

Abstract
BACKGROUND
In utero transplantation (IUT) has the potential to treat birth defects early before full development of the immune system. Relatively small grafts, that are not matched for major histocompatibility antigens, can be delivered even before onset of disease symptoms. IUT of hematopoietic stem cells is usually performed via intraperitoneal injection, yet the fate of donor cells in the peritoneal cavity is not fully understood. We review our recent work and present new data demonstrating that the peritoneum can be a site of ectopic hematopoiesis with implications for IUT and immune tolerance induction.
STUDY DESIGN AND METHODS
Haplogeneic and allogeneic fetal transplants were performed in mice and engraftment tracked by flow cytometry. Immune tolerance was studied by mixed lymphocyte reactions and skin transplantation. Adult syngeneic murine transplants and xenogeneic human into immunodeficient-mouse transplants were performed to follow hematopoietic retention in the peritoneum and engraftment of the bone marrow.
RESULTS
Although most transplanted cells rapidly clear the peritoneum, hematopoietic cells and cells with the phenotype of hematopoietic precursors can remain in the peritoneal cavity for months after transplant. The presence of donor cells in the peritoneum can contribute to donor-specific tolerance but sufficient peripheral blood chimerism is required to ensure acceptance of donor skin grafts.
CONCLUSION
Ectopic hematopoiesis and the survival of stem cells in the peritoneum offers the possibility of better using the peritoneal cavity to delivery stem cells and foster the development of immune tolerance to alloantigens or other foreign antigens.
Keywords: Fetus, Intraperitoneal, Peritoneal Cavity, Transplantation Chimera, Bone Marrow Transplantation
INTRODUCTION
Cellular therapy, and stem cell transplantation in particular, offers the possibility of providing a life-long cure for inherited diseases. In many cases the ideal therapy treats a disease as early as possible, perhaps even before clinical symptoms of the disease have manifested. This idea is among the reasons that have spurred consideration of prenatal transplantation as a way to treat genetic or developmental birth defects. A number of successful attempts at treating immunodeficiencies have been reported, but for many other diseases, such as hemoglobinopathies and enzyme storage diseases, in utero transplantation (IUT) has failed to generate the levels of donor cell chimerism that can significantly improve the clinical course of the disease.1 This has spurred those of us interested in developing fetal cell-therapy to gain a better understanding of the development of the human immune system and re-evaluate our approaches and assumptions surrounding the transplantation of foreign cells into a developing fetus.
There are a number of arguments in favor of prenatal transplantation, chief among these is the immaturity of the fetal immune system that should allow for transplantation allogeneic cells. Thymic maturation around 14 weeks’ of gestation marks the time of a rapid rise in peripheral T-cell numbers. 2, 3 Thus, transplantation prior to this time should avoid any possible rejection by host T-cells. As genetic testing by chorionic villous sampling is first feasible at 10 weeks’ gestation, this presents a few weeks during which IUT may be performed. As the fetal patient weighs >100g during this time period, it is possible to deliver a relatively much larger dose of stem cells than one can later in ontogeny.4 In the case of hematopoietic stem cell transplants, the beginning of the second trimester is a time when stem cells seed the bone marrow and hematopoiesis begins to shift from the liver to the bone marrow.5 The growth of the hematopoietic system results in a high frequency of circulating stem cells and it is believed that donor stem cells could simply join this procession of host cells to engraft new sites of hematopoiesis as they develop. Stem cell engraftment at such an early stage of development would also allow the donor cells to participate in the dramatic growth of tissues and organs that occurs in the second and third trimesters of development. Ideally, this leads to sufficient levels of chimerism in the target tissue to eliminate or alleviate clinical symptoms of disease. Additionally, the presence of donor cells during the period of immune development can also lead to immune tolerance towards the donor cells. For diseases such as immunodeficiencies, hemoglobinopathies and some metabolic diseases, the period of engraftment and expansion of donor cells would take place while the patient remains protected from infection and on maternal life-support. Thereby, the impact of the disease is lessoned during the early stages of donor engraftment.
In practice, IUT has only been successful in cases were the donor stem cells have a profound proliferative and survival advantage over host stem cells and/or their progeny. Patients transplanted for X-linked severe combined immunodeficiency (SCID) show evidence of T-cell engraftment but very little, or no, myeloid or B-cell engraftment.6 Referred to as mixed chimerism, this outcome points to a very low incidence of donor stem cell engraftment that only yields clinically significant chimerism because of the extensive proliferation that the donor cells undergo as hematopoietic progenitors and mature T-cells.
Clinical experience with IUT points to deficiencies in our understanding of the fate of cells transplanted into fetal recipients. Are these cells destroyed by immune mechanisms or do they simply fail to engraft appropriate stem cell niches that support their survival and growth? It is important to emphasize that prenatal transplants are performed without any cytoablation or immune suppression. Either of these factors or some combination must be preventing engraftment of transplanted cells. The focus of our National Blood Foundation grant was on the fate of the transplanted cells in the peritoneal cavity, the site of injection, and the effects of chimerism levels on the induction of immune tolerance.
MATERIALS AND METHODS
Mice and animal husbandry
All mice were obtained from The Jackson Laboratory (Bar Harbor, ME or Sacramento, CA). Adult female C57BL/6J mice were obtained for use as recipients of murine cell transplants. Transgene-homozygous breeding pairs of C57BL/6-Tg(UBC-GFP)30Scha/J, expressing enhanced green fluorescent protein (eGFP) under the direction of the human ubiquitin C promoter,7 and immunodeficient NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice8 were bought and adult (≥8 weeks old) experimental animals bred at our institute. Animals were maintained in microisolator cages (Innovive Inc., San Diego, CA) under specific-pathogen free conditions as previously described.9 Research was performed under approval of the Institutional Animal Care and Use Committee at ISIS Services LLC (San Carlos, CA).
