

Abstract
The cannabinoid receptor 2 (CB2) plays a pleiotropic role in innate immunity and is a crucial mediator of liver disease. In the present study, we investigated the impact of CB2 receptors on the regenerative process associated with liver injury. Following acute hepatitis induced by carbon tetrachloride (CCl4), CB2 were induced in the non-parenchymal cell fraction and remained undetectable in hepatocytes. Administration of CCl4 to CB2−/− mice accelerated liver injury, as shown by increased ALT/AST levels and hepatocyte apoptosis, and delayed liver regeneration, as reflected by a retarded induction of hepatocyte PCNA expression; PCNA induction was also delayed in CB2−/− mice undergoing partial hepatectomy. Conversely, following treatment with the CB2 agonist JWH-133, CCl4-treated WT mice displayed reduced liver injury and accelerated liver regeneration. CCl4-treated CB2−/− mice showed a decrease in inducible nitric oxide synthase and tumor necrosis-α (TNF-α expression, and administration of the nitric oxide donor SIN-1 to these animals reduced hepatocyte apoptosis, without affecting liver regeneration. Impaired liver regeneration was consecutive to an interleukin-6 (IL-6)-mediated decrease in matrix metalloproteinase-2 (MMP-2) activity. Indeed, CCl4-treated CB2−/− mice displayed lower levels of hepatic IL-6 mRNA and increased MMP-2 activity. Administration of IL-6 to these mice decreased MMP-2 activity and improved liver regeneration, without affecting hepatocyte apoptosis. Accordingly, administration of the MMP inhibitor CTTHWGFTLC to CCl4-treated CB2−/− mice improved liver regeneration. Finally, in vitro studies demonstrated that incubation of hepatic myofibroblasts with JWH-133 increased TNF-α and IL-6 and decreased MMP-2 expressions.
Conclusion
CB2 receptors reduce liver injury and promote liver regeneration following acute insult, via distinct paracrine mechanisms involving hepatic myofibroblasts. These results suggest that CB2 agonists display potent hepatoprotective properties, in addition to their antifibrogenic effects.
Keywords: Alanine Transaminase; metabolism; Animals; Apoptosis; physiology; Aspartate Aminotransferases; metabolism; Cannabinoids; pharmacology; Carbon Tetrachloride; adverse effects; Cells, Cultured; Disease Models, Animal; Drug-Induced Liver Injury; metabolism; pathology; physiopathology; Hepatectomy; Hepatocytes; drug effects; metabolism; pathology; Interleukin-6; metabolism; Liver Regeneration; drug effects; physiology; Matrix Metalloproteinase 2; metabolism; Mice; Mice, Inbred C57BL; Mice, Knockout; Paracrine Communication; physiology; Proliferating Cell Nuclear Antigen; metabolism; Receptor, Cannabinoid, CB2; agonists; genetics; physiology; Tumor Necrosis Factor-alpha; metabolism
INTRODUCTION
The endocannabinoid system comprises two G-protein coupled receptors, CB1 and CB2, a family of lipidic ligands, known as endocannabinoids and a machinery dedicated to endocannabinoid degradation (1, 2). CB1 receptors are highly expressed in the central nervous system and in a variety of peripheral tissues; in contrast, CB2 receptors show a more restricted distribution, predominating in immune cells (2). Several recent data suggest that the non-psychoactive cannabinoid receptor CB2 is up-regulated in inflammatory disorders and may represent a critical target for regulation of inflammation, with either pro- or anti-inflammatory properties according to pathophysiological settings. Thus, we have shown that CB2 receptors promote adipose tissue inflammation associated with obesity (3). In contrast, anti-inflammatory properties of CB2 receptors have been established in experimental models of multiple sclerosis, atherosclerosis, inflammatory bowel disease or liver ischemia reperfusion injury (4–6). CB2 receptors have also been identified in non-immune cells, such as osteoblasts, myocytes and cardiac fibroblasts, leading to the characterization of non immune beneficial effects of CB2 agonists on osteoporosis or post-ischemic heart failure (7, 8).
Accumulating data indicate that the cannabinoid system is a crucial mediator in the pathogenesis of a variety of liver diseases (1, 3, 9). We have shown that CB1 receptors promote the progression of liver fibrogenesis and that CB1 antagonism is an efficient antifibrotic strategy (10, 11). Moreover, CB1 receptors have also been implicated in the pathogenesis of alcoholic and non alcoholic fatty liver disease (12–14). Finally, CB1 receptors promote the development of portal hypertension and ascites in cirrhotic animals (15, 16). Unfortunately, exciting potential therapeutic openings derived from these findings have been put to a hold with the withdrawal of the CB1 receptor antagonist rimonabant, due to central adverse effects. Nonetheless, mounting evidences identify CB2 receptors as an alternative target for the management of liver diseases. Thus, we have shown that endogenous activation of CB2 receptors in hepatic myofibroblasts reduces the progression of experimental fibrosis (17) and a subsequent study established the curative properties of a CB2 agonist in cirrhotic rats (18). Recent data also indicate that CB2 receptors decrease the extent of liver injury in models of acute insult, as induced by ischemia-reperfusion or concanavalin-A administration (6, 19). However, their impact on the regenerative process associated with liver injury has not been investigated, as yet. In the present study we show that CB2 receptors reduce liver injury and accelerate liver regeneration via distinct pathways.
METHODS
Materials
CCl4, mineral oil (MO) and SIN-1 were from Sigma (France), IL-6 from Peprotech (France), JWH-133 from Tocris (ThermoFisher, France), CTTHWGFTLC from Merck (UK).
Animals and experimental design
Animals
Mice invalidated for CB2 receptor (CB2−/−) were generated as in (20) and wild type (WT) C57BL/6J mice were obtained from Janvier (France). Animals were housed in temperature and humidity controlled rooms, kept on a 12-h light/dark cycle and provided unrestricted amounts of food and water. Male mice aged 8–12 weeks were used. Animal procedures were conducted in accordance with French government policies (Services Vétérinaires de la Santé et de la Production Animale, Ministère de l’Agriculture).
CCl4-induced liver injury
Acute liver injury was induced by a single intraperitoneal (ip) injection of CCl4 (0.5 ml/kg body weight, 1:5 dilution in MO), as in (21). Control animals received MO. When indicated, mice were treated either with respective vehicle, IL-6 (0.5 mg/kg, subcutaneous), the CB2 agonist JWH-133 (3 mg/kg, ip), the NO donor SIN-1 (10 mg/kg, ip), or the MMP-2/MMP-9 inhibitor CTTHWGFTLC (13 mg/kg, ip), administered before CCl4 administration. No mortality was observed throughout treatments. Liver samples were taken from several lobes and either fixed in buffered formalin or snap frozen in liquid nitrogen and stored at −80°C until use. Experiments were performed on 4–9 animals/group.
Partial hepatectomy
Two-third hepatectomy was performed as previously described (22), while animals were under isoflurane anesthesia. After ventral laparotomy, the left lateral, left median and right median lobes were ligated, and excised. The removed liver specimens were weighed and snap frozen in liquid nitrogen to serve as control.
Liver function tests
Alanine aminotransferase activity was measured on an automated analyzer in the Biochemistry Department of Mondor Hospital. Results are the mean from 15 animals/group.
