

Abstract
Purpose
To compare physiochemical properties of mono-, di- and triglycerides of medium chain fatty acids for development of oral pharmaceutical dosage forms of poorly water-soluble drugs using phase diagrams, drug solubility, and drug dispersion experiments.
Methods
Phase diagrams were prepared using a monoglyceride (glycerol monocaprylocaprate: Capmul MCM® EP), a diglyceride (glycerol dicaprylate) and two triglycerides (glycerol tricaprylate: Captex 8000®; caprylic/capric triglycerides: Captex 355 EP/NF®) in combination with a common surfactant (PEG-35 castor oil: Cremophor EL®) and water. Psuedoternary phase diagrams using mixtures of monoglyceride with either diglyceride or triglyceride were constructed to determine any potential advantage of using lipid mixtures.
Result
The monoglyceride gave microemulsion (clear or translucent liquid) and emulsion phases, whereas di- and triglycerides exhibited an additional gel phase. Among individual mono-, di- and triglycerides, the oil-in-water microemulsion region was the largest for the diglyceride. Gel phase region within diglyceride and triglyceride phase diagrams could be practically eliminated and microemulsion regions expanded by mixing monoglyceride with di- or tri-glycerides (1:1). Addition of a model drug, danazol, had no effect on particle sizes of microemulsions formed. Dispersion of drug in aqueous media from mixtures of mono- and diglyceride or mono- and triglyceride was superior to individual lipids.
Conclusion
Systematic study on comparison of mono-, di- and triglyceride of medium chain fatty acids will help formulators select components for optimal lipid-based formulation.
KEY WORDS: danazol, diglyceride, drug dispersion, drug solubility, lipid-based drug delivery, medium chain lipid, monoglyceride, phase diagram, triglyceride
INTRODUCTION
Due to the application of combinatorial chemistry and high-throughput screening in drug discovery, the majority of new drug candidates have become extremely insoluble in water making the development of bioavailable clinical dosage forms very challenging (1). Two-thirds of compounds emerging from the drug discovery pipeline in recent years have an aqueous solubility <100 μg/mL (0.1 mg/mL) (2). This would classify them as insoluble or practically insoluble according to the definition of the United States Pharmacopeia. Although many drug candidates are categorized as poorly water-soluble, there is no limit how low aqueous solubility could be, since approximately one-third of newly discovered compounds have an aqueous solubility of <10 μg/mL. Even aqueous solubility of <1 μg/mL is increasingly more common. A lipid-based drug delivery system, in which the drug is solubilized by lipids or lipid-like excipients, has been recognized as an attractive approach for increasing the bioavailability of these compounds (3–16). Despite the abundance of poorly water-soluble drugs and the potential benefit of lipid-based drug delivery systems, the application of this technology has been rather limited. According to Strickley (14), a total of 31 drugs, employing 41 lipid-based products for oral administration, were marketed in the US in 2005. However, in most of these cases, lipids were not used to increase bioavailability of poorly water-soluble drugs; instead, they were used to retard drug release. In many of the products, lipids were not even the primary excipient. Based upon a survey of drug products available in the US in 2004, Gursoy and Benita (15) reported that there were only four major lipid-based products for poorly water-soluble drugs available. Of these, two were for cyclosporine A and two for ritonavir and sequinavir. The situation prompted Hauss (16) to ask whether it is a case of implementation failing to keep up with innovation.
According to Porter et al. (8), a rational basis for the selection of lipid-based excipients is ‘elusive’ and the prediction of in vivo performance of these excipients is poor. Thus, the widespread adoption of lipid-based strategies for enhancing drug exposure is limited. Therefore, it is not likely that implementation is failing to keep up with innovation, but rather the lipid-based drug delivery technology requires further development.
