

SUMMARY
Many tumor cells rely on aerobic glycolysis instead of oxidative phosphorylation for their continued proliferation and survival. Myc and HIF-1 are believed to promote such a metabolic switch by, in part, upregulating gene expression of pyruvate dehydrogenase (PDH) kinase 1 (PDHK1), which phosphorylates and inactivates mitochondrial PDH and consequently pyruvate dehydrogenase complex (PDC). Here we report that tyrosine phosphorylation enhances PDHK1 kinase activity by promoting ATP and PDC binding. Functional PDC can form in mitochondria outside of matrix in some cancer cells and PDHK1 is commonly tyrosine phosphorylated in human cancers by diverse oncogenic tyrosine kinases localized to different mitochondrial compartments. Expression of phosphorylation-deficient, catalytic hypomorph PDHK1 mutants in cancer cells leads to decreased cell proliferation under hypoxia and increased oxidative phosphorylation with enhanced mitochondrial utilization of pyruvate, and reduced tumor growth in xenograft nude mice. Together, tyrosine phosphorylation activates PDHK1 to promote the Warburg effect and tumor growth.
INTRODUCTION
Normal cells produce ATP in the mitochondria through oxidative phosphorylation, whereas under hypoxia, glucose is converted to lactate to produce ATP. In contrast, the Warburg effect describes that cancer cells take up more glucose than normal tissue and favor aerobic glycolysis (Kroemer and Pouyssegur, 2008; Vander Heiden et al., 2009; Warburg, 1956). In normal cells, pyruvate dehydrogenase (PDH) A1 catalyzes the conversion of pyruvate to acetyl-CoA, which, along with the acetyl-CoA from the fatty acid β-oxidation, enters into the Krebs cycle to produce ATP and electron donors including NADH. PDHK1 is a Ser/Thr kinase that negatively regulates PDHA1 activity by phosphorylating PDHA1. This occurs in the pyruvate dehydrogenase complex (PDC) (Roche et al., 2001). PDC is organized around a 60-meric dodecahedral core formed by acetyltransferase (E2p) and E3-binding protein (E3BP) (Hiromasa et al., 2004), which binds PDH (aka E1p), PDHK, dihydrolipoamide dehydrogenase (E3) and pyruvate dehydrogenase phosphatase (PDP) (Read, 2001). There are four PDHK isoforms (1–4) identified in humans. PDHKs are recruited to PDC through binding to the inner lipoyl (L2) domain of the E2p subunit in the E2p/E3BP core (Liu et al., 1995). This enhances PDHK activity by granting PDHK access to its substrate PDH, which binds to the E1-binding domain that is immediately downstream of L2 of E2p. Phosphorylation of PDH by PDHK results in the inactivation of PDC, while dephosphorylation by PDP restores PDC activity (Roche et al., 2001).
In cancer cells, however, pyruvate is converted to lactate regardless of the presence of oxygen. This may be in part due to upregulation of PDHK activity and/or inhibition of PDH in cancer cells. PDHK1 is believed to be upregulated by Myc and HIF-1 to achieve functional inhibition of mitochondria by phosphorylating and inactivating PDH in cancer cells (Kim et al., 2007; Kim et al., 2006; Papandreou et al., 2006). Recent studies revealed that targeting PDHK by dichloroacetate (DCA) shifts cancer cell metabolism from glycolysis to oxidative phosphorylation and inhibits tumor growth (Bonnet et al., 2007). This finding suggests that the PDHK/PDH axis may contribute to cancer cell metabolism and tumor growth.
However, how oncogenic signals activate PDHK1 to regulate cancer cell metabolism still remains unclear. Here we report that oncogenic tyrosine kinases are localized to the mitochondria in cancer cells, where they phosphorylate and activate the mitochondrial Ser/Thr kinase PDHK1 to promote cancer cell metabolism and tumor growth.
RESULTS
Mitochondrial PDHK1 is tyrosine phosphorylated and activated by FGFR1 in cancer cells
To better understand how tyrosine kinase signaling, commonly upregulated in tumors, regulates the Warburg effect, we examined whether oncogenic FGFR1 phosphorylates and regulates PDHK1 (Figure 1A). We found that active, recombinant FGFR1 (rFGFR1) effectively phosphorylates purified GST-tagged PDHK1 in an in vitro kinase assay (Figure 1B). Further mass spectrometric analysis identified three tyrosine residues of PDHK1, including Y136, Y243 and Y244, that are phosphorylated by FGFR1 (Figure 1A; numbering of PDHK1 is as per Swiss Prot entry Q15118). In addition, GST-tagged PDHK1 was tyrosine phosphorylated in 293T cells transiently co-transfected with FGFR1 wild type (WT), but not in cells co-expressing a kinase dead (KD) form of FGFR1 (Figure 1C lower and 1D). Moreover, in an in vitro PDHK1 kinase assay, tyrosine-phosphorylated GST-PDHK1 from cells co-expressing FGFR1 WT but not FGFR1 KD demonstrated enhanced kinase activity and effectively phosphorylated recombinant PDHA1 as a substrate (Figure 1C upper).
Figure 1. FGFR1 directly phosphorylates and activates PDHK1.
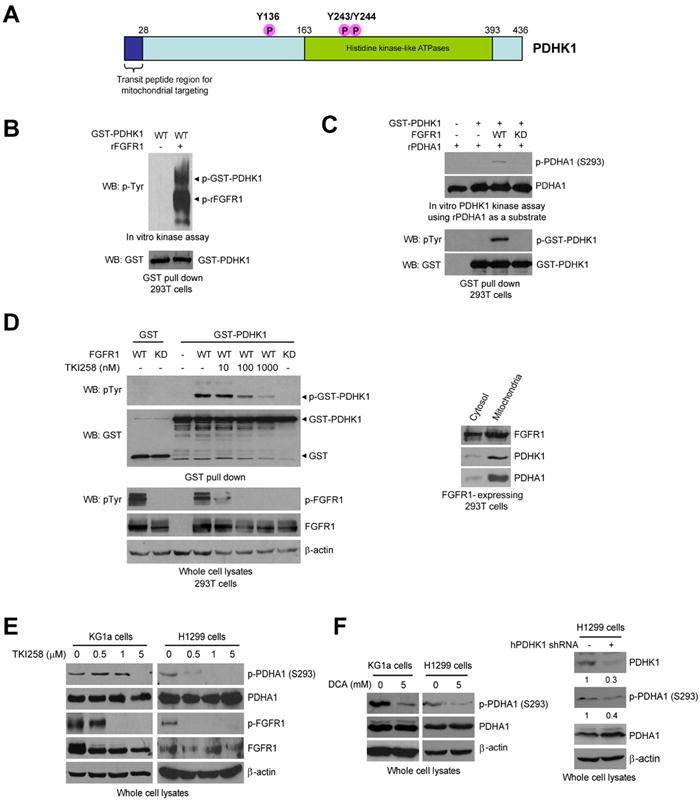
(A) Schematic representation of PDHK1. Three identified FGFR1-direct tyrosine phosphorylation sites are shown, including Y136, Y243 and Y244.
(B) Active recombinant FGFR1 (rFGFR1) directly phosphorylates purified GST-tagged PDHK1 at tyrosine residues in an in vitro kinase assay. Phosphorylated rFGFR1 and GST-PDHK1 were detected by a specific phospho-Tyr antibody (pY99).
(C) Purified GST-PDHK1 from cells co-expressing FGFR1 wild type (WT), but not an FGFR1 kinase dead form (KD) shows increased kinase activity in an in vitro PDHK1 kinase assay using recombinant pyruvate dehydrogenase A (rPDHA) as a substrate (upper). Western blot revealed that GST-PDHK1 was tyrosine phosphorylated in cells with co-expression of FGFR1 WT but not KD mutant (lower).
(D) Inhibition of FGFR1 by TKI258 (2 hours) results in decreased tyrosine phosphorylation levels of PDHK1 in cells co-expressing FGFR1 WT (left). Cells co-expressing FGFR1 KD and GST-PDHK1 were included as a negative control. Western blot result shows that FGFR1 is co-localized with PDHK1 and its substrate PDHA1 in mitochondria (right).