Liver cell fractionation
Liver cells were digested by two-step collagenase perfusion. Cell suspension was centrifugated at 500 rpm for 2 min. The pellet contained hepatocytes and non-parenchymal cells were purified from the supernatant by density gradient centrifugation with 25%–50% Percoll.
TUNEL assay
TUNEL staining was performed on paraffin-embedded tissue sections, using the In Situ Cell Death Detection Kit, POD (Roche). TUNEL-positive area from 2–3 fields (X100)/animal were quantified with Image J. Results are expressed as % of total area, and were quantified from 7–8 animals/group.
Immunohistochemistry
Immunohistochemistry was carried out on paraffin-embedded liver tissue sections as previously described (10). Immunohistochemical detection of PCNA was performed using the MOM immunodetection kit (Vector) and a mouse monoclonal anti-PCNA (1:1,750, Santa Cruz). The number of labeled hepatocytes/2000 hepatocytes was quantified in tissue sections (X200) from 4–6 mice/group. Immunohistochemical detection of F4/80 and myeloperoxydase was performed using rat anti-mouse F4/80 (1:20, Serotec) and rabbit anti-human myeloperoxydase (1:750, Dako) respectively, followed by biotinylated secondary antibody (anti-rat, 1/50, Serotec or anti-rabbit, 1/100, Santa Cruz). The signal was amplified with alkaline phosphatase–conjugated streptavidin (1/20, Serotec), and revealed using liquid permanent red chromogen system (Dako).
Western blot analysis
Western blot analysis was performed as previously described (10). Primary antibodies were mouse monoclonal anti-PCNA (1:1,000) or anti-cyclin D1 (1:500) and secondary antibody was peroxidase-conjugated goat anti-mouse IgG antibody (1:5,000), all from Santa Cruz. Proteins were visualized by an enhanced chemiluminescence assay kit (ECL Plus, GE Healthcare). Signals were quantified using ImageJ. Quantification relative to β-actin (mouse monoclonal 1:100,000, Sigma) was performed on 8–10 animals/group.
RNA preparation and RT-PCR
Total RNA was extracted using RNeasy Mini kit (Qiagen). Real-time PCR was carried out on a LightCycler (Roche), using Quantitech SYBR Green PCR kit (Qiagen), with oligonucleotide primers from MWG Biotech (see supplementary data) The PCR amplified products were analyzed on a 2% agarose gel, and sequenced. Data are from 6–10 animals/group,
Cell culture
Hepatic myofibroblasts
Hepatic myofibroblasts were obtained by outgrowth of explants prepared from surgical specimens of human normal liver, as previously described (23). This procedure was performed in accordance with ethical regulations imposed by the French legislation. Experiments were performed on confluent cells that were made quiescent by a 48 h incubation in serum-free medium.
Bone-marrow derived macrophages (BMDM)
BMDM were isolated from bone marrow obtained from posterior leg bones of WT mice, following differenciation in HBSS completed with supernatant from L-cells for 5 days. BMDM were collected, allowed to adhere on 6-well dishes and further treated with 5 μM JWH-133 for 7h. The purity of BMDM was > 95%.
Results are the mean of triplicate determinations on 5 wells/condition.
Gelatin zymography
Proteins (50 μg) from liver homogenates were obtained as described (23) and separated on a 10% polyacrylamide gel containing 1 mg/ml of bovine skin gelatin (Sigma). After washing for 2 h in 2.5% Triton X-100, gels were incubated for 18 h at 37°C in 50 mM Tris pH 7.8 containing 5 mM CaCl2, stained with Coomassie blue, destained in methanol/acetic acid/water (5/2/13) and fixed in methanol/glycerol/water (2/1/17).
Statistics
Values represent means ± SEM. Results were analyzed by either Mann and Whitney test, one way or two way ANOVA followed by multiple comparison test, as appropriate. p<0.05 was taken as the minimum level of significance.
RESULTS
Induction of CB2 receptors following acute CCl4-induced liver damage
Administration of CCl4 was associated with a 10-fold induction of CB2 mRNA expression at 24 h that was maintained after 48 h (Fig 1A). CB2 receptors were not detected in hepatocytes isolated from either control or CCl4-treated animals (Fig 1B). In contrast, non-parenchymal cells showed basal expression of CB2 receptors and marked induction following CCl4 administration (Fig 1B).
Figure 1. Expression of CB2 receptor mRNA following CCl4-induced liver injury.
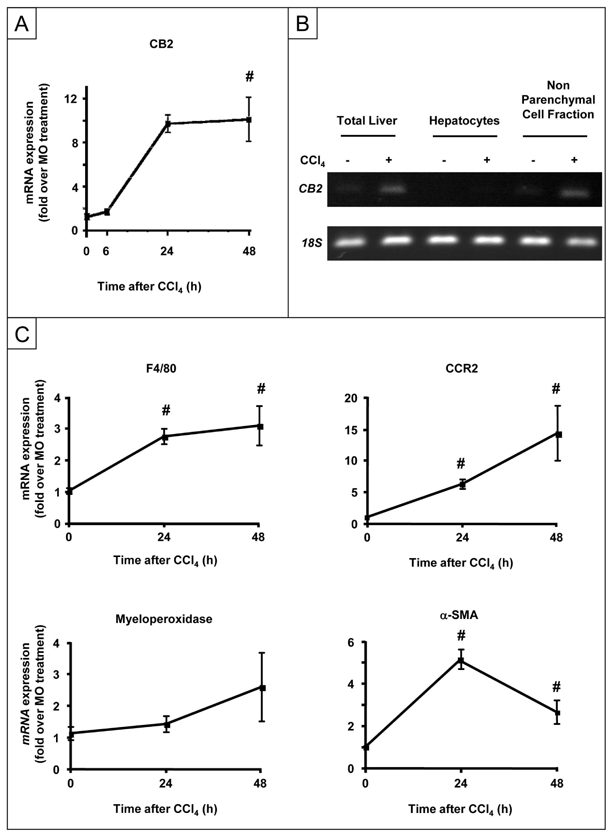
(A) RT-PCR analysis of hepatic CB2 mRNA expression. (B) CB2 receptor mRNA expression in the total liver, parenchymal and non parenchymal liver cell fractions at 24 h following CCl4 or MO administration. (C) RT-PCR analysis of hepatic F4/80, CCR2, MPO and α-SMA mRNA expression following CCl4 administration. #P<0.05 for CCl4 vs MO.
Toxic damage induced by CCl4 is associated with activation of Kupffer cells and hepatic myofibroblasts, and promotes infiltration of the liver by inflammatory cells (monocytes/macrophages and neutrophils) all of which express CB2 receptors. The mRNA expression of F4/80 (resident and recruited macrophage marker) and chemokine (C-C motif) receptor 2 (CCR2, recruited macrophages) was induced as of 24 h after CCl4 administration (Fig 1C), while induction of myeloperoxidase (MPO) mRNA, a marker of polymorphonuclear leukocytes, occurred only after 48 h (Fig 1C). Smooth muscle alpha-actin (α-SMA) expression was also induced after 24 h in CCl4–treated mice, reflecting activation of hepatic myofibroblasts (Fig 1C).
These data indicate that CCl4-induced liver injury is associated with an early induction of CB2 receptors in non-parenchymal cells, including hepatic myofibroblasts and macrophages at 24 h, although polymorphonuclear leukocytes may also contribute to CB2 induction after 48 h.