To identify a few critical factors influencing the successful development of lipid-based formulations, one may look to the examples of Neoral® and Sandimmune®, both marketed by Novartis, where Neoral is the reformulation of Sandimmune. In these products, a poorly water-soluble drug (cyclosporine A) was solubilized in different mixtures of lipid, surfactant, cosurfactant and organic solvent. In a comparative study of biopharmaceutical properties, it was observed that Neoral dispersed in aqueous media as extremely fine particles (<150 nm) and gave the appearance of a clear or transluscent solution. In contrast, Sandimmune phase separated after dilution with water as a milky emulsion containing large oil globules (17,18). As a result, Neoral provided better bioavailability than Sandimmune. It was demonstrated by Kovarik et al. (19) that the bioavailability of cyclosporine A from a 180-mg dose of Neoral was essentially similar to that of a 300-mg dose of Sandimmune when both were administered as soft gelatin capsules. Of additional benefit, the individual variation in the pharmacokinetic parameters, such as Cmax (maximum concentration), tmax (time to reach maximum concentration) and AUC (area under the curve), was much lower for Neoral. When administered with a fatty meal, Neoral did not alter the pharmacokinetics of cyclosporine A in humans, demonstrating practically no food effect (20,21). These advantages of the reformulated product were attributed to its spontaneous self-microemulsification (particle or globule size <150 nm) within the GI tract. Food generally increases the bioavailability of poorly soluble drugs through increased bile secretion and prolonged gastric retention time. It is possible that the formation of a microemulsion maximized the absorption of the drug, leaving no room for further improvement in the presence of food. Such a lack of food effect in humans was also observed for other self-emulsifying lipid-based formulations (22). Consistency of drug bioavailability, whether in the fed or fasted state, is an important consideration for clinical efficacy and commercial success of drug products. That is to say, elimination of the food effect may be an attainable goal using lipid-based formulations. For this purpose, the active pharmaceutical ingredient must first dissolve in the carrier, and then after oral administration, finely disperse within the fluids of the gastrointestinal tract before it is adequately absorbed.
There are numerous lipids consisting of triglycerides of fatty acids commercially available to formulators as excipients for lipid-based drug delivery systems. Many synthetic lipids are also available in which the glycerol backbone has been replaced by propylene glycol and/or polyethylene glycols. Additionally, the degree of esterification of the fatty acid moiety may vary, forming mono-, di- and tri-glyceride forms as well as different ester forms of propylene glycol and polyethylene glycols. The fatty acids are not necessarily long chain (C14–C20); they can be medium chain (C6–C12), short chain, unsaturated or branched. Due to these differences in chemical nature, there are numerous lipids or lipid-like excipients available commercially, all of which are colloquially called ‘lipids’ in the pharmaceutical field. Indeed, Cannon and Long (23) included the following categories of excipients as lipids for the purpose of developing oral lipid-based formulations: long-chain triglycerides, medium-chain triglycerides, propylene glycol esters, fatty acids, monoglycerides, diglycerides, and lipid mixtures. Due to such chemical differences, the physical properties of ‘lipids’ also differ. They are available as liquids, pastes, solids or combinations of these. If solid, they could be amorphous and/or crystalline. Strictly speaking, lipids are triglycerides of long chain fatty acids, and some purists are reluctant to call medium-chain glycerides as well as mono- and di-glycerides to be lipids. Irrespective of whether they are strictly lipids or not, all lipids and lipid-like excipients used in the development of lipid-based formulations require careful characterization. In agreement with Cannon and Long (23), we will also consider mono-, di- and tri-glycerides of medium-chain fatty acids as ‘lipids’ in the present investigation, the primary objective of various studies performed being the comparative evaluation of these three excipients for their suitability in the development of pharmaceutical dosage forms.
Pouton (24,25) proposed a classification system for various lipid-based drug delivery systems according to their general composition and ability to disperse in aqueous media (as fine particles usually <250 nm). To date, no systematic approach for the selection of lipids for particular dosage forms has been reported in the literature. A cursory look at the composition of marketed lipid-based drug products reveals that most of the formulations are rather complex. In addition to lipids, surfactants, co-surfactants, etc., many of these products contain alcohol and/or organic solvents, apparently to enhance drug solubility beyond what could be achieved by using lipids alone. Many of these products appear to be the result of trial-and-error experiments. There are no reports on how the individual components of a formulation are selected.