Isolation and transplantation of murine primitive hematopoietic precursors
Femoral bone marrow was extracted from C57/BL/6-Tg(UBC-GFP)30Scha/J mice. Light density cells were collected by 25 minute centrifugation at 600x g over a layer of 1.077 g/mL Lymphoprep (Axis-Shield PLC AS, Oslo, Norway) and suspended in blocking buffer consisting of phosphate buffered saline, calcium and magnesium free, (PBS; HyClone Laboratories, Inc., Logan, UT) containing 5% normal mouse serum, 2 μg/ml mouse CD16/CD32 and 0.01% NaN3 (Sigma Chemical Co., St. Louis, MO). All monoclonal antibodies (mAbs) recognizing murine antigens were purchased from BioLegend (San Diego, CA). Cells were stained with saturating levels of pacific blue (PB)-conjugated anti-mouse TER-119 (clone TER-119), anti-mouse Ly-6G/Ly-6C (Gr-1) (clone RB6-8C5), anti-mouse CD3 (clone 17A2), anti-mouse CD11b (clone M1/70) and anti-mouse CD45R/B220 (clone RA3-6B2) mAbs for at least 30 minutes. Collectively, these antigens are referred to as lineage (Lin) markers. Stained cells were washed twice with PBS. Magnetic sheep anti-rat IgG Dynabeads (Invitrogen, Carlsbad, CA) were used to deplete Lin+ cells. Linlow cells were collected, suspended in blocking buffer and stained with allophycocyanin (APC) anti-mouse CD117 (clone 2B8) and phycoerythrin (PE) anti-mouse CD150 (clone TC15-12F12.2). Cells were washed with PBS and suspended in PBS and propidium iodide (PI) buffer for fluorescence activated cell sorting (FACS). CD117+CD150+Linlow cells were isolated using a FACSAria (BD Biosystems, San Jose, CA) flow cytometer. Sorted cells were counted using a hemacytometer. 1.1×104 cells in 0.2mL of PBS were injected into the peritoneal cavity of C57BL/6J mice using a 28g insulin syringe (BD Biosystems, San Jose, CA).
Isolation and transplantation of human fetal bone marrow
Human fetal bone marrow was obtained from elective abortions at San Francisco General Hospital under a protocol approved by the University of California San Francisco Committee on Human Research and with permission of the women undergoing the abortion. The age of the tissues ranged from 23 to 24 weeks of gestation and was estimated based on the foot-length of the fetus. The bone marrow was harvested as previously described.10 Light-density cells were isolated and cells counted, as for the murine bone marrow, and suspended in PBS for i.p. injection in a volume of 0.2 ml.
Analysis of engraftment
Cells were harvested at the indicated times after transplantation. Peritoneal cells were recovered by flushing the peritoneal cavity with 10mL of PBS using a syringe and 18g needle. The volume recovered was measured to allow estimation of the overall number of cells retained in the peritoneal cavity. Additionally, hematopoietic tissues such as the peripheral blood, femoral bone marrow, spleen and liver were also harvested and light-density cells isolated. Spleens and livers were passed through 40 μm and 100 μm nylon sieves (BD Biosystems, San Jose, CA), respectively, and liver cells washed once prior to centrifugation over a layer of Lymphoprep medium. Cells were suspended in blocking buffer and enumerated.
Cells harvested from C57BL/6J mice transplanted with CD117+CD150+Lin− cells were stained with PB-labeled anti-Lin mAbs, CD117-APC, CD150-PE and PE-cyanine dye 7 (PE-Cy7) anti-mouse CD48 (clone HM48-1). Cells harvested from NSG mice transplanted with human cells were stained with a panel of antibodies recognizing stem cells, progenitors and mature blood elements as previously described.9 Only samples from healthy appearing mice with no obvious signs of ill health such as tumor formation were considered in the study. Samples were suspended in buffer containing PI to label dead cells and analyzed using LSR II flow cytometer (BD Biosystems). Data were analyzed using FlowJo software, version 9.2 (Tree Star, Inc., Ashland, OR).
RESULTS AND DISCUSSION
Resident hematopoietic cells in the peritoneum
IUT has been performed primarily by intraperitoneal (i.p.) injection of the graft because this route of administration is considered to be the simplest and safest. The peritoneum is a site of a resident population of hematopoietic cells that in adults includes mostly macrophages and T-lymphocytes but also small populations of B-cells, NK-cells, dendritic cells and can include neutrophils that infiltrate the peritoneal cavity in response to infection.11 The main sources of these cells are milky spots in the omentum representing clusters of immune and mesenchymal cells that are fed by peripheral blood.12 Hematopoietic cells transplanted into the peritoneum enter the venous blood circulation together with the drainage of peritoneal serous fluid into the right lymphatic duct and, less so, the thoracic duct (Fig. 1). Indeed, the passage of blood from the peritoneal cavity into the circulation is efficient enough that blood transfusions in fetuses and children has in the past been performed using this route of administration.13 Accordingly, not much attention has been focused on cells found in the peritoneal cavity after IUT as it was generally assumed that the transplanted cells are rapidly cleared into the peripheral circulation after transplant.
Fig. 1.
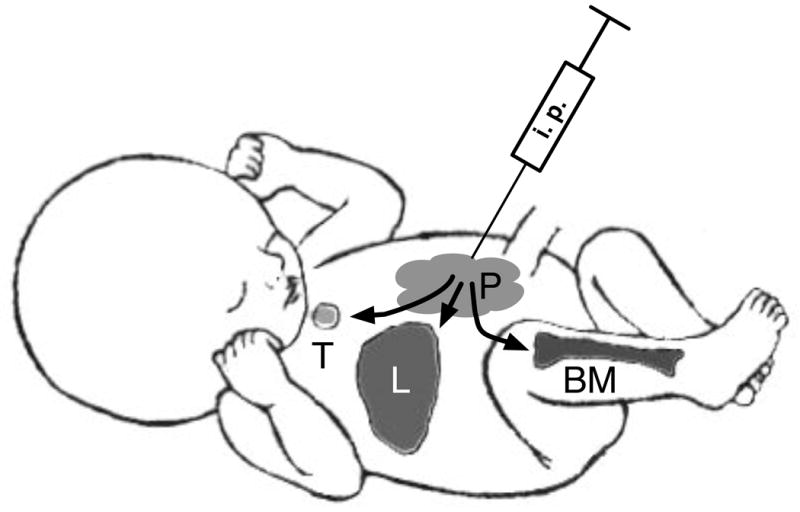
Overview of cell migration after IUT. IUT via the i.p route results in a population of donor cells residing in the peritoneum (P), whereas most transplanted cells rapidly leave the peritoneum and enter the circulation from were the can seed the liver (L), bone marrow (BM) and thymus (T).
