

Abstract
Anthropogenic practices and recycling in the environment through natural processes result in release of potentially harmful levels of mercury into the biosphere. Mercury, especially organic forms, accumulates in the food chain. Mercury reacts readily with sulfur-containing compounds and often exists as a thiol S-conjugate, such as the L-cysteine (Cys)-S-conjugate of methylmercury (CH3Hg-S-Cys) or inorganic mercury (Cys-S-Hg-S-Cys). These S-conjugates are structurally similar to L-methionine and L-cystine/L-cystathionine, respectively. Bovine and rat glutamine transaminase K (GTK) catalyze transamination of sulfur-containing amino acids. Recombinant human GTK (rhGTK) has a relatively open catalytic active site, and we report here that this enzyme, like the rat and bovine enzymes, can also utilize sulfur-containing L-amino acids, including L-methionine, L-cystine, and L-cystathionine as substrates. The current study extends this list to include mercuric S-conjugates, and shows that CH3Hg-S-Cys and Cys-S-Hg-S-Cys are substrates and reversible inhibitors of rhGTK. The homocysteine S-conjugates, Hcy-S-Hg-S-Hcy and CH3Hg-S-Hcy, are also inhibitors. Finally, we show that HgCl2, CH3Hg-S-Cys and Cys-S-Hg-S-Cys are potent irreversible inhibitors of rat cystathionine γ-lyase. The present study broadens our knowledge of the biochemistry of mercury compounds by showing that Cys S-conjugates of mercury interact with enzymes that catalyze transformations of biologically important sulfur-containing amino acids.
Keywords: Cystathionine γ-lyase, Glutamine transaminase K, Kynurenine aminotransferase isozyme I, Mercury cysteine S-conjugate, Methylmercury cysteine S-conjugate, Sulfur-containing amino acids
Environmental mercury may exist in elemental (Hg0), inorganic (Hg2+) or organic (Hg(I), Hg(II)) forms [1]. When mercury vapor (Hg0) and Hg(II) compounds in the atmosphere settle into bodies of water, organic mercury (primarily monomethylmercury) is formed by cobalamin-dependent methylation, which is mediated by a variety of microorganisms [1]. For simplicity, monomethylmercury will be referred to as methylmercury in the current manuscript; however, it is important to note that methylmercury, in aqueous solutions, often reacts with anions such as Cl− to form covalent bonds [2].
Methylmercury accumulates readily in tissues of numerous aquatic organisms, especially large, predatory fish [1]. It is the predominant form of organic mercury in the environment and humans are exposed to this organometallic species primarily via consumption of contaminated fish and/or water [1–3]. Inasmuch as methylmercury exposure can be detrimental to multiple tissues and organs, including the brain and kidneys, it is recommended that certain species of fish, particularly those at the top of the food chain, be consumed sparingly, especially by pregnant women and young children [4–8]. Despite this recommendation, 6–8% of women of childbearing age in the US may have unacceptably high levels of mercury [7,8]. Because of high lipid solubility, methylmercury can readily cross the placenta and accumulate in fetal tissues [1], thus raising the possibility that a sizable population of newborns may be exposed to mercury in utero.
Because of concerns about the toxicity of mercury, it is important to identify the forms of mercury capable of being transported by target organs and cells. Mercuric ions have a strong bonding affinity for reduced sulfur atoms [9]. Consequently, mercuric ions within biological systems are converted primarily to conjugates of one or more sulfur (thiol)-containing biomolecules, such as glutathione (GSH),1 L-cysteine (Cys), and L-homocysteine (Hcy). Some of these mercury conjugates are similar structurally to certain endogenous amino acids [10–12]. For example, the structure of the Cys S-conjugate of methylmercury (i.e. CH3Hg-S-Cys) is similar to that of methionine (Fig. 1). CH3Hg-S-Cys may be formed by direct reaction of Cys with methylmercury. Alternatively, methylmercury may react with GSH to form a glutathione S-conjugate (i.e. CH3Hg-S-G), which can then be catabolized to CH3Hg-S-Cys by the sequential actions of γ-glutamyltransferase and cysteinylglycinase/aminopeptidase M located in the luminal membranes of intestinal and renal epithelial cell [11,13] (Fig. 2). Due to the presence of a large hydrophobic side group and structural similarity to methionine, CH3Hg-S-Cys may be transported into cells via the amino acid transporter, system L [13]. This transporter has an affinity for large hydrophobic amino acids, including L-methionine [13].
Fig. 1.

Space-filled models showing the structural similarity of two mercuric S-conjugates with commonly occurring sulfur-containing amino acids. Structural similarity exists between methionine and the cysteine (Cys)-S-conjugate of methylmercury (CH3Hg-S-Cys), and between cystine and the Cys-S-conjugate of Hg2+ (Cys-S-Hg-S-Cys). The models were generated with MolPOV 2.0 and POV Ray 3.5. Atoms: O, red; N, blue; C, dark gray (small); H, light gray; S, yellow; Hg, dark gray (large).
Fig. 2.
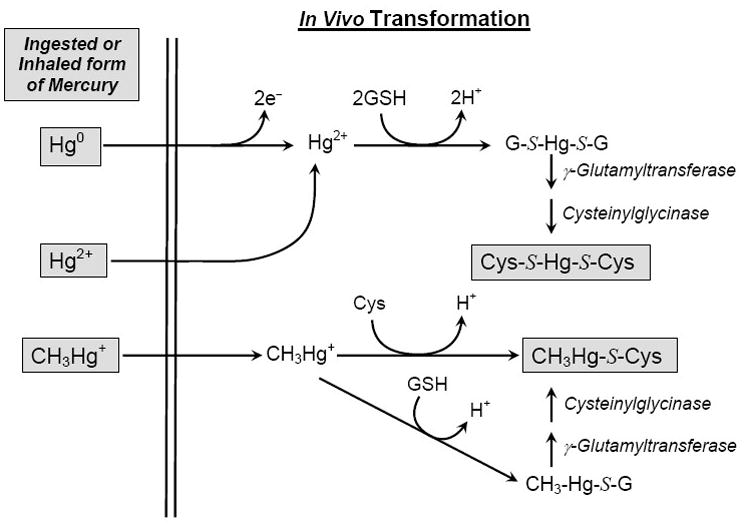
Transformation of mercury species in mammalian tissues. Mercuric ions that are ingested, absorbed or inhaled may bond with glutathione (GSH) and/or cysteine (Cys) to form G-S- and Cys-S-conjugates of Hg2+ (G-S-Hg-S-G, Cys-S-Hg-S-Cys, respectively) and methylmercury (CH3Hg-S-G, CH3Hg-S-Cys, respectively). G-S-Hg-S-G and CH3Hg-S-G are processed further by γ-glutamyltransferase and cysteinylglycinase to yield Cys-S-Hg-S-Cys and CH3Hg-S-Cys, respectively. Based on endogenous concentrations of intracellular glutathione (1–10 mM), these cysteine S-conjugates of mercury are likely to be among the major species of Hg present in cellular systems. It should be noted that inhaled Hg0 is oxidized rapidly in blood and tissues [18]. For convenience, methylmercuric species are depicted here as CH3Hg+ even though it is recognized that methylmercuric species are normally found bonded to an anion or sulfur-containing molecule.
The similarity between CH3Hg-S-Cys and methionine has been suggested by a number of authors [e.g. 14–16] to be an example of “molecular mimicry” whereby CH3Hg-S-Cys is a mimic of methionine at the site of the system L transporter. Molecular mimicry has been defined as the phenomenon whereby one molecule/compound can act as a structural and/or functional molecule of another endogenous molecule/compound [11,15]. However, the concept that cysteine S-conjugates of mercury are molecular mimics of sulfur-containing amino acids has been criticized by Hoffmeyer and colleagues [17]. These authors’ definition of molecular mimicry is restricted to isosteric compounds with similar electronic distributions [17]. Here, we simply point out the similarities between the cysteine S-conjugates of mercury and other amino acids, including sulfur-containing amino acids.
Following ingestion of methylmercury, there is evidence indicating that some of this species undergoes biotransformation in intracellular or extracellular compartments to yield inorganic mercury [3]. Thus, thiol-S-conjugates of both Hg2+ and methylmercury may be present in target cells and organs of biological systems. Interestingly, the Cys-S-conjugate of Hg2+ (i.e. Cys-S-Hg-S-Cys), which is similar structurally to L-cystine (Fig. 1) (and L-cystathionine) and Hcy-S-Hg-S-Hcy (similar to L-homocystine) is taken up at the luminal membrane of renal proximal tubular cells by the amino acid transporter, system b0,+ [18–20]. System b0,+ is an important absorptive transporter of L-cystine. In addition, the glutathione S-conjugate of Hg2+ (G-S-Hg-S-G) is similar structurally to glutathione disulfide (GSSG) and may be a substrate/inhibitor of proteins that interact with GSSG. GSSG is critical for post-translational modification of proteins involved in redox signaling [21,22].