To develop a rational basis for the selection of lipid-based excipients, we have initiated a series of studies in which the physicochemical and biopharmaceutical properties of various lipids are compared. In this paper, results of the combinations of mono-, di- and triglycerides of medium chain fatty acids with a common surfactant and water are presented. The physical effect of combining multiple excipients was investigated through the use of phase diagrams. The particle sizes from various regions of these phase diagrams were measured to determine emulsification efficiency of the different lipids employed. The viscosity of any gel phase formed was also determined. Phase diagrams have been used for the development of lipid-based formulations by several investigators in the past. However, many of those phase diagrams are partial in nature. Several investigators used phase diagrams to identify regions of specific lipid/surfactant mixtures giving oil-in-water microemulsions, or nanoemulsions, upon dilution with water (26–30). Shafiq et al. (31) used oil/surfactant/water phase diagrams to determine oil-in-water microemulsion regions, while Kang et al. (32) identified regions of both microemulsions and coarse emulsions. On the other hand, Constantinides and Scalart (33) used phase diagrams to identify water-in-oil microemulsions. While these studies established the importance of phase diagrams as a tool for developing lipid-based drug delivery systems, complete identification and characterization of all the different phases formed are lacking. Ping et al. (34) identified various phases in lipid/surfactant/water blends. However, only one lipid was used in the study as the primary focus of the researchers was to determine the effect of surfactant mixtures and not the lipid. In addition to the formation of oil-in-water and water-in-oil microemulsions, lipid/surfactant mixtures form gels or liquid crystalline phases upon dilution with water (35,36). The formation of a gel is more predominant when the surfactant concentration is relatively high (>50%). However, there are only limited reports on characterization of the gel phase and especially on its significance in the development of oral dosage forms. In the present study, the multiple phases formed in various regions of the phase diagrams, including gel phases, were evaluated. Further, the solubility of a model drug, danazol, in different drug/surfactant mixtures was determined and the rates of drug dispersion from systems containing individual lipids as well as lipid/lipid mixtures were compared.
MATERIALS AND METHODS
Materials
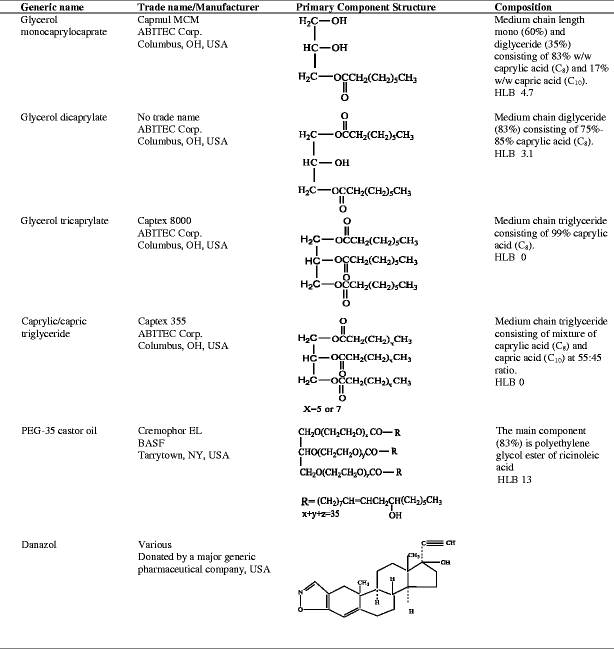
Lipids (mono-, di- and triglycerides), surfactant (PEG-35 castor oil) and drug used (danazol), along with their trade names, suppliers, chemical structures and compositions, are listed in Table I. It should be noted that the lipids studied do not exist as pure species, but are mixtures of glycerides, with differing degrees of esterification and different fatty acid compositions. However, only the chemical structure of the predominant component of each excipient is given in the table. The dicaprylate was specifically prepared by ABITEC Corporation for the present study and is not commercially available. Distilled water was used for all experiments as necessary. All other reagents used were of analytical grade or better.
Table I.
Chemical Name, Trade Name, Structure and Composition of Lipids, Surfactant and Drug
Construction of Phase Diagrams
Phase diagrams were constructed following the general procedure outlined by Li et al. (34). Mixtures (4 g each) of lipid/surfactant, in the following ratios, were prepared in separate 100-mL volumetric flasks: 1:9, 2:8, 3:7, 4:6, 5:5, 6:4, 7:3, 8:2, 9:1. Water was then added to each flask at 5% w/w, intervals. In certain phase diagrams, two lipids (fixed ratios) were used instead of a single lipid to create the phase diagram (a pseudoternary phase diagram). In such cases, the weight of lipid represented the total weight of both lipids. The phase boundaries were established by visual observation after equilibration (shaken 15 min in a 25°C water bath in the case of low viscosity, and shaken 30 min in a 25°C water bath in the case of a gel). Depending upon the components used, four different phases were observed in the phase diagrams: (1) a clear liquid region that included clear or translucent solutions; (2) cloudy liquids apparently consisting of coarse emulsions; (3) a viscous gel; or (4) a phase-separated mixture where the lipid separated from the aqueous phase to form a separate layer. Selected mixtures were analyzed for particle size and viscosity. To determine the effect of drug upon the phase diagrams, the clear region of the specific phase diagram prepared with glyceryl dicaprylate (as lipid) was reconstructed in the presence of danazol (2% w/w).