(E) Inhibition of FGFR1 by TKI258 (2 hours) results in decreased S293 phosphorylation levels of PDHA1 in leukemia KG1a cells (FOP2-FGFR1) (left) and lung cancer H1299 cells (FGFR1) (right).
(F) Left: Targeting PDHK1 by the PDHK inhibitor DCA results in decreased S293 phosphorylation levels of PDHA1 in KG1a and H1299 cells. Right: Inhibition of PDHK1 by specific shRNA decreases S293 phosphorylation levels of PDHA1 in H1299 cells. The numbers represent the relative intensity of the specific bands of PDHK1 (upper) or phospho-PDHA1 (S293; lower), which are normalized to the value of control sample without treatment with human PDHK1 (hPDHK1) shRNA.
Overexpression of FGFR1 or its mutational activation have been implicated in various human solid tumors, including breast cancer, pancreatic adenocarcinoma, and malignant astrocytoma (Kobrin et al., 1993; Luqmani et al., 1992; Morrison et al., 1994; Penault-Llorca et al., 1995). In addition, recurrent chromosomal translocations involving the FGFR1 gene on 8p11.2-11.1 are associated with stem cell myeloproliferative disorder (MPD), which result in expression of active FGFR1 fusion tyrosine kinases. We found that inhibition of FGFR1 by a small molecule inhibitor TKI258 results in decreased tyrosine phosphorylation levels of GST-PDHK1 in cells co-expressing FGFR1 WT (Figure 1D left). Similar results were obtained in TKI258-treated cells co-expressing GST-PDHK1 and ZNF198-FGFR1 4ZF, which is an active fusion tyrosine kinase associated with the t(8;13)(p11;q12) MPD (Xiao et al., 1998) (Figure S1). Interestingly, immunoblotting results show that FGFR1 as a receptor tyrosine kinase is co-localized with PDHK1 and its substrate PDHA1 in mitochondria (Figure 1D right). Moreover, TKI258 treatment significantly decreased phosphorylation levels of PDHA1 at S293 in human myeloid leukemia KG1a cells harboring a FOP2-FGFR1 fusion protein (Gu et al., 2006) (Figure 1E; left) and lung cancer NCI-H1299 cells overexpressing FGFR1 (Marek et al., 2009) (Figure 1E; right). In consonance with this, targeting PDHK1 by a PDHK inhibitor, dichloroacetate (DCA), or shRNA results in decreased S293 phosphorylation levels of PDHA1 in FGFR1-expressing cancer cells (Figure 1F).
FGFR1 activates PDHK1 through direct phosphorylation at multiple tyrosine sites that promotes ATP and PDC binding to PDHK1
We next found that incubation with rFGFR1 results in increased tyrosine phosphorylation levels and kinase activity of GST-PDHK1 WT. However, although rFGFR1 phosphorylates all of the Y→F mutants, substitution of Y136, Y243 or Y244 attenuates the FGFR1-dependent increase of PDHK1 kinase activity assessed by S293 phosphorylation levels of PDHA1 proteins, while double mutation of both Y243 and Y244 abolishes the enhanced activation of PDHK1 by FGFR1 (Figure 2A).
Figure 2. FGFR1-dependent tyrosine phosphorylation activates PDHK1 by promoting ATP and PDC binding.
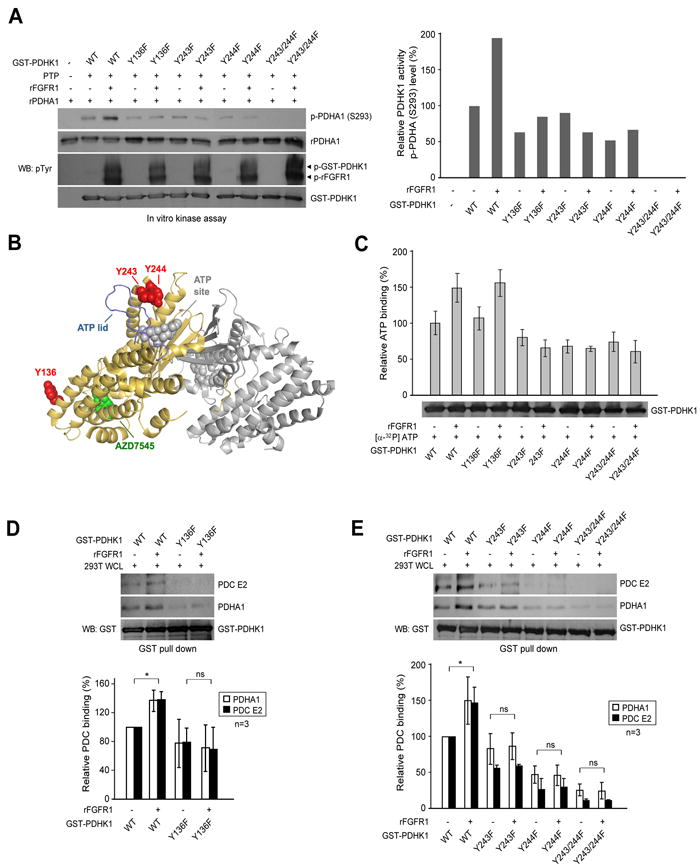
(A) GST-PDHK1 proteins were pulled down by beads and treated with protein tyrosine phosphatase (PTP), followed by treatment with activated rFGFR1. The beads were then incubated with recombinant PDHA1 in an in vitro kinase assay. S293 phosphorylation of PDHA1 was detected by immunoblotting (left). Right panel represents the relative intensity of the specific bands of phospho-PDHA1 E1α, which are normalized to the value of control sample (GST-PDHK1 WT) without treatment with rFGFR1.
(B) Cartoon representation of PDHK1 structure (PDB ID: 2Q8G) (Kato et al., 2007) made using Pymol (www.pymol.org). Y243 and Y244 are proximal to the ATP lid (in blue), and predicted location of ATP binding (modeled on the AMP-PNP-bound dimeric PDHK4 structure PDB ID: 2E0A in grey). Y136 is more proximal to AZD7545 (in green), which is a PDHK1 inhibitor that disrupts formation of functional pyruvate dehydrogenase complex (PDC).
(C) Phosphorylation at Y243 or Y244, but not Y136 by FGFR1 leads to increased ATP binding to PDHK1. GST-PDHK1 variants were pulled down by beads from transfected 293T cell lysates and incubated with active rFGFR1, followed by incubation with [α-32P]ATP. The unbound [α-32P]ATP was washed away. Retained [α-32P]ATP on PDHK1 was measured with a scintillation counter.
(D-E) FGFR1-dependent phosphorylation at Y136 (D), Y243 and Y244 (E) promotes PDHK1 binding to PDC. GST-PDHK1 variants were pulled down by beads and incubated with active rFGFR1, followed by incubation with whole cell lysates of 293T cells. The bound PDC E2 protein and PDHA1 E1α proteins were detected by immunoblotting. Upper panel shows immunoblotting results from one representative experiment. Lower panel shows summarized results from two independent experiments that represent the relative intensity of the specific bands of PDHA1 E1α and PDC E2 that bind to PDHK1, which are normalized to the value of control sample without treatment with rFGFR1. The error bars represent mean values +/- SD.
As shown in Figure 2B, crystal structures of PDHK1 reveal that Y243 and Y244 are close to the ATP lid and ATP binding site, suggesting that phosphorylation of these residues could affect ATP binding and PDHK1 catalytic activity. In contrast, Y136 is distal to both ATP and substrate binding sites of PDHK1, but is closer to the binding site of a small molecule drug, AZD7545, which inhibits PDHK1 and 3 by aborting the kinase binding to the pyruvate dehydrogenase complex (PDC) scaffold (Kato et al., 2007). Indeed, incubation with rFGFR1 led to increased [α-32P]ATP binding to PDHK1 WT and Y136F mutant. In contrast, substitution of Y243 and/or Y244 abolished the FGFR1-dependent enhancement of ATP binding to PDHK1 (Figure 2C). To determine whether Y136 phosphorylation affects the binding of PDHK1 to PDC, we incubated active rFGFR1 with purified GST-PDHK1 WT or Y→F mutants in an in vitro kinase assay, followed by incubation with whole cell lysates from 293T cells. Phosphorylation of PDHK1 WT by FGFR1 resulted in increased binding between PDHK1 and PDC E2 protein, as well as enhanced association between PDHK1 and its substrate PDHA1 that exists in PDC. In contrast, substitution of Y136 abolished the enhanced association of PDHK1 to PDC E2 protein or PDHA1 in the presence of rFGFR1 (Figure 2D).