Persistence of bone marrow cells in the peritoneum
We were surprised when we examined cells recovered from the peritoneal cavity of mice that had been transplanted in utero with haplogeneic cells (H-2b/d B6D2F1 donor cells into H-2b C57BL/6 recipients)to discover that in some 3–5 month old recipients donor cells comprising as high as 25% of all cells could be recovered.14, 15 Although in most animals the levels of chimerism were ≤1%, the frequency of donor cells in the peritoneum was higher than in the bone marrow, peripheral blood or spleen. The peritoneal cells consisted of B-cells, T-cells, F4/80-antigen+ macrophages and Gr1+ myeloid cells. We observed a correlation between the presence of chimerism in the peritoneum and the presence of donor cells in the circulation, spleen and bone marrow. Moreover, donor cells in the bone marrow consisted of mature blood cells, not hematopoietic precursors, suggesting that hematopoietic reconstitution of the bone marrow was not the source of circulating donor cells. This further indicated that the peritoneum was acting as a reservoir, and/or potential source of newly formed hematopoietic cells, that could enter the systemic circulation leading to observed chimerism in the spleen, blood and bone marrow.
These observations led us to investigate in greater detail the fate of transplanted cells in the peritoneum with support of a grant from the NBF. We focused on early time points after transplantation to determine to what degree donor cells remain in the peritoneum after fetal transplantation.16 Light-density bone marrow cells were transplanted into embryonic day 13 mice. The haplogeneic transplant model used allowed for graft rejection by host T-cells, but not graft-versus host disease. Donor cells were followed for 4 weeks after transplant and were found in the peritoneal cavities of recipients for at least 2 weeks after transplant, after which levels declined. The levels of donor cells in the peripheral blood, spleen, liver and bone marrow trended with peritoneal chimerism, also peaking at 2 weeks post transplant. Donor cells were nearly undetectable 2 weeks after analogous transplants into adult recipients, indicating that rejection of mismatched cells in the peritoneum occurs relatively quickly in immune competent animals.
Multiple cell types were retained short-term in the peritoneum including B-cells, macrophages and several subsets of Gr-1 expressing myeloid cells.16 T-cells appeared to be cleared rapidly from the peritoneum and represented less than 0.02% of donor cells. The presence of Gr-1 antigen on multiple cell populations, including cells with the light-scatter properties of progenitor cells, suggested the interesting possibility that hematopoietic precursors may survive and grow in the peritoneum.
Ectopic hematopoiesis in the peritoneum
To test the hypothesis that transplanted hematopoietic precursors may survive, grow and differentiate within the peritoneum, i.p. injections of 1.1×04 CD117+CD150+ donor cells into adult mice were performed and peritoneal fluid and other hematopoietic tissues were harvested from recipient mice after 24 hours and one week (Fig. 2). Donor cells were obtained from syngeneic mice expressing an eGFP transgene in all tissues. Peritoneal cells and light density cells from peripheral blood, femoral bone marrow and spleens were counted and analyzed by flow cytometry. Although variable, all tissues at both time points had detectable levels of eGFP+ cells. It has been reported by Kiel et al.17 that both CD117 (c-kit) and CD150 (signaling lymphocyte activation molecule) are markers for hematopoietic stem cells. CD117 is a cytokine receptor on hematopoietic stem cells whereas CD150 is best known for regulating the proliferation and activation of lymphocytes. Analysis of the donor cells after isolation confirmed the purity of the isolated CD117+CD150+Linlow cells and enriched for hematopoietic stem cells and early progenitor cells. The migration and evolution of cell differentiation of the i.p transplanted cells was followed over time.
Fig. 2.

Short-term retention of eGFP+ early progenitor cells in the murine peritoneum. 1.1×04 CD117+CD150+ donor cells from transgenic mice expressing eGFP in all tissues, were phenotyped by flow cytometry and transplanted i.p. into C57BL/6 mice. Peritoneum cells and hematopoietic cells were harvested, counted and phenotyped the same way after 24 hours and one week. Analysis of donor cells, peritoneum cells at 24 hours and one week, spleen at one week and peripheral blood at one week are shown. Data in the two columns on the left represent expression of CD150 and either CD117 or CD48 by viable cell (PI−), eGFP+ events. Data in the middle column represent the frequency of lineage expression from viable, eGFP+ events. Data in the two columns on the right represent CD150 and either CD117 or CD48 expression by viable, eGFP+ Linlow cells.
Assessment of lineage antigen expression at both 24 hours and one week showed that differentiation of eGFP+ cells had occurred, indicating that the donor cells both survived and gave rise to mature cells (Fig. 2). A higher abundance of lineage positive cells was observed in all tissues at one week than at 24 hours including the peritoneum, indicative of active hematopoiesis in the peritoneum. Additionally, the presence of lineage positive cells observed after 24 hours shows that activation and differentiation began quickly.
The first and perhaps the most obvious observation was the elevated concentration of eGFP+ cells in the peritoneum at both time points. Because the donor cells were injected i.p., it was hypothesized that a population of the donor cells would remain localized in the peritoneum with a trend of slowly diffusing to other tissues as well as starting to produce mature cells. The latter part of the expectation was verified by the increased frequency of lineage positive cells in the peritoneum. The former part of the expectation was evaluated by using the calculated number of cells per tissue and the numbers obtained from the flow cytometric analyses to formulate the frequency of eGFP+ distributed to each of the tissues. The average percentage of total eGFP+ cells found in the peritoneum at 24 hours was 62.0% (n=2) and decreased to an average of 56.4% (n=2) at one week (Fig. 3). The decrease in donor cells from 24 hours to one week indicates that overtime cells likely continued to exit the peritoneum.
Fig. 3.

Tissue distribution of eGFP+ cells 24 hours and one week after transplantation. 1.1×04 eGFP+CD117+CD150+ murine donor cells were transplanted i.p. into Balb/c mice. Peritoneal cells, bone marrow, spleen and blood were harvested at the indicated time points. Total numbers of viable eGFP+ cells in each of these tissues, based on cell recoveries and flow cytometric analyses, were used to calculate the distribution of donor cells. Results represent the mean of n = 2 at each time point.