Evaluation of Shake Time
After each addition of water during the determination of phase diagrams, the mixture was shaken on a wrist-action shaker (Burrell Wrist Action Shaker, Burrell Scientific, Pittsburgh, PA, USA) at the highest speed setting for 15 min (at 25°C, water bath). If the mixture was viscous and appeared to form a gel after the addition of water, shaking was continued for 30 min (25°C, water bath). These experimental conditions were established based upon preliminary experiments. When no change in visual appearance for both clear and cloudy liquid regions was observed, the shake time was set at 15 min. In the case of gel formation, non-uniformity in the mixtures was observed following mixing for 15 min. Therefore, shaking was continued for 30 min, when a uniform gel was observed. In some preliminary experiments, flasks were shaken overnight, with no observable difference in appearance or phase boundaries compared to the developed procedure.
Particle Size Determination
A DelsaNano C particle size analyzer (Beckman Coulter Inc., Brea, CA) was used to measure the particle size of selected lipid/surfactant mixtures at ca. 70%, 80%, 90% and 99% (w/w) water content. Approximately 2–3 mL of sample was added to a disposable plastic cuvette (Beckman Coulter disposable cell, Beckman Coulter Inc., Brea, CA) for the particle size determination at every data point using the dynamic light scattering technique at 25°C. The sample was placed back into the equilibrating flask after each analysis. As mentioned earlier, the flask was shaken (15 min) after each aliquot of water was added. Before the determination of particle size, the flask was equilibrated for 10 min. The shaking was resumed only after returning the sample to the flask, followed by addition of more water. To ascertain the effect of equilibration time on particle size, the particle size measurements of selected samples were performed at 15, 30, 60 and 120 min. To determine the effect of the presence of dilute acid or pharmaceutical API on particle size, the particle sizes of several lipid/surfactant mixtures, with and without drug, were determined after a 250-fold dilution (1 g of sample solution added to 250 mL water or 250 mL of 0.01N HCl), where the temperature of diluents was maintained at 37°C to mimic the gastrointestinal environment.
Viscosity Measurement
The viscosities of selected gel regions were determined using a Brookfield RVDV III Ultra CP (Brookfield Engineering Laboratories, Inc. Middleboro, MA, USA) viscometer equipped with a CPE-52 cone (150 rpm). Fresh samples with appropriate lipid, surfactant and water contents were prepared specifically for viscosity measurements. Typically, lipid/surfactant mixtures at three different ratios (5:5, 4:6 and 2:8) were prepared, 20% (w/w) water added, and the sample equilibrated at 25°C using a wrist action shaker. After equilibration, a sample (~ 0.5 mL) was taken and the viscosity determined (25°C).
Solubility Studies
Danazol, a neutral compound with an aqueous solubility of 0.59 μg/mL and log P value of 4.53 (37), was selected as the model drug to compare different lipids and lipid/surfactant mixtures for drug solubility. An excess amount of drug was added to individual lipids, the surfactant or the lipid/surfactant mixtures. The mixtures were vortexed to disperse suspended drug in the solvents, and the suspensions were then shaken using a wrist-action shaker at maximum shaking speed (Burrell Wrist Action Shaker, Burrell Scientific, Pittsburgh, PA, USA) for 24 h at 25°C. Aliquots were filtered (0.45 μm polypropylene filter), diluted and assayed for drug content by HPLC (reverse phase C18 150 x 4.6 mm x 3.5 μm against acetonitrile/H2O (65:35) @ 0.5 mL/min, λ = 280). Each solubility determination was conducted in duplicate. It was determined by continuing various solubility experiments for a period up to 7 day that an equilibration was achieved in 24 h.
Dispersion Test
As the preconcentrates containing drugs dissolved in lipid/surfactant mixtures are expected to disperse in the gastrointestinal tract upon oral administration, the dispersion test was performed for two different lipid/surfactant ratios (1:1 and 3:7) with drug (ca. 80% of equilibrium solubility). Similar lipid/surfactant mixtures without the drug served as controls. The dispersion test was performed using USP apparatus II (paddle method, 37°C, 250 mL of 0.01 N HCL, 50 rpm) with model drug. Approximately 1 g of preconcentrate was filled into a hard gelatin capsule (00 size) and the same procedure repeated for the control (without drug). Aliquots from dispersion vessels, 3 mL each, were withdrawn at 10, 15, 30, 60 and 120 min time points for particle size analysis. The drug present in the aliquots was analyzed after appropriate dilution with the mobile phase using the HPLC method described earlier. The samples collected from similar dispersion tests of the control were also analyzed by HPLC to determine if the gelatin capsule interfered with the analysis. All experiments were carried out in triplicate. The aliquots were filtered (0.45 μm) before analyzing for particle size and drug concentration. However, when it was suspected that the particle size might be larger than 0.45 μm, samples were analyzed with and without filtration to investigate the effect of filtration on the drug concentration. When any precipitation of the drug was observed, the aliquots were centrifuged and the supernatant liquids analyzed for drug concentration.