Given that select thiol-S-conjugates of Hg2+ and methylmercury are similar structurally to endogenous compounds, we hypothesized that these conjugates may also function as substrates of certain intracellular enzymes (in addition to the membrane-bound external enzymes γ-glutamyltransferase and cysteinylglycinase/aminopeptidase M). To investigate this possibility we chose to study the interaction of Cys- and Hcy-S-conjugates of Hg2+ and methylmercury with kidney glutamine transaminase K [GTK; also known as kynurenine aminotransferase isozyme I (KAT I)] and cystathionine γ-lyase (γ-cystathionase). The rationale for these investigations evolved from the observation that rat kidney GTK utilizes α-amino acid and α-keto acid substrates of the general structure Y(CH2)nCH(NH3+)CO2− and Z(CH2)nC(O)CO2−, respectively, where n = 1 or 2 and Y (or Z) is generally a hydrophobic or uncharged moiety that can be relatively large [23,24] (Fig. 3, Eq. 1). [An exception is cystine and some other sulfur-containing amino acids where Y possesses a terminal charged grouping.] Thus, for example, rat kidney GTK catalyzes transamination with L-glutamine (as the name implies), L-methionine and L-phenylalanine, and their corresponding α-keto acids [23,24]. Glutamine transaminases purified from bovine kidney [25] and brain [26] exhibit activity toward L-glutamine, L-methionine and L-phenylalanine, but also toward large sulfur-containing amino acids [e.g. L-cystine, L-homolanthionine and L-cystathionine]. Highly purified rat kidney GTK exhibits activity toward L-cystine, L-homocystine, L-cystathionine and L-lanthionine when phenylpyruvate is used as the co-substrate (amine acceptor) [27].
Fig. 3.

Specificity of glutamine transaminase K (GTK) and cystathionine γ-lyase. Transamination reactions between amino acids and α-keto acids catalyzed by GTK are depicted in equation 1, where n = 1 or 2, and Y and Z can range in size from H- (e.g. α-ketobutyrate, n = 2) to relatively large hydrophobic moieties such as CH3S-(KMB, n = 2; L-methionine, n = 2) or C6H5- (phenylpyruvate, n = 1; L-phenylalanine, n = 1). Cystine [Y or Z =-SSCH2CH(NH3+)CO2−, n = 1] and some other sulfur-containing amino acids, in which Y or Z possesses a terminal charged grouping, are also substrates. Cystathionine γ-lyase catalyzes several γ-elimination, β-elimination and replacement reactions. Some of the more important reactions that are pertinent to the current study are shown here. The enzyme catalyzes a γ-elimination reaction with L-cystathionine (equation 2) and with L-homoserine (equation 3). In both cases, the α-keto acid product is α-ketobutyrate. Cystathionine γ-lyase also catalyzes a β-elimination reaction with L-cystine (equation 4). In this case, the α-keto acid product is pyruvate.
X-ray crystallographic studies of recombinant human GTK/KAT I (rhGTK) have shown that this enzyme has a remarkably open configuration at the active site. The enzyme substrate binding site possesses a striking crown of aromatic residues that adorns the relatively large active site [28]. Although turnover is somewhat slow, rhGTK was shown to catalyze transamination of two positional isomers of β-naphthyl-L-alanine [29], consistent with a large, relatively open active site. Figure 4 depicts a ribbon model of hGTK monomer (the active enzyme is a homodimer) showing the position of the pyridoxal 5′-phosphate (PLP) coenzyme bound in Schiff base linkage (internal aldimine) to a lysine residue in the active site. Surface representations of the monomer reveal that the active site is spacious (Fig. 4). Figure 4 also illustrates the positions of all six cysteine moieties within the monomer. It is readily apparent that the active site does not contain a cysteine residue. One cysteine residue (C127) is 6.8 Å away from the PLP coenzyme; other cysteine residues are 12.5–20.4 Å away from the coenzyme epicenter.
Fig. 4.

Ribbon model of rhGTK monomer showing pyridoxal 5′-phosphate in the active site. The diagram shows a relatively large active site. Cysteine residues are emphasized. Atoms: S, yellow; H, light gray. The model was produced using the UCSF Chimera program ( http://www.cgl.ucsf.edu/chimera/).
Transamination of L-cystathionine, L-lanthionine, and L-cystine in vitro yields α-keto acids that can non-enzymatically cyclize to ketimines [reviewed in 24,30]. Some of these compounds and their reduced forms (cyclic amines) have been reported to be present in brain, plasma and/or urine and may be neuroactive (discussed in [24]). Furthermore, GTK activity is present in human brain [31]. Therefore, GTK may be responsible for generating sulfur-containing neuroactive cyclic ketimines in human brain. Because the active site of rhGTK is large [28], it is also possible that the enzyme will catalyze transamination in human tissues with thiol-S-conjugates of Hg [Fig. 3, Eq. 1 where n = 1 or 2, and Y = CH3HgS- or Cys-S-HgS-]. This hypothesis is supported by the finding that Se-methyl-L-selenocysteine is a substrate of rhGTK [29] and this conjugate and other selenocysteine Se-conjugates, many of which contain large aromatic moieties, are substrates of rat GTK [32]. On the other hand, whereas Se-methyl-L-selenocysteine and L-methionine are relatively good substrates of rhGTK, the closely related amino acid, L-selenomethionine, is an extremely poor substrate (<0.1% as affective as L-methionine) [29]. Thus, due to these subtleties in binding geometries it was not certain a priori that the enzyme would catalyze transamination of thiol-S-conjugates, despite the relatively open active site. The present work, however, does indeed show that rhGTK catalyzes transamination of large sulfur-containing amino acids as well as the Cys-S-conjugates of Hg2+ and methylmercury.
We next turned our attention to cystathionine γ-lyase. This enzyme catalyzes a γ-lyase reaction with L-cystathionine, generating α-ketobutyrate, cysteine (eliminated fragment) and ammonia (Fig. 3, Eq. 2). L-Homoserine is also a γ-lyase substrate, generating α-ketobutyrate and ammonia (Fig. 3, Eq. 3). In this case the hydrolysis reaction is balanced by elimination of H2O. Thus, the net reaction does not include an H2O term. The enzyme can also catalyze a β-lyase reaction with L-cystine (Fig. 3, Eq. 4), generating pyruvate, the persulfide analogue of cysteine (thiocysteine) and ammonia [33,34].
Rat liver cystathionine γ-lyase has a cysteine residue within the active site [35] that is highly susceptible to inactivation by a number of sulfhydryl reagents including p-chloromercuribenzoate, iodosobenzoate and N-ethylmaleimide [36,37]. Therefore, we considered the possibility that Cys-S-conjugates of mercury, particularly Cys-S-Hg-S-Cys, would interact with this sulfhydryl in the active site of cystathionine γ-lyase. The present work shows that Cys-S-Hg-S-Cys is a strong irreversible inhibitor of cystathionine γ-lyase.
Materials and methods
Reagents
Methylmercuric chloride (CH3HgCl) and HgCl2 were obtained from Aldrich, Milwaukee, WI. Mercuric S-conjugates were generated by mixing HgCl2 with Cys or Hcy in a 1:2.5 ratio. Methylmercury S-conjugates were generated by mixing CH3HgCl with Cys or Hcy in a 1:1.25 ratio. The conjugates were incubated for five minutes at room temperature prior to use. Ammediol (2-amino-2-methyl-1,3-propanediol), dithiothreitol (DTT), GSH, sulfur-containing amino acids, L-glutamine, L-phenylalanine, L-homoserine, phenylpyruvate, sodium α-keto-γ-methiolbutyrate [sodium α-keto-(γ-methylthio)butyrate; KMB] were obtained from Sigma, St. Louis, MO. 2,4-Dinitrophenylhydrazine was purchased from MP Biochemicals, Irvine, CA.
Enzymes
Highly purified rhGTK [1.8 mg/ml in 20% glycerol, 10 mM potassium phosphate buffer (pH 7.4); specific activity 18 U/mg in the standard reaction assay mixture, see below] was obtained by the method of Han et al. [38]. Cystathionine γ-lyase (specific activity 24 U/mg, 20 mM L-homoserine as substrate) was purified from rat liver as described by Pinto et al. [39]. One unit of enzyme activity is the amount of enzyme that generates 1 μmol of product per min at 37°C under standard reaction conditions.