The prevalence of eGFP+ donor cells in hematopoietic tissues generally increased from 24 hours to one week after transplant. The frequency of eGFP+ cells relative to the number of injected cells present in the blood increased from 15.3% to 19.1%; 15.1% to 23.8% in the bone marrow; and decreased from 7.6% to 6.7% in the spleen (Fig. 3, n=2 for all tissues and time points). The increase observed in the peripheral blood and bone marrow fits with the hypothesized continued migration and expansion of cells from the peritoneum. However, a slight decrease in eGFP+ cells from 24 hours to one week in the spleen was observed, which may or may not be representative of the distribution trend due to the small sample size. The prevalence of donor cells in the peritoneum and the distribution of donor cells to peripheral tissues seem to be inversely related, suggesting that primitive hematopoietic precursors injected i.p. tend to have slow efflux from the peritoneum to the peripheral tissues.
Intraperitoneal transplantation of human cells in immunodeficient mice
To understand the behavior of human cells transplanted into the peritoneal cavity, we examined the fate of human bone marrow cells transplanted into immunodeficient NSG mice. Adult mice approximate the size of a human fetus at the end of the first trimester, when most fetal transplants have been performed, and their complete lack of lymphoid cells prevents rejection of the donor cells. Thus, this model approximates, in size and immune function, the human fetus before maturation of the thymus.
The graft consisted of the light-density cell fraction of fetal bone marrow containing a range of hematopoietic precursors and mature blood cells (Fig. 4). The frequency and number of cells recovered from 3 transplanted animals were measured to evaluate the retention of bone marrow cells in the peritoneum. Eighteen hours after transplantation mean cell-enrichments of 1.7-fold for CD19+ B-cells, 3.2-fold for CD3+ T-cells, 2.0-fold for CD15+ granulocytes, 1.9-fold for CD14+ monocytes was observed. CD235a+ erythrocytes were the only defined cell population observed to dimmish in frequency. However, overall the number of transplanted cells recovered from the peritoneum was only 1.2% (range 0.7 – 1.9%), indicating that most donor cells rapidly exit the peritoneal cavity after transplant. It is uncertain how many cells were not recovered because they firmly adhered to the mesothelial layer that lines the peritoneum, but it is unlikely that such adherence could account for the majority of transplanted cells. Therefore, the bulk of transplanted cells rapidly exit the peritoneal cavity, including all the cell types that were preferentially retained in the peritoneum.
Fig. 4.

Short-term retention of human hematopoietic cells in the murine peritoneum. Donor cells consisted of human bone marrow cells, 23 weeks of gestation, which were phenotyped by flow cytometry and 7.5 × 107 cells transplanted i.p. into NSG mice. Eighteen hours later, cells were recovered from the peritoneum, counted and subjected to the same phenotypic analysis. Analysis of fresh bone marrow cells and representative results of 3 transplanted mice are shown. Results show live cell (PI−) events expressing human CD59 and lacking high-level murine CD45 and TER-119 expression. Additionally, the analysis of CD3, CD56 and CD19 expression was limited to gated events with a typical lymphoid forward and side light-scatter. Percentages reflect the number of events found in the associated region among all PI− human cells. Percentages are derived from a single measurement of donor cells and the mean of measurements from 3 transplanted mice.
Hematopoietic precursors were among the cell types found in the peritoneum as evinced by the spectrum of cells at different stages of differentiation indicated by their different levels of CD34 and CD38 expression (Fig. 4).18–20 Human hematopoietic stem cells are enriched among cells expressing high levels of CD34 and express CD133 but low, or negligible, levels of CD38.21 Cells with this phenotype were also observed in the peritoneum 18 hours after transplant at a slightly higher frequency than in the fresh graft. In a second similar experiment, the frequency of CD133+CD34++ cells was approximately 3 times higher 20 hours after transplant (data not shown). Nonetheless, the notable clearance from the peritoneum of most transplanted cells in less than a day indicates that most hematopoietic stem cells rapidly clear the peritoneum.
Despite the rapid emigration of hematopoietic cells from the peritoneum, we were interested in determining if some human cells remain in the peritoneum as suggested by the murine transplant studies. Mice were analyzed at a number of time points up to 321 days after transplantation (Fig. 5). At least some recipient mice at all time points had evidence of CD133+CD34++ cells present in the peritoneum. At 85 days after transplant, the total number of CD133+CD34++ cells recovered from 5 transplanted mice represented a mean 4.6% of the number of CD133+CD34++ cells in the original graft. Until 105 days after transplantation all mice (n = 4 or 5) had CD133+CD34++ cells present. However, by 223 days after transplant only half of the 8 mice transplanted continued to show evidence of hematopoietic stem cell engraftment in the peritoneum. At the final time point, 2 of 4 healthy mice had any detectable events that could be defined as CD133+CD34++ cells, although the number of events detected was too few to conclusively demonstrate continued engraftment by these cells. Thus, CD133+CD34++ cells persist in the peritoneum for at least 3 months after transplant along with evidence of intermediate hematopoietic progenitor populations, which express various levels of CD34 but don’t express CD133, also evident thereby suggesting ongoing hematopoiesis. In some cases the hematopoietic engraftment of the peritoneum lasted over 7 months.
Fig. 5.

Long-term retention of human hematopoietic precursors in the murine peritoneum. 2 × 107 donor bone marrow cells, ranging from 23–24 weeks of gestation, were transplanted i.p. into NSG mice. Peritoneal cells were harvest from untransplanted mice (top row) and transplanted mice (bottom row) at the indicated time points. Data shown represent gated human CD59+ events lacking PI and murine CD45 and TER119 antigen expression. As staining with murine leukocyte and erythrocyte markers doesn’t strongly stain all murine cells and CD59 expression on human cells is moderate, a small number of murine cells are captured by the gating strategy represented by the events shown for untransplanted mice. Arrows indicate candidate CD133+CD34++ hematopoietic stem cells.
Despite the lack of any preconditioning before transplant, NSG mice transplanted by the i.p. route of administration with human fetal bone marrow could result in bone marrow engraftment. Three of 4 mice analyzed 321 days after transplantation had evidence of human cells in their bone marrow and spleen. Multilineage reconstitution was confirmed by the observation of CD34+ progenitors, B-cells (including CD34+CD19+ B cell progenitors), CD14+ monocytes, CD7++CD56+ NK-cells, CD41+CD42b+ platelets, CD235a+ erythrocytes, and single positive CD3+ T-cells expressing either CD4 or CD8 (Fig. 6).