Interestingly, substitution of Y243 and/or Y244 also led to abolishment of the FGFR1-dependent increased PDHK1/PDC E2 and PDHK1/PDHA1 associations (Figure 2E). These results together suggest that phosphorylation at both Y243 and Y244, but not Y136 may be required to promote ATP binding to PDHK1, which consequently facilitates PDHK1 binding to PDC scaffold to access substrate PDHA1. In contrast, Y136 phosphorylation may only function to enhance binding between PDHK1 and PDC. Figure S2 shows the results of analysis of the relative stability of proteins to limited proteolytic digestion with chymotrypsin (Kang et al., 2007), which suggests the global structure of each mutant protein was not altered, and decreased kinase activation (Figure 3B) or ATP/PDC binding ability of PDHK1 Y→F mutants are not due to structural alterations.
Figure 3. FGFR1 phosphorylates mitochondrial PDHK1 in cancer cells.
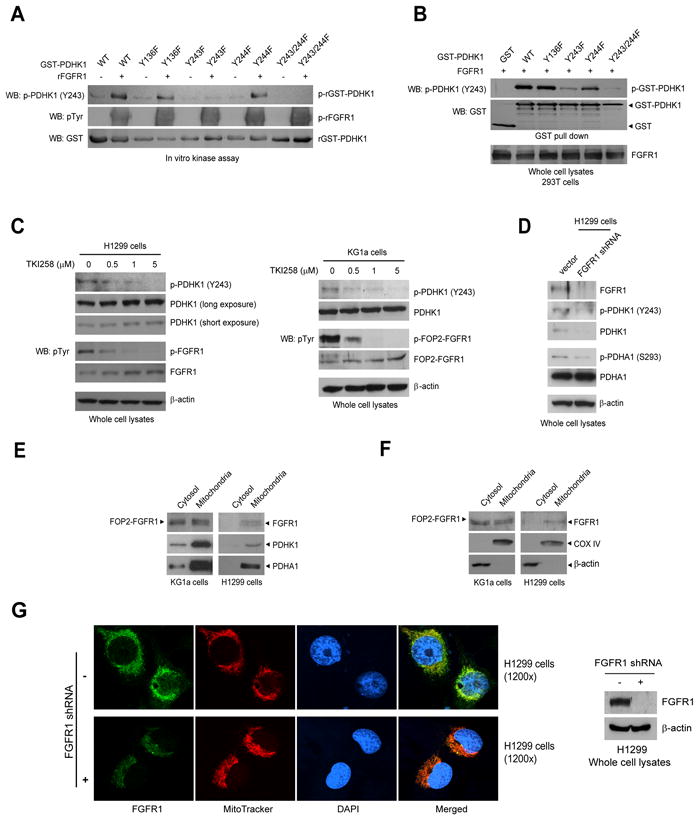
(A-B) PDHK1 Y243 is specifically phosphorylated by rFGFR1 in an in vitro kinase assay (A) and in cells expressing FGFR1 (B), detected by an antibody that specifically recognizes phospho-Y243 of PDHK1.
(C) Immunoblotting shows that targeting FGFR1 by TKI258 in lung cancer H1299 cells (left) or leukemia KG1a cells (right) decreases phosphorylation of PDHK1 Y243.
(D) Knockdown of FGFR1 by lentiviral shRNA for 48 hours in H1299 cells results in decreased phosphorylation levels of PDHK1 Y243 and PDHA1 S293.
(E) Western blot result shows that FGFR1 is co-localized with PDHK1 and its substrate PDHA1 in mitochondria in KG1a (left) and H1299 (right) cells.
(F) Immunoblotting result shows that FGFR1 is localized in mitochondria, with mitochondrial COX IV and cytosolic β-actin as control markers in KG1a (left) and H1299 (right) cells.
(G) Immunofluorescence assay reveals that FGFR1 is co-localized with mitochondrial marker MitoTracker® Mitochondrion-Selective dyes, but not with nuclear marker DAPI in H1299 cells (upper left). Knockdown of FGFR1 by shRNA (right panel) leads to decreased mitochondrial localization of FGFR1 (lower left).
Oncogenic FGFR1 is localized in mitochondria in cancer cells, where it phosphorylates PDHK1
Using an antibody that specifically recognizes PDHK1 phospho-Y243, we observed that rFGFR1 and FGFR1 WT phosphorylated GST-PDHK1 WT, Y136F and Y244F mutants, but not the Y243F and Y243/244F mutants, at Y243 in an in vitro kinase assay using recombinant proteins (Figure 3A) and in 293T cells co-expressing these PDHK1 variants (Figure 3B), respectively. We also observed that inhibiting FGFR1 by TKI258 decreased PDHK1 Y243 phosphorylation in FGFR1-expressing H1299 lung cancer cells (Figure 3C, left) and KG1a leukemia cells (Figure 3C, right). TKI258 does not inhibit PDHK1 kinase activity in an in vitro kinase assay (Figure S3A). Moreover, knockdown of FGFR1 by shRNA in H1299 cells for 48 hours results in decreased phosphorylation levels of PDHK1 Y243 and PDHA1 S293 (Figure 3D). Further discussion regarding decreased PDHK1 protein levels in H1299 cells with long term knockdown of FGFR1 is provided in the Discussion section.
We also detected a fraction of FOP2-FGFR1 fusion and FGFR1 in mitochondria along with mitochondrial PDHK1 and PDHA1 (Figure 3E) in KG1a and H1299 cells, respectively, with mitochondrial COX IV and cytosolic β-actin as control markers (Figure 3F). Figure S3B and 3C show no plasma membrane or nuclear contamination in the mitochondrial fractions. In consonance with these observations, immunofluorescence assay results show that FGFR1 is co-localized with the control mitochondrial marker MitoTracker® Mitochondrion-Selective dyes, but not with nuclear marker DAPI in H1299 cells (Figure 3G and Figure S4A). Figure S4B shows no cross-reaction between FGFR1 antibody and MitoTracker® Mitochondrion-Selective dyes. In addition, knockdown of FGFR1 by shRNA leads to decreased mitochondrial localization of FGFR1 in H1299 cells (Figure 3G), while stimulation of cells with FGFR1 ligand bFGF does not significantly alter the FGFR1 mitochondrial localization (Figure S4C).
We further investigated the ultrastructural localization of FGFR1 by immunogold transmission electron microscopy (TEM). Specific immunogold particles were found in mitochondria of H1299 cells. This immunogold particle distribution was not observed when the primary anti-FGFR1 antibody was omitted (Figure 4A, left) and was greatly reduced by FGFR1 shRNA (Figure 4A, right). To localize FGFR1 within mitochondria biochemically, we performed sub-fractionation of highly purified mitochondria from H1299 and KG1a cells. Western blot analyses of the sub-fractions show that both full length FGFR1 and FOP2-FGFR1 fusion tyrosine kinase are present predominately in the mitochondrial outer membrane (Om) (Figure 4B). We also observed that both PDHK1 and PDHA1 are present in the mitochondrial matrix (Ma), as well as in the intermembrane space (IMS) and Om. Detailed discussion is provided in the Discussion section. To determine whether FGFR1 and FOP2-FGFR1 fusion are integrated in Om, we performed a proteinase K protection assay using purified mitochondria from H1299 and KG1a cells, respectively. Outer membrane marker Tom 40 and inner membrane marker COX IV were included as controls. As shown in Figure 4C, Tom 40 was completely digested by proteinase K treatment in both the presence and absence of Triton X-100, whereas complete digestion of COX IV by proteinase K only occurred after solubilization of mitochondria with Triton X-100 treatment. We found that, similar to Tom 40, full length FGFR1 was completely digested by proteinase K in both presence and absence of Triton X-100. In contrast, FOP2-FGFR1 was only partially digested by proteinase K in the absence of Triton X-100 and complete digestion of FOP2-FGFR1 was observed after solubilization of mitochondria with Triton X-100. Together, mitochondrial FGFR1 and a portion of FOP2-FGFR1 may be integrated in the mitochondrial Om, whereas the rest of mitochondrial FOP2-FGFR1 may be located inside the mitochondria, probably in the IMS, but associated with certain unknown Om protein.