Enzyme activity measurements
The standard GTK assay mixture (50 μl) contained 100 mM ammediol buffer (pH 9.0), 5 mM KMB and 20 mM L-phenylalanine [40]. After incubation at 37°C (5–10 min), 150 μl of 1 M NaOH was added and the absorbance at 322 nm due to phenylpyruvate enol (ε322nm = 16,000 M−1.cm−1) was determined within 5 min against a blank consisting of complete reaction mixture lacking enzyme. The activity of rhGTK in a reaction mixture containing 100 mM potassium phosphate buffer (pH 7.4) in place of ammediol buffer in the standard reaction mixture is ~50% that exhibited at pH 9.0 [29]. The standard cystathionine γ-lyase assay mixture (50 μl) contained 100 mM potassium phosphate buffer (pH 7.4) and 20 mM L-homoserine [39]. After incubation at 37°C (10 min), the reaction was stopped by addition of 20 μl of 5 mM 2,4-dinitrophenylhydrazine in 2 M HCl. After further incubation at 37°C for 10 min, 130 μl of 1 M NaOH was added and the absorbance was read within 5 min at 430 nm against a blank consisting of reaction mixture lacking L-homoserine (or enzyme) (ε430nm α-ketobutyrate 2,4-dinitrophenylhydrazone, 15,000 M−1.cm−1). To measure aminotransferase activity of GTK toward various amino acids (other than the mercury S-conjugates) the reaction mixture (50 μl), except where noted, contained 100 mM potassium phosphate buffer (pH 7.4), 1.0 mM L-amino acid (50 nmol) and 0.4 mM phenylpyruvate (20 nmol) and GTK. In the case of L-cystine the concentration was 0.4 mM and the buffer was 50 mM sodium pyrophosphate, pH 9.2. The reaction mixture was incubated at 37°C and the amount of phenylpyruvate remaining was determined (as described above) against a blank containing complete reaction mixture lacking enzyme. A similar procedure was used to measure the rhGTK-catalyzed transamination of mercury S-conjugates, except that the concentration of phenylpyruvate was 0.2 mM. Blanks contained the complete reaction mixture (including mercuric conjugates) without enzyme.
Spectrophotometric determinations were conducted with a Tecan Infinite M1000 96-well plate spectrophotometer (Tecan, Durham, NC) or with a SpectraMax 96-well plate spectrophotometer (Molecular Devices, Sunnyvale, CA).
HPLC measurement of L-methionine and KMB
The HPLC system consisted of a liquid chromatograph equipped with an 8-channel coulometric array (CoulArray) detector (ESA, Inc., Chelmsford, MA) [39,41]. The enzyme activity in the reaction mixtures (50 μl) was terminated by addition of 15 μl of 25% w/v metaphosphoric acid (MPA). The resulting 5% w/v MPA homogenates were injected directly onto a Bio-Sil ODS-5S, 5-μm particle size, 4.0 × 250 mm, C18 column (Bio-Rad, Life Science Research Group, Hercules, CA) and eluted with a mobile phase consisting of 50 mM NaH2PO4, 50 μM octane sulfonic acid, and 1% (v/v) acetonitrile (pH 2.52) at a flow rate of 1 ml/min. All buffers, following preparation, were routinely degassed, filtered through a 0.2-μm Millipore nylon filter, and the pH re-adjusted, if necessary. PEEK™ (polyetheretherketone) tubing was used throughout the HPLC system, and a 0.2-μm PEEK™ filter was placed pre- and post-column to protect both column and flow cells, respectively, from any particulate matter. A Rheodyne injection valve with a 5-μl sample loop was used to manually introduce samples. The 8-channels of the CoulArray detector were set at 100, 200, 300, 400, 500, 600, 700 and 800 mV, respectively. Elution times (min) and detection potential ranges (mV) are: MPA (1.6), L-methionine (9.6, 600–800 mV) and KMB (11.1, 600–800 mV). Other sulfur-containing compounds eluted at times that did not interfere with analysis of L-methionine or KMB.
Data Analyses
For measurements of the effect of HgCl2 and CH3HgCl on the rhGTK-catalyzed transamination of L-phenylalanine with KMB, differences between two means were analyzed using the Mann-Whitney U test. For measurements of inactivation of cystathionine γ-lyase by various mercury-containing compounds, differences among means were analyzed using a two-way Analysis of Variance, followed by Tukey’s post-hoc testing. Each set of respective data was analyzed first with the Kolmogorov-Smirnov test for normality, followed by the Levene test for homogeneity of variances. A p-value of < 0.05 was considered statistically significant. Data are presented as the mean ± SD. Except where noted, measurements were carried out at least in triplicate.
Results
Sulfur-containing amino acids are substrates of rhGTK
Figure 5 shows that the sulfur-containing amino acids, L-methionine, L-cystine, L-cystathionine, and L-lanthionine are substrates of rhGTK when 0.4 mM phenylpyruvate is used as an amine acceptor. Except as noted, the buffer was 100 mM potassium phosphate (pH 7.4). After incubation of the reaction mixture containing 2.15 mU of enzyme at 37°C for 1 h the amount of phenylpyruvate remaining was determined. Under the conditions of the assay, L-methionine (1.0 mM), L-cystine (0.4 mM; 50 mM sodium pyrophosphate buffer, pH 9.2), L-cystathionine (1.0 mM) and L-lanthionine (1.0 mM) are about 12% – 30% as effective as L-glutamine (1.0 mM) as substrates. (Cystine has very limited solubility at pH 7.4, but is more soluble at higher pH values. Even at pH 9.2, however, the maximum concentration in the assay mixture is about 0.4 mM. The higher pH is not a major limitation for enzyme activity as GTK has a pH optimum of ~8.5 – 9.0) [23,38].
Fig. 5.

rhGTK-catalyzed transamination of phenylpyruvate with glutamine and various sulfur-containing amino acids. Except where indicated, the reaction mixture (50 μl) contained 1 mM amino acid, 0.4 mM phenylpyruvate, 100 mM potassium phosphate buffer (pH 7.4) and enzyme (2.15 mU). In the case of L-cystine the concentration was 0.4 mM and the buffer was 50 mM sodium pyrophosphate (pH 9.2). After incubation for 1 h at 37°C the amount of phenylpyruvate remaining in solution was determined relative to a blank reaction mixture lacking enzyme; n = 3.
In order to provide additional evidence for the ability of rhGTK to catalyze transamination of sulfur-containing amino acids, 0.5 mM KMB was used as an α-keto acid substrate in place of phenylpyruvate, and the L-methionine generated by transamination was determined by HPLC with CoulArray detection. The advantage of this technique is that redox-active compounds can be characterized directly without the need to derivatize them and they can be identified by both their column retention times and the voltage required to produce a signal [i.e. the voltage required to oxidize (remove an electron from) the analyte]. As a result of the presence of a sulfur ether moiety, which is readily oxidizable under the HPLC conditions, the substrate (KMB) and product (L-methionine) are both redox responsive and detectable by coulometry. Except in the case of L-cystine, the reaction mixture (50 μl) contained 100 mM potassium phosphate buffer (pH 7.4), 1.0 mM amino acid, 0.5 mM KMB and 2.15 mU rhGTK. After incubation for 60 min at 37°C the reaction was terminated by the addition of 12.5 μl of 25% w/v MPA. The L-methionine content of the mixture was measured relative to mixtures that lacked enzyme (blanks). [The concentration of L-cystine was 0.4 mM and the buffer used was 50 mM sodium pyrophosphate, pH 9.2.] The amount of L-methionine generated (nmol; average of two determinations) with various amino substrates was as follows: L-glutamine (16.0), L-cystine (1.4), L-cystathionine (0.9), L-lanthionine (0.6). The formation of L-methionine in reaction mixtures containing L-glutamine was accompanied by an approximately equimolar loss of KMB.
The values for rhGTK-catalyzed transamination of L-cystine and L-cystathionine with KMB are somewhat lower than those obtained with phenylpyruvate as an α-keto acid substrate. As was previously demonstrated for bovine [26] and rat kidney [27] glutamine transaminases, the critical issue here is that rhGTK catalyzes transamination of L-methionine, L-cystine and L-cystathionine with suitable α-keto acid acceptors. Thus, because CH3Hg-S-Cys and Cys-S-Hg-S-Cys are similar to L-methionine and L-cystine/L-cystathionine insofar as both conjugates contain sulfur and a relatively large side grouping, we considered the possibility that CH3Hg-S-Cys and Cys-S-Hg-S-Cys would be substrates and/or inhibitors of rhGTK.
Inhibition of rhGTK by mM concentrations of HgCl2 and CH3HgCl
As discussed below, cystathionine γ-lyase is strongly inhibited by μM (or less) concentrations of mercury-containing compounds. By contrast our results show that rhGTK, despite the fact that both enzymes contain cysteine residues, is much more resistant to inhibition. Figure 6 shows the time course for phenylpyruvate production at 37°C in a reaction mixture (50 μl) containing 2.6 mU rhGTK, 5 mM KMB, 20 mM L-phenylalanine, and 100 mM potassium phosphate buffer (pH 7.4). The transamination reaction is freely reversible (Fig. 3, Eq. 1). Thus, the slower reaction rate at 60 minutes relative to the initial rate is presumably a result of the back reaction competing with the forward reaction. When 10 μM HgCl2 or 10 μM CH3HgCl was included in the reaction mixture no difference in the rate of phenylpyruvate formation compared to the control was noted (data not shown). However, when 1 mM HgCl2 or 1 mM CH3HgCl was included in the reaction mixture, transamination of L-phenylalanine was inhibited at all time points relative to the control that lacked mercury compound (Fig. 6).
