

Abstract
This study reveals a new complexity in the cellular response to DNA damage: activation of interferon (IFN) signaling. The DNA damage response involves the rapid recruitment of repair enzymes, and the activation of signal transducers that regulate cell cycle checkpoints and cell survival. To understand the link between DNA damage and innate cellular defense that occurs in response to many viral infections, we evaluated the effects of agents such as etoposide that promote double-stranded DNA breaks. Treatment of human cells with etoposide led to the induction of IFN-stimulated genes, and the IFN-α and IFN-λ genes. The nuclear factor-κB (NF-κB), known to be activated in response to DNA damage, was shown to be a key regulator of this IFN gene induction. Expression of an NF-κB subunit, p65/RelA was sufficient for induction of the human IFN-λ1 gene. In addition, NF-κB was required for the induction of the IFN regulatory factors-1 and -7 that are able to stimulate expression of the IFN-α and IFN-λ genes. Cells that lack the NF-κB essential modulator (NEMO), lack the ability to induce the IFN genes following DNA damage. Breaks in DNA are generated during normal physiological processes of replication, transcription, and recombination, as well as by external genotoxic agents or infectious agents. The significant finding of IFN production as a stress response to DNA damage provides a new perspective on the role of IFN signaling.
Introduction
An effective DNA damage response is critical for maintaining genomic integrity and preventing mutations that can lead to cancer. Double strand breaks (DSB) are the most severe lesions, and they can occur during DNA replication, lymphocyte V(D)J gene rearrangement, meiosis, viral infection, and in response to naturally occurring ionizing radiation (1–5). These DNA breaks are sensed rapidly, and accurate repair is essential to prevent permanent genomic damage. However, the cellular response to DNA damage engages more than just DNA repair machinery, it engages complex signaling pathways that can promote cell survival or cell death (6, 7). In this report the activation of an additional network is revealed, the interferon (IFN) signal pathway.
A primary transducer of the response to DSBs is the nuclear kinase ataxia-telangiectasia mutated (ATM) (8). ATM belongs to a family of phosphatidylinositol 3-kinase (PI3K)-related kinase family, several of which are involved in the DNA-damage response including ATM and Rad3 related (ATR) and DNA-dependent protein kinase (DNA-PK) (9). ATM transduces the DNA damage response signal by phosphorylating downstream effectors such as the checkpoint kinases Chk1 and Chk2, and the p53 tumor suppressor. These effectors in turn establish cell cycle arrest to allow repair of damaged DNA, or promote damage-induced apoptosis. Major alterations in gene expression occur during this time, and this reflects the action of not only p53, but other transcription factors (10). One transcription factor that is activated in response to DNA damage is nuclear factor-kappa B (NF-κB) (11). NF-κB regulates the expression of diverse genes involved in cellular responses that include survival, proliferation, tissue remodeling, inflammation, immunity, and stress. We have found that NF-κB activation in response to DNA damage directs the induction of the IFN system, a stress pathway best known for its ability to confer viral resistance.
IFNs play vital roles in both innate and adaptive immunity and consist of three families of cytokines that bind to distinct cell surface receptors and are designated type I, II, and III (12). The genes encoding type I IFN (primarily α and β) and type III (λ) IFN are induced in response to viral or bacterial infection (13). The single type II (γ) IFN gene is induced primarily following receptor activation of T cells and NK cells. The regulated expression of the IFN-β gene in response to viral infection is a paradigm for cooperativity of DNA binding factors (14). NF-κB and interferon regulatory factors (IRFs) function along with activating transcription factor-2/c-Jun in the IFN-β enhancer. The IRFs were first characterized as regulators of type I IFN genes and IFN-stimulated genes (ISGs), and are now known to have diverse roles in immunity (15). Activation of ubiquitous IRF-3 during viral infection supports induction of a subset of ISGs and the IFN-β and IFN-α genes (16, 17). The IRF-1 and IRF-7 genes are induced in response to secreted IFN and can play a role in the secondary response to IFN (18). IFNs bind to cell surface receptors that activate Janus kinases and the tyrosine phosphorylation of signal transducers and activators of transcription, STAT1 and STAT2 (19, 20). We report here that signaling via the DNA damage response in human cells primarily induces the IFN-λ and IFN-α genes. The promoters of the IFN-λ genes have been found to possess both IRF and NF-κB binding sites (21, 22). In this study we demonstrate that NF-κB activation in response to DNA damage is sufficient and necessary to induce human IFN-λ.
This study identifies IFN signaling as part of the DNA damage response. IFNs are essential components of innate immunity, and are well recognized for their ability to inhibit viral infection and activate immune effector cells (23). In addition, they are known for their anti-tumor effects by inhibiting proliferation of cancerous cells and promoting apoptosis (24–26). The IFN arm of the DNA damage response may have evolved as an antiviral mechanism in reaction to DNA damage induced by viruses, as a mechanism that reduces cellular proliferation to allow DNA repair, or as a mechanism to promote the death of cells with irreparable damage.
Materials and Methods
Cell culture, transfections, and infections
HeLa S3, HT1080 and THP-1 cells were obtained from American Type Culture Collection. THP-1 cells were maintained in RPMI with 8% FBS, other cells were maintained in DMEM with 8% FBS. Primary human monocytes were isolated from blood cells of healthy donors (Long Island Blood Services, NY) using the Monocyte Isolation Kit II (Miltenyi Biotec Inc., Auburn, CA) and maintained in RPMI with 10% FBS. Stable HT1080 transfectants with tetracycline-inducible expression of IRF-7 were generated according to manufacturers’ instructions (T-Rex system; Invitrogen, Carlsbad, CA) and the gene was induced with 2μg/ml doxycycline. Murine embryo fibroblasts (MEFs) from NEMO/IKKγ knockout mice were a gift of Dr. Kenneth Marcu (Stony Brook University) (27), MEFs from IRF3 knockout mice were a gift of Dr. Tadatsugu Tanaguchi (University of Tokyo) (16, 17), MEFs from IRF1 knockout mice were a gift of Dr. Tak Mak (University of Toronto) (28), and MEFs from IRF7 knockout mice were a gift of Dr. Michael Gale (University of Washington)(29). DNA transfections were performed using FuGENE 6 (Roche Diagnostics Corp., Indianapolis, IN) or TransIt (Mirus, Madison, WI). Newcastle Disease Virus (NDV) (NJ- LaSota-1946) was a gift from Dr. Paula Pitha-Rowe (Johns Hopkins University, Baltimore, MD) and was propagated as described previously (30). Infections were performed at 200 hemagglutination units/ml.
Plasmids and luciferase assays
IRF-3, STAT1 and STAT2 constructs have been described (31–33). The dominant negative IκBα plasmid (S32A/S36A) was a gift of Dr. Dean Ballard (Vanderbilt University, Nashville, TN)(34). The HA tagged ubiquitin-K63-only (HA-Ub0R63K) plasmid was a gift of Dr. Dafna BarSagi (New York University, NY) (35). The reporter plasmid encoding the IFNλ1 promoter regulating expression of the firefly luciferase gene [pλ1(−554/+14)Luc] was a gift of Dr. Takashi Fujita (Kyoto University, Kyoto, Japan) (21). The human IRF-7A gene was obtained from Dr. Joseph S. Pagano (University of North Carolina, Chapel Hill, NC) (36), and subcloned into the pcDNA4/TO/Myc-His vector (T-Rex system; Invitrogen, Carlsbad, CA). The Dual-Luciferase reporter assay system was used for luciferase assays with the Renilla luciferase construct pRL-null as an internal control (Promega, Madison, WI).
Reagents
Etoposide was used at 40μg/ml and camptothecin, adriamycin, mitomycin, were used at the concentrations indicated (Sigma-Aldrich, St. Louis, MO). NF-κB inhibitor, BAY 11-7085, was obtained from Alexis Biochemicals (San Diego, CA) and used at 5μM, and the IKKβ inhibitor ML120B was a gift from Millennium Pharmaceuticals (Cambridge, MA) and used at 20μM. The ATM inhibitor, AZ12622702/KU55933, was a gift from AstraZeneca (Cheshire, UK) and used at 10μM. Antibodies recognizing STAT2 (C-20), c-Myc epitope (sc-40), CBP (A-22), and normal rabbit IgG were purchased from Santa Cruz Biotech, Inc. (Santa Cruz, Calif.). Antibody to STAT2 phosphorylated on tyrosine 689 was obtained from Upstate Biotechnology Inc. (Lake Placid, NY). Antibodies to p65 phosphorylated on serine 536, and STAT1 phosphorylated on tyrosine 701 were obtained from Cell Signaling (Beverly, MA). Antibody against HA epitope (12CAS) was purchased from Roche (Indianapolis, IN). Antibodies derived against IRF-3 and STAT1 were described previously (30, 33). Antibody to IRF-7 was raised in rabbits against the 246–432 amino acid region of IRF-7. Anti-mouse or anti-rabbit secondary antibodies conjugated to IRDye800 or 700 were obtained from Rockland Immunochemicals (Gilbertsville, PA,) TNFα was obtained from Invitrogen (Carlsbad, CA).
