

SUMMARY
The energy-sensing AMP-activated protein kinase (AMPK) is activated by low nutrient levels. Functions of AMPK, other than its role in cellular metabolism, are just beginning to emerge. Here we use a chemical genetics screen to identify direct substrates of AMPK in human cells. We find that AMPK phosphorylates 28 previously unidentified substrates, several of which are involved in mitosis and cytokinesis. We identify the residues phosphorylated by AMPK in vivo in several substrates, including protein phosphatase 1 regulatory subunit 12C (PPP1R12C) and p21 -activated protein kinase (PAK2). AMPK-induced phosphorylation is necessary for PPP1R12C interaction with 14-3-3 and phosphorylation of myosin regulatory light chain. Both AMPK activity and PPP1R12C phosphorylation are increased in mitotic cells and are important for mitosis completion. These findings suggest that AMPK coordinates nutrient status with mitosis completion, which may be critical for the organism’s response to low nutrients during development, or in adult stem and cancer cells.
INTRODUCTION
The ability to rapidly respond to changes in energy levels is essential for cells and organisms. AMPK plays a central role in the maintenance of energy homeostasis (Kahn et al., 2005), and has been implicated in longevity and tumor suppression (Apfeld et al., 2004; Greer et al., 2007a; Mair et al., 2011; Shackelford and Shaw, 2009). AMPK is a conserved heterotrimeric serine/threonine protein kinase composed of a catalytic alpha subunit, a scaffolding beta subunit, and a regulatory gamma subunit. AMPK is activated by a range of stimuli, including nutrient deprivation, exercise, anti-diabetic drugs, and cellular stresses, which lead to an increase in the AMP:ATP ratio (Kahn et al., 2005). AMP binding to the gamma subunit activates AMPK by allosterically activating the kinase and facilitating phosphorylation by upstream kinases (Hardie et al., 1999; Hawley et al., 2005), and by inhibiting dephosphorylation by protein phosphatases (Sanders et al., 2007b).
Once activated, AMPK phosphorylates a number of substrates involved in metabolic regulation, including acetyl-CoA carboxylase 1 (ACC1), to induce ATP-production and restore energy levels (Witters and Kemp, 1992; Woods et al., 1994). AMPK also phosphorylates several proteins in the TOR signaling pathway, including TSC2 (Inoki et al., 2003) and Raptor (Gwinn et al., 2008), resulting in the inhibition of protein translation, a high energy-consuming pathway. AMPK regulates gene expression through the phosphorylation of transcription factors (e.g. FOXO3 (Greer et al., 2007b)), co-activators (e.g. CRTC2 (Koo et al., 2005; Shaw et al., 2005)), histone deacetylases (Mihaylova et al., 2011), and histones (Bungard et al., 2010). AMPK has been proposed to promote cell cycle arrest at the G1 phase via phosphorylation of tumor suppressors such as p53 (Imamura et al., 2001; Jones et al., 2005), Rb (Dasgupta and Milbrandt, 2009), and p27Kip1 (Liang et al., 2007), although the phosphorylation site in some of these substrates diverges from the AMPK consensus motif (Gwinn et al., 2008). Emerging evidence suggests that AMPK might also regulate mitosis in Drosophila and human cells (Bettencourt-Dias et al., 2004; Dasgupta and Milbrandt, 2009; Lee et al., 2007; Vazquez-Martin et al., 2009a; Vazquez-Martin et al., 2011; Vazquez-Martin et al., 2009c). However, the exact nature of AMPK’s role in mitotic progression, and the mechanisms by which AMPK might control mitosis are not known.
Identifying substrates of AMPK in a systematic manner is a key step in understanding the cellular processes controlled by this energy-sensing protein kinase. Here we used a chemical genetic screen to identify direct in vivo substrates of one of the catalytic subunits of AMPK, AMPKα2, in human cells. We discovered 28 previously unidentified AMPK substrates that are enriched for proteins involved in chromosomal segregation, mitosis, cytokinesis, and cytoskeletal reorganization. We focused on two substrates, phosphatase 1 regulatory subunit 12C (PPP1R12C) and p21-activated protein kinase (PAK2) because they are both involved in the regulation of myosin regulatory light chain (MRLC), a crucial protein for mitotic progression. We found that AMPK is important for the phosphorylation of PPP1R12C and PAK2 in cells. Phosphorylation of PPP1R12C by AMPK is required for 14-3-3 binding and complete induction of MRLC phosphorylation. Both AMPK activity and phosphorylation of PPP1R12C are elevated during mitosis, and are important for mitotic progression. Thus, AMPK coordinates a network of proteins involved in mitosis completion, which may be essential for normal development, stem cell self-renewal, and cancer progression.
RESULTS
An analog-specific mutant of AMPKα2 can use bulky ATP analogs
To identify direct substrates of AMPKα2 in vivo, we used a chemical genetics approach (Alaimo et al., 2001). This approach is based on the fact that the ATP-binding pocket of protein kinases contains a conserved “gatekeeper residue” in close contact with the N6 position of the adenine ring of ATP. Replacement of this gatekeeper residue with a smaller amino-acid enables the mutant protein kinase (termed “analog-specific”) to use ATP analogs containing bulky groups at the N6 position (Allen et al., 2007) (Figure 1A). In contrast, the bulky ATP analogs are poor substrates for wild-type kinases due to the steric hindrance of the gatekeeper residue. N6-modified ATPγS nucleotides are also accepted by the analog-specific kinase and the transferred thiophosphate can be alkylated and recognized by a specific monoclonal antibody (Figure 1A) (Allen et al., 2005; Allen et al., 2007). This chemical genetic approach allows specific labeling of direct substrates of a protein kinase in cells.
Figure 1. Generation of an analog-specific version of AMPKα2.
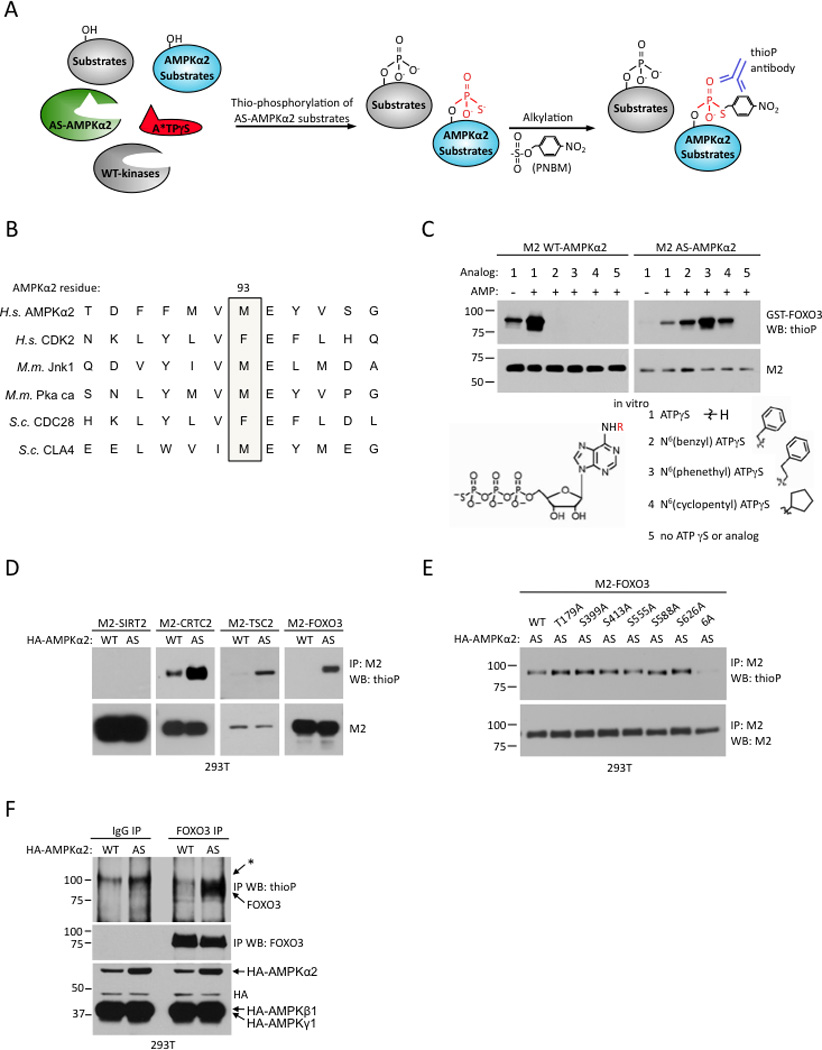
(A) Strategy for labeling substrates of analog-specific AMPKα2 (AS-AMPKα2). A*TPγS: bulky ATP analog. PNBM: p-nitrobenzyl mesylate.