Fig. 6.

Hematopoietic reconstitution after i.p. transplantation of human cells in NSG mice. Results are shown for a mouse transplanted with 2 × 107 fetal bone marrow cells and hematopoietic engraftment evaluated after 321 days. Data shown represent gated PI-CD59+ single cells lacking expression of murine CD45, H-2Kb and TER-119. CD4 and CD8 expression in the spleen is shown on CD3+CD45+ cells defined by the indicated region.
There was no evidence for recirculation of CD133+CD34++ cells from the bone marrow back into the peritoneum. Among 5 mice transplanted by intravenous (i.v.) injection only 2 mice each had a single event detected by flow cytometry within the region defining CD133+CD34++ cells (data not shown). Thus, there is no convincing evidence that mice transplanted by standard i.v. administration yield engraftment of the peritoneum by cells with the phenotypic properties of hematopoietic stem cells. These results are in line with expectations that the peritoneum is not naturally a site of extramedullary hematopoiesis beyond the proliferation of some mature blood cells that can occur in the peritoneum.
Peritoneal chimerism and immune tolerance
Long-term acceptance of a foreign graft requires the host immune system to be tolerant of the graft. One of the lures of prenatal transplantation is that immune tolerance may be achieved, without any enforced immune suppression, if the graft can take during the early development of the immune system when self-tolerance normally develops. Natural examples of prenatal tolerance induction include the classic studies of immune tolerance in freemartin cattle performed by Owen.22 These animals share blood circulation in utero and are exposed to each other’s antigens resulting in immune tolerance towards each other’s tissues. Another example is the case of maternal chimerism that occurs when maternal cells cross over into the fetus during gestation.23 Chimerism resulting from maternal cells is orders of magnitude lower than from placental cross-circulation and its effects on immune development can differ. Transplant studies that have evaluated the effects of noninherited maternal antigens (NIMAs) have shown that maternal chimerism can lead to immune tolerance or at least hypo-responsiveness towards NIMAs.24–26
The development of the cellular immune system in humans is only partially understood. Many studies have been performed on umbilical cord blood cells harvested at birth under the presumption that these fetal cells are representative of immunologically naïve and unactivated cells found throughout fetal development. That this assumption was incorrect was documented by Byrne et al.27 when they demonstrated a higher frequency of CD45RO+ ‘memory’ T-cells in tissues from early gestation specimens than from the blood harvested at term births. Fetal CD4+ T-cells expressed CD25, a component of the interleukin-2 receptor, and the authors speculated that these represented anergic auto-reactive T-cells. Our own study confirmed the unique properties of fetal T-cells demonstrating the expression of a number of T-cell markers associated with T-cell activation at frequencies much higher than at term and, sometimes, even higher than found in adult peripheral blood.28 We speculated that the presence of a high number of T-cells expressing interleukin-2 receptors might be indicative of the presence of CD4+ T-regulatory cells (Tregs), which could play a role in the development of fetal self-tolerance. Indeed, several groups soon went on to demonstrate an early wave of Treg production in the human fetus that likely plays a key role in the establishment of peripheral tolerance.29–31
Understanding the development of the human immune system is important to identify and overcome the barriers to successful prenatal cellular therapy. How the presence of a high frequency of Tregs in the human fetus may affect the outcome of IUT is hinted at by the response of fetal and neonatal T-cells towards maternal cell antigens. Study of term umbilical cord blood lymphocytes revealed a reduced response by T-cells in mixed lymphocyte reactions (MLRs) towards NIMAs.32 Thus, exposure to maternal cells during fetal development is likely responsible for promoting specific immune tolerance towards NIMAs. With support of the National Blood Foundation, we collaborated with Mold and his colleagues to demonstrate that human fetal Tregs can specifically inhibit responses towards NIMAs, a response that can persist throughout at least childhood.33 These results demonstrated that prenatal exposure to foreign antigens can result in specific immune tolerance that is in part conferred by Tregs. T-cells are not the only branch of the cellular immune system to show diminished responses towards foreign antigens that were present during gestation as reduced antibody responses by B cells towards NIMAs have also been reported.34
Donor-specific tolerance induction mediated by IUT, used to expose the developing immune system to antigens, is envisioned as a method to prepare for secondary transplantation after birth of either hematopoietic stem cells, to boost engraftment, or other tissues or organs. We studied this approach in mice, which although differing from humans in the kinetics of immune system development because of the short murine gestational period, have been shown to share many similarities in immune function. For instance, mice also show a specific tolerance towards NIMAs owing to prenatal exposure just as in humans.35, 36 However, murine studies have also shown that prenatal exposure to NIMAs can also induce sensitization to maternal antigens.37, 38 The specific question we were interested in was if persistent hematopoietic chimerism in the peritoneum following IUT could benefit immune tolerance induction.
Our initial observations using the haplogeneic mouse transplant model (H-2b/d into H-2b)demonstrated a correlation between peritoneal chimerism and T-cell tolerance.14, 15 MLRs were performed 3–5 months after IUT and responses categorized as normal responsiveness (relative to 3rd party allogeneic stimulator cells), hyper-responsiveness or hypo-responsiveness. Donor chimerism in the bone marrow, blood and spleen in this set of transplants was low (<1%) or absent. Peritoneal chimerism was also generally low, with some exceptions, but the presence of donor cells in the peritoneum correlated with hypo-responsive T-cell reaction towards donor cells. Some transplanted mice also exhibited hyper-responsiveness, suggesting that low or transient chimerism owing to IUT may act to sensitize mice to donor antigen, similar to what was reported in some studies of prenatal exposure to NIMAs. Thus, in the absence of significant chimerism, the effect of IUT on the host’s T-cell response to donor cells may either be tolerance or sensitization.
We further evaluated the relationship of chimerism and tolerance in an allogeneic mouse model in which C57BL/6 (H-2Kb) cells were transplanted into FVB/N (H-2Kq) recipients. In this model, clinically relevant levels of chimerism (>10%) could be achieved in some recipients with transplantation of a high dose of donor cells (≥5 × 106 cells/embryo).39 Nonetheless, a spectrum of chimerism levels was achieved among recipients, which allowed us to determine the effects of chimerism levels in different tissues on long-term hematopoietic engraftment and tolerance. A threshold level of ≥2% donor cells in the peripheral blood at 1 month of age was correlated with lasting hematopoietic chimerism. Less than this level of chimerism and the graft would be at risk of failure. A focus on the importance of peripheral blood chimerism levels was made because clinically the blood is the easiest and safest tissue to measure.