RESULTS
Phase Diagrams with Individual Lipids
As the primary objective of this study was to compare different medium chain glycerides relevant to dosage form development, all phase diagrams were constructed using a common surfactant, PEG-35 castor oil (Cremophor EL). Since the lipids and surfactant were nonionic in nature and preliminary studies showed that there was no effect of a change in pH on the phase diagrams, distilled water was used as the aqueous medium. A preliminary experiment showed that there was no change in phase diagram if 0.01 M HCl was used as the dilution medium instead of water. The lipid/surfactant/water phase diagrams of four medium chain lipids are shown in Fig. 1. A description of these phase diagrams follows.
Fig. 1.
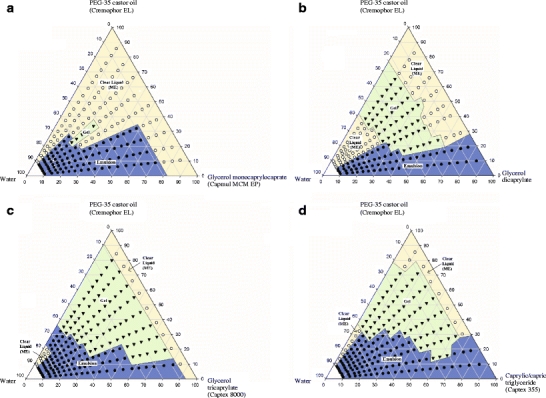
Phase diagrams of (a) glycerol monocaprylocaprate, (b) glycerol dicaprylate, (c) glycerol tricaprylate and (d) caprylic/capric triglycerides in combination with PEG-35 castor oil and water. ME in the figure indicates microemulsion.
Monoglyceride/Surfactant/Water Phase Diagram
Presented in Fig. 1a is the phase diagram of a monoglyceride/surfactant/water system, where glycerol monocaprylocaprate (Capmul MCM EP; ABITEC) and PEG-35 castor oil (Cremophor EL; BASF) were used as lipid and surfactant, respectively. Since Capmul MCM used in this phase diagram contains a mixure of monoglyceride (60%) and diglyceride (35%) as indicated in Table I, it is indeed a psuedoternary phase diagram. Phase boundaries were first identified by visual observation upon dilution with water. The particle size was then determined in the region of higher water content (>70% w/w). If a gel phase was observed, the viscosity was measured. It is evident in Fig. 1a that a clear liquid solution was observed at all compositions of lipid/surfactant containing up to 15% water. As discussed later under DISCUSSION, the clear region represent water-in-oil (w/o) microemulsion at low water content and oil-in-water (o/w) microemulsion at high water content. Upon further dilution with water, this clear region turned to a milky-white emulsion when the lipid comprised 60% or more of the lipid/surfactant blend. In contrast, the solutions remained clear throughout aqueous dilution up to 90% when the initial lipid content was 20% or less. At the intermediate lipid content of 30 and 50%, the solutions remained clear up to 65% dilution with water; after which, a milky white emulsion resulted. Furthermore, when the initial lipid content was in the middle of this range (40%), the clear solution transformed into a gel at water concentrations between 45 and 60%. If the mixtures turned milky-white, the term “emulsion” was used, while the term “clear” was used for both transparent and translucent solutions.