(B) Identification of the gatekeeper amino acid in the ATP-binding pocket of human AMPKα2 by alignment with other kinases for which analog-specific mutants have been created (Allen et al., 2007; Niswender et al., 2002; Polson et al., 2001; Ubersax et al., 2003; Ventura et al., 2006; Weiss et al., 2000). H.s.: Homo sapiens; M.m.: Mus musculus; S.c: Saccharomyces cerevisiae.
(C) In vitro kinase assay using WT- or AS-AMPKα2 immunoprecipitated from 293T cells with GST-FOXO3 as a substrate and the indicated N6-substituted ATPγS analog as a phosphodonor. Red R: site of N6-alkyl group attachment 1–4 indicated at right. Western blots representative of 2 independent experiments.
(D) AS-AMPKα2 phosphorylates known AMPK substrates in cells in the presence of N6-(phenethyl) ATPγS. Substrates were immunoprecipitated with the antibodies to M2. Western blots representative of 2 independent experiments.
(E) AS-AMPKα2 no longer phosphorylates M2-FOXO3 with the point mutations in the AMPK phosphorylation sites in cells. 6A: T179A/S399A/S413A/S55A/S588/S626A.
(F) AS-AMPKα2 can phosphorylate endogenous FOXO3 in 293T cells. *: background band present in all samples. Western blots representative of 3 independent experiments.
Alignment of the amino-acid sequence of human AMPKα2 with that of other protein kinases for which analog-specific versions have been successfully generated revealed that methionine 93 is likely the gatekeeper residue of AMPKα2 (Figure 1B). To generate an analog-specific version of AMPKα2 (AS-AMPKα2), we mutated methionine 93 to a glycine residue. We then tested the in vitro kinase activity of wild-type (WT) and AS-AMPKα2 by incubating these kinases with recombinant GST-FOXO3, a known AMPK substrate (Greer et al., 2007b), in the presence of ATPγS or a variety of N6-substituted forms of ATPγS. WT-AMPKα2 uses ATPγS to phosphorylate GST-FOXO3, but is unable to use N6-modified bulky forms of ATPγS (Figure 1C). In contrast, AS-AMPKα2 phosphorylates FOXO3 to a lesser extent than WT-AMPKα2 using ATPγS, but is able to use the N6-substituted bulky forms of ATPγS, particularly N6-(phenethyl) ATPγS, to phosphorylate FOXO3 in vitro (Figure 1C). Thus, methionine 93 is the gatekeeper residue in AMPKα2 and mutation of this residue to a glycine allows AMPKα2 to use bulky analogs of ATP in vitro.
In vivo labeling of known AMPK substrates by AS-AMPKα2
We asked if AS-AMPKα2 could directly phosphorylate substrates in cells under relatively physiological conditions. To this end, we expressed HA-tagged versions of WT- or AS-AMPKα2, together with the β1 and γ1 subunits, as well as several known M2 (Flag)-tagged AMPK substrates in 293T cells and stimulated AMPK activity by serum starvation and addition of 2-deoxyglucose (2DG). N6-(phenethyl) ATPγS was added in the presence of digitonin, a mild detergent that permeabilizes the plasma membrane to allow the N6-(phenethyl) ATPγS to enter cells while largely preserving other intracellular compartments. Importantly, these conditions of in vivo labeling preserved endogenous AMPK signaling (Figure S1A), although they led to higher AMPK activation than glucose starvation, a physiological way of activating AMPK (Figure S1B). To test if substrates could be directly phosphorylated by AS-AMPKα2 in cells, known AMPK substrates were immunoprecipitated using an antibody to the M2 tag and the immune-complexes were analyzed by western blot using the thioP antibody. These experiments confirmed that AS-AMPKα2 phosphorylates known substrates of AMPK such as CRTC2, TSC2, and FOXO3 in cells (Figure 1D). Not all proteins that are highly overexpressed were phosphorylated by AS-AMPKα2 (e.g. SIRT2) (Figure 1D), indicating that AS-AMPKα2 is relatively specific for its substrates. Some substrates (e.g. CRTC2) were also labeled to a minor extent in cells expressing WT-AMPKα2 (Figure 1D), suggesting that either WT-AMPKα2 or an endogenous kinase is able to use N6-(phenethyl) ATPγS to phosphorylate some substrates. Mutating the gate-keeper site in AMPKα2 does not significantly alter AMPKα2 site-specificity, as a form of FOXO3 in which all six residues known to be phosphorylated by AMPK were mutated to alanines (6A FOXO3) (Greer et al., 2007b) was no longer phosphorylated by AS-AMPKα2 in cells (Figure 1E). Moreover, endogenous FOXO3 was also phosphorylated by AS-AMPKα2 (Figure 1F). Finally, specific labeling of FOXO3 was observed in a stable cell line with more physiological levels of AS-AMPKα2 and with endogenous levels of β and γ subunits (Figure S1C). Thus, this chemical genetic approach is an effective method for identifying direct endogenous substrates of AMPKα2 in a relatively physiological cellular context.
Screen for direct AMPKα2 substrates in vivo using AS-AMPKα2
To identify previously unidentified AMPKα2 substrates, we expressed WT- or AS-AMPKα2 together with the β1 and γ1 subunits in 293T cells (Figure 2A). After activation of AMPK by serum starvation and 2DG, in vivo phosphorylation with N6-(phenethyl) ATPγS was performed. We used the thioP antibody to immunoprecipitate the alkylated protein products of the in vivo phosphorylation. The immunoprecipitated proteins were resolved by SDS-PAGE and analyzed by western blot. More proteins were labeled in cells expressing the AS-AMPKα2 than WT-AMPKα2, both before and after immunoprecipitation (Figure 2B), indicating that a number of proteins were specifically phosphorylated by AS-AMPKα2. The entire lanes of proteins immunoprecipitated from cells expressing WT- and AS-AMPKα2 were cut, the proteins extracted from these strips were digested with trypsin, and the identity of the resulting peptides was determined by tandem mass spectrometry (Figure 2A). Due to the abundance of peptides from the heavy and light chains of IgG, only proteins with a molecular weight higher than the IgG heavy chain (55 kDa) were analyzed. Two independent biological replicates were performed (Figure 2C). We focused on proteins for which at least 10-fold more peptides were identified in cells expressing AS-AMPKα2 compared to cells expressing WT-AMPKα2. Using this stringent cut-off, we identified 98 different proteins in the first experiment and 177 different proteins in the second experiment that were labeled specifically by AS-AMPKα2 (Figure 2C; Table S1). Thirty-two proteins were identified in both biological replicates (Table 1), thereby representing the most likely direct substrates of AMPKα2 in this screen. Among these 32 proteins are three previously known substrates of AMPK: ACC1 (Witters and Kemp, 1992; Woods et al., 1994), CRTC2 (Koo et al., 2005; Shaw et al., 2005), and kinesin light chain 1 (McDonald et al., 2009) (Table 1). We also identified AMPKα2 as an autophosphorylation substrate, which has been previously reported (Woods et al., 2003), although it may also be due to the fact that AS-AMPKα2 was overexpressed. Not all known AMPK substrates were found (e.g. TSC2, FOXO3, Raptor), possibly due to low abundance of some proteins or to molecular weights lower than 55 kDa. Overall, these results indicate that this chemical genetic screen is sensitive enough to identify previously known AMPK substrates.
Figure 2. Identification of direct substrates of AMPKα2 in cells by a chemical genetic screen.