We evaluated tolerance using skin grafts originating from host, donor and allogeneic 3rd party mouse strains.39 Survival of skin grafts is considered one of the most stringent tests of immune tolerance. Survival time of donor-type skins was clearly correlated with both peripheral blood and peritoneal chimerism. Higher levels of donor cells in the peripheral blood at the time of skin transplant predicted graft acceptance. Transient peripheral blood chimerism that had faded by the time of skin grafting sometimes correlated with prolonged skin graft survival, but didn’t predict long-term skin tolerance. For those animals that lacked detectable donor cells in peripheral blood, the presence of donor cells in the peritoneum correlated with increased skin survival, whereas rapid skin rejection occurred in untransplanted animals and animals with no blood or peritoneal chimerism. Thus, even when donor cells are restricted to the site of injection, the presence of hematopoietic cells in the peritoneum alone contributes to a hypo-responsive state.
We explored the relationship further between chimerism and the timing of skin grafts in a recently published study using the same murine allogeneic prenatal transplant model.40 Skin transplants were performed at three different time points to test if grafting sooner after the fetal hematopoietic transplant would improve graft survival over later transplants. Mice were grouped based on their levels of peripheral blood cell chimerism at 1 month of age. The survival of skin grafts was observed when peripheral blood chimerism was observed at the time of skin transplantation. The initial exposure to donor antigens from the fetal transplant or donor chimerism measured at an early time point that faded before skin transplantation did not promote acceptance of the skin graft. Thus the presence of donor hematopoietic cells at the time of transplant was a critical factor in tolerance induction. At ≥3% blood chimerism, acceptance of donor-type skin was assured. At this level of chimerism, donor cells were also found in the thymus, likely helping to promote central tolerance. There was about a 1 in 3 chance of skin graft acceptance if blood chimerism was between 0.2% and 3%.
The utility of MLRs in predicting skin tolerance as well as the role of peritoneal chimerism in skin tolerance was also studied.40 Lack of response in MLRs did not fully predict skin graft survival. Mice with donor cells present in the blood that demonstrated tolerance by MLR did not always accept donor skin grafts. Likewise, the correlation between peritoneal chimerism and lack of an MLR response was confirmed, but peritoneal chimerism alone did not ensure skin graft acceptance. These findings point to a hierarchy of immune tolerance as summarized in Fig. 7. T-cell tolerance as measured by MLR can occur after transient or persistent low-level chimerism. Persistence of donor cells in the peritoneum helps to promote tolerance or hypo-responsiveness. However, sufficient blood chimerism is needed for full tolerance that allows for skin grafts. The mechanism for this tolerance is likely complex involving both central and peripheral tolerance.
Fig. 7.

Relationship between chimerism and immune tolerance. Results of IUT studies performed on mice were used to summarize the relationship between donor-cell chimerism levels, location of chimerism and immune responses measured by MLR and donor-skin grafting.
Summary
IUT remains an alluring frontier in the realm of cellular therapy. Over a decade ago encouraging results from animal studies led to a number of attempts at transplanting highly purified stem cells in human fetuses with some success in treating severe immune deficiencies,6, 41, 42 but other ailments failed to be significantly affected by IUT. Our own attempt at treating a patient with chronic granulomatous disease with purified stem cells failed to detect any chimerism.43 These results underscored the difficulties in achieving clinically significant engraftment with a simple stem cell transplant. More research was, and still is, required to understand the development of the human immune system to better design cellular therapies that can exploit the opportunity of IUT and donor-specific tolerance induction to treat diseases of the hematopoietic system as well as diseases that require post-natal tissue or organ transplantation. Support of the National Blood Foundation has helped us continue to study this unique approach to the treatment of birth defects. Our work has shown the benefit of high-dose transplants containing mature and immature blood cell elements as opposed to the highly purified grafts tried in humans. Engraftment of multiple cells types in the peritoneum and their gradual dissemination to the peripheral circulation benefits the establishment of immune tolerance and offers the possibility engineering grafts to further use the peritoneal cavity as way station towards successful IUT.
Acknowledgments
SOURCES OF SUPPORT: This work was supported by a grant from the National Blood Foundation (MOM) as well as support from Blood Systems (MOM), NIH grant DK59301 (MOM), NHRI-EX97-9743SI (JCC), and CMRPG460021 (JCC). AIB was supported by a Bridges to Stem Cell Training grant from the California Institute of Regenerative Medicine.
We thank the staff and faculty at San Francisco General Hospital Women’s Options Center for assistance in the collection of human fetal tissues. We also thank Dale Hirschkorn for assistance with equipment, training and management of the flow cytometry core. We are also indebted to the administrative staff at our institute, in particular JoAnn Yates, Jerry Michaelson, Michelle Quintos, Abigail Schrock, Anne Eickelberg, Fredda Assenzio and Barbara ‘BJ’ Johnson. We appreciate Dr. Alicia Bárcena’s critical review of the manuscript.
Footnotes
CONFLICT OF INTEREST: The authors declare that they have no conflicts of interest relevant to the work presented in this manuscript.