CB2 receptors reduce acute liver injury
Acute exposure to CCl4 induces apoptosis of hepatocytes following cytochrome P450 2E1 (CYP-2E1)-dependent generation of hepatotoxic metabolites. We found that CYP-2E1 mRNA expression was similar in CCl4-treated CB2−/− and WT mice, ruling out an impact of CB2 receptors on CCl4 metabolism (not shown). Hepatocyte apoptosis was monitored by TUNEL staining and showed time-dependent increase in CCl4-treated WT mice, achieving 20% of parenchymal area after 48 h (Fig 2A). Administration of CCl4 to CB2−/− mice resulted in a faster progression of TUNEL staining, reaching a peak of 20% after 24 h, higher than the corresponding 10% value in WT counterparts (Fig 2A). Moreover, serum ALT and AST underwent earlier and higher elevation in CCl4-injected CB2−/− animals compared to WT mice (Fig 2B). Conversely, there was a reduction in the density of TUNEL-positive hepatocytes in WT mice treated with the specific CB2 receptor agonist, JWH-133, compared to vehicle-treated animals (Fig 2C). Accordingly, peak values of ALT and AST levels were lower in the JWH-133-treated group as compared to vehicle-treated animals (Fig 2D). Overall, these data indicate that CB2 receptors reduce liver injury.
Figure 2. Hepatoprotective properties of CB2 receptors.
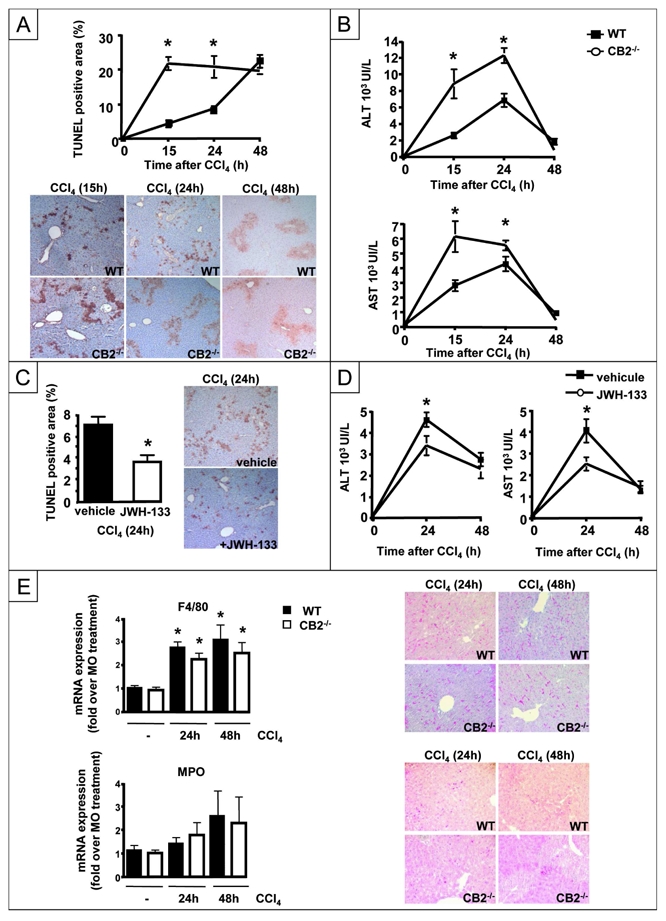
(A) Representative TUNEL staining and kinetics of mean TUNEL staining (*P<0.05 for CB2−/− vs WT mice; # P<0.05 for CCl4 vs MO). (B) ALT and AST levels. (C) Representative TUNEL staining of WT mice treated with JWH-133 or vehicle and sacrified 24 h after CCl4 administration.*P<0.05 for JWH-133 vs vehicle. (D) ALT and AST levels. (E) RT-PCR analysis of hepatic F4/80 and MPO expressions. #P<0.05 for CCl4 vs MO. Representative F4/80 and MPO immunostaining of CCl4-treated CB2−/− and WT liver tissue sections.
We also investigated whether CB2 receptor invalidation affects the extent of inflammatory infiltrate following CCl4 exposure. RT-PCR analysis showed that WT and CB2−/− mice did not differ in F4/80 and MPO mRNA expression. Accordingly, there was no difference in the density of F4/80 and MPO immuno-positive cells between both groups of mice (Fig 2E). These data indicate that CB2 receptors do not modulate inflammatory infiltration of the liver in response to CCl4.
CB2 receptors accelerate liver regeneration
We investigated the impact of CB2 receptors on the regenerative response arising from CCl4-induced injury. Cyclin D1 expression was induced in the liver of WT mice, peaking at 24 h and declining thereafter. In contrast, CB2−/− mice showed a lower level of hepatic cyclin D1 induction (Fig 3A). Accordingly, hepatocyte proliferation was delayed in CB2−/− animals compared to WT counterparts, as shown by western blot analysis and immunohistochemistry of PCNA expression (Fig 3B). Conversely, JWH-133 pretreatment accelerated the onset of PCNA induction in CCl4-treated WT mice (Fig 3C). These results were further confirmed in the partial hepatectomy model. CB2 mRNA expression was induced 48 and 72 h after partial hepatectomy (Fig 3D), and CB2−/− mice also showed a retarded regenerative response, as shown by Western blot analysis and immunohistochemistry of PCNA expression (Fig 3E) or BrDU staining (not shown). Altogether, these data indicate that CB2 receptors accelerate liver regeneration.
Figure 3. Beneficial effects of CB2 receptor on liver regeneration.
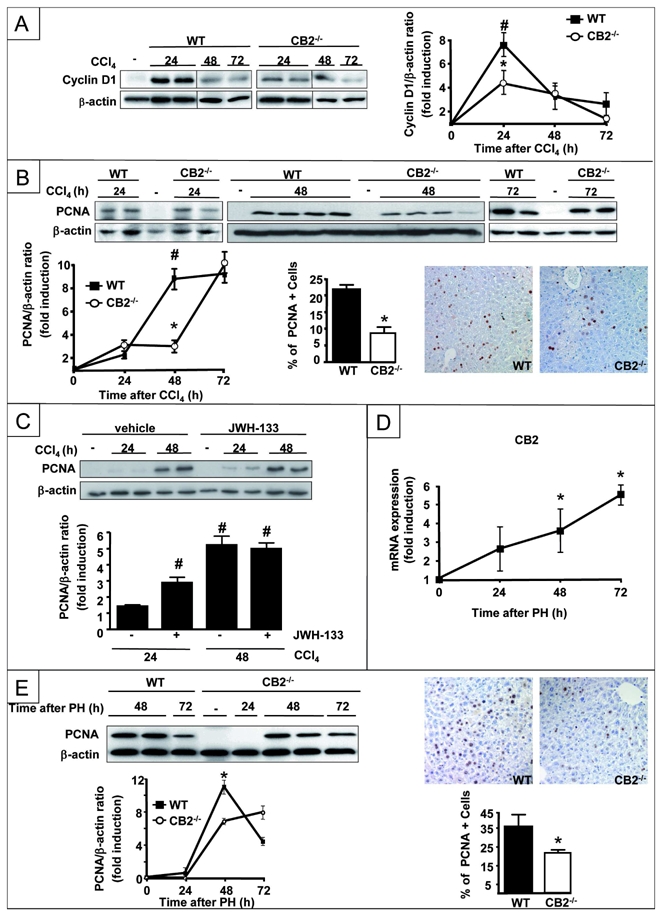
Representative western blot of (A) hepatic cyclin D1 expression and (B) hepatic PCNA expression after CCl4 injection. Immunohistochemical detection of PCNA in hepatocyte nuclei from WT and CB2−/− mice 48 h after CCl4 injection, and mean PCNA-positive cells. *P<0.05 for CB2−/− vs WT mice; # P<0.05 for CCl4 vs MO; (C) Representative western blot of hepatic PCNA expression in mice treated with JWH-133 or vehicle.*P<0.05 for JWH-133 vs vehicle; # P<0.05 for CCl4 vs MO. (D) RT-PCR analysis of hepatic CB2 mRNA expression following partial hepatectomy; #P<0.05 for hepatectomized vs control mice. (E) Representative western blot of hepatic PCNA expression after partial hepatectomy. Immunohistochemical detection of PCNA in nuclei of hepatocytes 48 h after partial hepatectomy and mean PCNA-positive cells. *P<0.05 for WT vs CB2−/− mice.