Polymerase Chain Reaction (PCR)
RNA was isolated from cells using SV Total RNA Isolation Kit (Promega, Madison, WI) and cDNA was generated using random hexamer primers and SuperScriptII Reverse Transcriptase (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol. Reverse transcription (RT)-PCR was performed using the indicated primers and Taq polymerase (Invitrogen, Carlsbad, CA). Alternatively, quantitative real-time PCR was performed using the indicated primers at their optimal conditions as suggested by the manufacturer’s instructions for the LightCycler-FastStart DNA Master SYBR Green I kit (Roche Molecular Biochemicals). Data was analyzed using the LightCycler software and values were normalized to actin mRNA levels. Human primers used in the studies corresponded to: IFNλ1 (37), IFNα6 (38), IFNα1/13 (38), actin (39), pan IFNα 5′-cacacaggcttccaggcattc-3′ and 5′-tcttcagcacaaaggactcatctg-3′; ISG54 (+1608) 5′-attctatcaccaagcccgtgg-3′ and (+1370) 5′-tggagtctggaagcctcatcc-3′; IFNα7 (+194) 5′-tcctcctccgggaatctgaat-3′ and (+106) 5′-agggccttgatactcctgg-3′; IFNα14 (+315) 5′-taggagggtctcatcccaagc-3 ′ and (+ 65) 5 ′-tgggctgtaatctgtctcaaac-3′; IFNβ (+27) 5′-tgctctcctgttgtgcttctccac-3′ and (+243) 5′-atagatggtcaatgcggcgtcc-3′; IRF-7 (+1476) 5′-gatgtcgtcatagaggctgttgg-3′ and (+1343) 5′-tggtcctggtgaagctggaa-3′; IRF-1 (+397) 5′-tcttagcatctcggctggacttc-3′ and (+190) 5′-cgatacaaagcaggggaaaagg-3′; GAPDH (+1007) 5′-tactccttggaggccatgtg-3′ and (+528) 5′-cacagtccatgccatcactg-3′. Murine primers corresponded to: pan mIFNα 5′-cctgagagagaagaaacacagcc-3′ and 5′-tctgctctgaccacytcccag -3′(40); mIFNλ2 (41); and mActin (42).
Immunoprecipitation and Western blot
For immunoprecipitation, cells were lysed in 50mM Hepes pH 7.2, 250mM NaCl, 0.5% Nonidet P-40, 5mM EDTA, 1mM dithiothreitol, 1mM NaF, 0.1mM Na3VO4, and protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO). Lysates were cleared by centrifugation for 5 min at 15,000g and reacted with antibodies for 3hr at 4 °C. Immunocomplexes were collected with Protein G-conjugated agarose (Invitrogen, Carlsbad, CA). For direct Western blot, cells were lysed in 50mM Tris pH 7.5, 400mM NaCl, 0.5% Nonidet P-40, 5mM EDTA, 10% glycerol, 50mM NaF, 0.1mM Na3VO4, and protease inhibitors. The lysates were cleared by centrifugation and directly added to SDS sample buffer. Proteins were separated on 8% SDS-PAGE and transferred to nitrocellulose membrane (Pierce Biotechnology, Rockford, IL). Membranes were reacted with indicated antibodies and images were detected using the Odyssey infrared imaging system (Li-COR Biosciences, Lincoln, NE, USA). Alternatively, secondary anti-rabbit or anti-mouse antibodies linked to HRP (Amersham/GE Healthcare, Piscataway, NJ) were used and the membrane was incubated in enhanced chemiluminescence reagents and exposed to film.
Fluorescence imaging
Cells were seeded on coverslips, fixed in 4% paraformaldehyde and either visualized directly for GFP fluorescence, or permeabilized in 0.2% Triton X-100 before reaction with anti-Myc antibody. Secondary antibodies were conjugated to Rhodamine (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA.). Coverslips were mounted in anti-fade solution (Vectashield, Vector Laboratories, Inc. Burlingame, CA.). Images were captured with a Zeiss Axiovert 200 M digital deconvolution microscope or Zeiss LSM 510 META NLO two-photon laser scanning confocal microscope.
Ubiquitination assay
The HA-Ub0R63K expression plasmid was transfected into stable cell line expressing tetracycline repressor and responsive Myc-His-IRF-7 gene as described above. Doxycycline was added 24 hours post transfection with or without etoposide as indicated. Cells were lysed by sonication in 6 M guanidine HCl, 0.1 M Na2HPO4/NaH2PO4(pH 8.0) and 10 mM imidazole). Lysates were incubated with Ni-NTA agarose beads (Qiagen, Chatsworth, CA), proteins were eluted with SDS loading buffer and analyzed by Western blot (43).
Results
Etoposide activation of IFN signaling
Cells respond to viral infection with the induction of IFN stimulated genes (ISGs), either directly by activation of the IRF-3 transcription factor, or as an indirect response to autocrine IFN and STAT activation (15, 19, 30). Since the genes induced by IRF-3 or STATs can promote cellular apoptosis, we examined gene expression during the DNA damage response to identify shared pro-apoptotic induced genes (24, 44–46). Etoposide, an anticancer drug, was used to initiate the DNA damage response since it inhibits the ability of topoisomerase II to religate cleaved DNA (47, 48). The inhibition results in an accumulation of double-stranded breaks in DNA, particularly during DNA replication and S/G2 phases of the cell cycle. The increase of double-stranded DNA breaks with time leads to the DNA damage response and cell cycle arrest or apoptosis. Following cell treatment with etoposide, we noted induction of several of the ISGs. The response of primary human monocytes was examined and induction of a representative gene, ISG54/Ifit2, is shown in Figure 1A. The effect of etoposide gradually increases the number of double strand DNA breaks, and accordingly the levels of ISG54 mRNA increased with time. To ensure this response was not specific to cell type, we evaluated the response of HeLa cells to etoposide. These cells also responded with induction of the ISG54 gene.
Figure 1. Induction of ISG54 mRNA and activation of STAT signaling in response to etoposide.

A) Primary (1°) human monocytes were untreated or treated with etoposide for 0, 18, or 24 hrs. RNA was extracted and real-time RT-PCR was performed with primers specific for ISG54 and quantified (left). HeLa cells were untreated or treated with etoposide for 24 hrs. RNA was extracted and real-time RT-PCR was performed with primers specific for ISG54 and quantified (right). B) HeLa cells were transfected with STAT1-GFP or STAT2-GFP and untreated (−) or treated with etoposide for 24 hours. Fluorescent images are shown. C) HeLa cells were treated with etoposide for the hours indicated and cell lysates were evaluated for endogenous STAT1 (top) or STAT2 (bottom) tyrosine phosphorylation by Western blot. Results represent duplicate determinations in three independent experiments.
To access whether IFN production was responsible for ISG54 induction, we evaluated the activation of the STAT1 and STAT2 transcription factors in etoposide-treated cells. Binding of type I IFNs to specific receptors leads to tyrosine phosphorylation and nuclear accumulation of STAT1 and STAT2 (49). Cells expressing GFP tagged STAT1 or STAT2 were examined for their response, and nuclear accumulation was evident by 24 hours of etoposide treatment (Figure 1B). In addition, the tyrosine phosphorylation of endogenous STAT1 and STAT2 was clearly evident (Figure 1C).
These results suggested that etoposide treatment could induce the production and action of IFN, and accordingly we evaluated the induction of IFN-α, IFN-β, and IFN-λ genes in primary human monocytes. There was little response of the IFN-β gene to DNA damage, although the gene was robustly induced in response to infection by Newcastle Disease Virus (Figure 2A). In contrast, IFN-α mRNA expression was evaluated with pan-specific primers and clearly displayed an induction in response to etoposide (Figure 2B). To determine the induction of IFN-α gene family subtypes, we tested the expression of individual genes. Various IFN-α genes tested were induced (Figure 2C). We next tested expression of the newest family of IFN, IFN-λ. Specific induction of the IFN-λ1 gene clearly increased with etoposide treatment (Figure 2D).