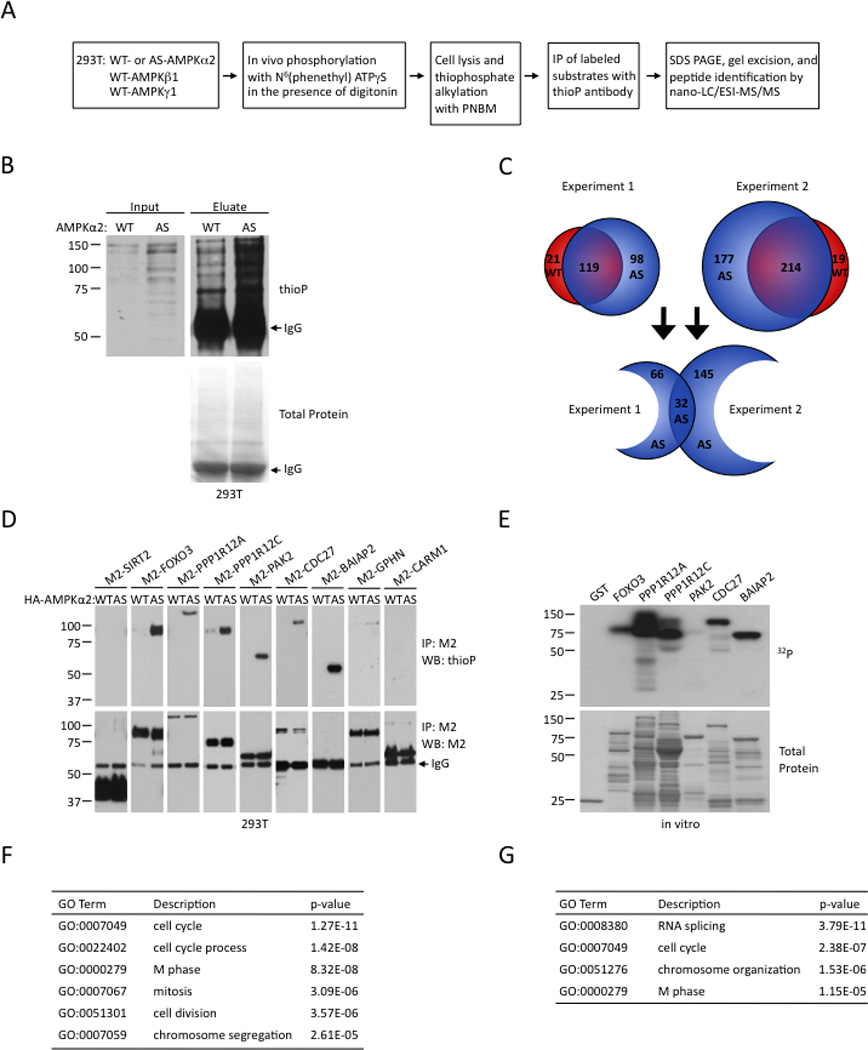
(A) Strategy for identifying endogenous direct substrates of AS-AMPKα2 in cells.
(B) Phosphorylation of endogenous AMPKα2 substrates in 293T cells expressing WT- or AS-AMPKα2. Left panel: western blot of lysates run before immunoprecipitation (input). Right panel: western blot of proteins released from the beads after the immunoprecipitation (eluate).
(C) Results of two independent labeling and substrate isolation experiments. Red and blue circles represent proteins found in cells expressing WT- and AS-AMPKα2 respectively. The numbers indicate the number of proteins identified in each category. The full list of proteins identified in each biological replicate is provided in Table S1.
(D) Validation of six AMPKα2 substrates identified in the chemical genetic screen by in vivo labeling with AS-AMPKα2. Note that BAIAP2 has a molecular weight similar to the IgG heavy chain. Western blots representative of at least 2 independent experiments.
(E) In vitro phosphorylation of five AMPKα2 substrates identified in the chemical genetic screen by purified WT-AMPK in the presence of radiolabeled γ32P-ATP. Western blots representative of 2 independent experiments.
(F) Selected Gene Ontology (GO) terms for AMPK substrates identified by the chemical genetic screen. The full list of enriched GO terms is in Table S2 and Table S3.
(G) Selected Gene Ontology (GO) terms for AMPK substrates when compared to proteins with molecular weights greater than 55 kDa. The full list of enriched GO terms is in Table S4.
Table 1. List of AMPKα2 substrates.
Numbers indicate the number of peptides found for each protein in each biological replicate. Proteins are listed in descending order according to the number of peptides identified. Bold: substrates validated in this study. Italics: previously identified substrates of AMPK. The full list of proteins identified in each biological replicate is provided in Table S1.
| Exp 1 | Exp 2 | Protein Name |
|---|---|---|
| 32 | 23 | Protein phosphatase 1 regulatory subunit 12A |
| 13 | 12 | Acetyl-CoA carboxylase 1 |
| 7 | 14 | Anaphase-promoting complex subunit 1 |
| 11 | 9 | Cell division cycle protein 27 homolog |
| 7 | 10 | Golgin subfamily A member 4 |
| 5 | 10 | Protein phosphatase 1 regulatory subunit 12C |
| 10 | 4 | Brain-specific angiogenesis inhibitor 1-associated protein 2-like protein 1 |
| 6 | 8 | Cation-independent mannose-6-phosphate receptor precursor |
| 7 | 6 | Transportin-1 |
| 11 | 1 | Brain-specific angiogenesis inhibitor 1-associated protein 2 |
| 10 | 2 | Rab11 family-interacting protein 1 |
| 6 | 6 | TRAF2 and NCK-interacting protein kinase |
| 4 | 5 | Nck-associated protein 1 |
| 5 | 4 | Sodium bicarbonate cotransporter 3 |
| 2 | 5 | Gephyrin |
| 2 | 5 | Misshapen-like kinase 1 |
| 4 | 2 | Protein transport protein Sec24A |
| 2 | 4 | Splicing factor 3 subunit 1 |
| 3 | 2 | Apoptosis-stimulating of p53 protein 2 |
| 2 | 3 | Kinesin light chain 1 |
| 2 | 3 | Protein KIAA1219 |
| 3 | 2 | Serine/threonine-protein kinase PAK2 |
| 2 | 2 | 5'-AMP-activated protein kinase catalytic subunit alpha-2 |
| 1 | 3 | AP-2 complex subunit beta-1 |
| 2 | 2 | CREB-regulated transcription coactivator 2 |
| 3 | 1 | Histone-arginine methyltransferase CARM1 |
| 1 | 3 | Hypoxia up-regulated protein 1 precursor |
| 2 | 2 | Putative eukaryotic translation initiation factor 3 subunit |
| 1 | 1 | General vesicular transport factor p115 |
| 1 | 1 | Glucosamine-fructose-6-phosphate aminotransferase [isomerizing] 1 |
| 1 | 1 | Neurofilament light polypeptide |
| 1 | 1 | Protein phosphatase 1 regulatory subunit 12B |
To confirm that the proteins identified in the chemical genetic screen were indeed substrates of AMPK, we tested seven of them (protein phosphatase 1 regulatory subunit 12 A (PPP1R12A), PPP1R12C, PAK2, CDC27, brain-specific angiogenesis inhibitor 1-associated protein 2 (BAIAP2), gephyrin (GPHN), and CARM1) using the candidate-based approach in cells described in Figure 1D. Six of these seven proteins were specifically labeled by AS-AMPKα2 (Figure 2D), indicating that they are direct substrates of AMPKα2 in human cells. CARM1 was the only protein that was not labeled by AS-AMPKα2 in the validation of the screen (Figure 2D), possibly because it was co-precipitated with a yet-to-be identified substrate of AMPKα2.
To confirm that these substrates are phosphorylated directly by AMPK, we performed in vitro kinase assays using WT-AMPK (a mixture of AMPKα1 and α2 containing complexes) purified from rat liver, γ32P-ATP, and recombinant substrates purified from E. coli. PPP1R12A, PPP1R12C, PAK2, CDC27, and BAIAP2 were all phosphorylated in vitro by WT-AMPK (Figures 2E, 3E, and 3G), although CARM1 was not (Figure S2), providing additional evidence that PPP1R12A, PPP1R12C, PAK2, CDC27, and BAIAP2 are direct substrates of AMPK. The fact that these proteins are phosphorylated by WT-AMPK also confirms that their identification in the chemical genetic screen is unlikely to have resulted from altered substrate specificity of AS-AMPKα2. Thus, the chemical genetic screen is an effective approach for the identification of direct substrates of AMPKα2 in cells, and could be used for other protein kinases.
Figure 3. Identification of AMPK phosphorylation sites in four AMPKα2 substrates.