Fig. 6.

Effect of HgCl2 and CH3HgCl on the rhGTK-catalyzed transamination of L-phenylalanine with KMB. The reaction mixture (50 μl) contained 20 mM L-phenylalanine, 5 mM KMB, 2.6 mU of enzyme, 100 mM potassium phosphate buffer (pH 7.4) in the presence or absence of 1 mM HgCl2 or CH3HgCl. Samples were incubated at 37°C and phenylpyruvate formation was measured at the times shown (n = 3).
One possible explanation for the findings is that slow binding of Hg2+ and CH3HgCl to the enzyme occurred over a 20 minute period followed by establishment of a greatly slowed transamination rate between 30 and 60 min. However, due to the nature of the plot shown in Figure 6 it was difficult to discern whether there was still some residual enzyme activity at 60 min or at later time points. Therefore, to investigate the possibility of residual activity in enzyme exposed to Hg2+ or CH3HgCl, rhGTK (~2.6 mU) was incubated in complete reaction mixture (50 μl) lacking KMB in the presence or absence of 1 mM CH3HgCl. After incubation for 1 hour at 37°C, transamination was initiated by addition of 2.5 μl of 100 mM KMB. After a further incubation for 30 min at 37°C, the formation of phenylpyruvate was measured. The amount of phenylpyruvate formed by rhGTK that was exposed to 1 mM CH3HgCl for 1 h was 11.7 ± 2.1% relative to the control [100 ± 13%] (n = 6). A similar experiment was carried out with HgCl2. In this case, the amount of phenylpyruvate formed by rhGTK that was exposed to 1 mM HgCl2 for 1 h was 6.9 ± 1.5% relative to the control [100 ± 10%] (n = 6).
There are at least two possible explanations for the findings of residual activity in rhGTK exposed to CH3HgCl or Hg2+. First, binding of the mercury compound causes 100% irreversible inactivation, but 5–10% of the enzyme population is resistant to binding of the mercury compound and remains fully active. Secondly, 100% of the enzyme is strongly modified within about 20 min and all modified enzyme species are active, albeit at a reduced efficiency. To distinguish between the two possibilities, 0.6 mU of enzyme was incubated for 1 h at 37°C in 40 μl of 100 mM potassium phosphate buffer (pH 7.4) containing 0, 0.5, 1 or 2 mM HgCl2. Thereafter, L-phenylalanine and KMB were added to yield final concentrations of 20 mM and 5 mM, respectively (final volume 50 μl). After an additional 30-min incubation at 37°C, the amounts of phenylpyruvate formed were 8.96 ± 0.66, 1.42 ± 0.42, 0.76 ± 0.26, and 0.52 ± 0.20 nmol, respectively (n = 3). In another series of experiments, 0.6 mU of enzyme was incubated for an hour at 37°C in 40 μl of 100 mM potassium phosphate buffer (pH 7.4) containing 2 mM HgCl2. Thereafter, 5 μl of 100 mM DTT was added and the mixture was incubated for 30 min at 37°C. Then, 10 μl of 100 mM L-phenylalanine and 5 μl of 100 mM KMB were added (final volume 55 μl). After an additional 30-min incubation at 37°C, phenylpyruvate was measured. The amount of phenylpyruvate formed was 7.12 ± 0.16 nmol (n = 3). Thus, after addition of a 6.25 fold molar excess of DTT over that of HgCl2, the inhibition by Hg2+ was largely (~80%) reversed.
The data suggest that reaction with mM concentrations of Hg2+ ions or CH3HgCl is a relatively slow process, but that the interaction eventually substantially lowers the activity of rhGTK. The activity, however, can be largely restored by treatment with DTT. Possibly, covalent binding of the mercury species to one or more cysteine sulfhydryls alters the conformation of the enzyme so that the enzyme cannot bind substrate or binds substrate much less effectively. The DTT restores the original sulfhydryl groups with recovery of activity. Alternatively, S-Hg exchange involving DTT results in an Hg conjugate that can no longer bind to the enzyme. As shown below, the results are in marked contrast to those obtained with cystathionine γ-lyase, where the enzyme is rapidly and irreversibly inactivated by μM (or lower) concentrations of HgCl2, Cys-S-Hg-S-Cys and CH3Hg-S-Cys. It should be noted that the use of mM concentrations of mercuric species is not biologically relevant. The experiments were designed to show the markedly different susceptibility of cystathionine γ-lyase to various forms of mercury compared to GTK despite the fact that both enzymes utilize sulfur-containing amino acids as substrates and possess cysteine residues.
rhGTK-catalyzed transamination of CH3Hg-S-Cys and Cys-S-Hg-S-Cys
As noted above, CH3Hg-S-Cys and Cys-S-Hg-S-Cys were hypothesized to be substrates of GTK. In order to provide evidence for this hypothesis, enzyme-catalyzed disappearance of phenylpyruvate was measured in a reaction mixture containing 2.6 mU rhGTK, 0.2 mM phenylpyruvate, 1 mM mercury S-conjugate and 100 mM potassium phosphate buffer (pH 7.4). The corresponding homocysteine S-conjugates were included for comparison. Lower concentrations of phenylpyruvate were included in this experiment compared to those used to determine the substrate specificity toward physiologically important sulfur-containing amino acids (Fig. 5). Significant disappearance of absorbance of phenylpyruvate (enol) relative to the blank should be more readily apparent at lower concentrations of phenylpyruvate. The low phenylpyruvate concentration in this experiment (0.2 mM) should not be limiting for enzyme activity because rat kidney GTK (and presumably rhGTK) has a high affinity for phenylpyruvate [23].
No significant rhGTK activity was detected with 1 mM Hcy-S-Hg-S-Hcy (Fig. 7). The enzyme appears to have some activity with the homocysteine derivative, CH3Hg-S-Hcy, but the activity value did not quite reach significance. On the other hand, significant enzymatic activity was found with the cysteinyl derivatives, Cys-S-Hg-S-Cys and CH3Hg-S-Cys (Fig. 7).
Fig. 7.

Substrate specificity of rhGTK toward Cys- and Hcy-S-conjugates of Hg2+ and methylmercury. The reaction mixture (50 μl) contained 0.2 mM phenylpyruvate, 1.0 mM mercury S-conjugate, 100 mM potassium phosphate buffer (pH 7.4) and 2.6 mU of enzyme. After incubation for 2 h at 37°C the disappearance of phenylpyruvate was measured in comparison to a blank lacking enzyme; ND not detectable. a Not significantly different from the blank values (n = 5).
Inhibition of rhGTK activity by Cys-S-Hg-S-Cys, Hcy-S-Hg-S-Hcy, CH3Hg-S-Cys and CH3Hg-S-Hcy
Because CH3Hg-S-Cys and Cys-S-Hg-S-Cys have similarities with methionine and cysteine/cystathionine, respectively, and are substrates of rhGTK, they were predicted to be inhibitors of the standard L-phenylalanine – KMB transaminase reaction catalyzed by rhGTK. We initially carried out experiments to determine the Km for L-phenylalanine exhibited by rhGTK under conditions used for the inhibition studies. Fitting of data to the Michaelis-Menten equation showed that the Km exhibited by rhGTK for KMB in the presence of 40 mM L-phenylalanine (in which the KMB concentrations were varied between 1 and 20 mM) and 100 mM potassium phosphate buffer (pH 7.4) at 37°C is 1.8 ± 0.6 mM (Sigma plot; n = 3 for each value of v). A similar analysis showed that the Km exhibited toward L-phenylalanine in the presence of 5 mM KMB (in which the L-phenylalanine concentration was varied between 5 and 20 mM) was 7.3 ± 1.3 mM (average of four separate experiments in which n ≥ 3 for the determination of each value of v). The Km for L-phenylalanine noted here is higher than that reported previously for rhGTK at pH 7.5 (1.7 mM) [38]. It should be noted that the previous study utilized 16 mM α-ketobutyrate as the α-keto acid substrate. Aminotransferases catalyze a ping-pong reaction. A feature of this type of reaction is that the apparent Km value of one of a pair of substrates is strongly dependent on the nature and concentration of the other substrate.