Figure 2. Specific IFN species are induced in response to etoposide.
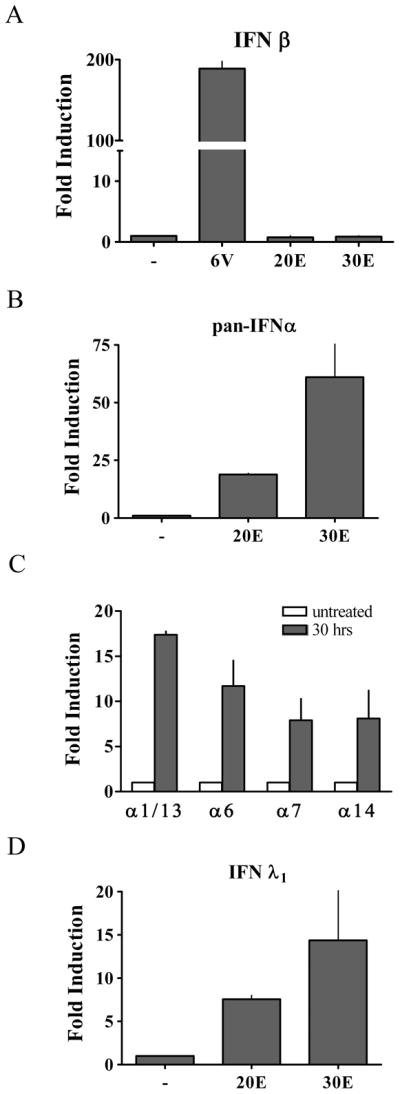
A) Primary human monocytes were untreated (−) or treated with etoposide for 20 hours (20E) or 30 hours (30E), or infected with Newcastle Disease Virus (NDV) for 6 hours. mRNA levels were quantified by real-time RT-PCR corresponding to the IFN-β gene in response to etoposide or NDV. B) mRNA levels quantified by pan-specific primers to IFN-α from primary monocytes treated with etoposide as in (A). C) mRNA levels were quantified to specific subspecies IFN-α1/α13, IFN–α6, IFN-α7, and IFN-α14 following 30 hr etoposide treatment of primary monocytes. D) mRNA levels corresponding to the IFN-λ1 gene from primary monocytes treated with etoposide as in (A). Values are means of duplicate determinations in two or three independent experiments.
Induction of ISG54, IFN-α, and IFN-λ in response to various DNA damaging agents
Etoposide elicits double strand DNA breaks by forming an inactive ternary complex with topoisomerase II and inhibits the ability of the enzyme to religate cleaved DNA. Effectiveness can vary with proliferation of cell cultures, but 40μg/ml etoposide was usually optimal for the IFN response (Supplemental Figure 1). To determine whether IFN signaling is a general response to DNA damage or specific to etoposide, three diverse agents were tested, camptothecin, mitomycin C, and adriamycin. Camptothecin elicits single strand DNA breaks by forming a ternary complex with topoisomerase I; mitomycin C is a DNA alkylating agent; and adriamycin causes DNA strand breaks by intercalation (50). Cells were treated with these agents and evaluated for ISG54 protein expression, and mRNA levels of IFN-α and IFN-λ1 (Figure 3). Despite the different mechanisms that elicit DNA damage by these agents, they all induced ISG54 and the IFN genes. The differences in fold induction may be a consequence of various peak response times due to different mechanisms of DNA damage. The results indicate that the production of IFN in etoposide treated cells is a general DNA damage response and not one specific to a single drug or DNA insult.
Figure 3. Various DNA damaging agents activate IFN signaling.

THP-1 cells were untreated (−) or treated with 40μg/ml etoposide (E), 5μM camptothecin (C), 20μg/ml mitomycin (M), or 20μg/ml adriamycin (A) for 24 hours. A) Cell lysates were prepared and ISG54 protein levels were evaluated by Western blot. B) IFN-α mRNA induction was evaluated with pan-specific primers and real-time RT-PCR. C) IFN-λ1 specific mRNA expression was evaluated by real-time RT-PCR. Values are means of duplicate determinations in two independent experiments.
DNA damage activates IRF-1 and IRF-7, but not IRF-3
IRF-3 is expressed constitutively in cells and in response to viral infection it is a critical transcription factor for the induction of the IFN-β gene, a subset of IFN-α genes, and the direct induction of a subset of ISGs (15, 16, 51, 52). It exists in a latent state primarily in the cytoplasm and is modified by specific serine phosphorylation following viral infection. Phosphorylation promotes IRF-3 nuclear accumulation, DNA binding, and association with CREB-binding protein (CBP). We evaluated these parameters for the activation of IRF-3 in response to DNA damage. Cells expressing IRF-3 tagged with GFP were treated with etoposide and visualized microscopically to access the cellular localization of IRF-3. IRF-3 remained primarily cytoplasmic and did not show evidence of nuclear accumulation (Figure 4A). The phosphorylated forms of IRF-3 can be detected by their reduced migration during SDS-PAGE. This can be easily observed following viral infection, but it was not evident is response to DNA damage elicited with camptothecin or etoposide (Figure 4B). The ability of activated IRF-3 to form complexes with CBP was evaluated by co-immunoprecipitation and Western blot. Although IRF-3 can be readily detected in immunocomplexes with CBP during viral infection, there was no evidence of association with CBP following etoposide treatment.
Figure 4. IRF-3 is not activated in response to DNA damage.

A) HeLa cells were transfected with IRF-3-GFP and left untreated (−) or were treated with etoposide (Etop) for 24 hours. Cellular localization was evaluated by fluorescence microscopy. Images represent random sampling from three independent experiments. B) Cells were untreated or infected with NDV for 6 or 12 hours, treated with camptothecin (Camp) for 12 or 20 hours, or treated with etoposide for 20 or 40 hours. Endogenous IRF-3 was detected in cell lysates by Western blot. C) CBP was immunoprecipitated (IP) from lysates of untreated cells or cells infected with NDV for 6 hours or treated with camptothecin or etoposide for 36 and 50 hours. Western blots (WB) were performed with antibodies to IRF-3.
The IRF-7 transcription factor is a key regulator of the IFN-α genes in response to viral or bacterial infection (18, 29, 51, 53). It is expressed at low levels in lymphoid cells, but it is induced in all cell types by IFN and can function in a secondary wave of IFN production. We evaluated induction of the IRF-7 gene in response to DNA damage, and found mRNA levels were clearly induced following etoposide (Figure 5A). mRNA levels increased by more than 10-fold estimated by real-time PCR in 24 hours (S.B-R and N.C.R., unpublished data). IRF-7 can be activated in response to viral infection by serine phosphorylation. Although this modification may occur, it was not evident by a reduced migration in SDS-PAGE (S.B-R and N.C.R., unpublished data). (54). Regulation of IRF-7 activity has also been demonstrated to occur by lysine-63-linked ubiquitination, and this modification was evaluated (55). A stable cell line was generated expressing a tetracycline/doxycycline inducible IRF-7 gene tagged with the Myc epitope and polyhistidine (His). These cells were transfected with a gene encoding ubiquitin in which all of the lysines were mutated except lysine 63, and therefore polyubiquitination could only occur via isopeptide linkage with lysine 63 (35). Cells were untreated or treated with etoposide, and IRF-7 protein was captured on nickel charged resins and evaluated by Western blot (Figure 5B). Following etoposide treatment, IRF-7 displayed slow migrating species indicative of its polyubiquitination and activation. Lysine 63-linked ubiquitin chains have been shown to regulate cellular processes including protein-protein interactions, and to play a critical role in the DNA damage response (56).
Figure 5. Evidence for a role of IRF-7 in the IFN response to DNA damage.