CB2 deficiency reduces expression of TNF-α, IL-6 and iNOS expression
Further experiments aimed at delineating the molecular mechanisms underlying the beneficial effect of CB2 receptors on hepatocyte survival and regeneration (24, 25). We first investigated the impact of CB2 receptor deficiency on hepatic expression of factors with known effects on hepatocyte survival and/or regeneration (24, 25). The induction of iNOS, TNF-α and IL-6 mRNA was attenuated in CCl4-treated CB2−/− mice as compared to WT counterparts (Fig 4A) whereas the induction profile of MCP-1, IL-10 and TLR4 was similar in both groups (Fig 4B).
Figure 4. Comparison of the expression profile of survival and mitogenic factors between CCl4-treated CB2−/− mice and WT counterparts.
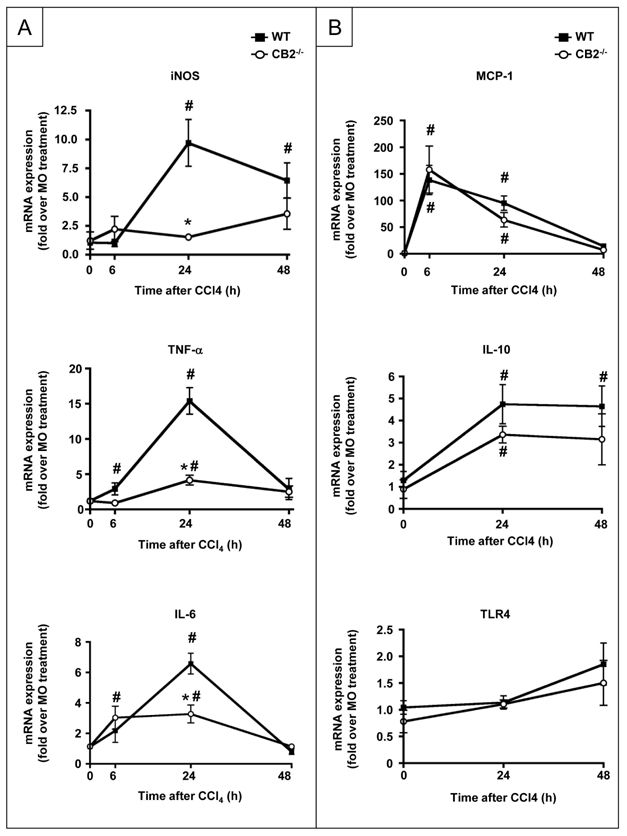
(A) RT-PCR analysis of iNOS TNF-α and IL-6 mRNA (B) MCP-1, IL-10, TLR4 mRNA following CCl4 administration. *P<0.05 for CB2−/− vs WT mice; # P<0.05 for CCl4 vs MO.
Defective hepatic iNOS induction is responsible for enhanced hepatocyte apoptosis in CB2−/− mice
Following CCl4 administration, up-regulation of iNOS in hepatocytes is a compensatory protective pathway with respect to hepatocyte apoptosis (26–29). Given the impairment of iNOS induction in the liver of CB2−/− mice exposed to CCl4, we investigated whether an NO donor would attenuate the exacerbation of liver injury in these mice. Treatment with SIN-1 reduced the rate of TUNEL-positive hepatocytes in CCl4-exposed CB2−/− mice, whereas liver injury was unchanged in CCl4-injected WT animals (Fig 5A). However, SIN-1 did not significantly improved liver regeneration in WT mice (not shown) or in CB2−/− animals, although a trend to increase was noted in the latter group. (p=0.13, Fig 5B). These results suggest that CB2 inactivation leads to defective induction of hepatic iNOS, thereby enhancing hepatocyte death following acute liver injury. In contrast, the impairment of liver regeneration found in CCl4-treated CB2−/− mice may result from additional mechanisms.
Figure 5. Effect of the NO donor SIN-1 on TUNEL staining and cyclin D1 expression in CCl4–treated CB2−/− and WT mice.
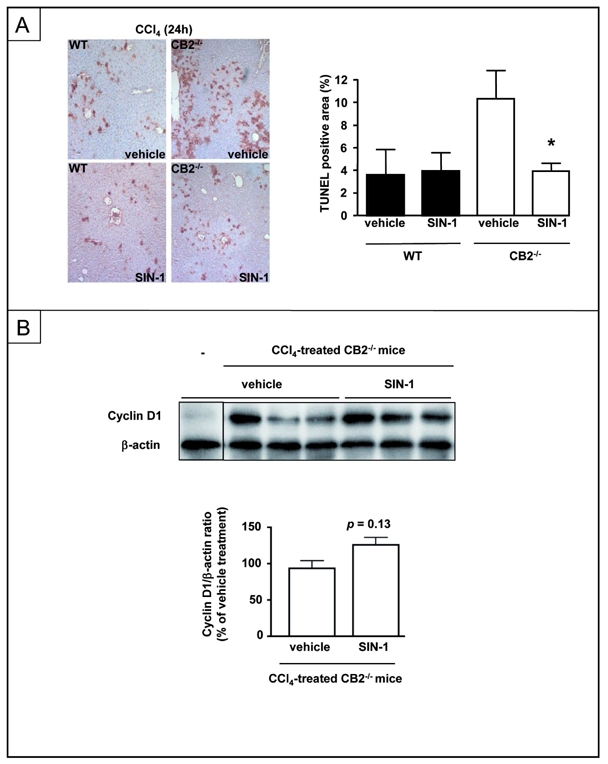
Mice were injected with SIN-1 or vehicle and sacrified 24 h after CCl4 injection. (A) Representative TUNEL staining and mean TUNEL-positive area. (B) Representative immunoblot and mean densitometric analysis of cyclin D1 expression. *P<0.05 for SIN-1 vs vehicle.
CB2 inactivation impairs liver regeneration via an IL-6 dependent inhibition of MMP-2
IL-6 plays a major role in hepatocyte survival and regeneration (24, 25, 30). Given the impairment of IL-6 induction in the liver of CCl4-treated CB2−/− mice, we explored whether defective liver regeneration would be restored by IL-6 administration. IL-6 did not affect TUNEL staining in CCl4-treated WT or CB2−/− mice at 24 h (Fig 6A). In contrast, IL6 restored PCNA expression in CCl4-treated CB2−/− animals to 75% of its level in CCl4-treated WT animals (Fig 6B). These findings suggest that IL-6 deficiency is a key event in the impairment of liver regeneration observed in CB2−/− animals.