The particle size analysis was performed only on mixtures deemed to be relevant to the development of oral dosage forms where the addition of water to the lipid/surfactant preconcentrate produced a microemulsion or an emulsion (see Table II under the subtitle Glycerol monocaprylocaprate). The mixtures of lipid/surfactant with ratios from 9:1 to 3:7 gave emulsions upon dilution with water (≥ 70%). Although the particle sizes of these mixtures decreased with increasing water content (70 to 99%), the particle sizes were nonetheless large (1 to 7 μm at 70% water and 0.3 to 2 μm at 99% water). As will be discussed later in this paper, we called the solutions “microemulsions” when the average particle size was found to be less than 200 nm (0.2 μm). It was apparent that glycerol monocaprylocaprate formed microemulsion only at high surfactant concentrations (>80%, giving particle sizes of 13 to 30 nm). For milky emulsions formed upon dilution, the particle size increased with time (data not shown). No increase in particle size vs. time was observed in solutions with an initial lipid content of 20% or less upon dilution with water. These results indicated that emulsions were thermodynamically unstable, whereas, the microemulsions at relatively higher surfactant concentrations were thermodynamically stable.
Table II.
Particle Size (nm) with Dilution with Water at Different Lipid: Surfactant Ratios. (n = 2)
| % Water | Lipid : Surfactant ratio | |||||
|---|---|---|---|---|---|---|
| 9:1 | 7:3 | 1:1 | 3:7 | 2:8 | 1:9 | |
| Glycerol monocaprylocaprate (Capmul MCM EP) | ||||||
| 70 | 6559 | 6226 | 1287 | 3093 | 30 | 15 |
| 80 | 6599 | 3410 | 1421 | 1715 | 24 | 12 |
| 90 | 2427 | 2582 | 705 | 994 | 22 | 14 |
| 99 | 1960 | 819 | 383 | 284 | 15 | 13 |
| Glycerol dicaprylate | ||||||
| 70 | 2788 | 683 | 106 | 54 | 90 | 35 |
| 80 | 2007 | 494 | 58 | 35 | 63 | 14 |
| 90 | 1844 | 419 | 45 | 29 | 21 | 14 |
| 99 | 1903 | 262 | 41 | 29 | 21 | 16 |
| Glycerol tricaprylate (Captex 8000) | ||||||
| 70 | 474 | 490 | 414 | 480 | 647 | 293 |
| 80 | 260 | 428 | 213 | 216 | 230 | 43 |
| 90 | 366 | 347 | 134 | 118 | 95 | 40 |
| 99 | 368 | 262 | 94 | 72 | 46 | 23 |
| Caprylic/capric triglyceride (Captex 355 EP/NF) | ||||||
| 70 | 1730 | 770 | 1296 | 632 | 826 | 103 |
| 80 | 1756 | 469 | 634 | 379 | 636 | 60 |
| 90 | 1890 | 358 | 388 | 274 | 393 | 39 |
| 99 | 3582 | 269 | 178 | 163 | 149 | 20 |
Diglyceride/Surfactant/Water Phase Diagram
Presented in Fig. 1b is the phase diagram of a medium chain diglyceride (glyceryl dicaprylate), PEG-35 castor oil (Cremophor EL) and water. Clear regions representing water-in-oil (w/o) microemulsion were observed with all lipid/surfactant blends containing up to 20% water except when the initial lipid content was greater than 80%. When lipid/surfactant mixtures with an initial diglyceride content of 70% or less were further diluted with water (25 to 60%), they transitioned to a gel phase. Upon still further dilution, this gel phase transitioned into a microemulsion or emulsion (o/w). The particle size data for mixtures diluted with water ranging from 70 to 99% are given in Table II under the subheading Glycerol dicaprylate. Microemulsions (<100 nm) formed when the initial lipid content was 50% or less, and emulsions formed at higher lipid/surfactant ratios (≥70% lipid). At 90% initial lipid concentration, the particle size was in the micron range (2 to 3 μm) upon dilution with water (≥ 99%). There was no change in particle size with respect to time when the clear solutions were left standing. In the case of milky emulsions, the droplet size increased with time, again indicating that the solutions were thermodynamically stable and the coarse emulsions were not.
Triglyceride/Surfactant/Water Phase Diagrams
Figure 1c and d represent the phase diagrams of two different triglycerides in combination with PEG-35 castor oil and water. The triglycerides used in these figures were glycerol tricaprylate (Captex 8000) and caprylic/capric triglycerides (Captex 355), the major difference between the two being Captex 8000 was prepared from caprylic acid (99%) while Captex 355 was prepared from caprylic/capric acid (55:45). Phase diagrams of the two triglycerides were qualitatively similar, although the clear region representing o/w microemulsion (starting at 60% water) appeared to be slightly larger for Captex 8000 than Captex 355. It should, however be noted, that Captex 355 is slightly more hydrophobic than Captex 8000 because of the higher capric acid (C10) content but this difference may not be significant. Furthermore, the gel phase regions of the triglycerides in Fig. 1c and d were larger than that of the diglyceride in Fig. 1b.