A) THP-1 cells were untreated (−) or treated with etoposide (+) for 24 hours. IRF-7 mRNA induction was evaluated by RT-PCR and displayed on agarose gels. Faint band in untreated sample is non-specific. mRNA levels of GAPDH are shown as controls. B) HT1080 stable cell line expressing tetracycline inducible IRF-7-Myc-His was transfected with the HA-Ub0R63K ubiquitin (K63Ub), and doxycycline (Dox) was used to induce IRF-7 expression in the absence or presence of etoposide for 24 hours. IRF-7 was collected on nickel charged resins and samples were analyzed by Western blot with antibody to IRF-7. Lower panel shows input before resin with anti-myc antibody. C) Expression of IRF-7-Myc-His was induced with doxycycline in the stable cell line in the absence or presence of etoposide for 15 hours before immunostaining with antibodies to Myc. Imaging analysis of three independent experiments indicate nuclear accumulation of IRF-7 at this time in >50% of the cells. D) Pan-specific primers were used to quantify IFN-α mRNA levels (top) and specific primers were used to assess IFN-λ1 mRNA levels (bottom) in the IRF-7-Myc-His inducible cells by real-time PCR. Cells were untreated (−), treated with etoposide (E), doxycycline (D), or etoposide and doxycycline for 24 hours. Values are means of duplicate determinations in two independent experiments.
In a latent state IRF-7 resides primarily in the cytoplasm and accumulates in the nucleus following activation. The tetracycline inducible cell line was used to evaluate IRF-7 localization by immunofluorescence during DNA damage (Figure 5C). Following induction of IRF-7 expression, the protein was clearly cytoplasmic in untreated cells. However IRF-7 accumulated in the nucleus following etoposide treatment, suggesting its activation. To evaluate the effects of inducible IRF-7 on IFN-α and IFN-λ1, their mRNA levels were measured (Figure 5D). Cells were either treated with etoposide, treated with tetracycline to induce IRF-7, or treated with both etoposide and tetracycline. Pan-specific primers were used to quantify expression of the IFN-α family. IFN-α was found to be induced in response to etoposide, but there was minimal effect of tetracycline-induced IRF-7 alone. However, etoposide and tetracycline potently increased etoposide-induced IFN-α expression. The results indicate modification of IRF-7 protein in response to DNA damage contributes to induction of the IFN-α genes. Expression of IFN-λ1 mRNA was similarly evaluated and IRF-7 activation by etoposide also was found to contribute to induction of the IFN-λ1 gene.
Another member of the IRF family, IRF-1, has diverse roles in response to pathogens, development of the immune system, growth arrest, and apoptosis (15, 57, 58). The protein is constitutively nuclear but only expressed following induction by cytokines such as IFN. We evaluated IRF-1 mRNA expression and found mRNA levels increased by more than 12-fold estimated by real-time PCR following 24 hours of etoposide treatment (Figure 6A, S.B-R and N.C.R., unpublished data). To evaluate the possible role of IRF-1 in induction of the IFN genes during DNA damage, we tested its effect on expression of the IFN-λ1 promoter driving a luciferase reporter gene. The promoter of IFN-λ1 possesses IRF binding sites and a bona fide NF-κB site (21, 22). The IFN-λ1 promoter activity was induced following etoposide treatment, or by co-transfection with a plasmid encoding IRF-1 (Figure 6B). Combined IRF-1 expression with etoposide induced significant expression of the IFN-λ1 promoter. IRF-1 could also be demonstrated to bind to a site in the IFN-λ1 promoter, suggesting direct action on the IFN-λ1 gene (Supplemental Figure 2).
Figure 6. Activation and inhibition of IRF and IFN genes.

A) THP-1 cells were untreated (−) or treated with etoposide (+) for 24 hours. IRF-1 mRNA levels were evaluated by RT-PCR and displayed on agarose gels. mRNA levels of actin are shown as controls. B) The IFN-λ1 luciferase reporter was expressed in HeLa cells untreated or treated with etoposide (Etop) for 24 hours. Empty vector (c) or IRF-1 expression plasmid (IRF-1) were co-transfected where indicated with or without etoposide treatment and luciferase activity was measured. C) Effects of the ATM inhibitor AZ12622702 (AZ) on IFN-α and IFN-λ gene expression. HeLa cells were untreated or treated with AZ for one hour followed by etoposide (E) for 24 hours as indicated. Real-time PCR was used to quantify IFN-α (left) or IFN-λ (right) mRNA expression. D) Effects of IKKβ inhibitors BAY117085 (BAY) or ML120B (ML120) on IRF-1 and IRF-7 gene expression. HeLa cells were untreated or treated with the inhibitors for one hour followed by etoposide for five hours as indicated. Real-time PCR was used to quantify endogenous IRF-1 (left) or IRF-7 (right) mRNA expression. Quantitative results are means of duplicate determinations in three independent experiments.
Double strand DNA breaks that occur in response to etoposide lead to the recruitment and activation of the ATM kinase. To evaluate the potential role of ATM in the induction of IFN gene expression, we tested the effects of a pharmacological inhibitor, AZ12622702. Treatment of cells with the ATM inhibitor decreased the ability of etoposide to induce the IFN-λ1 gene and the IFN-α genes (Figure 6C). The results indicate ATM is a significant signaling kinase upstream of IFN gene induction, and the residual response may be due to other PI3K-related kinases that are activated during DNA damage.
Role of NF-κB in the response of IFN genes to DNA damage
IRF-1 and IRF-7 are both induced in response to IFN signaling and in response to DNA damage. Studies have also indicated that the promoters of both of these genes can be activated by the NF-κB transcription factor (59–61). Since NF-κB activation is a well characterized response to DNA damage and ATM activity, we determined whether induction of IRF-1 and IRF-7 by etoposide is a direct response to NF-κB (11, 62). Inactive NF-κB is localized in the cytoplasm due to binding the inhibitor of NF-κB (IκB) (63). IκB is released following its phosphorylation by an IκB kinase (IKK) complex composed of IKKα, IKKβ, and NF-κB essential modulator (NEMO/IKKγ). To determine the role of NF-κB in induction of IRF-1 and IRF-7 by DNA damage, we tested the effect of several specific IKKβ inhibitors (Figure 6D). Cells were treated with etoposide in the absence or presence of BAY117085 or ML120B and real-time PCR was used to quantify the endogenous expression of IRF-1 and IRF-7 mRNAs. Both inhibitors effectively blocked the induction of IRF-1 and IRF-7 expression, indicating activation of NF-κB in response to DNA damage is necessary to induce these genes.
A major species of NF-κB is the p50 and p65/RelA heterodimer (63). Since serine phosphorylation of the carboxyl terminus of p65 has been shown to increase its transcriptional activity and stability, we evaluated the phosphorylation of p65 following etoposide treatment (64). Etoposide stimulated the serine 536 phosphorylation of p65 indicating the NF-κB is transcriptionally active (Figure 7A). The activation of NF-κB can also be detected by its ability to accumulate in the nucleus. For this reason, we performed immunofluorescence staining of the endogenous p65 subunit before and after etoposide treatment. Cells treated with etoposide clearly displayed p65 nuclear accumulation (Figure 7B). These properties accompanied the ability of NF-κB to bind a consensus DNA site in electrophoretic mobility shift assays (S.B-R and N.C.R., unpublished data). The data support NF-κB activation in our system of DNA damage, as reported previously (11).
Figure 7. NF-κB is activated and required for IFN gene induction in response to etoposide.

A) HeLa cells were treated with etoposide for 15 hours or 5ng/ml tumor necrosis factor-α (TNF- α) for 1 hour and p65 was immunoprecipitated (IP) from lysates. Specific antibody to p65 phosphoserine 536 was used for the Western blot (WB). Lower panel displays Western blot with antibody to p65. B) HeLa cells were untreated or treated with etoposide for 2 hours before immunostaining with antibodies to p65. C) HeLa cells were untreated or treated with etoposide (E) in absence or presence of ML120B. left: Real-time PCR was used with pan-specific primers to quantify the endogenous levels of IFN-α mRNA. right: Real-time PCR was used to quantify the endogenous levels of IFN-λ1 mRNA. D) left: The IFN-λ1 luciferase reporter plasmid was co-transfected with empty vector or with a plasmid encoding the dominant negative IκBS32A/S36A (IκBSS/AA) gene. Cells were untreated or treated with etoposide for 24 hours prior to the luciferase assays. right: The IFN-λ1 luciferase reporter plasmid was co-transfected with empty vector (c) or with a plasmid encoding the p65/RelA gene. Cells were untreated or treated with etoposide for 24 hours prior to luciferase assays. Quantitative results are means of duplicate determinations in three independent experiments.