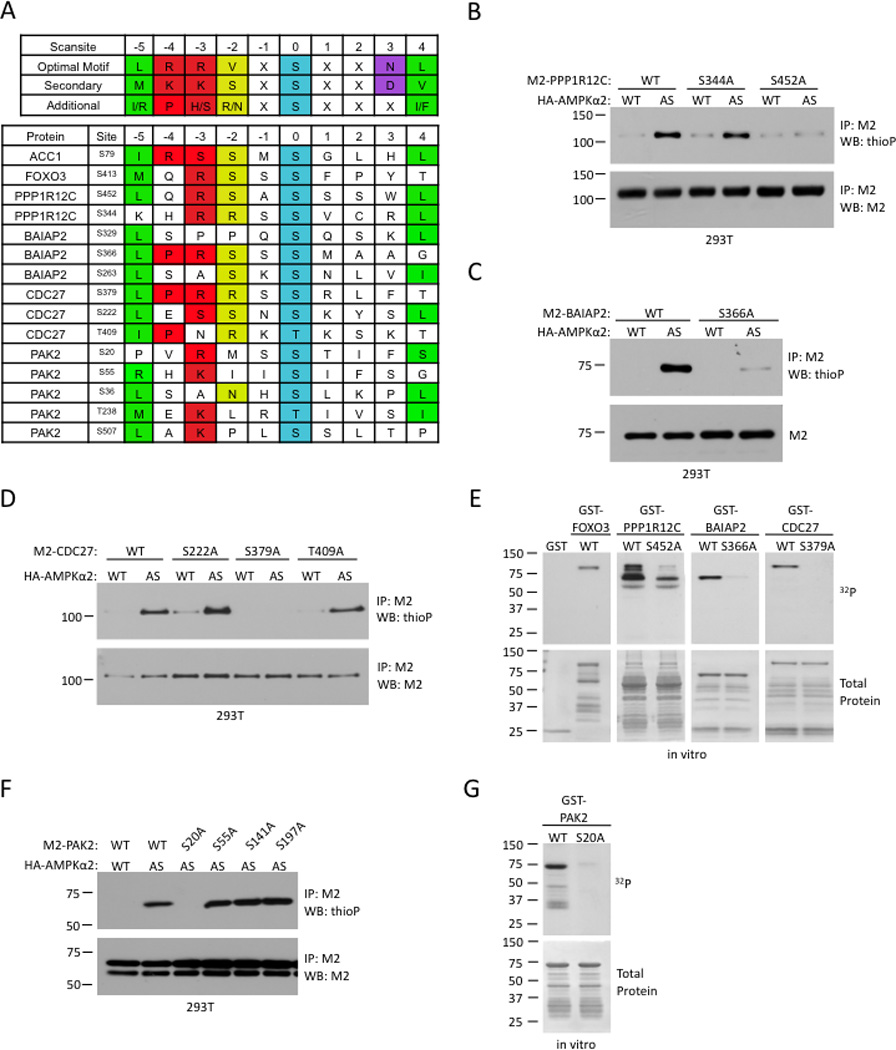
(A) Scansite prediction of potential AMPK phosphorylation sites. The AMPK consensus phosphorylation motif, derived from Gwinn et al (2008), is shown on the top in single letter amino acid code. Positions denoted as ‘X’ showed some additional modest selectivities among amino acids, but lack the strong discrimination shown in the specified positions. Known AMPK phosphorylation sites in ACC1 and FOXO3 are shown as comparisons.
(B)–(D) In vivo identification of AMPKα2 phosphorylation sites in PPP1R12C (B), BAIAP2 (C), and CDC27 (D). Analysis was carried out as in Figure 1D.
(E) In vitro validation of AMPK phosphorylation sites in PPP1R12C, BAIAP2, and CDC27. Kinase assays were carried out as in Figure 2E.
(F) In vivo testing of putative phosphorylation sites identified by tandem mass spectrometry for PAK2. Analysis was carried out as described in Figure 1D.
(G) In vitro validation of AMPK phosphorylation sites in PAK2. Kinase assay was carried out as in Figure 2E. All panels are representative of 2 independent experiments.
AMPKα2 substrates are enriched for proteins involved in mitosis
Global analysis of AMPKα2 substrates identified in either biological replicate of our screen using GO, GSEA and PANTHER revealed that these AMPK substrates displayed a significant enrichment for proteins involved in cell cycle, particularly mitosis and chromosomal segregation (Figure 2F; Tables S2 and S3). The enrichment for “M-phase” was still highly significant even when the comparison list was restricted to proteins larger than 55 kDa (Figure 2G; Table S4). These observations raise the intriguing possibility that AMPKα2 may coordinate different aspects of mitosis in cells. For example, APC1 and CDC27 are two components of the anaphase-promoting complex (Peters, 2006). PPP1R12A/MYPT1, PPP1R12B/MYPT2, and PPP1R12C/MBS85 are known to inhibit the activity of the myosin regulatory light chain (MRLC) (Ito et al., 2004), a protein that regulates cytokinesis (Komatsu et al., 2000). The protein kinase PAK2 activates MRLC by direct phosphorylation and is involved in cytoskeletal reorganization (Tuazon and Traugh, 1984). Finally, BAIAP2 is a protein required for actin-myosin dependent processes and motility (Chauhan et al., 2009). Thus, AMPKa2 may regulate a network of substrates to coordinate chromosomal segregation, mitosis, cytokinesis, and cytoskeletal reorganization.
Identification of AMPK phosphorylation sites in four AMPKα2 substrates
We identified the specific residues phosphorylated by AMPKα2 in PPP1R12C, BAIAP2, CDC27, and PAK2. Analysis of these proteins using Scansite with an optimized AMPK phosphorylation consensus motif derived from (Gwinn et al., 2008) revealed residues that conformed to the AMPK consensus motif in each substrate (Figure 3A). We mutated these sites to non-phosphorylatable alanines and determined the extent of phosphorylation each mutant using the candidate-based in vivo labeling method. Using this approach for PPP1R12C, BAIAP2, and CDC27, we found that mutation of a single serine to alanine (S452, S366, and S379 respectively) resulted in almost a complete loss of AMPK phosphorylation in these proteins (Figures 3B–3D). The mutation of these sites also diminished phosphorylation of PPP1R12C, BAIAP2, and CDC27 by WT-AMPK in vitro (Figure 3E), indicating that the specificity of AMPK for these sites is not affected by the gatekeeper methionine to glycine mutation at position 93 in AS-AMPKα2. Thus, S452, S366, S379, which are part of an AMPK consensus motif, are the major AMPKα2 phosphorylation sites in PPP1R12C, BAIAP2, and CDC27 respectively.
PAK2 has several potential AMPK phosphorylation sites (Figure 3A). Tandem mass spectrometry analysis of PAK2 phosphorylated in vitro by WT-AMPK revealed that S20, S55, S141, and S197 of PAK2 were phosphorylated in vitro by AMPK (Table S5). The mutation of serine 20 to an alanine abolished the phosphorylation of PAK2 by AS-AMPKα2 in cells and by WT-AMPK in vitro, whereas the mutation of the other serines had no or little impact (Figures 3F and 3G). Thus, S20, which is part of an AMPK consensus motif, is the major site of AMPK phosphorylation on PAK2.
AMPK phosphorylates PPP1R12C and PAK2 in human cells
We focused on AMPK substrates potentially involved in mitosis. The PPP1R12 family of protein phosphatase regulatory subunits stood out because three members of this family were identified in the screen. PPP1R12C limits the phosphorylation of MRLC (Ito et al., 2004), a protein involved in mitosis (Komatsu et al., 2000). Intriguingly, the kinase PAK2, which was also found in the screen, promotes MRLC phosphorylation (Chew et al., 1998; Tuazon and Traugh, 1984). Thus, AMPK might regulate a network of antagonistic signaling molecules to coordinate mitotic progression.
To determine if AMPK promotes the phosphorylation of PPP1R12C in cells, we generated a phospho-specific antibody to S452 of PPP1R12C (P-S452). This P-S452 antibody was specific to the phosphorylated form of PPP1R12C in that it recognized WT-PPP1R12C in the presence of AMPK, but did not recognize the PPP1R12C S452A mutant or WT-PPP1R12C that had not been incubated with AMPK (Figure S3A). The P-S452 antibody was also specific to the endogenous phosphorylated form of PPP1R12C in human cells, as shown using siRNA or shRNA to PPP1R12C (Figure S3B, Figure 4F). Using this antibody, we showed that phosphorylation of M2-PPP1R12C at S452 was enhanced when 293T cells were treated with the AMPK activator 2DG and decreased when cells were treated with compound C, a chemical inhibitor of AMPK (Zhou et al., 2001) (Figure 4A). The phosphorylation of endogenous PPP1R12C at S452 in 293T cells was increased in response to 2DG or the highly specific AMPK activator A-769662 (Cool et al., 2006; Goransson et al., 2007; Sanders et al., 2007a) (Figure 4B). The phosphorylation of M2-PPP1R12C at S452 was also increased in response to 2DG and A-769662 in U2OS cells, and this phosphorylation was reduced when both catalytic subunits of AMPK were knocked-down with siRNA (Figure 4C). Similar results were observed with endogenous PPP1R12C (Figure 4D; Figure S3C). Importantly, endogenous PPP1R12C phosphorylation at S452 was increased in response to glucose or nutrient starvation (Figures 4E and 4F), which are physiological ways of activating AMPK. Similar results were obtained for PAK2 (Figure S4). Together, these results indicate that AMPK phosphorylates endogenous PPP1R12C at S452 and PAK2 at S20 in human cells.
Figure 4. AMPK promotes the phosphorylation of PPP1R12C in human cells.

(A) Phosphorylation of M2-PPP1R12C at S452 is enhanced in response to the AMPK activator 2-deoxyglucose (2DG) and inhibited by the AMPK inhibitor compound C (CC) in 293T cells. Cell lysates were analyzed by western blot.