References
- 1.Muench MO. In utero transplantation: baby steps towards an effective therapy. Bone Marrow Transplant. 2005;35:537–47. doi: 10.1038/sj.bmt.1704811. [DOI] [PubMed] [Google Scholar]
- 2.Lobach DF, Hensley LL, Ho W, Haynes BF. Human T cell antigen expression during the early stages of fetal thymic maturation. J Immunol. 1985;135:1752–9. [PubMed] [Google Scholar]
- 3.Haynes BF, Heinly CS. Early human T cell development: analysis of the human thymus at the time of initial entry of hematopoietic stem cells into the fetal thymic microenvironment. J Exp Med. 1995;181:1445–58. doi: 10.1084/jem.181.4.1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guihard-Costa AM, Larroche JC, Droulle P, Narcy F. Fetal Biometry. Growth charts for practical use in fetopathology and antenatal ultrasonography. Introduction Fetal Diagn Ther. 1995;10:215–78. doi: 10.1159/000264241. [DOI] [PubMed] [Google Scholar]
- 5.Charbord P, Tavian M, Humeau L, Péault B. Early ontogeny of the human marrow from long bones: an immunohistochemical study of hematopoiesis and its microenvironment. Blood. 1996;87:4109–19. [PubMed] [Google Scholar]
- 6.Flake AW, Roncarolo MG, Puck JM, Almeida-Porada G, Evans MI, Johnson MP, Abella EM, Harrison DD, Zanjani ED. Treatment of X-linked severe combined immunodeficiency by in utero transplantation of paternal bone marrow. N Engl J Med. 1996;335:1806–10. doi: 10.1056/NEJM199612123352404. [DOI] [PubMed] [Google Scholar]
- 7.Schaefer BC, Schaefer ML, Kappler JW, Marrack P, Kedl RM. Observation of antigen-dependent CD8+ T-cell/ dendritic cell interactions in vivo. Cell Immunol. 2001;214:110–22. doi: 10.1006/cimm.2001.1895. [DOI] [PubMed] [Google Scholar]
- 8.Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, Kotb M, Gillies SD, King M, Mangada J, Greiner DL, Handgretinger R. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol. 2005;174:6477–89. doi: 10.4049/jimmunol.174.10.6477. [DOI] [PubMed] [Google Scholar]
- 9.Varga NL, Bárcena A, Fomin ME, Muench MO. Detection of human hematopoietic stem cell engraftment in the livers of adult immunodeficient mice by an optimized flow cytometric method. Stem Cell Stud. 2010;1:e1. doi: 10.4081/scs.2010.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Golfier F, Bárcena A, Harrison MR, Muench MO. Fetal bone marrow as a source of stem cells for in utero or postnatal transplantation. Br J Haematol. 2000;109:173–81. doi: 10.1046/j.1365-2141.2000.02009.x. [DOI] [PubMed] [Google Scholar]
- 11.Broche F, Tellado JM. Defense mechanisms of the peritoneal cavity. Curr Opin Crit Care. 2001;7:105–16. doi: 10.1097/00075198-200104000-00009. [DOI] [PubMed] [Google Scholar]
- 12.Heel KA, Hall JC. Peritoneal defences and peritoneum-associated lymphoid tissue. Br J Surg. 1996;83:1031–6. doi: 10.1002/bjs.1800830804. [DOI] [PubMed] [Google Scholar]
- 13.Khanna R, Mactier R, Twardowski ZJ, Nolph KD. Peritoneal cavity lymphatics. Perit Dial Int. 1986;6:113–21. [Google Scholar]
- 14.Chen JC, Chang ML, Lee H, Muench MO. Haploidentical donor T cells fail to facilitate engraftment but lessen the immune response of host T cells in murine fetal transplantation. Br J Haematol. 2004;126:377–84. doi: 10.1111/j.1365-2141.2004.05040.x. [DOI] [PubMed] [Google Scholar]
- 15.Chen JC, Chang ML, Lee H, Muench MO. Prevention of graft rejection by donor type II CD8(+) T cells (Tc2 cells) is not sufficient to improve engraftment in fetal transplantation. Fetal Diagn Ther. 2005;20:35–43. doi: 10.1159/000081367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen JC, Chang ML, Muench MO. Persistence of allografts in the peritoneal cavity after prenatal transplantation in mice. Transfusion. 2008;48:553–60. doi: 10.1111/j.1537-2995.2007.01570.x. [DOI] [PubMed] [Google Scholar]
- 17.Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121:1109–21. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 18.Terstappen LW, Huang S, Safford M, Lansdorp PM, Loken MR. Sequential generations of hematopoietic colonies derived from single nonlineage-committed CD34+CD38- progenitor cells. Blood. 1991;77:1218–27. [PubMed] [Google Scholar]
- 19.Muench MO, Cupp J, Polakoff J, Roncarolo MG. Expression of CD33, CD38, and HLA-DR on CD34+ human fetal liver progenitors with a high proliferative potential. Blood. 1994;83:3170–81. [PubMed] [Google Scholar]
- 20.DiGiusto D, Chen S, Combs J, Webb S, Namikawa R, Tsukamoto A, Chen BP, Galy AH. Human fetal bone marrow early progenitors for T, B, and myeloid cells are found exclusively in the population expressing high levels of CD34. Blood. 1994;84:421–32. [PubMed] [Google Scholar]
- 21.Yin AH, Miraglia S, Zanjani ED, Almeida-Porada G, Ogawa M, Leary AG, Olweus J, Kearney J, Buck DW. AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood. 1997;90:5002–12. [PubMed] [Google Scholar]
- 22.Owen RD. Immunogenetic consequences of vascular anastomoses between bovine twins. Science. 1945;102:400–1. doi: 10.1126/science.102.2651.400. [DOI] [PubMed] [Google Scholar]
- 23.Gammill HS, Nelson JL. Naturally acquired microchimerism. Int J Dev Biol. 2010;54:531–43. doi: 10.1387/ijdb.082767hg. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smits JM, Claas FH, van Houwelingen HC, Persijn GG. Do noninherited maternal antigens (NIMA) enhance renal graft survival? Transpl Int. 1998;11:82–8. doi: 10.1007/s001470050109. [DOI] [PubMed] [Google Scholar]