Figure 6. Effect of IL-6 administration to CCl4–treated CB2−/− and WT mice on TUNEL staining and PCNA expression.
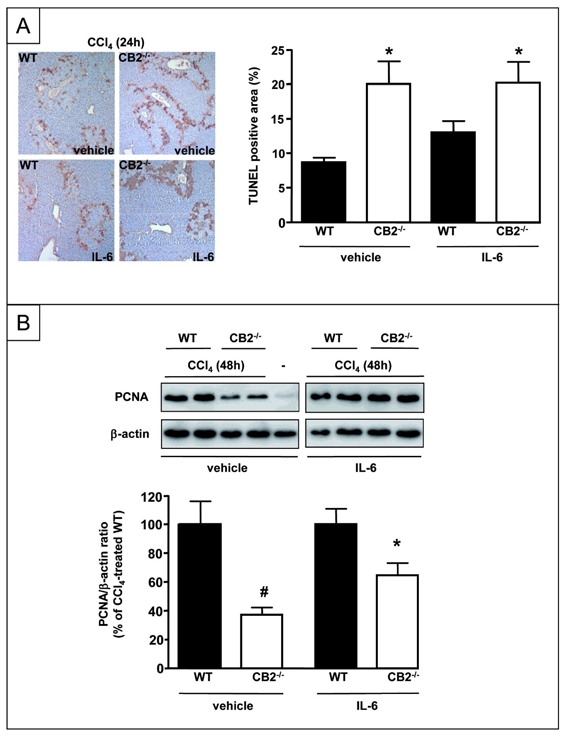
(A) Representative TUNEL staining and mean TUNEL-positive area 24 h after CCl4 treatment *P<0.05 vs vehicle-injected WT mice administered CCl4. (B) Representative immunoblot and mean densitometric analysis of PCNA expression 48 h after CCl4 exposure. *P<0.05 for IL-6 vs vehicle.
Matrix metalloproteinases contribute to liver injury and regeneration. IL-6 has been shown to down-regulate hepatic MMP-2 activity following acute CCl4 administration (31). We therefore investigated whether CB2 receptors modulate hepatic MMP activity via an IL-6-dependent mechanism. WT and CB2−/− mice showed no differences in hepatic MMP-9 activation following exposure to CCl4 (Fig 7A). In contrast, MMP-2 activation was enhanced in CB2−/− mice, as compared to WT counterparts (Fig 7A). Moreover, treatment of CB2−/− animals with IL-6 down-regulated MMP-2 activity to levels similar to those found in CCl4 treated-WT mice (Fig 7B). These data demonstrate that following acute liver injury, CB2 receptor inactivation enhances MMP-2 activity as a consequence of IL-6 down-regulation. In order to determine whether MMP-2 mediates the effects of CB2 on liver regeneration, WT and CB2−/− mice underwent an injection of the MMP-2/9 inhibitor CTTHWGFTLC (CTT) or vehicle before CCl4 administration. The defective induction of cyclin D1 associated with CB2 deficiency was fully restored in mice treated with CTT, whereas CTT had no effect in WT mice (Fig 7C).
Figure 7. CB2 receptor inactivation delays liver regeneration by a mechanism involving enhanced MMP-2 activity.
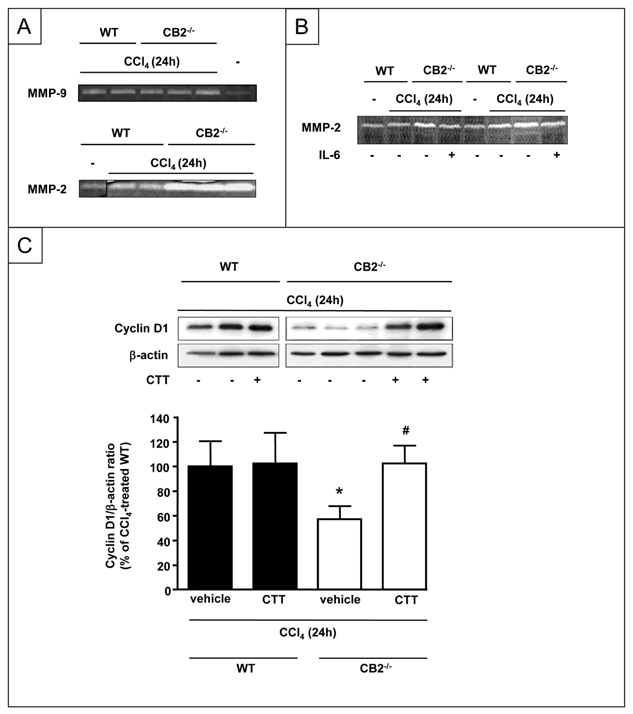
Gelatin zymography showing (A) MMP-9 and MMP-2 activities and (B) MMP-2 activities of liver homogenates from WT and CB2−/− mice injected with IL-6 or vehicle and sacrified 24 h after CCl4 administration. (C) Representative western blot of hepatic cyclin D1 expression 24 h after CCl4 injection, following CTT or vehicle treatment. *P<0.05 for CTT vs vehicle.
Altogether, our results indicate that impairment of liver regeneration in CB2−/− mice is consecutive to a defect in IL-6 production leading to an increase in MMP-2 activity.
CB2 receptors up-regulates TNF-α and IL-6 expression in cultured hepatic myofibroblasts
The absence of CB2 receptors in hepatocytes, and their predominant expression in non parenchymal cells (Fig 1) suggested that CB2-dependent regulation of hepatocyte injury and proliferation results from paracrine interactions between non parenchymal cells and hepatocytes. We therefore conducted experiments in primary cultures of macrophages and hepatic myofibroblasts. Indeed, both cell types express CB2 receptors (3, 17) and produce bioactive factors with antiapoptotic and mitogenic properties for hepatocytes, in particular TNF-α and IL-6. Moreover, hepatic myofibroblasts are the main source of MMP-2 during liver injury (32).
Cultured hepatic myofibroblasts showed a decrease in MMP-2 mRNA following treatment with JWH-133. Moreover, JWH-133 induced TNF-α and IL-6 mRNAs, that peaked after 6 h and declined to basal levels within 24 h (Fig 8A). In contrast, exposure of bone marrow-derived macrophages to JWH-133 did not affect TNF-α and down-regulated IL-6 mRNA expressions (Fig 8B). These findings suggest that CB2-dependent regulation of hepatocyte injury and regeneration may depend on paracrine effects of hepatic myofibroblasts.
Figure 8. Consequences of CB2 receptor activation in cultured macrophages and hepatic myofibroblasts.