(B) Phosphorylation of endogenous PPP1R12C at S452 is stimulated in response to 2DG or A-769662 (A) in 293T cells. Cell lysates were analyzed by western blot.
(C) Phosphorylation of M2-PPP1R12C at S452 in U2OS cells in response to activation of AMPK by 2DG or A-769662 (A) is diminished in the presence of siRNAs to both AMPKα1 and α2 (+) compared to scrambled control siRNAs (−). Cell lysates were analyzed by western blot.
(D) The phosphorylation of endogenous PPP1R12C at S452 in response to A-769662 (A) is diminished in U2OS cells expressing an shRNA to both AMPKα1 and α2 (AMPKα1/α2) compared to control cells (vector). Western blots representative of 2 independent experiments.
(E–F) Glucose and nutrient starvation promotes the phosphorylation of endogenous PPP1R12C at S452 and PAK2 at S20 in U2OS cells. Western blots representative of 2 independent experiments.
Phosphorylation by AMPK promotes the interaction between PPP1R12C and 14-3-3ζ
We examined the consequences of AMPK phosphorylation on PPP1R12C. PPP1R12A, another regulatory subunit of myosin phosphatase, has been shown to interact with 14-3-3 (Koga and Ikebe, 2008). We therefore asked if phosphorylation by AMPK affects a potential interaction between PPP1R12C and 14-3-3. Co-immunoprecipitation studies revealed that PPP1R12C interacted with 14-3-3ζ in 293T cells (Figure 5A). Interestingly, the interaction between PPP1R12C and 14-3-3ζ was abolished by mutation of serine 452 to alanine in PPP1R12C (Figure 5A). In contrast, this mutation did not affect the interaction between PPP1R12C and the catalytic phosphatase subunit PP1Cβ (Figure S5A). The interaction between PPP1R12C and 14-3-3ζ was decreased when AMPKα1 and α2 were both knocked-down (Figure 5B). Conversely, the interaction between PPP1R12C and 14-3-3ζ was increased in response to the specific AMPK activator A-769662 and to glucose starvation (Figure 5C). Collectively, these results show that phosphorylation of S452 promotes the interaction between PPP1R12C and 14-3-3ζ , but does not regulate the interaction with the catalytic subunit of the phosphatase (Figure 5D). The interaction between PPP1R12C and 14-3-3ζ may inactivate the PPP1R12C–containing phosphatase complex in vivo.
Figure 5. AMPK phosphorylation of PPP1R12C and PAK2 affects the interaction between PPP1R12C and 14-3-3ζ and MRLC phosphorylation.

(A) M2-14-3-3ζ co-immunoprecipitates with WT, but not S452A, T7-PPP1R12C in 293T cells stimulated with 2-deoxyglucose (2DG). Western blots representative of 3 independent experiments.
(B) M2-14-3-3ζ no longer co-immunoprecipitates with WT PPP1R12C in 293T cells expressing an shRNA to both AMPKα1 and α2 (AMPKα1/α2) in response to 2-deoxyglucose (2DG). Western blots representative of 2 independent experiments.
(C) The interaction between PPP1R12C and 14-3-3ζ is increased in response to AMPK activation by 2-deoxyglucose (2DG) for 5 min, A-769662 (A) for 1 hour, or glucose starvation (GS) for 2 hours. Immune-complexes were analyzed by western blot.
(D) Model summarizing the effect of AMPK phosphorylation on the interaction between PPP1R12C and 14-3-3ζ.
(E) AMPK activation with A-769662 (A) leads to an increase in MRLC phosphorylation at S19 and T18/S19 in U2OS cells. Western blots representative of 3 independent experiments.
(F) Phosphorylation of MRLC at S19 is slightly, but significantly reduced in U2OS cells expressing PPP1R12C S452A in response to A-769662 (A). Western blots representative of 4 independent experiments. Quantification is presented in Figure S5E.
(G) Phosphorylation of MRLC at S19 is reduced in U2OS cells expressing PAK2 S20A in response to A-769662 (A). Western blots representative of 2 independent experiments.
AMPK indirectly regulates MRLC phosphorylation, in part via PPP1R12C and PAK2
PPP1R12C and PAK2 directly regulate MRLC phosphorylation at S19 in an opposing manner (Matsumura and Hartshorne, 2008; Tuazon and Traugh, 1984). We asked whether AMPK affects MRLC phosphorylation in cells. Stimulation of cells with the AMPK activator A-769662 led to an increased phosphorylation of endogenous MRLC at S19, which can be visualized either by an antibody to phospho-S19 or to phospho-T18/S19 (Figure 5E). Conversely, knocking-down both AMPKα1 and α2 resulted in a modest but significant (p=0.0012) reduction in M2-MRLC phosphorylation at S19 in 293T cells (Figures S5B and S5C). MRLC was not phosphorylated directly by AMPK in vivo, as shown by in vivo labeling using AS-AMPKα2 (Figure S5D). These results support the notion that AMPK affects MRLC phosphorylation at S19 indirectly.
We next tested if AMPK regulates MRLC phosphorylation via PPP1R12C and PAK2. The increase in MRLC S19 phosphorylation in response to A-769662 was slightly, but reproducibly, reduced in cells expressing the non-phosphorylatable form of PPP1R12C, S452A (Figure 5F; Figure S5E). Expression of a mutant of PAK2 at the AMPK phosphorylation site (S20A) strongly reduced the phosphorylation of MRLC in response to AMPK activation (Figure 5G). Phosphorylation by AMPK did not affect PAK2’s activity toward MRLC in vitro (M.R.B. and A.B., unpublished data), although it may alter PAK2’s accessibility to substrates in cells. Glucose starvation, a physiological way of activating AMPK, led to an increase in the phosphorylation of endogenous AMPK, PPP1R12C, PAK2, and MRLC (Figure 4E). Together, these data suggest that AMPK indirectly regulates MRLC phosphorylation in part by phosphorylating PPP1R12C and PAK2.
AMPK activity and PPP1R12C phosphorylation are high in mitotic cells
Given the enrichment in AMPK substrates for proteins involved in mitosis, we examined the phosphorylation of endogenous AMPK, PPP1R12C, and PAK2 throughout mitosis. U2OS cells were synchronized with the prometaphase blockers monastrol or nocodazole, and cells were released from the block for different lengths of time. Phosphorylation of AMPK at T172 was high in prometaphase of mitosis and decreased one hour after release from the monastrol or nocodazole block (Figures 6A; Figures S6A and S6B). Phosphorylation of PPP1R12C at S452 was also high in prometaphase and tended to decrease one hour after the release from the monastrol or nocodazole block, before significantly increasing at the end of mitosis (Figures 6A; Figures S6A and S6B). In contrast, phosphorylation of PAK2 at S20 was not significantly altered during mitotic progression (Figures 6A; Figures S6A and S6B).
Figure 6. AMPK activity and PPP1R12C phosphorylation are elevated in mitosis and are important for mitotic progression.

(A) Phosphorylation of endogenous AMPK, PPP1R12C, and PAK2 in U2OS cells synchronized by thymidine followed by monastrol block and collected at different time points after release from the block (blue bar). Cell lysates were analyzed by western blot.
(B) AMPK FRET signal is high when cells enter mitosis and starts to decrease around the time the cells initiated cytokinesis in U2OS cells expressing AMPKAR, a fluorescent reporter for AMPK activity. AMPK activity was measured at 10 min intervals. Red: increased AMPK activity.
(C) U2OS cells were synchronized by thymidine followed by monastrol block and chemicals were added 1.5 hours after the release from the monastrol block (blue bar) and replating of mitotic cells. Cells were fixed 4.5 hours after the addition of chemicals. The number of multinucleated cells was normalized to the number observed in non-treated control cells for each experiment. Means +/− SEM of 3 independent experiments are shown, except for the LY-294002 condition, which represents 2 independent experiments. *p<0.05 by one-way ANOVA analysis. ns: not statistically significant.
(D) U2OS cells expressing WT or S452A PPP1R12C were synchronized by thymidine block. Cells were fixed at the indicated time post-release from the block (blue bar). Percent of multinucleated cells in each condition are shown. Means +/− SEM of 3 independent staining for a synchronization experiment. *p<0.05, **p<0.01, ***p<0.001 by 2-way ANOVA, with Bonferroni post-hoc tests.
(E) AMPK directly phosphorylates a number of substrates involved in coordinating different aspects of mitosis.
To independently follow AMPK activity in mitosis, we used fluorescence resonance energy transfer (FRET) in live cells with a fluorescence reporter for AMPK activity, AMPKAR (Tsou et al., 2011). AMPKAR fluorescence reached a peak just before U2OS cells divide (Figure 6B), indicating that AMPK is highly active in the early phases of mitosis. The levels of AMPK activity slightly decreased as U2OS cells undergo cytokinesis (Figure 6B). Similar results were observed in other cell types (Figures S6C and S6D). Together, these results indicate that AMPK activity and the phosphorylation of PPP1R12C are high in mitotic cells and decrease as cells progress through mitosis.
