gen Metab. 2003;79:288. doi: 10.1016/s1096-7192(03)00097-0. [DOI] [PubMed] [Google Scholar]
- 32.Lopez LC, Schuelke M, Quinzii CM, Kanki T, Rodenburg RJ, Naini A, Dimauro S, Hirano M. Am J Hum Genet. 2006;79:1125. doi: 10.1086/510023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.López-Martín JM, Salviati L, DiMauro S, Hirano M, Rodriguez-Hernandez A, Cordero M, Sanchez Alcazar JA, Santos-Ocaña C, Navas P. Hum Mol Genet. 2007;16:1091. doi: 10.1093/hmg/ddm058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mollet J, Delahodde A, Serre V, Chretien D, Schlemmer D, Lombes A, Boddaert N, Desguerre I, de Lonlay P, de Baulny HO, Munnich A, Rötig A. Am J Hum Genet. 2008;82:623. doi: 10.1016/j.ajhg.2007.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mollet J, Giurgea I, Schlemmer D, Dallner G, Chretien D, Delahodde A, Bacq D, de Lonlay P, Munnich A, Rotig A. J Clin Invest. 2007;117:765. doi: 10.1172/JCI29089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Montero R, Sáchez-Alcázar JA, Briones P, Rodríguez-Hernández MA, Cordero MD, Trevisson E, Salviati L, Pineda M, Garcia-Cazorla A, Navas P, Artuch R. Clin Biochem. 2008;41:697. doi: 10.1016/j.clinbiochem.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 37.Montero R, Sánchez-Alcázar JA, Briones P, Navarro-Sastre A, Gallardo E, Bornstein B, Herrero-Martín D, Rivera H, Martin MA, Marti R, García-Cazorla A, Montoya J, Navas P, Artuch R. Clin Biochem. 2009;42:742. doi: 10.1016/j.clinbiochem.2008.10.027. [DOI] [PubMed] [Google Scholar]
- 38.Montini G, Malaventura C, Salviati L. N Engl J Med. 2008;358:2849. doi: 10.1056/NEJMc0800582. [DOI] [PubMed] [Google Scholar]
- 39.Musumeci O, Naini A, Slonim AE, Skavin N, Hadjigeorgiou GL, Krawiecki N, Weissman BM, Tsao CY, Mendell JR, Shanske S, De Vivo DC, Hirano M, DiMauro S. Neurology. 2001;56:849. doi: 10.1212/wnl.56.7.849. [DOI] [PubMed] [Google Scholar]
- 40.Navas P, Fernández-Ayala DM, Martín SF, López-Lluch G, De Cabo R, Rodríguez-Aguilera JC, Villalba JM. Free Radic Res. 2002;36:369. doi: 10.1080/10715760290021207. [DOI] [PubMed] [Google Scholar]
- 41.Ogasahara S, Engel AG, Frens D, Mack D. Proc Natl Acad Sci USA. 1989;86:2379. doi: 10.1073/pnas.86.7.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Okada K, Minehira M, Zhu X, Suzuki K, Nakagawa T, Matsuda H, Kawamukai M. J Bacteriol. 1997;179:3058. doi: 10.1128/jb.179.9.3058-3060.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Padilla S, Santos-Ocaña JC, Jiménez-Hidalgo M, López-Martín JM, Martín-Montalvo A, Navas P. Cell Mol Life Sci. 2008 doi: 10.1007/s00018-008-8547-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Poon WW, Barkovich RJ, Hsu AY, Frankel A, Lee PT, Shepherd JN, Myles DC, Clarke CF. J Biol Chem. 1999;274:21665. doi: 10.1074/jbc.274.31.21665. [DOI] [PubMed] [Google Scholar]
- 45.Quinzii CM, DiMauro S, Hirano M. Neurochem Res. 2007;32:723. doi: 10.1007/s11064-006-9190-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Quinzii CM, Kattah AG, Naini A, Akman HO, Mootha VK, DiMauro S, Hirano M. Neurology. 2005;64:539. doi: 10.1212/01.WNL.0000150588.75281.58. [DOI] [PubMed] [Google Scholar]