Next, we determined the effect of various mercury S-conjugates on rhGTK-catalyzed transamination of L-phenylalanine with KMB. Accordingly, reaction mixtures (50 μl) containing enzyme (2 mU), various concentrations of L-phenylalanine, 5 mM KMB, and 100 mM potassium phosphate buffer (pH 7.4) were incubated for 30 min at 37°C in the presence or absence of mercury S-conjugate (i.e. Cys-S-Hg-S-Cys, Hcy-S-Hg-S-Hcy, CH3Hg-S-Cys or CH3Hg-S-Hcy) and the amount of phenylpyruvate determined. The relative amounts of phenylpyruvate formed at three concentrations of L-phenylalanine, ranging from 10 mM (a concentration slightly above the Km value) to 50 mM (a concentration considerably above the Km value) in the presence and absence of various mercury S-conjugates are shown in Table 1. This table shows that, under the conditions of the assay, the mercury S-conjugates, at a concentration of 0.5 – 1 mM, are strongly inhibitory. Because Cys-S-Hg-S-Cys and CH3Hg-S-Cys are substrates of rhGTK (Fig. 7) part of the inhibition noted with these two compounds must be competitive with respect to L-phenylalanine. If the inhibition, however, were strictly competitive then the relative inhibition would be less as the concentration of L-phenylalanine was increased relative to the concentration of conjugate. This was not the case for these two conjugates and also for Hcy-S-Hg-S-Hcy and CH3Hg-S-Hcy. In fact, within experimental error there was little change in the degree of inhibition exerted by the mercury S-conjugates from 10 to 50 mM L-phenylalanine (Table 1). This finding suggests a strong noncompetitive component. However, it was not possible to carry out kinetic experiments with concentrations of mercury S-conjugates at concentrations >1 mM because this is the limit of the solubility of these conjugates. Moreover, the non-linearity of the reaction over the 30 min incubation (Fig. 6) complicates interpretation of data. The exact type of inhibition remains to be determined, but it is worth noting that the mercury conjugates appear to bind more strongly to rhGTK (strong inhibition at ≤1 mM) than L-phenylalanine (Km ~7.6 mM).
Table 1.
Inhibition of rhGTK-catalyzed transamination of L-phenylalanine with KMB by Cys- and Hcy-S-conjugates of Hg2+ and CH3Hg-
| Inhibitor | L-Phenylalanine concentration
|
||
|---|---|---|---|
| 10 mM | 20 mM | 50 mM | |
| Relative phenylpyruvate formation (%) | |||
| None | [100] | [100] | [100] |
|
| |||
| CH3Hg-S-Cys (0.5 mM) | 72 ± 26 | 55 ± 11 | 63 ± 25 |
| CH3Hg-S-Hcy (0.5 mM) | 49 ± 11 | 40 ± 4 | 50 ± 3 |
| Cys-S-Hg-S-Cys (1 mM) | 31 ± 4 | 29 ± 8 | 33 ± 6 |
| Hcy-S-Hg-S-Hcy (0.5 mM) | 43 ±5 | 35 ± 4 | 50 ± 7 |
The reaction mixture contained varying concentrations of L-phenylalanine, 5 mM KMB, 100 mM potassium phosphate buffer (pH 7.4) and enzyme (2 mU) in a final volume of 50 μl in the presence or absence of mercury S-conjugate. After incubation for 30 min at 37 °C, phenylpyruvate formation was determined. N = 4 or 5.
Inhibition of rat liver cystathionine γ-lyase by μM concentrations of HgCl2, Cys-S-Hg-S-Cys and CH3Hg-S-Cys
As noted above, cystathionine γ-lyase catalyzes a β-elimination reaction with L-cystine in a reaction that generates pyruvate [33,34; Fig. 3, Eq. 4]. Therefore, we considered the possibility that the enzyme might be able to catalyze pyruvate formation from Cys-S-Hg-S-Cys and CH3Hg-S-Cys. However, we were unable to detect pyruvate formation (as the 2,4-dinitrophenylhydrazone derivative) within the sensitivity of the assay (~0.2 nmol), when 1 mU cystathionine γ-lyase was incubated for 1 h at 37°C in a reaction mixture (50 μl) containing 1 mM Cys-S-Hg-S-Cys (or CH3Hg-S-Cys) and 100 mM potassium phosphate buffer, pH 7.4.
As also noted above, rat liver cystathionine γ-lyase was previously shown to be highly susceptible to inactivation by a number of sulfhydryl reagents including p-chloromercuribenzoate. This reaction and that with HgCl2 are formally depicted below, where P = protein. Thus, it was predicted that HgCl2 would also be a strong inactivator of cystathionine γ-lyase and this was found to be the case (see below).
The previous findings with p-chloromercuribenzoate [36] and the present findings with HgCl2 indicate that the active site cysteine is reactive. It was then of interest to determine whether this reactive cysteine residue can interact with mercury cysteine conjugates that are substrate analogues. If the conjugates bind simply as substrate analogues then it is expected that they will be reversible inhibitors. However, if they can bind in such a manner as to come in close contact with this reactive cysteine residue then it was predicted that the mercury cysteine conjugates would be strong irreversible inhibitors of the enzyme. This was found to be the case.
When the effect of various mercury-containing compounds on the activity of purified rat liver cystathionine γ-lyase was determined, significant inhibition of cystathionine γ-lyase occurred when μM amounts of these compounds were added to the standard reaction mixture containing 20 mM L-homoserine (Fig. 8). [L-Homoserine (Km = 20 mM) [42] is used in routine analytical assays for cystathionine γ-lyase (Fig. 3, Eq. 3.] Interestingly, the extent to which inactivation occurred with the various mercury-containing compounds was as follows: Cys-S-Hg-S-Cys > HgCl2 > CH3Hg-S-Cys. Possibly, the difference between Cys-S-Hg-S-Cys and CH3Hg-S-Cys reflects the fact that Cys-S-Hg-S-Cys bears closer resemblance to the substrates L-cystathionine and L-cystine than does CH3Hg-S-Cys. CH3Hg-S-Cys resembles methionine, which is not a cystathionine γ-lyase substrate. It should be noted that the amount of mercury present in the 50-μl reaction mixture at the lowest concentration (0.2 μM) was 10 pmol. Based on the specific activity of cystathionine γ-lyase and the fact that the enzyme is a homotetramer (subunit Mr ~44,000), the amount of cystathionine γ-lyase monomers in the incubation mixture was 75 fmol. Thus, the mercury-containing compounds were in large excess over cystathionine γ-lyase in these experiments. Another point of concern is whether the mercury-containing compounds interfere with the 2,4-dinitrophenylhydrazone assay for the quantitation of pyruvate (β-lyase reaction) or α-ketobutyrate (standard cystathionine β-lyase reaction). In control experiments it was shown that the presence of 2 mM HgCl2 or 2 mM Cys-S-Hg-S-Cys had no effect on the yield of α-keto acid 2,4-dinitrophenylhydrazone when standard cystathionine γ-lyase reaction mixtures (0.05 ml) were spiked with 10 nmol pyruvate and incubated at 37°C for one hour followed by a further 10 min incubation with the 2,4-dinitrophenylhydrazine reagent.
Fig. 8.

Inactivation of cystathionine γ-lyase by various mercury-containing compounds. The reaction mixture (50 μl) contained 20 mM L-homoserine, 100 mM potassium phosphate (pH 7.4) and, where indicated, Hg-containing compound. Enzyme (0.08 mU; 75 fmol of enzyme subunit) was added last. After incubation for 1 h at 37°C, α-ketobutyrate was measured by the 2,4-dinitrophenylhydrazine method. The control contained complete reaction mixture plus enzyme, but no added Hg-containing compound. * = significantly different from the same treatment group exposed to 0.2 μM Hg-containing compound (10 pmol/50 μl reaction mixture) (P<0.05). ** = significantly different from the same treatment group exposed to 0.2 or 2.0 μM Hg-containing compound (P<0.05) (n ≥ 3).
The present findings suggest that positioning a mercuric S-conjugate as a substrate analogue in close proximity to the active site of cystathionine γ-lyase facilitates modification of the cysteinyl moiety within the enzyme active site. Despite the fact that the S-Hg bond has a high stability constant (~0.22 Kj/mol) [9,43], the coordination bonds of mercury are kinetically quite labile [43–45]. Thus, the greater potency of Cys-S-Hg-S-Cys compared to HgCl2 as an inactivator of cystathionine γ-lyase (Fig. 7) may be due to binding of Cys-S-Hg-S-Cys as an analogue of L-cystathionine/L-cystine followed by a rapid S-Hg exchange reaction with a protein-bound cysteine moiety within the active site of the enzyme.