To evaluate the role of NF-κB in the induction of IFN genes by etoposide, we tested the effects of IKKβ inhibition. Cells were treated with etoposide in the absence or presence of the IKKβ inhibitor ML120B, and IFN-α mRNA levels were quantified by real-time RT-PCR (Figure 7C). Inhibition of IKKβ was found to block expression of the IFN-α genes, indicating that NF-κB is required for induction. This is a significant finding since the IFN-α genes are not directly regulated by NF-κB and do not possess NF-κB binding sites, although they do possess IRF binding sites (51).
Induction of the IFN-λ1 gene by DNA damage was also was found to be dependent on NF-κB. Real-time RT-PCR was used to quantify IFN-λ1 mRNA levels following etoposide in the presence or absence of ML120B. Expression of IFN-λ1 was blocked with the inhibition of IKKβ and NF-κB. The effect of NF-κB inhibition could also be demonstrated with IFN-λ1 promoter driving the luciferase gene. The IFN-λ1 promoter reporter was expressed alone or with a dominant negative IκB mutant that lacks serine target phosphorylation sites (S32A/S36A)(34). Etoposide stimulated the expression of the IFN-λ1 luciferase reporter, but this induction was completely inhibited with co-expression of the NF-κB repressor, IκB(S32A/S36A) (Figure 7D).
Since the promoter of the IFN-λ1 gene contains an NF-κB binding site, the more direct question is whether NF-κB in the absence of viral infection or DNA damage can induce the IFN-λ1 gene (21). To determine whether NF-κB regulates induction of the IFN-λ1 gene, we tested the response of the IFN-λ1 promoter reporter to expression of p65/RelA, a potent activator of NF-κB target sites. The IFN-λ1 gene was induced directly by p65 co-expression, indicating NF-κB is sufficient to induce the human IFN-λ1 gene independent of viral infection or DNA damage (Figure 7D). This result has obvious implications for the involvement of IFN action in the many signaling pathways that activate NF-κB.
Response of murine embryo fibroblasts. Our studies with human primary cells or established human cell lines provide clear evidence that the IFN-λ and IFN-α genes are induced in response to DNA damage by etoposide, and that NF-κB is requisite for the induction. Although murine cells may not accurately reflect the human response, the murine system affords the ability to test cells from animals with specific gene knockouts. For this reason we obtained murine embryo fibroblasts (MEFs) from wt or gene knockout animals and tested their response to DNA damage. Wt MEFs or MEFs that lack NF-κB essential modulator (NEMO) were treated with etoposide and IFN gene induction was evaluated by RT-PCR (Figure 8). Since murine IFN-λ1 is a pseudogene, we assayed expression of murine IFN-λ2. Results showed etoposide treatment induced the genes encoding IFN-λ2 and IFN-α in wt cells, but not in NEMO knockout cells. The results are in accordance with our studies in human cells that demonstrated a requirement of NF-κB activation. In addition, MEFs from IRF3 knockout animals treated with etoposide induced the IFN-λ2 and IFN-α genes, supporting our studies with human cells that showed IRF3 did not play a major role in the DNA damage response. Analyses of MEFs that lack the IRF-1 gene indicated it was critical for IFN-λ2 gene expression, but not for IFN-α expression, whereas MEFs that lack the IRF-7 gene had a profound defect in the induction of both IFN-λ2 and IFN-α genes in response to etoposide. The promoters of the murine IFN-λ genes are not well characterized and therefore they may respond differently from the human IFN-λ genes during the DNA damage response. The levels of IFN induction in these spontaneously immortalized MEFs were modest but reproducible following etoposide treatment.
Figure 8.

Effect of etoposide on murine embryo fibroblasts. Murine embryo fibroblasts (MEFs) isolated from wt mice, or mice with targeted gene knockouts in NEMO (NEMO−/−), IRF-3 (IRF3−/−), IRF-1 (IRF1−/−), or IRF-7 (IRF7−/−) were untreated or treated with etoposide for 24 hours. Real-time RT-PCR was used to quantify endogenous levels of murine IFNλ2 (top) or pan- IFNα (bottom). Quantitative results are means of duplicate determinations in three independent experiments.
Expression of human IRF and IFN genes with time during the DNA damage response
Our studies with human cells indicate that NF-κB activated by DNA damage stimulates induction of the IFN-λ1, IRF-1, and IRF-7 genes. To determine the time course of expression of these genes, human THP-1 cells were treated with etoposide and real time PCR was used to quantify IRF and IFN mRNA levels. IRF-1 and IRF-7 mRNA levels displayed an initial peak of expression at 4 hours of etoposide treatment and reached steady state levels by approximately 12 hours (Figure 9A). Expression of the IFN-λ1 gene showed a small increase at 4 hours of etoposide treatment and peaked at 15 hours with a kinetic profile similar to that of the IRFs. Expression of IFN-α mRNA trailed that of IFN-λ1 by 3–4 hours, possibly indicating a greater dependency on IRF-7 induction and activation (Figure 9B).
Figure 9. Time course of IRF and IFN mRNA expression in the response to etoposide treatment.

THP-1 cells were treated with etoposide, and RNA was isolated from cells during a 24 hour period at the times indicated. Real-time RT-PCR with specific primers was used to quantify IRF-1 and IRF-7 mRNA (A) or IFN-α and IFN-λ1 mRNA (B). Results are means of two experiments performed in duplicate. C) Conceptual model of double strand DNA break response signaling to IFN gene expression. ATM phosphorylation of NEMO promotes its ability to bind and activate the IKK complexes. IKKs phosphorylate IκB releasing NF-κB to activate gene targets IFN-λ1, IRF-1, and IRF-7. The IRFs induce IFN-α genes and enhance IFN-λ1 expression.
Discussion
The DNA damage response rapidly engages multimeric protein complexes to repair DNA and activate transcriptional programs that regulate cell cycle checkpoints and cell survival (6, 8, 65, 66). The recruitment of ATM, ATR, and DNA-PK to DNA breaks initiates phosphorylation and ubiquitination events that lead to specific transcription factor activation and gene expression. ATM is activated primarily in response to double strand DNA breaks, followed by ATR and DNA-PK in response to DNA single strands and ends generated during break resolution. One of the known substrates of ATM is NEMO, and phosphorylation promotes its ubiquitination, nuclear export, and activation of IKK complexes (62, 67, 68) (Figure 9C). IKK phosphorylation of IκB leads to release of NF-κB dimers and their ability to translocate to the nucleus and bind DNA targets. Our studies demonstrate that NF-κB is sufficient to induce the human IFN-λ1 gene during the response to DNA damage. The promoter of the human IFN-λ1 gene has a bona fide NF-κB binding site as well as an IRF binding site (21). NF-κB also induces the IRF-1 and IRF-7 genes that can influence expression of the IFN-α and IFN-λ genes. The induced IFNs can additionally amplify expression of the IRFs.
These findings add a new dimension to the complexity of the DNA damage response. ATM appears to be primarily accountable for initial signal pathways that lead to human IFN gene expression by etoposide. Inhibition of ATM significantly reduces the induction of IFN-α and IFN-λ1 genes (Figure 6). The ATM deficiency responsible for the development of ataxia-telangiectasia results in an array of clinical manifestations including cerebellar ataxia, oculocutaneous telangiectasia, immunodeficiency, and susceptibility to cancer (69, 70). The lack of IFN production in response to DNA damage that occurs through physiological processes in ataxia-telangiectasia speculatively may contribute to the immunodeficiency and tumor formation in the disease. Results of a few studies have suggested a potential role of IFN signaling during DNA damage (71–74). One study observed ISG expression correlated with resistance to radiation therapy (74), and another study reported that STAT1 facilitated cell cycle checkpoint following DNA damage (72). IFN pathways stimulate not only Janus kinases and STAT factors but elicit a broad range of effects on transcription and translation. The contribution of IFN signaling in the response to DNA breaks may have multifaceted consequences.
The novel observation of the convergence of the DNA damage response with IFN signaling stimulates speculation as to the possible function of IFN in reaction to genotoxic stress. In our experimental system the addition of IFN did not block the apoptotic effects of etoposide or significantly contribute to cell death (S.B-R and N.C.R., unpublished data). But IFNs are well characterized for their ability to inhibit viral infection, and this may reflect the evolutionary link. Many viral infections are known to stimulate DNA damage response pathways. Viruses like human immunodeficiency virus have RNA genomes but integrate viral DNA into the host genome, creating DNA strand breaks (4). Viruses with DNA genomes can generate single strand and double strand DNA breaks during lytic replication. Epstein-Barr virus, herpes simplex virus 1, and adenovirus are a few examples of DNA viruses that have been documented to activate a DNA damage response (4, 75, 76). More significantly, some of these viruses have evolved mechanisms to inhibit activation or downstream function of the DNA damage response (77–79). Viruses may inhibit this pathway not only to block cell cycle arrest and apoptosis, but to block the antiviral functions of IFNs that are produced by DNA damage.