Perturbation of AMPK activity impairs mitosis progression
To test the function of AMPK activity in mitosis, we acutely perturbed AMPK activity after synchronizing cells in mitosis and counted the number of multinucleated cells. Compound C, an inhibitor of AMPK, led to a significant increase in the number of multinucleated cells, similar to BTO-1, an inhibitor of the mitotic kinase Polo-like kinase (PLK) (Figure 6C, Figure S7A). In contrast, an inhibitor of PI3K (LY-294002) had no effect on the number of multinucleated cells (Figure 6C). Because compound C has other targets in addition to AMPK (Bain et al., 2007; Vogt et al., 2011), we used alternative approaches to perturb AMPK. U2OS cells expressing shRNA to both AMPKα1 and α2 exhibited a slight increase in multinucleated cells at some time points after release from a mitotic block, although they did not display an overall increase in multinucleated cells (Figure S7B, B.E.S. and A.B., data not shown). Interestingly, acutely activating AMPK with A-769662, a highly specific activator of AMPK, significantly increased the number of multinucleated cells (Figure 6C). Activating AMPK with metformin has also been found to increase the number of multinucleated cells (Vazquez-Martin et al., 2009b). These observations are reminiscent of what has been shown for important regulators of mitosis (e.g. Mad2, PLK), whose inhibition or activation result in mitotic defects (Brennan et al., 2007; Conn et al., 2000; Michel et al., 2004; Sotillo et al., 2007). Collectively, these experiments suggest that proper AMPK activity is important for mitotic progression.
AMPK phosphorylation site in PPP1R12C affects mitotic progression
We asked if the regulation of PPP1R12C by AMPK is important for mitosis completion. U2OS cells stably expressing PPP1R12C S452A had a significant increase in the percent of multinucleated cells compared to cells expressing WT-PPP1R12C (Figure S7C; Figure 6D) and to cells expressing the empty vector (Figure S7D). These results suggest that the mutation of the AMPK phosphorylation site in PPP1R12C results in defects in mitotic progression and cytokinesis. WT PPP1R12C also led to an increase in the percentage of multinucleated cells, possibly because overexpressed WT PPP1R12C may no longer be phosphorylated by endogenous AMPK and may act as a ‘dominant-negative’. In contrast, expression of WT or S20A PAK2 did not result in an increase in multinucleated cells compared to control cells (Figure S7E). Thus, the phosphorylation of PPP1R12C by AMPKα2 appears to be more important for proper cytokinesis than phosphorylation of PAK2. Interestingly, cells expressing S452A PPP1R12C also had an overall slight, but significant increase in the percent of cells in anaphase with lagging chromosomes compared to cells expressing WT PPP1R12C (Figures S8A and S8B), which is likely a result of defects in chromosomal segregation. Thus, AMPK phosphorylation of PPP1R12C might coordinate chromosomal segregation and cytokinesis.
Our findings indicate that AMPKα2, a critical energy-sensing protein kinase, directly phosphorylates a series of substrates enriched for proteins involved in mitosis completion and chromosomal segregation. AMPK activity and the phosphorylation of one of its substrates, PPP1R12C, are elevated in mitosis. Altering AMPK activity during mitosis impairs mitotic completion. Cells expressing a mutant of PPP1R12C that can no longer be phosphorylated by AMPK also exhibit mitotic defects. Importantly, the residue phosphorylated by AMPK in PPP1R12C is not part of a consensus for the other kinases targeted by compound C or for well-known mitotic kinases (e.g. PLK, CDK1). Taken together, these data support the notion that AMPK regulates mitotic completion.
DISCUSSION
The chemical genetic screen presented here provides a rapid and physiological means of identifying direct in vivo substrates of a protein kinase in cells, and could be applied to other kinases. Our screen was performed in gently permeabilized cells, thus preserving, at least in part, the physiological content of cells. A potential limitation of this screen is that it did not identify all of the previously known AMPK substrates (e.g. TSC2, FOXO3). Improved immunoprecipitation efficiency with the thioP antibody or the use of a peptide capture approach (Blethrow et al., 2008) should help the identification of low-abundant and low molecular weight substrates. Another limitation is that the screen was performed in cells overexpressing the AS-AMPKα2, which could lead to artifactual phosphorylation. However, independent confirmation of the phosphorylation of two endogenous substrates (PPP1R12C and PAK2) in cells supports the notion that this chemical genetic approach is a valid way of identifying physiologically relevant substrates of a protein kinase.
While previously known substrates of AMPK have largely implicated this kinase in metabolism, G1 arrest, and transcriptional regulation, the substrates identified here show a significant enrichment for proteins involved in the regulation of mitosis. Phosphorylation of PPP1R12C at the AMPK site appears to be important for the normal completion of mitosis and chromosomal segregation in mammalian cells. We do not rule out the possibility that the effects of AMPK activity on cytokinesis are dependent on substrates other than PPP1R12C or that the effects of phosphorylation of S452 of PPP1R12C on mitosis are dependent on other kinases. For example, the network of AMPK substrates involved in mitosis may extend to APC1 and CDC27, two components of the anaphase promoting complex identified in our screen. Our results are consistent with the G2/M arrest observed in several cell types lacking AMPK, in drosophila and mice (Bettencourt-Dias et al., 2004; Dasgupta and Milbrandt, 2009; Gwinn et al., 2008), with the defects in chromosomal segregation in drosophila larvae lacking AMPK (Lee et al., 2007), and with the observation that AMPK is highly phosphorylated at the mitotic apparatus in cells (Vazquez-Martin et al., 2009a; Vazquez-Martin et al., 2011; Vazquez-Martin et al., 2009c). Our results further extend prior studies by proposing a mechanism of action for AMPK in the control of mitosis. Thus, AMPK may control a network of proteins involved in coordinating various aspects of mitotic completion, including anaphase and cytokinesis.
Energy deprivation, by regulating AMPK, could promote the completion of previously initiated mitosis to allow the cell to arrest at the G1/S checkpoint and await more favorable nutrient conditions. Alternatively, AMPK could play a role in mitosis regardless of the energy status of the cell. In fact, AMPK phosphorylation has been shown to increase in mitosis independently of the energy status of the cell (Vazquez-Martin et al., 2009a), and the percent of multinucleated cells did not appear to change when cells were deprived of glucose (M.R.B. and A.B., unpublished data). Phosphorylation of substrates involved in orchestrating mitosis and cytoskeletal rearrangements by AMPK could result in coordinating AMPK activity with other cellular fates tightly associated with the birth of new cells, such as polarity and migration. Coordinated completion of mitosis, polarization, and migration by AMPK could be critical for a number of physiological functions, including stem cell homeostasis, and for preventing pathological conditions, such as cancer.
EXPERIMENTAL PROCEDURES
Chemical genetic screen for AMPK substrates in cells
293T cells (2 × 106 cells/10-cm dish) were transfected with 6.67 µg of M2-WT- or AS-AMPKα2, 6.67 µg HA-AMPKβ1, and 6.67 µg HA-AMPKγ1 using the calcium phosphate method. Twenty-five 10-cm plates were used for each condition. Forty-eight hours after transfection, the cells were incubated for 2 hours in serum-free DMEM, prior to stimulation with 100 mM 2DG for 5 min. Following stimulation, 500 µl of phosphorylation buffer (20 mM HEPES [pH 7.3], 100 mM KOAc, 5 mM NaOAc, 2 mM MgOAc2, 1 mM EGTA, 10 mM MgCl2, 0.5 mM DTT, 5mM creatine phosphate (Calbiochem), 57 µg/ml creatine kinase (Calbiochem), 30 µg/ml digitonin, 5 mM GTP, 0.1 mM ATP, 0.1 mM N6-(phenethyl) ATPγS, 0.45 mM AMP, 1X phosphatase inhibitor cocktail I and II (Sigma), and 1X complete protease inhibitors, EDTA-Free [Roche]) was added. After shaking gently for 20 min at room temperature, the cells for each condition were pooled and EDTA was added to a concentration of 20 mM. The cells were then sonicated 6 times at 4.0 W for 30 sec and the cell pellets were removed by centrifugation. PNBM (p-nitrobenzyl mesylate, Epitomics) was added to the lysates to a final concentration of 2.5 mM, and the samples were then incubated for 1 hr at room temperature. PNBM was removed and samples were exchanged onto RIPA buffer (100 mM Tris pH8, 300 mM NaCl, 2% NP-40, 0.2% SDS, and 20 mM EDTA) using PD-10 columns (Amersham). The protein fractions were then pre-cleared by incubation with protein G agarose beads (Sigma) for 3 hrs at 4°C. The pre-cleared extracts were incubated with protein G agarose coupled to the thioP antibody and incubated at 4°C overnight. The beads were washed 5 times in RIPA buffer, and the bound proteins were eluted with SDS sample buffer. Proteins were resolved by SDS-PAGE and visualized by staining with colloidal blue (Invitrogen).