In a separate set of experiments, 2.1 mU of cystathionine γ-lyase was incubated for 1 h at 37°C in the presence of 100 mM potassium phosphate buffer (pH 7.4) and 20 μM of either HgCl2 or Cys-S-Hg-S-Cys (final volume 20 μl). At the end of the incubation 1 M potassium phosphate (pH 7.4) and 100 mM L-homoserine were added, such that the final volume was 50 μl and the concentrations of buffer and L-homoserine were 100 mM and 20 mM, respectively. After an additional 30-min incubation at 37°C, α-ketobutyrate formation was measured. Only 5% of the activity relative to a control (2.1 mU of enzyme incubated in phosphate buffer in the absence of mercury compound) was detected for enzyme that had been exposed to HgCl2, whereas no activity could be detected with enzyme that had been exposed to Cys-S-Hg-S-Cys. In both cases, addition of DTT to a final concentration of 5 mM did not restore any enzyme activity, even after incubation for 5 h at 37°C. However, the choice of DTT in this experiment may not have been optimal. For example, it is known that in the case of bacterial MerB [organomercurial lyase; catalyzes the reaction: RHg(I) → RH + Hg(II)], DTT binds to the Hg-S enzyme adduct to form a stable 3-coordinate Hg complex [46,47]. Therefore, the experiment was repeated to determine whether 20 mM cysteine or 20 mM GSH (final concentration) in place of DTT could reactivate enzyme that had been inactivated by HgCl2 or Cys-S-Hg-S-Cys. No activity was restored after incubation of HgCl2- or Cys-S-Hg-S-Cys-inactivated enzyme for two hours at 37°C in the presence of 20 mM L-cysteine or 20 mM GSH.
Discussion
As mentioned in the Introduction, L-methionine, L-cystine and L-cystathionine are substrates of rat kidney GTK. Consistent with this finding, the present work shows that these sulfur-containing amino acids are also substrates of the human counterpart (rhGTK) (Fig. 5). Moreover, this work also shows that the sulfur-containing mercury conjugates Cys-S-Hg-S-Cys and CH3Hg-S-Cys are transaminase substrates of this enzyme (Fig. 7). The ability to transaminate these conjugates presumably is a result in part of the relatively open active site and the ability of the enzyme to utilize large amino acid substrates. However, under the conditions of our assay these mercury conjugates are not as active as the “natural” in vivo substrates L-glutamine, L-cystine, L-methionine and L-phenylalanine.
The question arises as to whether these transamination reactions with Cys-S-Hg-S-Cys and CH3Hg-S-Cys are biologically relevant. Transamination of Cys-S-Hg-S-Cys and CH3Hg-S-Cys was demonstrated at 1 mM concentration, but it is doubtful that this concentration could be attained in vivo even after severe mercury poisoning. Moreover, there will be strong competition with endogenous amino acid substrates naturally present at much higher concentrations. However, aminotransferases generally exhibit Km values in the mM or tens of mM range and this is true of GTK [38; present work]. Thus, it is possible that GTK is not fully saturated with endogenous amino acid substrates and that some binding of Cys-S-Hg-S-Cys and CH3Hg-S-Cys will occur even when these conjugates are present at low concentrations. There is precedent for transamination of a metabolite at very low levels in vivo despite the presence of much higher levels of other endogenous amino acid substrates. For example, neuroactive kynurenate is obtained by transamination of kynurenine. Two aminotransferases have been extensively studied as contributing to the formation of kynurenate from kynurenine in vivo, namely KAT I and KAT II. As noted above, KAT I is identical to GTK. KAT II is identical to glutamate – α-aminoadipate aminotransferase (for a review of the role of KATs in the brain see [48]). The concentration of kynurenine and its transamination product kynurenate in the brain are in the 400 nM range and 1 nM range, respectively [48,49]. Thus, kynurenate can be generated in brain through transamination of kynurenine despite the low levels of precursor kynurenine and high levels of endogenous alternative substrates – e.g. glutamine (KAT I) and glutamate (KAT II). Thus, we suggest that it is entirely possible that Cys-S-Hg-S-Cys and CH3Hg-S-Cys even at nM concentrations could be transaminated in vivo.
Transamination of Cys-S-Hg-S-Cys and CH3Hg-S-Cys are predicted to yield the corresponding mercury-containing α-keto acids. However, it is not clear to what extent these α-keto acids might accumulate in vivo given the large pool of GSH (mM) in most tissues and the potential for Hg – S exchange. If the exchange is relatively slow, then it is possible that the α-keto acids derived from Cys-S-Hg-S-Cys and CH3Hg-S-Cys may contribute to the toxic effects in vivo in individuals exposed to organic mercury via food intake, especially since GTK is of relatively high specific activity in the kidney [23], and the kidney is a major site of accumulation of Cys-S-Hg-S-Cys and CH3Hg-S-Cys [13]. It is possible that if the α-keto acid analogues of Cys-S-Hg-S-Cys and CH3Hg-S-Cys are produced in vivo they may react with/inhibit α-keto acid-utilizing enzymes thereby contributing to the toxicity. However, the predicted mercury-containing α-keto acids have not yet been characterized and their biological and toxicological properties must await further studies. Finally, if there is some Hg – S exchange between GSH and the α-keto acid analogues of Cys-S-Hg-S-Cys and CH3Hg-S-Cys, mercaptopyruvate may be one of the products formed. Mercaptopyruvate is a donor of sulfane sulfur to a suitable acceptor in a reaction catalyzed by mercaptopyruvate sulfurtransferase [50]. Addition of sulfane sulfur to cysteine residues has the potential to modify protein structure and function [50].
In addition, our data show that Cys-S-Hg-S-Cys and CH3Hg-S-Cys (and the corresponding Hcy conjugates) are strongly inhibitory relative to the substrate phenylalanine. However, the exact kinetic mechanism remains to be elucidated. One possibility is that the mercury S-conjugates react covalently (participate in Hg-S exchange) with one or more cysteine residues in rhGTK. Reaction with the cysteinyl moiety (C127) near the active site (or possibly with more distant cysteines) may account for the apparent noncompetitive type of inhibition observed during our kinetic studies. Planned X-ray crystallographic studies of rhGTK crystallized in the presence of either HgCl2 or Cys-S-Hg-S-Cys should provide additional information.
It is important to note that the activity of many enzymes, including, for example, certain aminotransferases [51,52] and dehydrogenases [53,54], contain cysteine residues that, when modified, can significantly alter enzyme activity. Thiol/disulfide bonds are critical for maintaining the structural, catalytic and allosteric integrity of a vast number of enzymes and signal proteins. In addition, they are key components involved in the maintenance of redox balance and redox-sensitive reaction pathways, and in mediating protein signaling events [21,22,55–57]. Binding of mercury compounds to sulfhydryl centers of exposed cysteine residues may lead to reversible and/or irreversible inhibition of the enzyme, enzyme denaturation and/or protein aggregation. For example, a recent study provides data suggesting that HgCl2 and CH3HgCl irreversibly inhibit the activity of arylamine N-acetyltransferase-1, an intracellular enzyme involved in the biotransformation of aromatic and heterocyclic amines [58]. Similarly, the current study indicates that HgCl2 and CH3HgCl are able to inhibit the intracellular enzymes, GTK and, much more potently and irreversibly, cystathionine γ-lyase. In previous work it was shown that after inactivation of crystalline rat liver cystathionine γ-lyase by p-chloromercuribenzoate, activity could only be partially restored by addition of 2,3-dimercaptopropionate [36].
Although more detailed studies are required to firmly establish the mechanism, the present findings of inhibition of rhGTK by HgCl2 and CH3HgCl have the characteristics of a slow, but reversible noncompetitive inhibition. There is a good precedent for such an occurrence. Frasco et al. [59] showed that human butyrylcholinesterase, which does not possess a cysteine residue sensitive to thiol reagents, is slowly, but reversibly, inhibited (minutes) by mM amounts of HgCl2 in a noncompetitive manner. Human butyrylcholinesterase crystallized in the presence of HgCl2 was found to contain two Hg binding sites with variable Hg occupancy. No Hg was found in the active site or was bound to cysteine sulfur [59]. As noted above, the crystal structure of rhGTK is known [28]. Subsequent studies from our group will focus on rhGTK crystallized in the presence of HgCl2 and mercury S-conjugates. As also noted above, mM concentrations of CH3HgCl and HgCl2 are not biologically relevant. Nevertheless, the results are helpful because they indicate that the compounds will be useful in X-ray crystallographic studies of rhGTK. Moreover, they highlight the different susceptibilities of rhGTK and cystathionine γ-lyase to mercury-containing compounds as discussed below.