Etoposide and IFN both have been used clinically for years for their anti-tumorigenic effects. IFNs produced in response to DNA damage may contribute to the anti-tumorigenic effects of etoposide. IFNs are recognized for their ability to cause growth arrest and/or apoptosis in neoplastic cells, although they can stimulate proliferation of healthy cells (24, 25, 46, 80–83). They also have vital immunoregulatory functions that include direct and indirect effects on activation of Natural Killer cells, macrophages, dendritic cells, T cells and B cells (84, 85). The anti-proliferative effects of IFN and the potential enhanced clearance of tumor cells may play a role in the in vivo DNA damage response pathway.
The profile of IFN gene expression in response to DNA damage in human cells is distinct from that induced by viral infection. The existence of multiple IFN genes with distinct promoter elements appears to have evolved as a response to different cellular stresses. A significant finding of our study is the ability of NF-κB to stimulate the expression of IFN genes in the absence of viral infection. NF–κB is required for the induction of IRF-1, IRF-7, IFN-λ and IFN-α in response to etoposide. Although the promoter of the human IFN-λ1 gene possesses both an NF-κB binding site and an IRF binding site, the promoters of the IFN-α genes possess only IRF binding sites. Results with the knockout MEFs indicate a critical function of IRF-7 activated in response to etoposide for induction of both murine IFN–λ2 and IFN–α genes. Future studies are needed to provide additional insight on the impact of IRF-1 and IRF-7 on the IFN genes during the DNA damage response and whether there are significant mechanistic differences in the human versus murine response. NF-κB is activated not only by DNA damage but by a wide array of cellular stimuli and it plays a major role in inflammation, immunity, cell survival, and cancer (63). The intimate link of NF-κB to IFN gene induction during the DNA damage response may reflect a potential role of IFN in other biological responses to NF-κB in the absence of infections. Our study adds a significant finding of IFN signaling to the complexity of pathways that are orchestrated by the response to genotoxic stress.
Supplementary Material
Acknowledgments
This work was supported by NIAID R21AI067885, PO1AI0555621 and NIHGM T32GM008444.
The authors would like to extend appreciation to all members of the NCR lab for their support. We also thank Dr. Martha Furie, Gregory Sabino, and Indralatha Jayatilaka for their helpfulness in preparation of primary monocytes.
References
- 1.Vilenchik MM, Knudson AG. Endogenous DNA double-strand breaks: production, fidelity of repair, and induction of cancer. Proc Natl Acad Sci U S A. 2003;100:12871–12876. doi: 10.1073/pnas.2135498100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fugmann SD, Lee AI, Shockett PE, Villey IJ, Schatz DG. The RAG proteins and V(D)J recombination: complexes, ends, and transposition. Annu Rev Immunol. 2000;18:495–527. doi: 10.1146/annurev.immunol.18.1.495. [DOI] [PubMed] [Google Scholar]
- 3.Richardson C, Horikoshi N, Pandita TK. The role of the DNA double-strand break response network in meiosis. DNA Repair (Amst) 2004;3:1149–1164. doi: 10.1016/j.dnarep.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 4.Sinclair A, Yarranton S, Schelcher C. DNA-damage response pathways triggered by viral replication. Expert Rev Mol Med. 2006;8:1–11. doi: 10.1017/S1462399406010544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mahaney BL, Meek K, Lees-Miller SP. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem J. 2009;417:639–650. doi: 10.1042/BJ20080413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Su TT. Cellular responses to DNA damage: one signal, multiple choices. Annu Rev Genet. 2006;40:187–208. doi: 10.1146/annurev.genet.40.110405.090428. [DOI] [PubMed] [Google Scholar]
- 8.Shiloh Y. The ATM-mediated DNA-damage response: taking shape. Trends Biochem Sci. 2006;31:402–410. doi: 10.1016/j.tibs.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 9.Abraham RT. PI 3-kinase related kinases: ‘big’ players in stress-induced signaling pathways. DNA Repair (Amst) 2004;3:883–887. doi: 10.1016/j.dnarep.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 10.Elkon R, Rashi-Elkeles S, Lerenthal Y, Linhart C, Tenne T, Amariglio N, Rechavi G, Shamir R, Shiloh Y. Dissection of a DNA-damage-induced transcriptional network using a combination of microarrays, RNA interference and computational promoter analysis. Genome Biol. 2005;6:R43. doi: 10.1186/gb-2005-6-5-r43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li N, Banin S, Ouyang H, Li GC, Courtois G, Shiloh Y, Karin M, Rotman G. ATM is required for IkappaB kinase (IKKk) activation in response to DNA double strand breaks. J Biol Chem. 2001;276:8898–8903. doi: 10.1074/jbc.M009809200. [DOI] [PubMed] [Google Scholar]
- 12.Fensterl V, Sen GC. Interferons and viral infections. Biofactors. 2009;35:14–20. doi: 10.1002/biof.6. [DOI] [PubMed] [Google Scholar]
- 13.Yoneyama M, Fujita T. Recognition of viral nucleic acids in innate immunity. Rev Med Virol. 2010;20:4–22. doi: 10.1002/rmv.633. [DOI] [PubMed] [Google Scholar]
- 14.Panne D, Maniatis T, Harrison SC. An atomic model of the interferon-beta enhanceosome. Cell. 2007;129:1111–1123. doi: 10.1016/j.cell.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tamura T, Yanai H, Savitsky D, Taniguchi T. The IRF family transcription factors in immunity and oncogenesis. Annu Rev Immunol. 2008;26:535–584. doi: 10.1146/annurev.immunol.26.021607.090400. [DOI] [PubMed] [Google Scholar]
- 16.Andersen J, VanScoy S, Cheng TF, Gomez D, Reich NC. IRF-3-dependent and augmented target genes during viral infection. Genes Immun. 2008;9:168–175. doi: 10.1038/sj.gene.6364449. [DOI] [PubMed] [Google Scholar]
- 17.Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, Taniguchi T. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity. 2000;13:539–548. doi: 10.1016/s1074-7613(00)00053-4. [DOI] [PubMed] [Google Scholar]
- 18.Marie I, Durbin JE, Levy DE. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J. 1998;17:6660–6669. doi: 10.1093/emboj/17.22.6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stark GR, I, Kerr M, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Annu Rev Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 20.Levy DE, Darnell JE., Jr Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 21.Onoguchi K, Yoneyama M, Takemura A, Akira S, Taniguchi T, Namiki H, Fujita T. Viral infections activate types I and III interferon genes through a common mechanism. J Biol Chem. 2007;282:7576–7581. doi: 10.1074/jbc.M608618200. [DOI] [PubMed] [Google Scholar]
- 22.Osterlund PI, Pietila TE, Veckman V, Kotenko SV, Julkunen I. IFN regulatory factor family members differentially regulate the expression of type III IFN (IFN-lambda) genes. J Immunol. 2007;179:3434–3442. doi: 10.4049/jimmunol.179.6.3434. [DOI] [PubMed] [Google Scholar]
- 23.Borden EC, Sen GC, Uze G, Silverman RH, Ransohoff RM, Foster GR, Stark GR. Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discov. 2007;6:975–990. doi: 10.1038/nrd2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clemens MJ. Interferons and apoptosis. J Interferon Cytokine Res. 2003;23:277–292. doi: 10.1089/107999003766628124. [DOI] [PubMed] [Google Scholar]