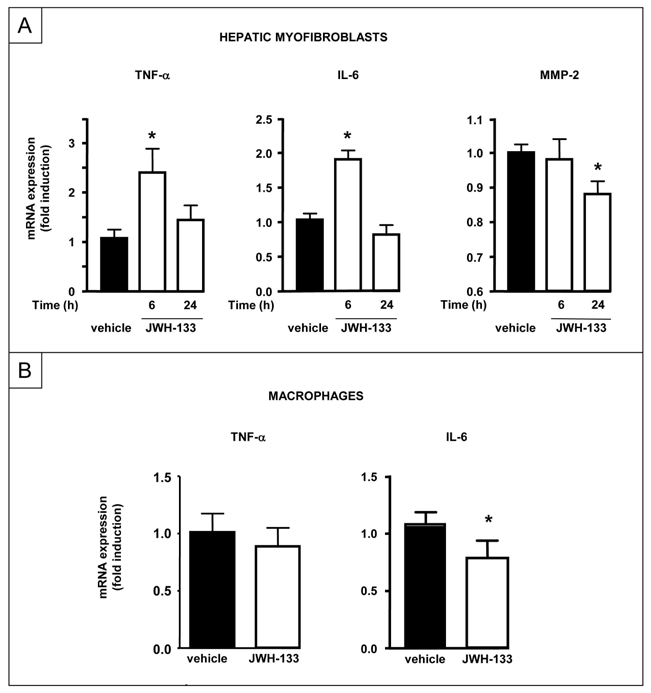
(A) RT-PCR analysis of TNF-α, IL-6 and MMP-2 mRNA on human hepatic myofibroblasts incubated with 5 μM JWH-133 or vehicle for 6 or 24 h. *P<0.05 for JWH-133 vs vehicle. (B) RT-PCR analysis of TNF-α and IL-6 mRNA on bone marrow-derived macrophages incubated with 5 μM JWH-133 or vehicle for 7 h. *P<0.05 for JWH-133 vs vehicle.
DISCUSSION
The present study shows that activation of CB2 receptors alleviates CCl4-induced hepatitis and accelerates liver regeneration, therefore identifying CB2 agonists as potential beneficial hepatoprotective agents.
We show that hepatic CB2 receptor expression is increased in the non parenchymal cell fraction during acute hepatitis triggered by CCl4. Moreover, our data suggest that early up-regulation of CB2 receptors may arise from macrophages and activated myofibroblasts, while other recruited inflammatory cells (i.e polymorphonuclear leucocytes) most probably also contribute to CB2 receptor induction at later time points. Interestingly, a recent study has reported increased production of the endogenous CB2 ligand 2-arachidonoylglycerol in the liver following a single injection of CCl4 (33). Altogether, these data indicate that acute liver injury is associated with an enhancement of the endogenous CB2 tone.
We show that in the CCl4 model, administration of the CB2 agonist JWH-133 reduces the extent of liver injury, whereas CB2−/− mice are more susceptible to the toxic insult. These findings corroborate previous studies demonstrating hepatoprotective properties of CB2 receptors in experimental models of acute liver injury elicited by ischemia/reperfusion injury, thioacetamide or concanavalin A (6, 19, 34). In addition, we identify iNOS as a central mediator in the beneficial effects mediated by CB2 receptors. Indeed, CCl4-treated CB2−/− mice show impaired induction of hepatic iNOS, and treatment of these mice with the NO donor SIN-1 reduces their exacerbated susceptibility to liver injury. These findings are in line with the reported protective effects of hepatocyte iNOS on liver injury. Thus, iNOS−/− mice display enhanced hepatocyte apoptosis when exposed to either CCl4 (26, 27) or to partial hepatectomy (28). In addition, cultured hepatocytes are more resistant to apoptosis in the presence of NO donors, or following induction of iNOS with cytokines, such as TNF-α (29). Interestingly, our data also indicate that CCl4-treated CB2−/− mice show decreased induction of TNF-α, a well-characterized inducer of iNOS expression. Whether TNF-α release triggered by CB2 receptors in non-parenchymal cells may contribute to the iNOS-dependent antiapoptic effects in hepatocytes remains to be determined.
It is well demonstrated that liver injury triggered by CCl4 is followed by a regenerative response orchestrated by the activation of multiple coordinated pathways, involving cross-talk between hepatocytes and non parenchymal cells (25). We demonstrate that CB2 receptors display beneficial effects on liver regeneration in this model, as well as in the partial hepatectomy model. We also demonstrate that beneficial effects of CB2 receptors are mediated by a pathway distinct from its protective effects against hepatocyte apoptosis, that involve IL-6. Thus, CCl4-treated CB2−/− mice display reduced hepatic expression of IL-6, and administration of IL-6 to these animals partially restores PCNA expression. Interestingly, CB2−/− mice and IL-6−/− mice behave similarly in response to acute and chronic liver injury, with increased liver damage, decreased liver regeneration and increased fibrogenesis (17, 35). However, our data indicate that although IL-6 mediates CB2 receptor impact on liver regeneration, the cytokine is not involved in CB2 receptor-dependent anti-apoptotic effect. These data are in line with the reported beneficial effects of IL-6 on liver regeneration (25), but contrast with studies reporting the protective role of IL-6 against liver damage (31, 36, 37). The mechanisms underlying these discrepancies, although not fully elucidated, may rely on the duration of IL-6 treatment, as suggested in a recent study (38). Our data also identify MMP-2 as a downstream target of the CB2/IL-6 pathway, as CCl4-treated CB2−/− mice display an increase in MMP-2 activity that is down-regulated following IL-6 administration. These findings are in line with the increase in MMP-2 activity reported in IL-6−/− mice upon CCl4 exposure, and the inhibitory effect of IL-6 on MMP-2 expression in hepatic myofibroblasts (31). Moreover, we also demonstrate that IL-6-dependent dysregulation of MMP2 activity is responsible for impaired liver regeneration, as shown by the beneficial effects of an MMP2/9 inhibitor on cyclin D1 expression in CB2−/− mice. Taken together, these data further argue for a central role of IL-6 in the regenerative response promoted by CB2 receptors, and identify MMP-2 as a downstream target.
Our data show that hepatocytes do not express CB2 receptors, indicating that paracrine interactions mediate the beneficial impact of these receptors on hepatocyte injury and regeneration. It is well established that following acute liver injury, Kupffer cells rapidly release pro-inflammatory mediators, such as TNF-α and IL-6, that regulate hepatocyte death and proliferation. Accumulating evidence suggest that, apart from their fibrogenic properties, hepatic myofibroblats are also central in the regulation of hepatocyte injury and regeneration (39–41). Indeed, at sites of injury, myofibroblasts produce bioactive mediators with antiapoptic and mitogenic effects on hepatocytes, including TNF-α and IL-6 (32). Macrophage culture experiments indicate that activation of CB2 receptors does not increase either TNF-α or IL-6 expression. These results suggest that macrophages are not responsible for the CB2-dependent production of these cytokines in the CCl4 model. In contrast, activation of CB2 receptors in cultured hepatic myofibroblasts leads to a concurrent increase in TNF-α and IL-6 expressions, associated with a down-regulation of MMP-2 expression. These data therefore suggest that production of TNF-α by hepatic myofibroblasts may contribute to iNOS-dependent hepatoprotective effects mediated by CB2 receptors following acute liver injury. Similarly, and since hepatic myofibroblasts are the major source of MMP-2 during liver injury (32), our data also suggest that hepatic myofibroblasts may also be key contributors in the IL-6/MMP-2-dependent regenerative effects of CB2 receptors. Our results indicate that CB2 receptors expressed in hepatic myofibroblasts elicit dual beneficial properties, by producing hepatoprotective factors, and by triggering antifibrogenic effects following growth inhibition and apoptosis of hepatic myofibroblasts (17). In keeping with our results, hepatic myofibroblasts have been shown to display similar hepatoprotective and antifibrogenic effects following stimulation of IGF-1 receptors (41) or neurotrophin p75NTR (39, 42).