- 47.Quinzii C, Naini A, Salviati L, Trevisson E, Navas P, DiMauro S, Hirano M. Am J Hum Genet. 2006;78:345. doi: 10.1086/500092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Quinzii C, López LC, Von-Moltke J, Naini A, Krishna S, Schuelke M, Salviati L, Navas P, DiMauro S, Hirano M. FASEB J. 2008;22:1874. doi: 10.1096/fj.07-100149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rahman S, Hargreaves I, Clayton P, Heales S. J Pediatr. 2001;139:456. doi: 10.1067/mpd.2001.117575. [DOI] [PubMed] [Google Scholar]
- 50.Rötig A, Appelkvist EL, Geromel V, Chretien D, Kadhom N, Edery P, Lebideau M, Dallner G, Munnich A, Ernster L, Rustin P. Lancet. 2000;356:391. doi: 10.1016/S0140-6736(00)02531-9. [DOI] [PubMed] [Google Scholar]
- 51.Saiki R, Lunceford AL, Shi Y, Marbois B, King R, Pachuski J, Kawamukai M, Gasser DL, Clarke CF. Am J Physiol Renal Physiol. 2008;295:F1535. doi: 10.1152/ajprenal.90445.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Salviati L, Sacconi S, Murer L, Zacchello G, Franceschini L, Laverda AM, Basso G, Quinzii C, Angelini C, Hirano M, Naini AB, Navas P, DiMauro S, Montini G. Neurology. 2005;65:606. doi: 10.1212/01.wnl.0000172859.55579.a7. [DOI] [PubMed] [Google Scholar]
- 53.Santos-Ocaña C, Do TQ, Padilla S, Navas P, Clarke CF. J Biol Chem. 2002;277:10973. doi: 10.1074/jbc.M112222200. [DOI] [PubMed] [Google Scholar]
- 54.Sobreira C, Hirano M, Shanske S, Keller RK, Haller RG, Davidson E, Santorelli FM, Miranda AF, Bonilla E, Mojon DS, Barreira AA, King MP, DiMauro S. Neurology. 1997;48:1238. doi: 10.1212/wnl.48.5.1238. [DOI] [PubMed] [Google Scholar]
- 55.Stenmark P, Grunler J, Mattsson J, Sindelar PJ, Nordlund P, Berthold DA. J Biol Chem. 2001;276:33297. doi: 10.1074/jbc.C100346200. [DOI] [PubMed] [Google Scholar]
- 56.Tran UC, Clarke CF. Mitochondrion. 2007;7(Suppl):S62. doi: 10.1016/j.mito.2007.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Turunen M, Olsson J, Dallner G. Biochim Biophys Acta. 2004;1660:171. doi: 10.1016/j.bbamem.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 58.Turunen M, Swiezewska E, Chojnacki T, Sindelar P, Dallner G. Free Rad Res. 2002;36:437. doi: 10.1080/10715760290021298. [DOI] [PubMed] [Google Scholar]
- 59.Vajo Z, King LM, Jonassen T, Wilkin DJ, Ho N, Munnich A, Clarke CF, Francomano CA. Mamm Genome. 1999;10:1000. doi: 10.1007/s003359901147. [DOI] [PubMed] [Google Scholar]
- 60.Van Maldergem L, Trijbels F, DiMauro S, Sindelar PJ, Musumeci O, Janssen A, Delberghe X, Martin JJ, Gillerot Y. Ann Neurol. 2002;52:750. doi: 10.1002/ana.10371. [DOI] [PubMed] [Google Scholar]
- 61.Villalba JM, Navarro F, Córdoba F, Serrano A, Arroyo A, Crane FL, Navas P. Proc Natl Acad Sci USA. 1995;92:4887. doi: 10.1073/pnas.92.11.4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zierz S, Jahns G, Jerusalem F. J Neurol. 1989;236:97. doi: 10.1007/BF00314404. [DOI] [PubMed] [Google Scholar]