Figure 4. FGFR1 is localized to outer membrane of mitochondria and functional PDC can form in mitochondria outside of matrix in cancer cells.
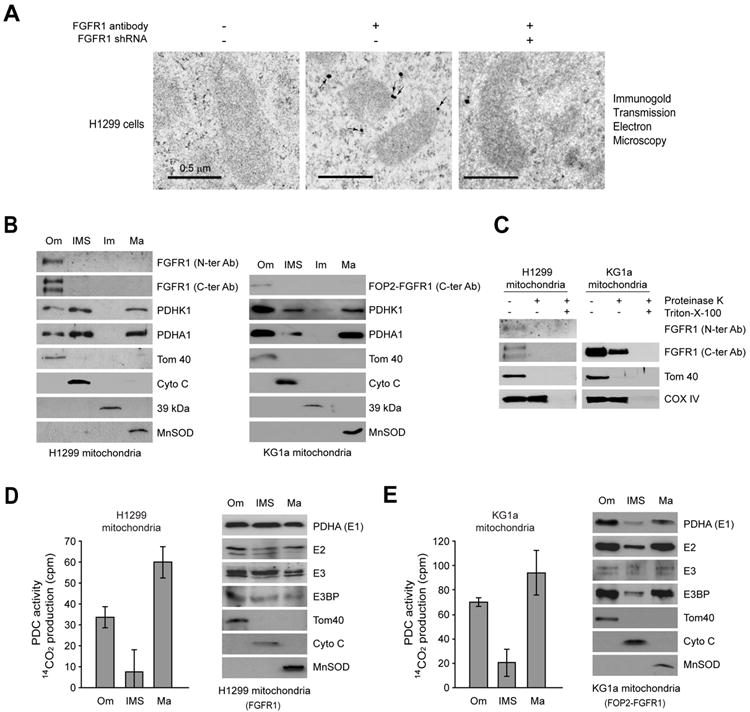
(A) Mitochondrial localization of FGFR1 in H1299 cells under transmission electron microscopy (TEM). Left panel shows the control without primary antibody. Middle panel shows the localization of FGFR1 in mitochondria of H1299 cells, with arrows indicating representative staining of mitochondrial FGFR1. Right panel shows the control with FGFR1 knockdown by shRNA. Scale bars: 0.5 μm.
(B) Localization of full length FGFR1 and FOP2-FGFR1 fusion tyrosine kinase in the outer membrane of mitochondria in lung cancer H1299 and leukemia KG1a cells, respectively. Tom40, Cyto C, complex I 39-kDa protein, and MnSOD are markers for mitochondrial outer membrane (Om), inter-membrane space (IMS), inner membrane (Im), and matrix (Ma), respectively.
(C) Proteinase K protection experiment where isolated mitochondria were treated with proteinase K in the presence and absence of Triton X-100, followed by Western blot.
(D-E) Left: Activity of PDC-mediated conversion of 14C-labeled pyruvate into 14C-labeled CO2 was detected in the mitochondrial subfractions including Om, IMS and Ma of H1299 cells (D) and KG1a cells (E). The error bars represent mean values +/- SD. Right: Western blotting results show expression levels of PDHA1 (E1), E2, E3 and E3BP in indicated mitochondrial subfractions. Tom40, Cyto C and MnSOD were included as markers.
Functional PDC can form in mitochondria outside of matrix in some cancer cells and PDHK1 is commonly phosphorylated in these cells at Y243 by various oncogenic tyrosine kinases
Furthermore, we detected PDC activity and various PDC components in the mitochondrial Om and IMS, in addition to the matrix in both H1299 and KG1a cells (Figure 4D-4E, respectively). These results suggest that oncogenic FGFR1 may inhibit PDC by phosphorylating PDHK1 in the Om in cancer cells. Detailed discussion is provided in the Discussion section.
We found that PDHK1 was phosphorylated at Y243 in diverse hematopoietic cancer cell lines associated with various constitutively activated tyrosine kinase mutants, including HEL (JAK2 Val617Phe mutant), KG1a (FOP2-FGFR1), K562 (BCR-ABL) and Mo91 (TEL-TrkC), whereas Y243 phosphorylation levels are relatively lower in FLT3-internal tandem duplication (ITD) mutant positive Molm14 and Mv4;11 cells, but not detected in EOL-1 (FIP1L1-PDGFRA) cells. Phosphorylation levels of PDHA1 at S293 in general correlated with PDHK1 Y243 phosphorylation levels in these leukemia cells (Figure 5A). Y243 phosphorylation of PDHK1 was also detected in various human solid tumor cell lines, including lung cancer H1299 (FGFR1; Figure 3C) and A549 cells and breast cancer MCF-7 cells, but not breast cancer MDA-MB435 cells and prostate cancer PC3 and DU145 cells (Figure 5A). However, the phosphorylation levels of PDHA1 at S293 in these solid tumor cells did not correlate with PDHK1 Y243 phosphorylation levels (Figure 5A). Detailed discussion is provided below.
Figure 5. Y243 of PDHK1 is commonly phosphorylated by oncogenic tyrosine kinases localized to mitochondria in cancer cells.
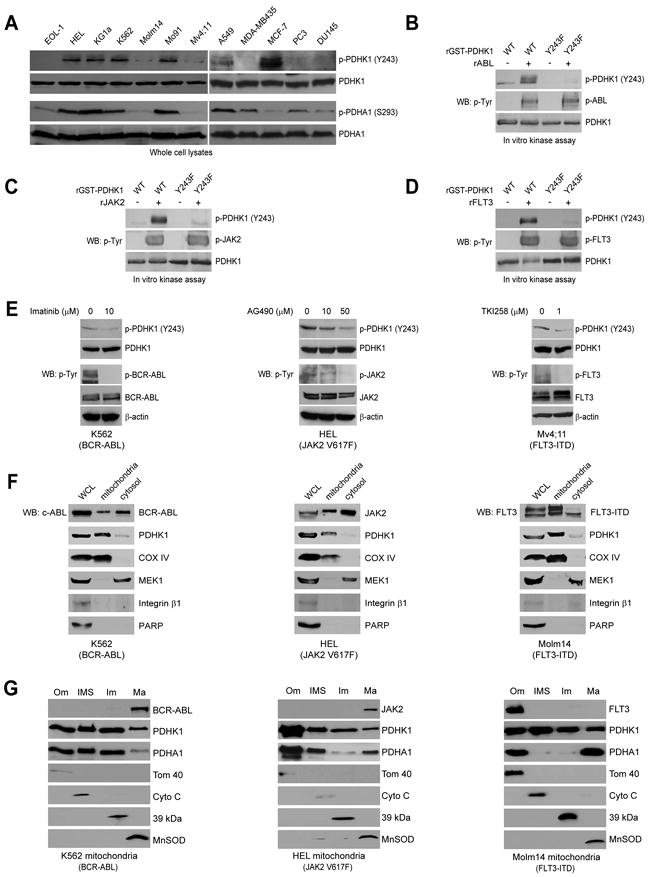
(A) Immunoblotting detected Y243 phosphorylation of PDHK1 in diverse cancer cells. Western blot results of phosphorylation levels of PDHA1 at S293 are shown.
(B-D) Active, recombinant ABL (B), JAK2 (C) and FLT3 (D) directly phosphorylate PDHK1 at Y243 in corresponding in vitro kinase assays.
(E) Immunoblotting shows that targeting BCR-ABL by imatinib in K562 cells (left), JAK2 by AG490 in HEL cells (middle) or FLT3-ITD by TKI258 in Mv4;11 cells (right) decreases phosphorylation of PDHK1 Y243.
(F) Immunoblotting results show that BCR-ABL (left), JAK2 (middle) and FLT3 (right) are localized in mitochondria with mitochondrial COX IV and cytosolic MEK1 as control markers in leukemia K562 (BCR-ABL), HEL (JAK2), and Molm14 (FLT3) cells, respectively. BCR-ABL and JAK2 but not FLT3 were also detected in the cytoplasm. Integrin β1 and PARP are markers for plasma membrane and nucleus, respectively.