In conclusion, our data demonstrate that CB2 receptors reduce liver injury and promote liver regeneration following acute insult, by distinct paracrine mechanisms on hepatocytes originating from hepatic myofibroblasts. These results suggest that CB2 agonists display potent hepatoprotective properties, in addition to their antifibrogenic effects.
Supplementary Material
Acknowledgments
This work was supported by the INSERM, the Université Paris-Est, and by grants (to SL) of the Agence Nationale de la Recherche and the Fondation pour la Recherche Médicale. MPB was supported by a fellowship from the Agence Nationale de la Recherche. We thank Mathilde Body-Malapel (Inserm U995) for her help in preparing bone-marrow-derived macrophages. We thank F Pecker for helpful and constant guidance, C Pavoine, F Lafdil for helpful comments, and S Balustre for her help during in vivo experiments.
References
- 1.Mallat A, Lotersztajn S. Endocannabinoids and Their Receptors in the Liver. Am J Physiol Gastrointest Liver Physiol. 2008;294:G9–G12. doi: 10.1152/ajpgi.00467.2007. [DOI] [PubMed] [Google Scholar]
- 2.Pacher P, Batkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. 2006;58:389–462. doi: 10.1124/pr.58.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lotersztajn S, Teixeira-Clerc F, Julien B, Deveaux V, Ichigotani Y, Manin S, Tran-Van-Nhieu J, et al. CB2 receptors as new therapeutic targets during liver diseases. Br J Pharmacol. 2008;153:286–289. doi: 10.1038/sj.bjp.0707511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klein TW. Cannabinoid-based drugs as anti-inflammatory therapeutics. Nat Rev Immunol. 2005;5:400–411. doi: 10.1038/nri1602. [DOI] [PubMed] [Google Scholar]
- 5.Steffens S, Veillard NR, Arnaud C, Pelli G, Burger F, Staub C, Karsak M, et al. Low dose oral cannabinoid therapy reduces progression of atherosclerosis in mice. Nature Medicine. 2005;434:782–786. doi: 10.1038/nature03389. [DOI] [PubMed] [Google Scholar]
- 6.Batkai S, Osei-Hyiaman D, Pan H, El-Assal O, Rajesh M, Mukhopadhyay P, Hong F, et al. Cannabinoid-2 receptor mediates protection against hepatic ischemia/reperfusion injury. Faseb J. 2007;21:1788–1800. doi: 10.1096/fj.06-7451com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ofek O, Karsak M, Leclerc N, Fogel M, Frenkel B, Wright K, Tam J, et al. Peripheral cannabinoid receptor, CB2, regulates bone mass. Proc Natl Acad Sci U S A. 2006;103:696–701. doi: 10.1073/pnas.0504187103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Defer N, Wan J, Souktani R, Escoubet B, Perier M, Caramelle P, Manin S, et al. The cannabinoid receptor type 2 promotes cardiac myocyte and fibroblast survival and protects against ischemia/reperfusion-induced cardiomyopathy. Faseb J. 2009;23:2120–2130. doi: 10.1096/fj.09-129478. [DOI] [PubMed] [Google Scholar]
- 9.Mallat A, Teixeira-Clerc F, Deveaux V, Lotersztajn S. Cannabinoid receptors as new targets of antifibrosing strategies during chronic liver diseases. Expert Opin Ther Targets. 2007;11:403–409. doi: 10.1517/14728222.11.3.403. [DOI] [PubMed] [Google Scholar]
- 10.Teixeira-Clerc F, Julien B, Grenard P, Tran-Van-Nhieu J, Deveaux V, Serriere-Lanneau, Li L, et al. CB1 cannabinoid receptor antagonism: a novel strategy for the treatment of liver fibrosis. Nature Medicine. 2006;12:671–676. doi: 10.1038/nm1421. [DOI] [PubMed] [Google Scholar]
- 11.Hezode C, Roudot-Thoraval F, Nguyen S, Grenard P, Julien B, Zafrani ES, Pawlotsky JM, et al. Daily cannabis smoking as a risk factor for fibrosis progression in chronic hepatitis C. Hepatology. 2005;42:63–71. doi: 10.1002/hep.20733. [DOI] [PubMed] [Google Scholar]
- 12.Jeong WI, Osei-Hyiaman D, Park O, Liu J, Batkai S, Mukhopadhyay P, Horiguchi N, et al. Paracrine Activation of Hepatic CB(1) Receptors by Stellate Cell-Derived Endocannabinoids Mediates Alcoholic Fatty Liver. Cell Metab. 2008;7:227–235. doi: 10.1016/j.cmet.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 13.Osei-Hyiaman D, Depetrillo M, Pacher P, Liu J, Radaeva S, Batkai S, Harvey-White J, et al. Endocannabinoid activation at hepatic CB(1) receptors stimulates fatty acid synthesis and contributes to diet-induced obesity. J Clin Invest. 2005;115:1298–1305. doi: 10.1172/JCI23057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hezode C, Zafrani ES, Roudot-Thoraval F, Costentin C, Hessami A, Bouvier-Alias M, Medkour F, et al. Daily cannabis use, a novel risk factor of steatosis severity in patients with chronic hepatitis C. Gastroenterology. 2008;134:432–439. doi: 10.1053/j.gastro.2007.11.039. [DOI] [PubMed] [Google Scholar]
- 15.Batkai S, Jarai Z, Wagner JA, Goparaju SK, Varga K, Liu J, Wang L, et al. Endocannabinoids acting at vascular CB1 receptors mediate the vasodilated state in advanced liver cirrhosis. Nat Med. 2001;7:827–832. doi: 10.1038/89953. [DOI] [PubMed] [Google Scholar]
- 16.Ros J, Claria J, To-Figueras J, Planaguma A, Cejudo-Martin P, Fernandez-Varo G, Martin-Ruiz R, et al. Endogenous Cannabinoids: A New System Involved in the Homeostasis of Arterial Pressure in Experimental Cirrhosis in the Rat. Gastroenterology. 2002;122:85–93. doi: 10.1053/gast.2002.30305. [DOI] [PubMed] [Google Scholar]
- 17.Julien B, Grenard P, Teixeira-Clerc F, Van Nhieu JT, Li L, Karsak M, Zimmer A, et al. Antifibrogenic role of the cannabinoid receptor CB2 in the liver. Gastroenterology. 2005;128:742–755. doi: 10.1053/j.gastro.2004.12.050. [DOI] [PubMed] [Google Scholar]
- 18.Munoz-Luque J, Ros J, Fernandez-Varo G, Tugues S, Morales-Ruiz M, Alvarez CE, Friedman SL, et al. Regression of fibrosis after chronic stimulation of cannabinoid CB2 receptor in cirrhotic rats. J Pharmacol Exp Ther. 2008;324:475–483. doi: 10.1124/jpet.107.131896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hegde VL, Hegde S, Cravatt BF, Hofseth LJ, Nagarkatti M, Nagarkatti PS. Attenuation of experimental autoimmune hepatitis by exogenous and endogenous cannabinoids: involvement of regulatory T cells. Mol Pharmacol. 2008;74:20–33. doi: 10.1124/mol.108.047035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buckley NE, McCoy KL, Mezey E, Bonner T, Zimmer A, Felder CC, Glass M. Immunomodulation by cannabinoids is absent in mice deficient for the cannabinoid CB(2) receptor. Eur J Pharmacol. 2000;396:141–149. doi: 10.1016/s0014-2999(00)00211-9. [DOI] [PubMed] [Google Scholar]