The particle sizes within the emulsion regions of Fig. 1c and d were less than 0.5 μm (≥ 80% water). The particle sizes when using Captex 355 at the lipid/surfactant ratio of 9:1 were the exception, ranging from 1.7 to 3.6 μm. It is also apparent from Table II that the particle size decreased with increasing water content (≥ 70%). Microemulsions (< 200 nm) formed upon dilution with water (99% w/w) when the initial lipid content was less than 50%. The biologically significant conclusion is that medium chain triglycerides may form microemulsions at very dilute concentrations (1 to 100 dilution).
To further illustrate the effect of dilution on particle size, a graphical representation of particle sizes of mono-, di- and triglycerides with surfactant at different ratios (7:3, 1:1 and 3:7) is given in Fig. 2. This is the dilution of 1 g of a lipid/surfactant mixture with 99 g of water and could be a typical situation in the gastrointestinal tract after ingestion of a lipid-based formulation. The graph shows that the particle size increased in the following order: diglyceride < triglyceride < monoglyceride. In all cases, the diglyceride provided a microemulsion with a particle size of ~200 nm or less, whereas, the particle sizes for mono- and triglycerides were somewhat larger.
Fig. 2.
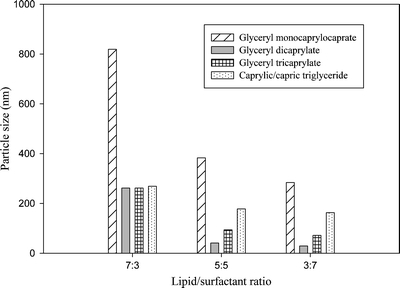
Comparison of particle size of all four lipids at different lipid/ surfactant ratios at 99% water.
As can be seen in Fig. 1, the triglycerides gave a larger gel phase region compared to the mono- or diglycerides (triglyceride > diglyceride>> monoglyceride). The clear region decreased in the order of monoglyceride > diglyceride > triglyceride. However, if we just compare the clear region corresponding to the formation of a microemulsion, the diglyceride provided the largest area.
Effect of the Presence of Drug on Phase Diagrams
Since lipids are formulated with drugs, it was of interest to investigate the effect of the presence of a drug on the phase diagram. This was determined using the clear oil-in-water microemulsion phase of the glycerol dicaprylate/PEG-35 castor oil system (Fig. 3). For this diglyceride/surfactant mixture, no significant difference was observed for the microemulsion produced upon dilution with water in the presence of danazol (2% w/w). The effect of the presence of drug could not be determined on emulsion and gel phases, as it was not possible to ascertain whether the drug remained in solution or not.
Fig. 3.
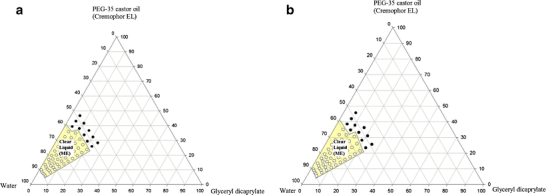
Ternary phase diagram of glycerol dicaprylate with PEG-35 castor oil and water indicating clear region (microemulsion, ME) (a) without drug and (b) with drug (2% w/w danazol).
Effect of Monoglyceride/Diglyceride Combination
As disclosed in Table I, the mono- and diglycerides used in the present investigation were primarily mixtures with varying ratios of mono- and diesters. Thus, the medium chain monoglyceride (Capmul MCM EP) contained 60% monoglyceride and 35% diglyceride, whereas, the diglyceride used contained ~17% monoglyceride and 83% diglyceride. As shown in Fig. 1a and b, as well as in Table II, the difference in composition of the monoglyceride and the diglyceride had a major influence on the phase diagrams generated. While the monoglyceride (60% actual monoglyceride content) produced only a minimal gel phase region, it had a larger emulsion region with larger particle sizes. In contrast, the dicaprylate (17% actual monoglyceride content) gave a larger gel phase region. The microemulsion region in its phase diagram was larger and the particle size upon dilution with water was smaller (Table II). Therefore, it was of interest to investigate the effect of mixing the monoglyceride and the diglyceride to generate a psuedoternary phase diagram. The phase diagram of Capmul MCM EP and glycerol dicaprylate (1:1) in combination with surfactant and water is given (Fig. 4) along with the particle size analysis (Table III). This phase diagram was constructed in the same way as those given in Fig. 1, except that two lipids (thus the term ‘pseudo’) were used instead of one. The phase diagram from the combination of mono- and diglycerides (Fig. 4) was significantly different than those of the monoglyceride (Fig. 1a) or diglyceride (Fig. 1b) alone. The area of the emulsion phase of Fig. 1a was reduced and the area of the gel phase of Fig. 1b was eliminated. Furthermore, the particle size, after dilution with water (70 to 99%), was much smaller than with monoglyceride alone and comparable to that of the diglyceride alone. The primary advantage observed in the phase diagrams of Fig. 4 over Fig. 1b (diglyceride alone) was that the gel phase was not formed. This could be significant for the rate of absorption and bioavailability in oral drug delivery systems as a gel may trap the drug, thereby delay or impede its release.