(G) Sub-fractionation results show that BCR-ABL and JAK2 are localized in the mitochondrial matrix in K562 and HEL cells (left and middle, respectively), while FLT3 is localized in the mitochondrial outer membrane in Molm14 cells (right). Tom40, Cyto C, complex I 39-kDa protein, and MnSOD are markers for mitochondrial outer membrane (Om), inter-membrane space (IMS), inner membrane (Im), and matrix (Ma), respectively.
We next found that active, recombinant ABL (Figure 5B), JAK2 (Figure 5C) and FLT3 (Figure 5D) also directly phosphorylated PDHK1 at Y243 in the in vitro kinase assays using recombinant proteins, whereas EGFR phosphorylated PDHK1 with less efficiency (Figure S5A). Inhibition of BCR-ABL by imatinib, JAK2 by AG490 and FLT3-ITD by TKI258 resulted in decreased Y243 phosphorylation of PDHK1 in the pertinent human cancer cell lines (Figure 5E; left, middle and right, respectively). In addition, immunoblotting results confirm the mitochondrial localization of BCR-ABL, JAK2 and FLT3 (Figure 5F), which are co-localized with PDHK1 and its substrate PDHA1 in mitochondria (Figure S5B) in the pertinent human leukemia cell lines. Moreover, Western blot analyses of the sub-fractions of highly purified mitochondria from K562, HEL and Molm14 leukemia cells (Figure 5G; left, middle and right, respectively) show that a portion of cytoplasmic BCR-ABL and JAK2 proteins are present predominately in the matrix of mitochondria, while a portion of receptor tyrosine kinase FLT3 is present predominately in the outer membrane of mitochondria. These oncogenic tyrosine kinases are also co-localized with PDHK1 and PDHA1 in the corresponding mitochondrial sub-fractions in the pertinent human leukemia cell lines, where these tyrosine kinases may phosphorylate PDHK1. Consistently, PDC activity was also detected in the mitochondrial Om and IMS, in addition to the matrix in all of these cells (Figure S5C-5E).
Presence of the catalytically less active mouse PDHK1 Y134F and Y239/240F mutants in cancer cells leads to decreased cell proliferation under hypoxia and increased oxidative phosphorylation
We next generated “rescue” cell lines as previously described (Hitosugi et al., 2009) by RNAi-mediated stable knockdown of endogenous human PDHK1 (hPDHK1) and rescue expression of Flag-tagged mouse PDHK1 (mPDHK1) WT, or the corresponding Y134F and Y239/240F mutants (Figure 6A). Both Flag-mPDHK1 WT and Y134F mutant, but not Y239/240F mutant, were phosphorylated at Y239 (corresponding to Y243 in human PDHK1 numbering) by FGFR1 in H1299 cells (Figure S6A). In addition, Y134F and Y239/240F mutants showed decreased kinase activity that led to reduced phosphorylation levels of PDHA1 at S293 in Y134F and Y239/240F rescue cells, respectively, compared to cells with mPDHK1 WT (Figure S6B). Moreover, although hypoxia results in increased PDHK1 expression and Y243 phosphorylation levels (Figure S6C), endogenous hPDHK1 protein levels were not detected in diverse rescue cells under normoxia nor hypoxia, suggesting that the efficacy of hPDHK1 knockdown mediated by shRNA is not altered under hypoxia (Figure S6D).
Figure 6. Expression of catalytically less active mouse PDHK1 mutants, including Y134F and Y239/240F, in H1299 cells leads to decreased proliferation under hypoxia and increased oxidative phosphorylation.
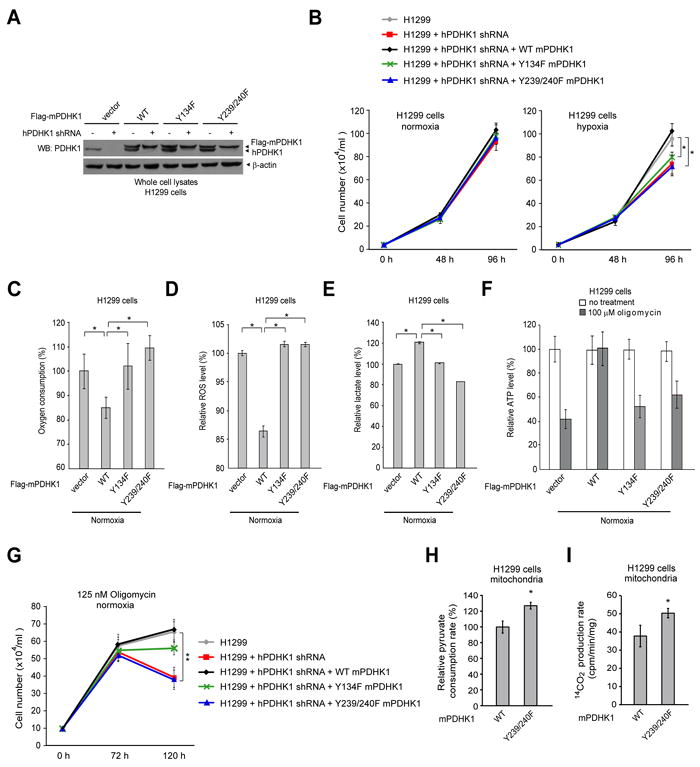
(A) Immunoblotting shows shRNA-mediated stable knockdown of endogenous human PDHK1 (hPDHK1) in H1299 cells using lentiviral transduction, and rescue expression of Flag-tagged mouse PDHK1 (mPDHK1) proteins including WT, Y134F mutant and Y239/240F double mutant. Y134, Y239 and Y240 of mPDHK1 are corresponding to Y136, Y243 and Y244 of hPDHK1, respectively.
(B) Rescue expression of mPDHK1 Y134F or Y239/240F mutants in H1299 cells results in reduced cell proliferation rate under hypoxic conditions (1% oxygen) but not at normal oxygen tension (normoxic; 17% oxygen) compared to cells expressing mPDHK1 WT. Cell proliferation was determined by the cell number of each cell line 48 and 96 hours after seeding (n=3; *: 0.01<p<0.05). The error bars represent mean values +/- SD.
(C-E) Y134F and Y239/240F rescue cells have a significantly elevated oxygen consumption rate (C), an increased intracellular ROS production under normoxia (D), and a significantly reduced lactate production under normoxia (E), compared to cells expressing mPDHK1 WT. Media were incubated with cells for 1 hour prior to lactate assay. The error bars represent mean values +/- SD.
(F-G) Treatment with oligomycin resulted in increased inhibition of ATP production (F) and decreased proliferation (G) in Y134F and Y239/240F rescue cells, compared to cells expressing mPDHK1 WT. The error bars represent mean values +/- SD.
(H-I) Isolated mitochondria from rescue cells expressing mPDHK1 Y239/240F show significantly increased rates of pyruvate consumption (H) and PDC-mediated conversion of 14C-labeled pyruvate into 14C-labeled CO2 (I), compared to mitochondria isolated from WT rescue cells. The error bars represent mean values +/- SD.
We also observed that, under normoxia, cells rescued with any of the mPDHK1 variants showed a comparable rate of proliferation that was greater than that of parental cells, in which endogenous hPDHK1 was stably knocked down. However, Y134F and Y239/240F rescue cells showed a significantly slower proliferation rate under hypoxic conditions than did cells rescued with mPDHK1 WT (Figure 6B). Moreover, compared to cells rescued with mPDHK1 WT, the Y134F and Y239/240F rescue cells and parental cells with stable knockdown of endogenous hPDHK1 had a higher rate of oxygen consumption (Figure 6C), an increased production of intracellular reactive oxygen species (ROS) (Figure 6D) and reduced lactate production (Figure 6E). In addition, treatment with oligomycin, a specific inhibitor of mitochondrial ATP synthase, resulted in an increased inhibition of ATP production and a decreased proliferation rate among parental control H1299 cells with stable knockdown of endogenous hPDHK1 and rescue cells expressing Y134F and Y239/240F mutants, compared to cells with mPDHK1 WT (Figure 6F and 6G, respectively). These results together suggest that cells expressing catalytically less active mPDHK1 mutants, including Y134F and Y239/240F, rely more on oxidative phosphorylation for ATP production and cell proliferation compared to cells with mPDHK1 WT.