- 25.Burlingham WJ, Grailer AP, Heisey DM, Claas FH, Norman D, Mohanakumar T, Brennan DC, de Fijter H, van Gelder T, Pirsch JD, Sollinger HW, Bean MA. The effect of tolerance to noninherited maternal HLA antigens on the survival of renal transplants from sibling donors. N Engl J Med. 1998;339:1657–64. doi: 10.1056/NEJM199812033392302. [DOI] [PubMed] [Google Scholar]
- 26.Ichinohe T, Uchiyama T, Shimazaki C, Matsuo K, Tamaki S, Hino M, Watanabe A, Hamaguchi M, Adachi S, Gondo H, Uoshima N, Yoshihara T, Hatanaka K, Fujii H, Kawa K, Kawanishi K, Oka K, Kimura H, Itoh M, Inukai T, Maruya E, Saji H, Kodera Y Japanese Collaborative Study Group for NIMA-Complementary Haploidentical Stem Cell Transplantation. Feasibility of HLA-haploidentical hematopoietic stem cell transplantation between noninherited maternal antigen (NIMA)-mismatched family members linked with long-term fetomaternal microchimerism. Blood. 2004;104:3821–8. doi: 10.1182/blood-2004-03-1212. [DOI] [PubMed] [Google Scholar]
- 27.Byrne JA, Stankovic AK, Cooper MD. A novel subpopulation of primed T cells in the human fetus. J Immunol. 1994;152:3098–106. [PubMed] [Google Scholar]
- 28.Muench MO, Pott Bärtsch EM, Chen JC, Lopoo JB, Bárcena A. Ontogenic changes in CD95 expression on human leukocytes: prevalence of T-cells expressing activation markers and identification of CD95-CD45RO+ T-cells in the fetus. Dev Comp Immunol. 2003;27:899–914. doi: 10.1016/s0145-305x(03)00081-8. [DOI] [PubMed] [Google Scholar]
- 29.Darrasse-Jèze G, Marodon G, Salomon BL, Catala M, Klatzmann D. Ontogeny of CD4+CD25+ regulatory/suppressor T cells in human fetus. Blood. 2005;105:4715–21. doi: 10.1182/blood-2004-10-4051. [DOI] [PubMed] [Google Scholar]
- 30.Cupedo T, Nagasawa M, Weijer K, Blom B, Spits H. Development and activation of regulatory T cells in the human fetus. Eur J Immunol. 2005;35:383–90. doi: 10.1002/eji.200425763. [DOI] [PubMed] [Google Scholar]
- 31.Michaëlsson J, Mold JE, McCune JM, Nixon DF. Regulation of T cell responses in the developing human fetus. J Immunol. 2006;176:5741–8. doi: 10.4049/jimmunol.176.10.5741. [DOI] [PubMed] [Google Scholar]
- 32.Tsafrir A, Brautbar C, Nagler A, Elchalal U, Miller K, Bishara A. Alloreactivity of umbilical cord blood mononuclear cells: specific hyporesponse to noninherited maternal antigens. Hum Immunol. 2000;61:548–54. doi: 10.1016/s0198-8859(00)00110-5. [DOI] [PubMed] [Google Scholar]
- 33.Mold JE, Michaëlsson J, Burt TD, Muench MO, Beckerman KP, Busch MP, Lee TH, Nixon DF, McCune JM. Maternal alloantigens promote the development of tolerogenic fetal regulatory T cells in utero. Science. 2008;322:1562–5. doi: 10.1126/science.1164511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Claas FH, Gijbels Y, van der Velden-de Munck J, van Rood JJ. Induction of B cell unresponsiveness to noninherited maternal HLA antigens during fetal life. Science. 1988;241:1815–7. doi: 10.1126/science.3051377. [DOI] [PubMed] [Google Scholar]
- 35.Andrassy J, Kusaka S, Jankowska-Gan E, Torrealba JR, Haynes LD, Marthaler BR, Tam RC, Illigens BM, Anosova N, Benichou G, Burlingham WJ. Tolerance to noninherited maternal MHC antigens in mice. J Immunol. 2003;171:5554–61. doi: 10.4049/jimmunol.171.10.5554. [DOI] [PubMed] [Google Scholar]
- 36.Akiyama Y, Caucheteux SM, Iwamoto Y, Guimezanes A, Kanellopoulos-Langevin C, Benichou G. Effects of noninherited maternal antigens on allotransplant rejection in a transgenic mouse model. Transplant Proc. 2005;37:1940–1. doi: 10.1016/j.transproceed.2005.02.115. [DOI] [PubMed] [Google Scholar]
- 37.Molitor-Dart ML, Andrassy J, Haynes LD, Burlingham WJ. Tolerance induction or sensitization in mice exposed to noninherited maternal antigens (NIMA) Am J Transplant. 2008;8:2307–15. doi: 10.1111/j.1600-6143.2008.02417.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Opiela SJ, Adkins B. The pendulum swings: Tolerance versus priming to NIMA. Chimerism. 2010;1:36–8. doi: 10.4161/chim.1.1.12880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen JC, Chang ML, Huang SF, Chang PY, Muench MO, Fu RH, Ou LS, Kuo ML. Prenatal tolerance induction: relationship between cell dose, marrow T-cells, chimerism, and tolerance. Cell Transplant. 2008;17:495–506. doi: 10.3727/096368908785095971. [DOI] [PubMed] [Google Scholar]
- 40.Chen JC, Kuo ML, Ou LS, Chang PY, Muench MO, Shen CR, Chang HL, Yu HY, Fu RH. Characterization of tolerance induction through prenatal marrow transplantation: the requirement for a threshold level of chimerism to establish rather than maintain postnatal skin tolerance. Cell Transplant. 2010;19:1609–22. doi: 10.3727/096368910X516583. [DOI] [PubMed] [Google Scholar]
- 41.Wengler GS, Lanfranchi A, Frusca T, Verardi R, Neva A, Brugnoni D, Giliani S, Fiorini M, Mella P, Guandalini F, Mazzolari E, Pecorelli S, Notarangelo LD, Porta F, Ugazio AG. In-utero transplantation of parental CD34 haematopoietic progenitor cells in a patient with X-linked severe combined immunodeficiency (SCIDXI) Lancet. 1996;348:1484–7. doi: 10.1016/s0140-6736(96)09392-0. [DOI] [PubMed] [Google Scholar]
- 42.Touraine JL, Raudrant D, Golfier F, Rebaud A, Sembeil R, Roncarolo MG, Bacchetta R, D’Oiron R, Lambert T, Gebuhrer L. Reappraisal of in utero stem cell transplantation based on long-term results. Fetal Diagn Ther. 2004;19:305–12. doi: 10.1159/000077957. [DOI] [PubMed] [Google Scholar]
- 43.Muench MO, Rae J, Bárcena A, Leemhuis T, Farrell J, Humeau L, Maxwell-Wiggins JR, Capper J, Mychaliska GB, Albanese CT, Martin T, Tsukamoto A, Curnutte JT, Harrison MR. Transplantation of a fetus with paternal Thy-1(+)CD34(+)cells for chronic granulomatous disease. Bone Marrow Transplant. 2001;27:355–64. doi: 10.1038/sj.bmt.1702798. [DOI] [PubMed] [Google Scholar]