Mitosis assay on stable cell lines
U2OS cells stably expressing empty vector, M2-PPP1R12C (WT or S452A), M2-PAK2 (WT or S20A) were plated at 40% confluence and blocked with thymidine (Sigma, 2.5 mM) for 18 hours followed by release from the block by washing twice in fresh media. Cells were fixed in 4% paraformaldehyde at various time points following release, and co-stained with DAPI and the DM1A antibody to stain microtubules or with wheat germ agglutinin (Alexa Fluor 594). The percent of multinucleated cells was determined for each condition using fluorescence microscopy by counting at least 300–400 cells per condition. A cell was considered multinucleated when it contained more than one nucleus and there was no visible membrane separation between nuclei.
Supplementary Material
ACKNOWLEDGMENTS
We thank the UCSF Mass Spectrometry Facility, supported by NIH NCRR Grants RR01614, and D. Maltby and R. Chalkley for assistance with data acquisition and analysis. We thank M. Bedford, M. Montminy, and E. Verdin for generously providing reagents. We thank J. Ferrell, S. Kim, J. Pringle, and A. Straight for helpful discussions and advice. We thank members of the Brunet lab for critical reading of the manuscript. This work was supported by NIH R01 grants AG026648 and AG31198 (A.B.), EB001987 (K.M.S.), ES153393 and GM068762 (M.B.Y.), GM56203 (L.C.C.), and HG3456 (S.P.G.). M.R.B. was supported by a Stanford Graduate Fellowship and by NIH graduate fellowship AG032837.
M.R.B. and A.B. conceived and planned the study and M.R.B. performed experiments. J.J.A., B.W., and K.M.S. developed the technique for in vivo substrate labeling, helped design the chemical genetic screen, and synthesized the (phenethyl)-ATPγS. J.J.A. helped with Figures 2B and 2C and performed the mass spectrometry to identify substrates. B.E.S. examined endogenous AMPK substrate phosphorylation (Figures 4E, 4F, 6A; Figures S1B, S6A, and S6B). E.W.W., S.R.K. and M.B.Y. performed the initial mitotic progression assays with PPP1R12C (Figure 6D; Figures S8A and S8B). P.T. and L.C.C. assessed AMPK activity in mitosis using FRET (Figure 6B; Figures S6C and S6D). J.L.W. helped with Figures 4D, S4C, 5C, and 5F. J.V. and S.P.G. identified phosphorylation sites in PAK2 and PPP1R12C by mass spectrometry (Table S5). K.S. provided the A-769662 compound and helpful advice. M.R.B. and A.B. wrote the paper. All authors discussed the results and commented on the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Extended Discussion, Extended Experimental Procedures, eight figures, and five tables and can be found with this article online at doi:xxxxxxxx.
REFERENCES
- Alaimo PJ, Shogren-Knaak MA, Shokat KM. Chemical genetic approaches for the elucidation of signaling pathways. Curr Opin Chem Biol. 2001;5:360–367. doi: 10.1016/s1367-5931(00)00215-5. [DOI] [PubMed] [Google Scholar]
- Allen JJ, Lazerwith SE, Shokat KM. Bio-orthogonal affinity purification of direct kinase substrates. J Am Chem Soc. 2005;127:5288–5289. doi: 10.1021/ja050727t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen JJ, Li M, Brinkworth CS, Paulson JL, Wang D, Hubner A, Chou WH, Davis RJ, Burlingame AL, Messing RO, et al. A semisynthetic epitope for kinase substrates. Nat Methods. 2007;4:511–516. doi: 10.1038/nmeth1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apfeld J, O'Connor G, McDonagh T, DiStefano PS, Curtis R. The AMP-activated protein kinase AAK-2 links energy levels and insulin-like signals to lifespan in C. elegans. Genes Dev. 2004;18:3004–3009. doi: 10.1101/gad.1255404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettencourt-Dias M, Giet R, Sinka R, Mazumdar A, Lock WG, Balloux F, Zafiropoulos PJ, Yamaguchi S, Winter S, Carthew RW, et al. Genome-wide survey of protein kinases required for cell cycle progression. Nature. 2004;432:980–987. doi: 10.1038/nature03160. [DOI] [PubMed] [Google Scholar]
- Blethrow JD, Glavy JS, Morgan DO, Shokat KM. Covalent capture of kinase-specific phosphopeptides reveals Cdk1-cyclin B substrates. Proc Natl Acad Sci U S A. 2008;105:1442–1447. doi: 10.1073/pnas.0708966105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan IM, Peters U, Kapoor TM, Straight AF. Polo-like kinase controls vertebrate spindle elongation and cytokinesis. PLoS One. 2007;2:e409. doi: 10.1371/journal.pone.0000409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bungard D, Fuerth BJ, Zeng PY, Faubert B, Maas NL, Viollet B, Carling D, Thompson CB, Jones RG, Berger SL. Signaling kinase AMPK activates stress-promoted transcription via histone H2B phosphorylation. Science. 2010;329:1201–1205. doi: 10.1126/science.1191241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan BK, Disanza A, Choi SY, Faber SC, Lou M, Beggs HE, Scita G, Zheng Y, Lang RA. Cdc42- and IRSp53-dependent contractile filopodia tether presumptive lens and retina to coordinate epithelial invagination. Development. 2009;136:3657–3667. doi: 10.1242/dev.042242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew TL, Masaracchia RA, Goeckeler ZM, Wysolmerski RB. Phosphorylation of non-muscle myosin II regulatory light chain by p21-activated kinase (gamma-PAK) J Muscle Res Cell Motil. 1998;19:839–854. doi: 10.1023/a:1005417926585. [DOI] [PubMed] [Google Scholar]
- Conn CW, Hennigan RF, Dai W, Sanchez Y, Stambrook PJ. Incomplete cytokinesis and induction of apoptosis by overexpression of the mammalian polo-like kinase, Plk3. Cancer Res. 2000;60:6826–6831. [PubMed] [Google Scholar]
- Cool B, Zinker B, Chiou W, Kifle L, Cao N, Perham M, Dickinson R, Adler A, Gagne G, Iyengar R, et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 2006;3:403–416. doi: 10.1016/j.cmet.2006.05.005. [DOI] [PubMed] [Google Scholar]
- Dasgupta B, Milbrandt J. AMP-activated protein kinase phosphorylates retinoblastoma protein to control mammalian brain development. Dev Cell. 2009;16:256–270. doi: 10.1016/j.devcel.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goransson O, McBride A, Hawley SA, Ross FA, Shpiro N, Foretz M, Viollet B, Hardie DG, Sakamoto K. Mechanism of action of A-769662, a valuable tool for activation of AMP-activated protein kinase. J Biol Chem. 2007;282:32549–32560. doi: 10.1074/jbc.M706536200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Dowlatshahi D, Banko MR, Villen J, Hoang K, Blanchard D, Gygi SP, Brunet A. An AMPK-FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. elegans. Curr Biol. 2007a;17:1646–1656. doi: 10.1016/j.cub.2007.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Oskoui PR, Banko MR, Maniar JM, Gygi MP, Gygi SP, Brunet A. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J Biol Chem. 2007b;282:30107–30119. doi: 10.1074/jbc.M705325200. [DOI] [PubMed] [Google Scholar]
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, Salt IP, Hawley SA, Davies SP. AMP-activated protein kinase: an ultrasensitive system for monitoring cellular energy charge. Biochem J. 1999;338(Pt 3):717–722. [PMC free article] [PubMed] [Google Scholar]
- Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Imamura K, Ogura T, Kishimoto A, Kaminishi M, Esumi H. Cell cycle regulation via p53 phosphorylation by a 5’-AMP activated protein kinase activator, 5-aminoimidazole- 4-carboxamide-1-beta-D-ribofuranoside, in a human hepatocellular carcinoma cell line. Biochem Biophys Res Commun. 2001;287:562–567. doi: 10.1006/bbrc.2001.5627. [DOI] [PubMed] [Google Scholar]
- Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- Ito M, Nakano T, Erdodi F, Hartshorne DJ. Myosin phosphatase: structure, regulation and function. Mol Cell Biochem. 2004;259:197–209. doi: 10.1023/b:mcbi.0000021373.14288.00. [DOI] [PubMed] [Google Scholar]
- Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18:283–293. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1:15–25. doi: 10.1016/j.cmet.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Koga Y, Ikebe M. A novel regulatory mechanism of myosin light chain phosphorylation via binding of 14-3-3 to myosin phosphatase. Mol Biol Cell. 2008;19:1062–1071. doi: 10.1091/mbc.E07-07-0668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu S, Yano T, Shibata M, Tuft RA, Ikebe M. Effects of the regulatory light chain phosphorylation of myosin II on mitosis and cytokinesis of mammalian cells. J Biol Chem. 2000;275:34512–34520. doi: 10.1074/jbc.M003019200. [DOI] [PubMed] [Google Scholar]
- Koo SH, Flechner L, Qi L, Zhang X, Screaton RA, Jeffries S, Hedrick S, Xu W, Boussouar F, Brindle P, et al. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature. 2005;437:1109–1111. doi: 10.1038/nature03967. [DOI] [PubMed] [Google Scholar]
- Lee JH, Koh H, Kim M, Kim Y, Lee SY, Karess RE, Lee SH, Shong M, Kim JM, Kim J, et al. Energy-dependent regulation of cell structure by AMP-activated protein kinase. Nature. 2007;447:1017–1020. doi: 10.1038/nature05828. [DOI] [PubMed] [Google Scholar]
- Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M, Kondo S, Dumont DJ, Gutterman JU, Walker CL, et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol. 2007;9:218–224. doi: 10.1038/ncb1537. [DOI] [PubMed] [Google Scholar]
- Mair W, Morantte I, Rodrigues AP, Manning G, Montminy M, Shaw RJ, Dillin A. Lifespan extension induced by AMPK and calcineurin is mediated by CRTC-1 and CREB. Nature. 2011;470:404–408. doi: 10.1038/nature09706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura F, Hartshorne DJ. Myosin phosphatase target subunit: Many roles in cell function. Biochem Biophys Res Commun. 2008;369:149–156. doi: 10.1016/j.bbrc.2007.12.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald A, Fogarty S, Leclerc I, Hill EV, Hardie DG, Rutter GA. Control of insulin granule dynamics by AMPK dependent KLC1 phosphorylation. Islets. 2009;1:198–209. doi: 10.4161/isl.1.3.9608. [DOI] [PubMed] [Google Scholar]
- Michel L, Diaz-Rodriguez E, Narayan G, Hernando E, Murty VV, Benezra R. Complete loss of the tumor suppressor MAD2 causes premature cyclin B degradation and mitotic failure in human somatic cells. Proc Natl Acad Sci U S A. 2004;101:4459–4464. doi: 10.1073/pnas.0306069101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihaylova MM, Vasquez DS, Ravnskjaer K, Denechaud PD, Yu RT, Alvarez JG, Downes M, Evans RM, Montminy M, Shaw RJ. Class IIa Histone Deacetylases Are Hormone-Activated Regulators of FOXO and Mammalian Glucose Homeostasis. Cell. 2011;145:607–621. doi: 10.1016/j.cell.2011.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender CM, Ishihara RW, Judge LM, Zhang C, Shokat KM, McKnight GS. Protein engineering of protein kinase A catalytic subunits results in the acquisition of novel inhibitor sensitivity. J Biol Chem. 2002;277:28916–28922. doi: 10.1074/jbc.M203327200. [DOI] [PubMed] [Google Scholar]
- Peters JM. The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat Rev Mol Cell Biol. 2006;7:644–656. doi: 10.1038/nrm1988. [DOI] [PubMed] [Google Scholar]
- Polson AG, Huang L, Lukac DM, Blethrow JD, Morgan DO, Burlingame AL, Ganem D. Kaposi’s sarcoma-associated herpesvirus K-bZIP protein is phosphorylated by cyclin-dependent kinases. J Virol. 2001;75:3175–3184. doi: 10.1128/JVI.75.7.3175-3184.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders MJ, Ali ZS, Hegarty BD, Heath R, Snowden MA, Carling D. Defining the mechanism of activation of AMP-activated protein kinase by the small molecule A-769662, a member of the thienopyridone family. J Biol Chem. 2007a;282:32539–32548. doi: 10.1074/jbc.M706543200. [DOI] [PubMed] [Google Scholar]
- Sanders MJ, Grondin PO, Hegarty BD, Snowden MA, Carling D. Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem J. 2007b;403:139–148. doi: 10.1042/BJ20061520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9:563–575. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, Cantley LC. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642–1646. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotillo R, Hernando E, Diaz-Rodriguez E, Teruya-Feldstein J, Cordon-Cardo C, Lowe SW, Benezra R. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell. 2007;11:9–23. doi: 10.1016/j.ccr.2006.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou P, Zheng B, Hsu CH, Sasaki AT, Cantley LC. A fluorescent reporter of AMPK activity and cellular energy stress. Cell Metab. 2011;13:476–486. doi: 10.1016/j.cmet.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuazon PT, Traugh JA. Activation of actin-activated ATPase in smooth muscle by phosphorylation of myosin light chain with protease-activated kinase I. J Biol Chem. 1984;259:541–546. [PubMed] [Google Scholar]
- Ubersax JA, Woodbury EL, Quang PN, Paraz M, Blethrow JD, Shah K, Shokat KM, Morgan DO. Targets of the cyclin-dependent kinase Cdk1. Nature. 2003;425:859–864. doi: 10.1038/nature02062. [DOI] [PubMed] [Google Scholar]
- Vazquez-Martin A, Lopez-Bonet E, Oliveras-Ferraros C, Perez-Martinez MC, Bernado L, Menendez JA. Mitotic kinase dynamics of the active form of AMPK (phospho-AMPKalphaThr172) in human cancer cells. Cell Cycle. 2009a;8:788–791. doi: 10.4161/cc.8.5.7787. [DOI] [PubMed] [Google Scholar]
- Vazquez-Martin A, Oliveras-Ferraros C, Cufi S, Menendez JA. Polo-like kinase 1 regulates activation of AMP-activated protein kinase (AMPK) at the mitotic apparatus. Cell Cycle. 2011;10:1295–1302. doi: 10.4161/cc.10.8.15342. [DOI] [PubMed] [Google Scholar]
- Vazquez-Martin A, Oliveras-Ferraros C, Lopez-Bonet E, Menendez JA. AMPK: Evidence for an energy-sensing cytokinetic tumor suppressor. Cell Cycle. 2009b;8:3679–3683. doi: 10.4161/cc.8.22.9905. [DOI] [PubMed] [Google Scholar]
- Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. The active form of the metabolic sensor: AMP-activated protein kinase (AMPK) directly binds the mitotic apparatus and travels from centrosomes to the spindle midzone during mitosis and cytokinesis. Cell Cycle. 2009c;8:2385–2398. doi: 10.4161/cc.8.15.9082. [DOI] [PubMed] [Google Scholar]
- Ventura JJ, Hubner A, Zhang C, Flavell RA, Shokat KM, Davis RJ. Chemical genetic analysis of the time course of signal transduction by JNK. Mol Cell. 2006;21:701–710. doi: 10.1016/j.molcel.2006.01.018. [DOI] [PubMed] [Google Scholar]
- Vogt J, Traynor R, Sapkota GP. The specificities of small molecule inhibitors of the TGFβ and BMP pathways. Cell Signal. 2011;23:1831–1842. doi: 10.1016/j.cellsig.2011.06.019. [DOI] [PubMed] [Google Scholar]
- Weiss EL, Bishop AC, Shokat KM, Drubin DG. Chemical genetic analysis of the budding-yeast p21-activated kinase Cla4p. Nat Cell Biol. 2000;2:677–685. doi: 10.1038/35036300. [DOI] [PubMed] [Google Scholar]
- Witters LA, Kemp BE. Insulin activation of acetyl-CoA carboxylase accompanied by inhibition of the 5’-AMP-activated protein kinase. J Biol Chem. 1992;267:2864–2867. [PubMed] [Google Scholar]
- Woods A, Munday MR, Scott J, Yang X, Carlson M, Carling D. Yeast SNF1 is functionally related to mammalian AMP-activated protein kinase and regulates acetyl-CoA carboxylase in vivo. J Biol Chem. 1994;269:19509–19515. [PubMed] [Google Scholar]
- Woods A, Vertommen D, Neumann D, Turk R, Bayliss J, Schlattner U, Wallimann T, Carling D, Rider MH. Identification of phosphorylation sites in AMP-activated protein kinase (AMPK) for upstream AMPK kinases and study of their roles by site-directed mutagenesis. J Biol Chem. 2003;278:28434–28442. doi: 10.1074/jbc.M303946200. [DOI] [PubMed] [Google Scholar]
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.