Tyrosine phosphorylation of PDHK1 is important for PDC-mediated pyruvate metabolism, probably through inhibitory phosphorylation of PDHA1
The pyruvate dehydrogenase complex (PDC) contributes to pyruvate decarboxylation, in which pyruvate is converted to acetyl-CoA and carbon dioxide. To determine the role of tyrosine phosphorylation of PDHK1 in regulation of PDC activity, we performed a pyruvate consumption experiment using highly purified mitochondria. We found that mitochondria isolated from rescue cells expressing mPDHK1 Y239/240F mutant show a significantly increased pyruvate consumption rate, compared to mitochondria isolated from WT rescue cells (Figure 6H). Moreover, we examined the PDC activity by assessing the rate of PDC-mediated conversion of pyruvate to CO2. As shown in Figure 6I, isolated mitochondria from cells expressing mPDHK1 Y239/240F mutant have a significantly increased rate of transforming 14C-labeled pyruvate to 14C-labeled CO2, compared to mitochondria isolated from WT rescue cells. These data suggest an important role for tyrosine phosphorylation of PDHK1 in regulation of PDC activity, probably by phosphorylating PDHA1.
We next sought to determine the role of PDHA1 in PDHK1-regulated cancer cell metabolism. As shown in Figure 7A, expression of PDHA1 WT or an active, phospho-deficient mutant S293A in H1299 rescue cells of either mPDHK1 WT or Y239/240F mutant resulted in an increased O2 consumption rate. In contrast, mPDHK1 WT rescue cells expressing a phospho-mimetic, catalytically less active form of PDHA1 (S293D) showed less increase in O2 consumption compared to cells expressing PDHA1 WT or S293A mutant, whereas expression of PDHA1 S293D mutant, but not PDHA1 WT or S293A mutant, resulted in a significant decrease in O2 consumption in H1299 rescue cells of mPDHK1 Y239/240F. Furthermore, expression of PDHA1 WT, S293A or S293D mutants in rescue cells of mPDHK1 Y239/240F did not affect cell proliferation under normoxia. However, expression of PDHA1 S293D mutant, but not WT or S293A mutant, resulted in decreased sensitivity of mPDHK1 Y239/240F rescue cells to hypoxia in regard to cell proliferation (Figure 7B). Moreover, rescue cells of mPDHK1 Y239/240F transfected with PDHA1 S293D mutant showed increased lactate production (Figure 7C) and decreased sensitivity to the treatment with oligomycin, an inhibitor of ATP synthase, in regard to ATP production and cell proliferation (Figure 7D and 7E, respectively), compared to cells transfected with PDHA1 WT or S293A mutant. These data together with data presented in Figure 6 suggest that rescue expression of mPDHK1 Y239/240F makes cells rely more on oxidative phosphorylation, whereas expression of the inactive, phospho-mimetic PDHA1 S293D mutant attenuates such a metabolic switch.
Figure 7. Phosphorylation-dependent activation of PDHK1 is mediated through phosphorylation and inactivation of PDHA1, which is important for tumor growth in xenograft nude mice.
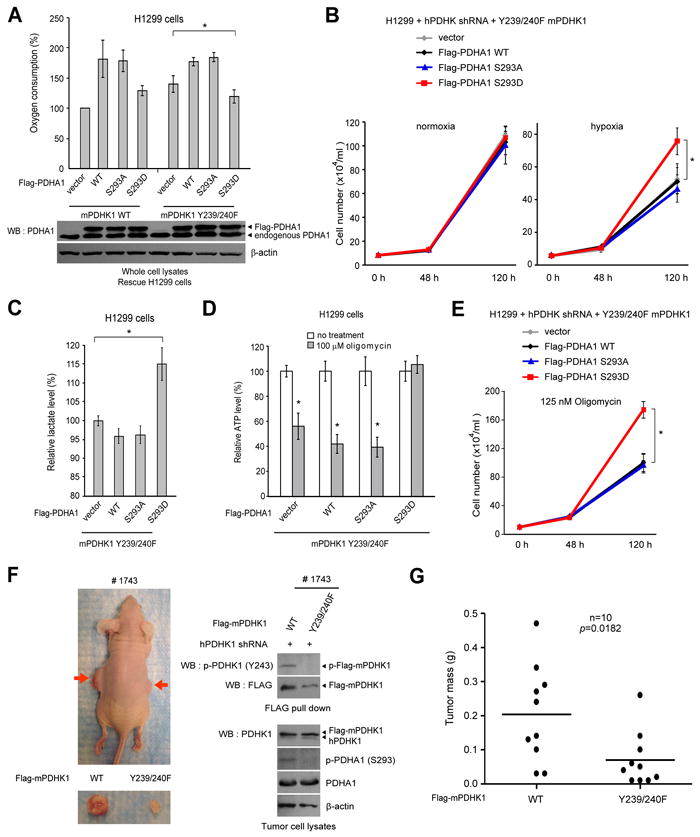
(A) Expression of PDHA1 S293D mutant, but not PDHA1 WT or S293A (lower), resulted in a significant decrease in O2 consumption in H1299 rescue cells of mPDHK1 Y239/240F but not in WT rescue cells (upper).
(B-E) Expression of PDHA1 WT, S293A or S293D mutant in rescue cells of mPDHK1 Y239/240F did not affect cell proliferation under normoxia (B; left); however, expression of PDHA1 S293D mutant, but not WT or S293A mutant resulted in decreased sensitivity of mPDHK1 Y239/240F rescue cells to hypoxia in regard to cell proliferation (B; right), increased lactate production (C) and decreased sensitivity to the treatment with oligomycin in regard to ATP levels (D) and cell proliferation (E). The error bars represent mean values +/-SD.
(F) Left: Dissected tumors (indicated by red arrows) in a representative nude mouse (#1743) injected with mPDHK1 WT H1299 cells on the left flank and Y239/240F H1299 cells on the right flank are shown. Right: Expression of Flag-mPDHK1 WT and Y239/240F, as well as phosphorylation levels of PDHK1 and PDHA1 in tumor lysates.
(G) Y239/240F rescue cells show significantly reduced tumor formation in xenograft nude mice compared to cells with mPDHK1 WT (p value was determined by the paired Student’s t test).
Expression of mPDHK1 Y239/240F mutant in cancer cells leads to reduced tumor growth in xenograft nude mice
We next performed xenograft experiments in which nude mice were injected with Flag-mPDHK1 WT and Y239/240F rescue H1299 cells (Figure S7A). Ten million cells each were injected (Flag-mPDHK1 WT rescue cells on the left flank and Y239/240F cells on the right flank; n=10), and the mice were monitored for tumor growth over a 5-week time period. The masses of tumors derived from Y239/240F rescue cells were significantly reduced compared to those of tumors formed by Flag-mPDHK1 WT rescue cells (Figure 7F-7G).
DISCUSSION
Our finding that tyrosine phosphorylation activates PDHK1 may, at least in part, explain the functional attenuation of mitochondria in cancer cells (Kim and Dang, 2006). This could represent a common, short-term molecular mechanism underlying the Warburg effect in both leukemias and solid tumors, in addition to the chronic changes, which include upregulation of PDHK1 gene expression to achieve mitochondrial inhibition, believed to be regulated by transcription factors including Myc and HIF-1.
An interesting observation is that a fraction of receptor tyrosine kinases including FGFR1 and FLT3 are predominantly present in the mitochondrial outer membrane, whereas a fraction of cytoplasmic tyrosine kinases BCR-ABL and JAK2 are found in the mitochondrial matrix. However, we also observed that a fraction of cytoplasmic fusion tyrosine kinase FOP2-FGFR1 is predominantly present in the mitochondrial Om. Surprisingly, the results of proteinase protection assay suggest that FGFR1 may be integrated in the Om of mitochondria, while FOP2-FGFR1 may also be presented inside the mitochondria, probably in the IMS but associated with certain Om proteins to demonstrate a predominant sub-localization to the mitochondrial Om. These findings together suggest a new role for oncogenic tyrosine kinases which may be localized to mitochondria and function by phosphorylating mitochondrial proteins. FOP2-FGFR1, BCR-ABL, FLT3-ITD and JAK2 V617F are constitutively active and ligand independent. We detected mitochondrial full length FGFR1 as tyrosine phosphorylated in H1299 cells, suggesting that mitochondrial FGFR1 has basal level of tyrosine kinase activity and may be ligand independent (Kang and Chen, unpublished data). Further studies are warranted to determine the regions of these oncogenic tyrosine kinases for mitochondrial localization as well as the molecular mechanism underlying the differential mitochondrial sub-localization and contribution to mitochondrial biology and cancer cell metabolism.