- 25.Li W, Lewis-Antes A, Huang J, Balan M, Kotenko SV. Regulation of apoptosis by type III interferons. Cell Prolif. 2008;41:960–979. doi: 10.1111/j.1365-2184.2008.00558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller CH, Maher SG, Young HA. Clinical Use of Interferon-gamma. Ann N Y Acad Sci. 2009;1182:69–79. doi: 10.1111/j.1749-6632.2009.05069.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Makris C, V, Godfrey L, Krahn-Senftleben G, Takahashi T, Roberts JL, Schwarz T, Feng L, Johnson RS, Karin M. Female mice heterozygous for IKK gamma/NEMO deficiencies develop a dermatopathy similar to the human X-linked disorder incontinentia pigmenti. Mol Cell. 2000;5:969–979. doi: 10.1016/s1097-2765(00)80262-2. [DOI] [PubMed] [Google Scholar]
- 28.Kimura T, Nakayama K, Penninger J, Kitagawa M, Harada H, Matsuyama T, Tanaka N, Kamijo R, Vilcek J, Mak TW, et al. Involvement of the IRF-1 transcription factor in antiviral responses to interferons. Science. 1994;264:1921–1924. doi: 10.1126/science.8009222. [DOI] [PubMed] [Google Scholar]
- 29.Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434:772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 30.Weaver BK, Kumar KP, Reich NC. Interferon regulatory factor 3 and CREB-binding protein/p300 are subunits of double-stranded RNA-activated transcription factor DRAF1. Mol Cell Biol. 1998;18:1359–1368. doi: 10.1128/mcb.18.3.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kumar KP, McBride KM, Weaver BK, Dingwall C, Reich NC. Regulated nuclear-cytoplasmic localization of interferon regulatory factor 3, a subunit of double-stranded RNA-activated factor 1. Mol Cell Biol. 2000;20:4159–4168. doi: 10.1128/mcb.20.11.4159-4168.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Banninger G, Reich NC. STAT2 nuclear trafficking. J Biol Chem. 2004;279:39199–39206. doi: 10.1074/jbc.M400815200. [DOI] [PubMed] [Google Scholar]
- 33.McBride KM, McDonald C, Reich NC. Nuclear export signal located within the DNA-binding domain of the STAT1 transcription factor. EMBO J. 2000;19:6196–6206. doi: 10.1093/emboj/19.22.6196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scherer DC, Brockman JA, Chen Z, Maniatis T, Ballard DW. Signal-induced degradation of I kappa B alpha requires site-specific ubiquitination. Proc Natl Acad Sci U S A. 1995;92:11259–11263. doi: 10.1073/pnas.92.24.11259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jura N, Scotto-Lavino E, Sobczyk A, Bar-Sagi D. Differential modification of Ras proteins by ubiquitination. Mol Cell. 2006;21:679–687. doi: 10.1016/j.molcel.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 36.Zhang L, Pagano JS. IRF-7, a new interferon regulatory factor associated with Epstein-Barr virus latency. Mol Cell Biol. 1997;17:5748–5757. doi: 10.1128/mcb.17.10.5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ank N, West H, Bartholdy C, Eriksson K, Thomsen AR, Paludan SR. Lambda interferon (IFN-lambda), a type III IFN, is induced by viruses and IFNs and displays potent antiviral activity against select virus infections in vivo. J Virol. 2006;80:4501–4509. doi: 10.1128/JVI.80.9.4501-4509.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Loseke S, Grage-Griebenow E, Wagner A, Gehlhar K, Bufe A. Differential expression of IFN-alpha subtypes in human PBMC: evaluation of novel real-time PCR assays. J Immunol Methods. 2003;276:207–222. doi: 10.1016/s0022-1759(03)00072-3. [DOI] [PubMed] [Google Scholar]
- 39.Medhurst AD, Harrison DC, Read SJ, Campbell CA, Robbins MJ, Pangalos MN. The use of TaqMan RT-PCR assays for semiquantitative analysis of gene expression in CNS tissues and disease models. J Neurosci Methods. 2000;98:9–20. doi: 10.1016/s0165-0270(00)00178-3. [DOI] [PubMed] [Google Scholar]
- 40.Gadberry MD, Malcomber ST, Doust AN, Kellogg EA. Primaclade--a flexible tool to find conserved PCR primers across multiple species. Bioinformatics. 2005;21:1263–1264. doi: 10.1093/bioinformatics/bti134. [DOI] [PubMed] [Google Scholar]
- 41.Ank N, Iversen MB, Bartholdy C, Staeheli P, Hartmann R, Jensen UB, Dagnaes-Hansen F, Thomsen AR, Chen Z, Haugen H, Klucher K, Paludan SR. An important role for type III interferon (IFN-lambda/IL-28) in TLR-induced antiviral activity. J Immunol. 2008;180:2474–2485. doi: 10.4049/jimmunol.180.4.2474. [DOI] [PubMed] [Google Scholar]
- 42.Cheng TF, Brzostek S, Ando O, Van Scoy S, Kumar KP, Reich NC. Differential activation of IFN regulatory factor (IRF)-3 and IRF-5 transcription factors during viral infection. J Immunol. 2006;176:7462–7470. doi: 10.4049/jimmunol.176.12.7462. [DOI] [PubMed] [Google Scholar]
- 43.Tansey WP. Detection of Ubiquitylated Proteins in Mammalian Cells. Cold Spring Harb Protocols. 2006 doi: 10.1101/pdb.prot4616. [DOI] [PubMed] [Google Scholar]
- 44.Weaver BK, Ando O, Kumar KP, Reich NC. Apoptosis is promoted by the dsRNA-activated factor (DRAF1) during viral infection independent of the action of interferon or p53. FASEB J. 2001;15:501–515. doi: 10.1096/fj.00-0222com. [DOI] [PubMed] [Google Scholar]
- 45.Heylbroeck C, Balachandran S, Servant MJ, DeLuca C, Barber GN, Lin R, Hiscott J. The IRF-3 transcription factor mediates Sendai virus-induced apoptosis. J Virol. 2000;74:3781–3792. doi: 10.1128/jvi.74.8.3781-3792.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chawla-Sarkar M, Lindner DJ, Liu YF, Williams BR, Sen GC, Silverman RH, Borden EC. Apoptosis and interferons: role of interferon-stimulated genes as mediators of apoptosis. Apoptosis. 2003;8:237–249. doi: 10.1023/a:1023668705040. [DOI] [PubMed] [Google Scholar]
- 47.Baldwin EL, Osheroff N. Etoposide, topoisomerase II and cancer. Curr Med Chem Anticancer Agents. 2005;5:363–372. doi: 10.2174/1568011054222364. [DOI] [PubMed] [Google Scholar]
- 48.Meresse P, Dechaux E, Monneret C, Bertounesque E. Etoposide: discovery and medicinal chemistry. Curr Med Chem. 2004;11:2443–2466. doi: 10.2174/0929867043364531. [DOI] [PubMed] [Google Scholar]
- 49.Reich NC, Liu L. Tracking STAT nuclear traffic. Nat Rev Immunol. 2006;6:602–612. doi: 10.1038/nri1885. [DOI] [PubMed] [Google Scholar]
- 50.Aoki R, Kavanagh JJ. Antineoplastic Agents: Classification and Mechanisms of Action. In: Roberto Angioli PBP, Kavanagh John J, Pecorelli Sergio, Penalver Manuel, editors. Chemotherapy for Gynecological Neoplasms: Current Therapy and Novel Approaches. 1. Marcel Dekker; New York, NY: 2004. pp. 1–12. [Google Scholar]
- 51.Genin P, Lin R, Hiscott J, Civas A. Differential regulation of human interferon A gene expression by interferon regulatory factors 3 and 7. Mol Cell Biol. 2009;29:3435–3450. doi: 10.1128/MCB.01805-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hiscott J. Triggering the innate antiviral response through IRF-3 activation. J Biol Chem. 2007;282:15325–15329. doi: 10.1074/jbc.R700002200. [DOI] [PubMed] [Google Scholar]
- 53.Au WC, Moore PA, LaFleur DW, Tombal B, Pitha PM. Characterization of the interferon regulatory factor-7 and its potential role in the transcription activation of interferon A genes. J Biol Chem. 1998;273:29210–29217. doi: 10.1074/jbc.273.44.29210. [DOI] [PubMed] [Google Scholar]
- 54.Caillaud A, Hovanessian AG, Levy DE, Marie IJ. Regulatory serine residues mediate phosphorylation-dependent and phosphorylation-independent activation of interferon regulatory factor 7. J Biol Chem. 2005;280:17671–17677. doi: 10.1074/jbc.M411389200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huye LE, Ning S, Kelliher M, Pagano JS. Interferon regulatory factor 7 is activated by a viral oncoprotein through RIP-dependent ubiquitination. Mol Cell Biol. 2007;27:2910–2918. doi: 10.1128/MCB.02256-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bennett EJ, Harper JW. DNA damage: ubiquitin marks the spot. Nat Struct Mol Biol. 2008;15:20–22. doi: 10.1038/nsmb0108-20. [DOI] [PubMed] [Google Scholar]