- 21.Serriere-Lanneau V, Teixeira-Clerc F, Li L, Schippers M, de Wries W, Julien B, Tran-Van-Nhieu J, et al. The sphingosine 1-phosphate receptor S1P2 triggers hepatic wound healing. Faseb J. 2007;21:2005–2013. doi: 10.1096/fj.06-6889com. [DOI] [PubMed] [Google Scholar]
- 22.Nicou A, Serriere V, Hilly M, Prigent S, Combettes L, Guillon G, Tordjmann T. Remodelling of calcium signalling during liver regeneration in the rat. J Hepatol. 2007;46:247–256. doi: 10.1016/j.jhep.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 23.Li L, Grenard P, Tran-Van-Nhieu JT, Julien B, Mallat A, Habib A, Lotersztajn S. Heme oxygenase-1 is an antifibrogenic protein in human hepatic myofibroblasts. Gastroenterology. 2003;125:460–469. doi: 10.1016/s0016-5085(03)00906-5. [DOI] [PubMed] [Google Scholar]
- 24.Taub R. Liver regeneration: from myth to mechanism. Nat Rev Mol Cell Biol. 2004;5:836–847. doi: 10.1038/nrm1489. [DOI] [PubMed] [Google Scholar]
- 25.Fausto N, Campbell JS, Riehle KJ. Liver regeneration. Hepatology. 2006;43:S45–53. doi: 10.1002/hep.20969. [DOI] [PubMed] [Google Scholar]
- 26.Morio LA, Chiu H, Sprowles KA, Zhou P, Heck DE, Gordon MK, Laskin DL. Distinct roles of tumor necrosis factor-alpha and nitric oxide in acute liver injury induced by carbon tetrachloride in mice. Toxicol Appl Pharmacol. 2001;172:44–51. doi: 10.1006/taap.2000.9133. [DOI] [PubMed] [Google Scholar]
- 27.Aram G, Potter JJ, Liu X, Torbenson MS, Mezey E. Lack of inducible nitric oxide synthase leads to increased hepatic apoptosis and decreased fibrosis in mice after chronic carbon tetrachloride administration. Hepatology. 2008;47:2051–2058. doi: 10.1002/hep.22278. [DOI] [PubMed] [Google Scholar]
- 28.Rai RM, Lee FY, Rosen A, Yang SQ, Lin HZ, Koteish A, Liew FY, et al. Impaired liver regeneration in inducible nitric oxide synthase-deficient mice. Proc Natl Acad Sci U S A. 1998;95:13829–13834. doi: 10.1073/pnas.95.23.13829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim YM, Talanian RV, Billiar TR. Nitric oxide inhibits apoptosis by preventing increases in caspase-3-like activity via two distinct mechanisms. J Biol Chem. 1997;272:31138–31148. doi: 10.1074/jbc.272.49.31138. [DOI] [PubMed] [Google Scholar]
- 30.Michalopoulos GK. Liver regeneration. J Cell Physiol. 2007;213:286–300. doi: 10.1002/jcp.21172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bansal MB, Kovalovich K, Gupta R, Li W, Agarwal A, Radbill B, Alvarez CE, et al. Interleukin-6 protects hepatocytes from CCl4-mediated necrosis and apoptosis in mice by reducing MMP-2 expression. J Hepatol. 2005;42:548–556. doi: 10.1016/j.jhep.2004.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lotersztajn S, Julien B, Teixeira-Clerc F, Grenard P, Mallat A. Hepatic fibrosis: molecular mechanisms and drug targets. Ann Rev Pharmacol Toxicol. 2005;45:605–628. doi: 10.1146/annurev.pharmtox.45.120403.095906. [DOI] [PubMed] [Google Scholar]
- 33.Siegmund SV, Qian T, de Minicis S, Harvey-White J, Kunos G, Vinod KY, Hungund B, et al. The endocannabinoid 2-arachidonoyl glycerol induces death of hepatic stellate cells via mitochondrial reactive oxygen species. Faseb J. 2007;21:2798–2806. doi: 10.1096/fj.06-7717com. [DOI] [PubMed] [Google Scholar]
- 34.Avraham Y, Zolotarev O, Grigoriadis NC, Poutahidis T, Magen I, Vorobiav L, Zimmer A, et al. Cannabinoids and capsaicin improve liver function following thioacetamide-induced acute injury in mice. Am J Gastroenterol. 2008;103:3047–3056. doi: 10.1111/j.1572-0241.2008.02155.x. [DOI] [PubMed] [Google Scholar]
- 35.Kovalovich K, DeAngelis RA, Li W, Furth EE, Ciliberto G, Taub R. Increased toxin-induced liver injury and fibrosis in interleukin-6-deficient mice. Hepatology. 2000;31:149–159. doi: 10.1002/hep.510310123. [DOI] [PubMed] [Google Scholar]
- 36.Gao B. Therapeutic potential of interleukin-6 in preventing obesity- and alcohol-associated fatty liver transplant failure. Alcohol. 2004;34:59–65. doi: 10.1016/j.alcohol.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 37.Luedde T, Trautwein C. Intracellular survival pathways in the liver. Liver Int. 2006;26:1163–1174. doi: 10.1111/j.1478-3231.2006.01366.x. [DOI] [PubMed] [Google Scholar]
- 38.Jin X, Zimmers TA, Perez EA, Pierce RH, Zhang Z, Koniaris LG. Paradoxical effects of short- and long-term interleukin-6 exposure on liver injury and repair. Hepatology. 2006;43:474–484. doi: 10.1002/hep.21087. [DOI] [PubMed] [Google Scholar]
- 39.Passino MA, Adams RA, Sikorski SL, Akassoglou K. Regulation of hepatic stellate cell differentiation by the neurotrophin receptor p75NTR. Science. 2007;315:1853–1856. doi: 10.1126/science.1137603. [DOI] [PubMed] [Google Scholar]
- 40.Kalinichenko VV, Bhattacharyya D, Zhou Y, Gusarova GA, Kim W, Shin B, Costa RH. Foxf1 +/− mice exhibit defective stellate cell activation and abnormal liver regeneration following CCl4 injury. Hepatology. 2003;37:107–117. doi: 10.1053/jhep.2003.50005. [DOI] [PubMed] [Google Scholar]
- 41.Sanz S, Pucilowska JB, Liu S, Rodriguez-Ortigosa CM, Lund PK, Brenner DA, Fuller CR, et al. Expression of insulin-like growth factor I by activated hepatic stellate cells reduces fibrogenesis and enhances regeneration after liver injury. Gut. 2005;54:134–141. doi: 10.1136/gut.2003.024505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Trim N, Morgan S, Evans M, Issa R, Fine D, Afford S, Wilkins B, et al. Hepatic stellate cells express the low affinity nerve growth factor receptor p75 and undergo apoptosis in response to nerve growth factor stimulation. Am J Pathol. 2000;156:1235–1243. doi: 10.1016/S0002-9440(10)64994-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.