We also observed that in the tested human cancer cells, both PDHK1 and PDHA1 are present in Om and IMS, and in some cases the inner membrane, in addition to the mitochondrial matrix. The Om sub-localization may be due to binding of PDHK1 and PDHA1 to some outer membrane proteins. In fact, we observed that PDHK1 interacts with FGFR1 WT that is catalytically active, while the PDHK1-FGFR1 interaction is decreased when PDHK1 is co-expressed with a FGFR1 kinase dead mutant (KD), suggesting that the tyrosine kinase activity may be required for the interaction between FGFR1 and PDHK1 (Figure S7B). Moreover, PDC activity and various PDC components were detected in the mitochondrial outer membrane and intermembrane space, in addition to the matrix in all of the tested cancer cell lines (Figure 4D-4E, Figure S5C-5E). This suggests that PDC can be functionally formed outside of the matrix. Moreover, cancer cells with oncogenic tyrosine kinases in the Om including H1299 (FGFR1), KG-1a (FOP2-FGFR1) and Molm 14 (FLT3-ITD) show relatively low levels of PDC activity in the Om compared to the matrix, whereas cells with oncogenic TKs in matrix including K562 (BCR-ABL) and HEL (JAK2 V617F) demonstrate lower PDC activity in the matrix compared to the Om. These data suggest that, although diverse oncogenic TKs differentially localize to different compartments of mitochondria in cancer cells, they may regulate cancer metabolism in a similar way by inhibiting PDC activity in the correlated mitochondrial compartments. However, how the attenuated PDC activity in these cells contributes to cancer cell metabolism may vary due to, for example, the different mitochondrial compartmentation of PDC and its products NADH and acetyl-coA. These findings also warrant further studies to determine how the functional PDC activity outside of matrix contributes to pyruvate decarboxylation and consequently mitochondrial function and metabolism.
The structural mechanisms by which phosphorylation affects PDHK1 function are still to be determined, but the proximity of Y243 and Y244 suggests altered dynamics for the ATP lid, and consequently ADP/ATP binding properties. It will be important to further investigate the atomic-level effects of phosphorylation of these residues using structural biology techniques. It is interesting that PDHA1 was detected to be S293 phosphorylated in EOL-1, MDA-MB435, PC3 and DU145 cells that have non-detectable PDHK1 Y243 phosphorylation. Since PDHK1 WT proteins treated with PTP still maintain basal levels of kinase activity (Figure 2A), this may suggest that these cells might rely on PDHK1 protein expression levels for PDHA1 phosphorylation more than tyrosine phosphorylation dependent activation of PDHK1. It is also possible that the pyruvate dehydrogenase phosphatase (PDP) activity is relatively low in these cells, which functions by dephosphorylating PDHA1, or isoforms of PDHK other than PDHK1 may be responsible for PDHA1 phosphorylation in these cells. In contrast, MCF-7 cells have high protein expression levels of PDP1 (Figure S7C) which explains why MCF-7 cells have high levels of PDHK1 phosphorylation but the phosphorylation level of PDHA1 is low.
Our findings suggest that PDHK1 may serve as a therapeutic target in cancer treatment, such that inhibition of PDHK1 may affect cancer cell metabolism and cause tumor regression. These findings are consistent with previous observations that targeting PDHK by small molecule inhibitor DCA attenuates cancer cell proliferation and tumor growth using A549 lung cancer cells (Bonnet et al., 2007). However, the authors reported that targeting PDHK2 by specific siRNA, but not the control scrambled siRNA, mimics DCA effects on A549 cells. Further studies to identify distinct roles of different PDHK isoforms in cancer cell metabolism and tumor growth are warranted.
We previously reported that oncogenic tyrosine kinases including FGFR1 phosphorylate and inhibit PKM2 to regulate cancer metabolism (Hitosugi et al., 2009). Both tyrosine phosphorylation-dependent regulation of PKM2 and PDHK1 contributes to the metabolic switch from oxidative phosphorylation to aerobic glycolysis in cancer cells. Interestingly, we found that Y105 phosphorylation of PKM2 is important in regulation of PDHK1 gene expression; the mRNA and protein levels of PDHK1 are significantly decreased in FGFR1-expressing H1299 lung cancer cells with stable knockdown of endogenous human PKM2 and rescue expression of mouse PKM2 Y105F mutant, compared to cells expressing mPKM2 WT (Hitosugi and Chen, unpublished data). This in part explains the decreased PDHK1 protein levels in H1299 cells with shRNA-mediated long-term knockdown of FGFR1 (Figure 3D). These findings suggest a dual role for FGFR1 in regulation of PDHK1 in cancer metabolism, which consists of a long-term mechanism by which FGFR1 may phosphorylate PKM2 to promote PDHK1 gene expression and a short-term mechanism by which FGFR1 activates PDHK1 through tyrosine phosphorylation at multiple sites. Y105 phosphorylation of PKM2, which is involved in FGFR1-regulated PDHK1 expression, appears to be an upstream event that precedes FGFR1-dependent phosphorylation and activation of PDHK1 in cancer cell metabolism.
EXPERIMENTAL PROCEDURES
Immunofluorescence microscopy, TEM, and mitochondrial sub-fractionation assays
For immunofluorescence microscopy, cells seeded on glass coverslips were stained for mitochondria with MitoTracker Red CMXRos (Invitrogen), followed by fixation in PHEMO buffer and then blocked with 10% goat serum (Invitrogen). Cells were incubated with FGFR1 primary antibody in PBS containing 5% goat serum, followed by incubation with goat anti rabbit IgG conjugated with Alexa Fluor 488 (Invitrogen) in PBS with 5% goat serum. Cells were washed in PBS, mounted, and imaged on a Zeiss LSM510 META confocal microscope. Immunogold labeling of FGFR1 was performed as previously described (Yi et al., 2001). Mitochondrial sub-fractionation was performed as described by She et al (She et al., 2011).
Mitochondrial pyruvate consumption and [1-14C]-pyruvate conversion assays
Pyruvate utilization by mitochondria was measured as described (Olsen, 1971). Briefly, isolated mitochondria were mixed with the mitochondria resuspension buffer, followed by centrifugation and pyruvate assay. The assay was carried out in reaction buffer containing 20 mM Hepes, pH 7.2, 0.05% BSA, 20 μM NADH, and recombinant LDH (0.01 mg/ml). Pyruvate concentration was determined by measuring the decrease in fluorescence from the oxidation of NADH with a spectrofluometer (ex.340nm; em.460nm). The [1-14C]-pyruvate conversion assay was performed as described (Pezzato et al., 2009). Briefly, 14CO2 production through PDC was measured using isolated mitochondria (1mg) in the mitochondria resuspension buffer (1ml) containing [1-14C]-pyruvate (0.1 μCi/ml). The incubation mixture was placed at the bottom of a vial with a rubber stopper and maintained in agitation. The 14CO2 produced during incubation was trapped by hyamine hydroxide placed in an eppendorf tube in the vial. The reaction was blocked with 0.5 ml of 50% TCA after 1h. Twenty minutes after the TCA injection, all the samples in hyamine hydroxide were transferred to mini-vials together with 5 ml of scintillator liquid, and radioactivity was assayed on a scintillation counter. The results were normalized based on mitochondrial protein levels assayed by Bradford assay using BSA as a standard.
Xenograft studies
Nude mice (nu/nu, female 6–8-week-old, Harlan Labs) were subcutaneously injected with 1 × 107 H1299 cells stably expressing mPDHK1 WT and Y239/240F mutant in conjunction with stable knock-down of endogenous PDHK1 on the left and right flanks, respectively. Tumor formation was assessed every 2 to 3 days. Tumor growth was recorded by measuring two perpendicular diameters of the tumors over a 5-week time course using the formula 4π/3 × (width/2)2 × (length/2). The tumors were harvested and weighed at the experimental endpoint, and the masses of tumors (g) in both flanks of each mouse were compared. Statistical analyses were performed using a paired Student’s t test.