- 57.Tamura T, Ishihara M, Lamphier MS, Tanaka N, Oishi I, Aizawa S, Matsuyama T, Mak TW, Taki S, Taniguchi T. An IRF-1-dependent pathway of DNA damage-induced apoptosis in mitogen-activated T lymphocytes. Nature. 1995;376:596–599. doi: 10.1038/376596a0. [DOI] [PubMed] [Google Scholar]
- 58.Tanaka N, Ishihara M, Lamphier MS, Nozawa H, Matsuyama T, Mak TW, Aizawa S, Tokino T, Oren M, Taniguchi T. Cooperation of the tumour suppressors IRF-1 and p53 in response to DNA damage. Nature. 1996;382:816–818. doi: 10.1038/382816a0. [DOI] [PubMed] [Google Scholar]
- 59.Sims SH, Cha Y, Romine MF, Gao PQ, Gottlieb K, Deisseroth AB. A novel interferon-inducible domain: structural and functional analysis of the human interferon regulatory factor 1 gene promoter. Mol Cell Biol. 1993;13:690–702. doi: 10.1128/mcb.13.1.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pine R. Convergence of TNFalpha and IFNgamma signalling pathways through synergistic induction of IRF-1/ISGF-2 is mediated by a composite GAS/kappaB promoter element. Nucleic Acids Res. 1997;25:4346–4354. doi: 10.1093/nar/25.21.4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lu R, Moore PA, Pitha PM. Stimulation of IRF-7 gene expression by tumor necrosis factor alpha: requirement for NFkappa B transcription factor and gene accessibility. J Biol Chem. 2002;277:16592–16598. doi: 10.1074/jbc.M111440200. [DOI] [PubMed] [Google Scholar]
- 62.Wu ZH, Shi Y, Tibbetts RS, Miyamoto S. Molecular linkage between the kinase ATM and NF-kappaB signaling in response to genotoxic stimuli. Science. 2006;311:1141–1146. doi: 10.1126/science.1121513. [DOI] [PubMed] [Google Scholar]
- 63.Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 64.Perkins ND. Post-translational modifications regulating the activity and function of the nuclear factor kappa B pathway. Oncogene. 2006;25:6717–6730. doi: 10.1038/sj.onc.1209937. [DOI] [PubMed] [Google Scholar]
- 65.Harrison JC, Haber JE. Surviving the breakup: the DNA damage checkpoint. Annu Rev Genet. 2006;40:209–235. doi: 10.1146/annurev.genet.40.051206.105231. [DOI] [PubMed] [Google Scholar]
- 66.Nyberg KA, Michelson RJ, Putnam CW, Weinert TA. Toward maintaining the genome: DNA damage and replication checkpoints. Annu Rev Genet. 2002;36:617–656. doi: 10.1146/annurev.genet.36.060402.113540. [DOI] [PubMed] [Google Scholar]
- 67.Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol. 2007;8:49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- 68.Stilmann M, Hinz M, Arslan SC, Zimmer A, Schreiber V, Scheidereit C. A nuclear poly(ADP-ribose)-dependent signalosome confers DNA damage-induced IkappaB kinase activation. Mol Cell. 2009;36:365–378. doi: 10.1016/j.molcel.2009.09.032. [DOI] [PubMed] [Google Scholar]
- 69.Chun HH, Gatti RA. Ataxia-telangiectasia, an evolving phenotype. DNA Repair (Amst) 2004;3:1187–1196. doi: 10.1016/j.dnarep.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 70.Lavin MF. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat Rev Mol Cell Biol. 2008;9:759–769. doi: 10.1038/nrm2514. [DOI] [PubMed] [Google Scholar]
- 71.Liu M, Hummer BT, Li X, Hassel BA. Camptothecin induces the ubiquitin-like protein, ISG15, and enhances ISG15 conjugation in response to interferon. J Interferon Cytokine Res. 2004;24:647–654. doi: 10.1089/jir.2004.24.647. [DOI] [PubMed] [Google Scholar]
- 72.Townsend PA, Cragg MS, Davidson SM, McCormick J, Barry S, Lawrence KM, Knight RA, Hubank M, Chen PL, Latchman DS, Stephanou A. STAT-1 facilitates the ATM activated checkpoint pathway following DNA damage. J Cell Sci. 2005;118:1629–1639. doi: 10.1242/jcs.01728. [DOI] [PubMed] [Google Scholar]
- 73.Tsai MH, Cook JA, Chandramouli GV, DeGraff W, Yan H, Zhao S, Coleman CN, Mitchell JB, Chuang EY. Gene expression profiling of breast, prostate, and glioma cells following single versus fractionated doses of radiation. Cancer Res. 2007;67:3845–3852. doi: 10.1158/0008-5472.CAN-06-4250. [DOI] [PubMed] [Google Scholar]
- 74.Weichselbaum RR, Ishwaran H, Yoon T, Nuyten DS, Baker SW, Khodarev N, Su AW, Shaikh AY, Roach P, Kreike B, Roizman B, Bergh J, Pawitan Y, van de Vijver MJ, Minn AJ. An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proc Natl Acad Sci U S A. 2008;105:18490–18495. doi: 10.1073/pnas.0809242105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shirata N, Kudoh A, Daikoku T, Tatsumi Y, Fujita M, Kiyono T, Sugaya Y, Isomura H, Ishizaki K, Tsurumi T. Activation of ataxia telangiectasia-mutated DNA damage checkpoint signal transduction elicited by herpes simplex virus infection. Journal of Biological Chemistry. 2005;280:30336–30341. doi: 10.1074/jbc.M500976200. [DOI] [PubMed] [Google Scholar]
- 76.Lilley CE, Chaurushiya MS, Weitzman MD. Chromatin at the intersection of viral infection and DNA damage. Biochim Biophys Acta. 1799:319–327. doi: 10.1016/j.bbagrm.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Evans JD, Hearing P. Relocalization of the Mre11-Rad50-Nbs1 complex by the adenovirus E4 ORF3 protein is required for viral replication. Journal of Virology. 2005;79:6207–6215. doi: 10.1128/JVI.79.10.6207-6215.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stracker TH, Carson CT, Weitzman MD. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature. 2002;418:348–352. doi: 10.1038/nature00863. [DOI] [PubMed] [Google Scholar]
- 79.Lilley CE, Chaurushiya MS, Boutell C, Landry S, Suh J, Panier S, Everett RD, Stewart GS, Durocher D, Weitzman MD. A viral E3 ligase targets RNF8 and RNF168 to control histone ubiquitination and DNA damage responses. EMBO J. 29:943–955. doi: 10.1038/emboj.2009.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Moiseeva O, Mallette FA, Mukhopadhyay UK, Moores A, Ferbeyre G. DNA damage signaling and p53-dependent senescence after prolonged beta-interferon stimulation. Mol Biol Cell. 2006;17:1583–1592. doi: 10.1091/mbc.E05-09-0858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Takaoka A, Hayakawa S, Yanai H, Stoiber D, Negishi H, Kikuchi H, Sasaki S, Imai K, Shibue T, Honda K, Taniguchi T. Integration of interferon-alpha/beta signalling to p53 responses in tumour suppression and antiviral defence. Nature. 2003;424:516–523. doi: 10.1038/nature01850. [DOI] [PubMed] [Google Scholar]
- 82.Thyrell L, Erickson S, Zhivotovsky B, Pokrovskaja K, Sangfelt O, Castro J, Einhorn S, Grander D. Mechanisms of Interferon-alpha induced apoptosis in malignant cells. Oncogene. 2002;21:1251–1262. doi: 10.1038/sj.onc.1205179. [DOI] [PubMed] [Google Scholar]
- 83.Gomez D, Reich NC. Stimulation of primary human endothelial cell proliferation by IFN. J Immunol. 2003;170:5373–5381. doi: 10.4049/jimmunol.170.11.5373. [DOI] [PubMed] [Google Scholar]
- 84.Le Bon A, Tough DF. Links between innate and adaptive immunity via type I interferon. Curr Opin Immunol. 2002;14:432–436. doi: 10.1016/s0952-7915(02)00354-0. [DOI] [PubMed] [Google Scholar]
- 85.Prchal M, Pilz A, Simma O, Lingnau K, von Gabain A, Strobl B, Muller M, Decker T. Type I interferons as mediators of immune adjuvants for T- and B cell- dependent acquired immunity. Vaccine. 2009;27(Suppl 6):G17–20. doi: 10.1016/j.vaccine.2009.10.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.