HgCl2, Cys-S-Hg-S-Cys and CH3HgCl strongly inhibit cystathionine γ-lyase at μM, or lower, concentrations (Fig. 8). This inhibition is irreversible for HgCl2 and Cys-S-Hg-S-Cys (and probably also for CH3Hg-S-Cys). These findings are markedly different from those noted for rhGTK, where strong inhibition by HgCl2 and CH3HgCl requires mM concentrations. Moreover, inhibition of rhGTK by HgCl2 was shown to be reversible by addition of DTT. [The reversibility of the inhibition by CH3HgCl was not investigated.] As noted above, the sensitivity of cystathionine γ-lyase to inhibition by HgCl2 is probably due to a reactive cysteine in the vicinity of the active site. Again, there is a good precedent in the work of Frasco et al. [59]. These authors showed that, unlike human butyrylcholinesterase, Torpedo californica acetylcholinesterase possesses a cysteine residue sensitive to sulfhydryl reagents, and is irreversibly inhibited in a pseudo-first-order process by μM amounts of HgCl2.
The earlier discovery that mercury species bond avidly with thiol-containing biomolecules to form S-conjugates such as Cys-S-Hg-S-Cys and CH3Hg-S-Cys [60] was instrumental in expanding our understanding of the toxicity of mercury compounds within target organs and cells. When these S-conjugates of mercury act as molecular analogues of endogenous, sulfur-containing amino acids, they broaden the scope of the toxicological consequences of mercury exposure. Indeed, the present study shows that Cys-S-Hg-S-Cys is a potent irreversible inhibitor of cystathionine γ-lyase, while both Cys-S-Hg-S-Cys and CH3Hg-S-Cys are aminotransferase substrates and reversible inhibitors of GTK. In rodents, the specific activity of cystathionine γ-lyase is highest in the liver followed by kidney, with much lower levels in other organs [61]. On the other hand, the specific activity of GTK is highest in kidney, but the enzyme is also notably active in liver and, to a lesser extent, in brain and other organs [23,24]. Thus, the differences in organ toxicity of Cys-S-Hg-S-Cys versus CH3Hg-S-Cys may be related, in part, to differences in the way the compounds interact with enzymes/transporters. For example, CH3Hg-S-Cys, but not Cys-S-Hg-S-Cys, has been shown to cross the blood-brain barrier via the amino acid transporter, system L [62,63]. Given that the human brain contains large amounts of cystathionine [64], the neurotoxicity of CH3Hg-S-Cys may be related, in part, to its transport through the blood-brain barrier and its subsequent targeting of cystathionine metabolism/turnover/function in the human brain. These mercury-containing organosulfur conjugates are the forms to which humans would be most often likely exposed following consumption of contaminated fish that are high in the food chain. Thus, the metabolic burden of processing inorganic or organic forms of mercury, such as monomethyl S-conjugates, would be expected to occur within tissues of autotrophic species and/or animals lower in the food web that are initially exposed to mercury from the atmosphere or that settled in water.
Clearly human exposure to mercury-containing compounds has numerous consequences, not only at the organ level, but also at the cellular/molecular level. Our results emphasize the fact that mercury not only binds covalently and indiscriminately to thiol moieties in proteins, but once conjugated with sulfur-containing amino acids and peptides, particularly cysteine and GSH, forms compounds that can interact with enzymes, thereby potentially broadening the profile associated with overall mercury toxicity. These compounds may represent the major portion of organomercury that contributes to the metabolic burden following consumption of mercury-contaminated foods.
In summary, we have demonstrated that cysteine S-conjugates of mercury in vitro are a) substrates/inhibitors of GTK, and b) participate in enzyme inactivation (e.g. cystathionine γ-lyase). However, given the high concentration of GSH in most tissues and the propensity of mercury to undergo exchange reactions with sulfhydryl-containing compounds, the α-keto acid products of the GTK reaction on Cys-S-Hg-S-Cys and CH3Hg-S-Cys may become diluted within the GSH pool. Nevertheless, we wish to emphasize that as a result of exposure to mercury-containing amino acids and subsequent in vivo transformation, mercury may be more mobile in its ability to distribute among various organic forms and to interact with more enzymes (as substrates/inhibitors) than previously appreciated. Future studies on the toxicity of mercury should take into account these biological transformations.
Acknowledgments
This work was supported by grants from the National Institutes of Health RO1 [ES8421] (to AJLC), RO1 [ES5980] (to RKZ), RO1 [ES11288] (to RKZ), NS062836 (to JL), RO3 [ES15511] (to CCB), R15 [ES19991] (to CCB) and CA111842 (to JTP).
Footnotes
Abbreviations used: CH3Hg-S-Cys, L-cysteine S-conjugate of methylmercury; CH3Hg-S-Hcy, L-homocysteine S-conjugate of methylmercury; Cys-S-Hg-S-Cys, L-cysteine S-conjugate of inorganic mercury; DTT, dithiothreitol; GTK, glutamine transaminase K; GSH, glutathione; G-S-Hg-S-G, glutathione S-conjugate of inorganic mercury; GSSG, glutathione disulfide; Hcy-S-Hg-S-Hcy, L-homocysteine S-conjugate of inorganic mercury; KAT I, kynurenine aminotransferase isozyme I; KMB, α-keto-γ-methiolbutyrate; MPA, metaphosphoric acid; PLP, pyridoxal 5′-phosphate; rhGTK, recombinant human GTK.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Agency for Toxic Substances and Disease Registry: Toxicological Profile for Mercury. TP-93/10. United States Department of Health and Human Services; 2010. [Google Scholar]
- 2.Harris HH, Pickering IJ, George GN. Science. 2003;301:1203. doi: 10.1126/science.1085941. [DOI] [PubMed] [Google Scholar]
- 3.Clarkson TW, Magos L. Crit Rev Toxicol. 2006;36:609–662. doi: 10.1080/10408440600845619. [DOI] [PubMed] [Google Scholar]
- 4.Schober SE, Sinks TH, Jones RL, Bolger PM, McDowell M, Osterloh J, Garrett ES, Canady RA, Dillon CF, Sun Y, Joseph CB, Mahaffey KR. J Am Med Assoc. 2003;289:1667–1674. doi: 10.1001/jama.289.13.1667. [DOI] [PubMed] [Google Scholar]
- 5.McDowell MA, Dillon CF, Osterloh J, Bolger PM, Pellizzari E, Fernando R, Montes de Oca R, Schober SE, Sinks T, Jones RL, Mahaffey KR. Environ Health Perspect. 2004;112:1165–1171. doi: 10.1289/ehp.7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mahaffey KR. Trans Am Climatol Assoc. 2005;116:127–153. [PMC free article] [PubMed] [Google Scholar]
- 7.Castoldi AF, Coccini T, Manzo L. Rev Environ Health. 2003;18:19–31. doi: 10.1515/reveh.2003.18.1.19. [DOI] [PubMed] [Google Scholar]
- 8.Trasande L, Landrigan PJ, Schecter C. Environ Health Perspect. 2005;113:590–596. doi: 10.1289/ehp.7743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fuhr B, Rabenstein DL. J Am Chem Soc. 1973;95:6944–6950. doi: 10.1021/ja00802a013. [DOI] [PubMed] [Google Scholar]
- 10.Clarkson TW. Ann Rev Pharmacol Toxicol. 1993;32:545–571. doi: 10.1146/annurev.pa.33.040193.002553. [DOI] [PubMed] [Google Scholar]
- 11.Bridges CC, Zalups RK. Toxicol Appl Pharmacol. 2005;204:274–308. doi: 10.1016/j.taap.2004.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Simmons-Willis TA, Koh AS, Clarkson TW, Ballatori N. Biochem J. 2002;367:239–246. doi: 10.1042/BJ20020841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zalups RK. Pharmacol Rev. 2000;52:113–143. [PubMed] [Google Scholar]