Supplementary Material
Highlights.
Tyrosine phosphorylation of PDHK1 enhances its kinase activity
Mitochondrial oncogenic tyrosine kinases phosphorylate PDHK1 in cancers cells
Functional PDC can form in mitochondria outside of matrix in some cancer cells
Tyrosine phosphorylation of PDHK1 promotes tumor growth
Acknowledgments
We gratefully acknowledge the critical reading of the manuscript by Shannon Elf. We thank Hong Yi for assistance with electron microscopy. This work was supported in part by NIH grants CA120272 and CA140515 (J.C.). Federal Funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E (H.F.). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. J.X., Q.G., T.-L.G., and R.D.P. are employees of Cell Signaling Technology, Inc. J.L.R. is an employee of Novartis Pharma AG. T.H. is a Fellow Scholar of the American Society of Hematology. Z.G.C., H.F., F.R.K., S.K. and J.C. are Georgia Cancer Coalition Distinguished Cancer Scholars. S. K. is a Robbins Scholar. S.K. and J.C. are American Cancer Society Basic Research Scholars. S. K. is a Special Fellow and J.C. is a Scholar of the Leukemia and Lymphoma Society.
Footnotes
SUPPLEMENTAL INFORMATION Supplemental Information includes detailed experimental procedures and seven figures.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11:37–51. doi: 10.1016/j.ccr.2006.10.020. [DOI] [PubMed] [Google Scholar]
- Gu TL, Goss VL, Reeves C, Popova L, Nardone J, Macneill J, Walters DK, Wang Y, Rush J, Comb MJ, et al. Phosphotyrosine profiling identifies the KG-1 cell line as a model for the study of FGFR1 fusions in acute myeloid leukemia. Blood. 2006;108:4202–4204. doi: 10.1182/blood-2006-06-026666. [DOI] [PubMed] [Google Scholar]
- Hiromasa Y, Fujisawa T, Aso Y, Roche TE. Organization of the cores of the mammalian pyruvate dehydrogenase complex formed by E2 and E2 plus the E3-binding protein and their capacities to bind the E1 and E3 components. J Biol Chem. 2004;279:6921–6933. doi: 10.1074/jbc.M308172200. [DOI] [PubMed] [Google Scholar]
- Hitosugi T, Kang S, Vander Heiden MG, Chung TW, Elf S, Lythgoe K, Dong S, Lonial S, Wang X, Chen GZ, et al. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci Signal. 2009;2:ra73. doi: 10.1126/scisignal.2000431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S, Dong S, Gu TL, Guo A, Cohen MS, Lonial S, Khoury HJ, Fabbro D, Gilliland DG, Bergsagel PL, et al. FGFR3 Activates RSK2 to Mediate Hematopoietic Transformation through Tyrosine Phosphorylation of RSK2 and Activation of the MEK/ERK Pathway. Cancer Cell. 2007;12:201–214. doi: 10.1016/j.ccr.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Li J, Chuang JL, Chuang DT. Distinct structural mechanisms for inhibition of pyruvate dehydrogenase kinase isoforms by AZD7545, dichloroacetate, and radicicol. Structure. 2007;15:992–1004. doi: 10.1016/j.str.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Dang CV. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 2006;66:8927–8930. doi: 10.1158/0008-5472.CAN-06-1501. [DOI] [PubMed] [Google Scholar]
- Kim JW, Gao P, Liu YC, Semenza GL, Dang CV. Hypoxia-inducible factor I and dysregulated c-myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Molecular and Cellular Biology. 2007;27:7381–7393. doi: 10.1128/MCB.00440-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Kobrin MS, Yamanaka Y, Friess H, Lopez ME, Korc M. Aberrant expression of type I fibroblast growth factor receptor in human pancreatic adenocarcinomas. Cancer Res. 1993;53:4741–4744. [PubMed] [Google Scholar]
- Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell. 2008;13:472–482. doi: 10.1016/j.ccr.2008.05.005. [DOI] [PubMed] [Google Scholar]
- Liu SJ, Baker JC, Roche TE. Binding of the Pyruvate-Dehydrogenase Kinase to Recombinant Constructs Containing the Inner Lipoyl Domain of the Dihydrolipoyl Acetyltransferase Component. J Biol Chem. 1995;270:793–800. doi: 10.1074/jbc.270.2.793. [DOI] [PubMed] [Google Scholar]
- Luqmani YA, Graham M, Coombes RC. Expression of basic fibroblast growth factor, FGFR1 and FGFR2 in normal and malignant human breast, and comparison with other normal tissues. Br J Cancer. 1992;66:273–280. doi: 10.1038/bjc.1992.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marek L, Ware KE, Fritzsche A, Hercule P, Helton WR, Smith JE, McDermott LA, Coldren CD, Nemenoff RA, Merrick DT, et al. Fibroblast growth factor (FGF) and FGF receptor-mediated autocrine signaling in non-small-cell lung cancer cells. Molecular pharmacology. 2009;75:196–207. doi: 10.1124/mol.108.049544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison RS, Yamaguchi F, Bruner JM, Tang M, McKeehan W, Berger MS. Fibroblast growth factor receptor gene expression and immunoreactivity are elevated in human glioblastoma multiforme. Cancer Res. 1994;54:2794–2799. [PubMed] [Google Scholar]
- Olsen C. An enzymatic fluorimetric micromethod for the determination of acetoacetate, -hydroxybutyrate, pyruvate and lactate. Clinica chimica acta; international journal of clinical chemistry. 1971;33:293–300. doi: 10.1016/0009-8981(71)90486-4. [DOI] [PubMed] [Google Scholar]
- Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3:187–197. doi: 10.1016/j.cmet.2006.01.012. [DOI] [PubMed] [Google Scholar]
- Penault-Llorca F, Bertucci F, Adelaide J, Parc P, Coulier F, Jacquemier J, Birnbaum D, deLapeyriere O. Expression of FGF and FGF receptor genes in human breast cancer. International journal of cancer. 1995;61:170–176. doi: 10.1002/ijc.2910610205. [DOI] [PubMed] [Google Scholar]
- Pezzato E, Battaglia V, Brunati AM, Agostinelli E, Toninello A. Ca2+ -independent effects of spermine on pyruvate dehydrogenase complex activity in energized rat liver mitochondria incubated in the absence of exogenous Ca2+ and Mg2+ Amino Acids. 2009;36:449–456. doi: 10.1007/s00726-008-0099-5. [DOI] [PubMed] [Google Scholar]
- Read RJ. Pushing the boundaries of molecular replacement with maximum likelihood. Acta Crystallogr D. 2001;57:1373–1382. doi: 10.1107/s0907444901012471. [DOI] [PubMed] [Google Scholar]
- Roche TE, Baker JC, Yan YH, Hiromasa Y, Gong XM, Peng T, Dong JC, Turkan A, Kasten SA. Distinct regulatory properties of pyruvate dehydrogenase kinase and phosphatase isoforms. Prog Nucleic Acid Re. 2001;70:33–75. doi: 10.1016/s0079-6603(01)70013-x. [DOI] [PubMed] [Google Scholar]
- She H, Yang Q, Shepherd K, Smith Y, Miller G, Testa C, Mao Z. Direct regulation of complex I by mitochondrial MEF2D is disrupted in a mouse model of Parkinson disease and in human patients. J Clin Invest. 2011;121:930–940. doi: 10.1172/JCI43871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Xiao S, Nalabolu SR, Aster JC, Ma J, Abruzzo L, Jaffe ES, Stone R, Weissman SM, Hudson TJ, Fletcher JA. FGFR1 is fused with a novel zinc-finger gene, ZNF198, in the t(8;13) leukaemia/lymphoma syndrome. Nat Genet. 1998;18:84–87. doi: 10.1038/ng0198-84. [DOI] [PubMed] [Google Scholar]
- Yi H, Leunissen J, Shi G, Gutekunst C, Hersch S. A novel procedure for pre-embedding double immunogold-silver labeling at the ultrastructural level. J Histochem Cytochem. 2001;49:279–284. doi: 10.1177/002215540104900301. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.