- 14.Roos DH, Puntel RO, Farina M, Aschner M, Bohrer D, Rocha JBT, Barbosa NBV. Toxicol Appl Pharmacol. 2011;252:28–35. doi: 10.1016/j.taap.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clarkson TW. Annu Rev Pharmacol Toxicol. 1993;33:545–571. doi: 10.1146/annurev.pa.33.040193.002553. [DOI] [PubMed] [Google Scholar]
- 16.Ballatori N. Environ Health Perspect. 2002;110(Suppl 5):689–694. doi: 10.1289/ehp.02110s5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoffmeyer RE, Singh SP, Doonan CJ, Ross AR, Hughes RJ, Pickering IJ, George GN. Chem Res Toxicol. 2006;19:753–759. doi: 10.1021/tx0503449. [DOI] [PubMed] [Google Scholar]
- 18.Clarkson TW, Vyas JB, Ballatori N. Am J Ind Med. 2007;50:757–764. doi: 10.1002/ajim.20476. [DOI] [PubMed] [Google Scholar]
- 19.Bridges CC, Bauch C, Verrey F, Zalups RK. J Am Soc Nephrol. 2004;15:663–673. doi: 10.1097/01.ASN.0000113553.62380.F5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bridges CC, Zalups RK. Am J Pathol. 2004;165:1385–1394. doi: 10.1016/S0002-9440(10)63396-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dalle-Donne I, Rossi R, Giustarini D, Colombo R, Milzani A. Free Radic Biol Med. 2007;43:883–898. doi: 10.1016/j.freeradbiomed.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 22.Cooper AJL, Pinto JT, Callery PS. Expert Opin Drug Metab Toxicol. 2011;7:891–910. doi: 10.1517/17425255.2011.577738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cooper AJL, Meister A. Comp Biochem Physiol. 1981;69B:137–145. [Google Scholar]
- 24.Cooper AJL. Neurochem Int. 2004;44:557–577. doi: 10.1016/j.neuint.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 25.Ricci G, Nardini M, Federici G, Cavallini D. Eur J Biochem. 1986;157:57–63. doi: 10.1111/j.1432-1033.1986.tb09637.x. [DOI] [PubMed] [Google Scholar]
- 26.Costa M, Pensa B, Di Costanzo B, Coccia R, Cavallini D. Neurochem Int. 1987;10:377–382. doi: 10.1016/0197-0186(87)90113-6. [DOI] [PubMed] [Google Scholar]
- 27.Cooper AJL, Anders MW. Ann N Y Acad Sci. 1990;585:118–127. doi: 10.1111/j.1749-6632.1990.tb28048.x. [DOI] [PubMed] [Google Scholar]
- 28.Rossi F, Han Q, Li R, Li R, Rizzi M. J Biol Chem. 2004;279:50214–50220. doi: 10.1074/jbc.M409291200. [DOI] [PubMed] [Google Scholar]
- 29.Cooper AJL, Pinto JT, Krasnikov BF, Niatsetskaya ZV, Han Q, Li J, Vauzour D, Spencer JPE. Arch Biochem Biophys. 2008;474:72–81. doi: 10.1016/j.abb.2008.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cavallini D, Ricci G, Duprè S, Pecci L, Costa M, Matarese RM, Pensa B, Antonucci A, Solinas SP, Fontana M. Eur J Biochem. 1991;202:217–223. doi: 10.1111/j.1432-1033.1991.tb16365.x. [DOI] [PubMed] [Google Scholar]
- 31.Cooper AJL, Gross MJ. J Neurochem. 1977;28:771–778. doi: 10.1111/j.1471-4159.1977.tb10626.x. [DOI] [PubMed] [Google Scholar]
- 32.Commandeur JNM, Andreadou I, Rooseboom M, Out M, de Leur LJ, Groot E, Vermeulen NPE. J Pharmacol Exp Ther. 2000;294:753–761. [PubMed] [Google Scholar]
- 33.Cavallini D, De Marco C, Mondovi B, Mori BG. Enzymologia. 1960;22:161–173. [PubMed] [Google Scholar]
- 34.Braunstein AE, Goryachenkova EV. Adv Enzymol Relat Areas Mol Biol. 1984;56:1–89. doi: 10.1002/9780470123027.ch1. [DOI] [PubMed] [Google Scholar]
- 35.Fearon CW, Rodkey JA, Abeles RH. Biochemistry. 1982;21:3790–3794. doi: 10.1021/bi00259a011. [DOI] [PubMed] [Google Scholar]
- 36.Matsuo Y, Greenberg DM. J Biol Chem. 1959;234:507–515. [PubMed] [Google Scholar]
- 37.Brown FC, DeFoor MC. Eur J Biochem. 1974;46:317–322. doi: 10.1111/j.1432-1033.1974.tb03623.x. [DOI] [PubMed] [Google Scholar]
- 38.Han Q, Li J, Li J. Eur J Biochem. 2004;271:4804–4814. doi: 10.1111/j.1432-1033.2004.04446.x. [DOI] [PubMed] [Google Scholar]
- 39.Pinto JT, Krasnikov BF, Cooper AJL. J Nutr. 2005;136:S835–S841. doi: 10.1093/jn/136.3.835S. [DOI] [PubMed] [Google Scholar]
- 40.Cooper AJL. Anal Biochem. 1978;89:451–460. doi: 10.1016/0003-2697(78)90374-3. [DOI] [PubMed] [Google Scholar]
- 41.Pinto JT, Khomenko T, Szabo S, McLaren GD, Denton TT, Krasnikov BF, Jeitner TM, Cooper AJL. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:3434–3441. doi: 10.1016/j.jchromb.2009.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Washtien W, Cooper AJL, Abeles RH. Biochemistry. 1977;16:460–463. doi: 10.1021/bi00622a019. [DOI] [PubMed] [Google Scholar]
- 43.Rabenstein DL. Acc Chem Res. 1978;11:100–107. [Google Scholar]
- 44.Erni I, Geier G. Helv Chim Acta. 1979;62:1007–1015. [Google Scholar]
- 45.Rabenstein DL, Fairhurst MT. J Am Chem Soc. 1975;97:2086–2092. doi: 10.1021/ja00841a015. [DOI] [PubMed] [Google Scholar]
- 46.Pitts KE, Summers AO. Biochemistry. 2002;41:10287–10296. doi: 10.1021/bi0259148. [DOI] [PubMed] [Google Scholar]
- 47.Di Lello P, Benison GC, Valafar H, Pitts KE, Summers AO, Legault P, Omichinski JG. Biochemistry. 2004;43:8322–8332. doi: 10.1021/bi049669z. [DOI] [PubMed] [Google Scholar]
- 48.Potter MC, Elmer GI, Bergeron R, Albuquerque EX, Guidetti P, Wu HQ, Schwarcz R. Neuropsychopharmacology. 2010;35:1734–1742. doi: 10.1038/npp.2010.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saito K, Fujigaki S, Heyes MP, Shibata K, Takemura M, Fujii H, Wada H, Noma A, Seishima M. Am J Physiol Renal Physiol. 2000;279:F565–F572. doi: 10.1152/ajprenal.2000.279.3.F565. [DOI] [PubMed] [Google Scholar]
- 50.Toohey JI. Anal Biochem. 2011;413:1–7. doi: 10.1016/j.ab.2011.01.044. [DOI] [PubMed] [Google Scholar]
- 51.Birchmeier W, Wilson KJ, Christen P. J Biol Chem. 1973;248:1751–1759. [PubMed] [Google Scholar]
- 52.Kalogerakos TG, Oikonomakos NG, Dimitropoulos CG, Karni-Katsadima IA, Evangelopoulos AE. Biochem J. 1977;167:53–63. doi: 10.1042/bj1670053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pamp K, Bramey T, Kirsch M, De Groot H, Petrat F. Free Radic Res. 2005;39:31–40. doi: 10.1080/10715760400023671. [DOI] [PubMed] [Google Scholar]
- 54.Chang GG, Hsu RY. Biochemistry. 1977;16:311–320. doi: 10.1021/bi00621a024. [DOI] [PubMed] [Google Scholar]
- 55.Moran LK, Gutteridge JM, Quinlan GJ. Curr Med Chem. 2001;8:763–772. doi: 10.2174/0929867013372904. [DOI] [PubMed] [Google Scholar]
- 56.Schmidt B, Ho L, Hogg PJ. Biochemistry. 2006;45:7429–7433. doi: 10.1021/bi0603064. [DOI] [PubMed] [Google Scholar]
- 57.Summa D, Spiga O, Bernini A, Venditti V, Priora R, Frosali S, Margaritis A, Di Giuseppe D, Niccolai N, Di Simplicio P. Proteins. 2007;69:369–378. doi: 10.1002/prot.21532. [DOI] [PubMed] [Google Scholar]
- 58.Ragunathan N, Busi F, Pluvinage B, Sanfins E, Dupret JM, Rodrigues-Lima F, Dairou J. FEBS Lett. 2010;584:3366–3369. doi: 10.1016/j.febslet.2010.06.022. [DOI] [PubMed] [Google Scholar]
- 59.Frasco MF, Colletier JP, Weik M, Carvalho F, Guilhermino L, Stojan J, Fournier D. FEBS J. 2007;274:1849–1861. doi: 10.1111/j.1742-4658.2007.05732.x. [DOI] [PubMed] [Google Scholar]
- 60.Fuhr B, Rabenstein DL. J Am Chem Soc. 1973;95:6944–6950. doi: 10.1021/ja00802a013. [DOI] [PubMed] [Google Scholar]
- 61.Ishii I, Akahoshi N, Yu XN, Kobayashi Y, Namekata K, Komaki G, Kimura H. Biochem J. 2004;381:113–123. doi: 10.1042/BJ20040243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yin Z, Jiang H, Syversen T, Rocha JB, Farina M, Aschner M. J Neurochem. 2008;107:1083–1090. doi: 10.1111/j.1471-4159.2008.05683.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Aschner M, Eberle NB, Goderie S, Kimelberg HK. Brain Res. 1990;521:221–228. doi: 10.1016/0006-8993(90)91546-s. [DOI] [PubMed] [Google Scholar]
- 64.Tallan HH, Moore S, Stein WH. J Biol Chem. 1958;230:707–716. [PubMed] [Google Scholar]