Fig. 4.
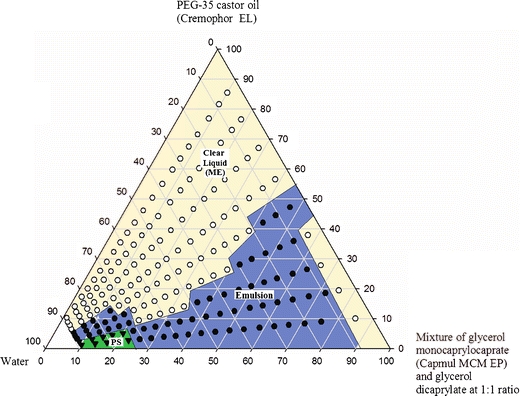
Phase diagram of mixtures of glycerol monocaprylocaprate and glyceryl dicaprylate (1:1) with PEG-35 castor oil and water. PS and ME in the figure indicates phase separation and microemulsion respectively.
Table III.
Droplet Size (nm) with Dilution with Water for Mixture of Lipids: Surfactant at Different Ratios. (n = 2)
| % Water | Lipid : Surfactant ratio | |||||
|---|---|---|---|---|---|---|
| 9:1 | 7:3 | 1:1 | 3:7 | 2:8 | 1:9 | |
| Glycerol monocaprylocaprate : Glycerol dicaprylate (1:1) | ||||||
| 70 | ND | 1029 | 1079 | 150 | 82 | 80 |
| 80 | ND | 1063 | 442 | 22 | 18 | 17 |
| 90 | ND | 870 | 252 | 18 | 16 | 17 |
| 99 | ND | 50 | 78 | 20 | 16 | 16 |
ND Not determined due to phase separation of lipid
Based upon the compositions of lipids given in Table I, the concentrations of monoglyceride and diglyceride in a 1:1 mixture of Capmul MCM and glycerol dicaprylate were ca. 40 and 60%, respectively. Thus, in the three lipid systems used in the present study containing both mono- and diglycerides (1, Capmul MCM, 2, Capmul MCM/glycerol dicaprylate, 1:1; and 3, glycerol dicaprylate), the concentration of the monoglyceride was ca. 60, 40 and 17% and the concentration of diglyceride was ca. 35, 60 and 83%, respectively. From the major differences observed in the phase diagrams (Figs. 1a, 4 and 1b, respectively) and the particle size data for the three systems, it was apparent that lipids used in a drug delivery system may be optimized, according to formulation requirements, by adjusting their composition through admixture.
Effects of Monoglyceride/Triglyceride Combinations
Figure 5a, b and c represent psuedoternary phase diagrams where the lipid phase was 1:3, 1:1 and 3:1 mixtures of the monoglyceride (Capmul MCM) to triglyceride (Captex 355), respectively. The particle size analyses corresponding to these figures are given in Table IV. Comparing Fig. 5a to b, the area of the clear liquid phase progressively increased, while the gel phase progressively decreased. Indeed, the area of microemulsion for the combination Capmul MCM/Captex 355 (1:1) was very large. No further enlargement in the clear region was observed when the ratio of monoglyceride/triglyceride was increased to 3:1. Rather, the emulsion region remained large, and as can be seen (Fig. 5c), a phase separation of lipid was observed upon dilution with water (>75% w/w and initial lipid concentration ≥ 80%). For the microemulsion phase, the particle size was smaller in the case of the combination compared to the individual monoglyceride and triglyceride. Thus, the mixing of monoglyceride with triglyceride had a major impact in reducing gel formation and promoting microemulsion formation. These effects are similar to those observed for mixtures of monoglyceride and diglyceride. It is observed in Table IV that the particle size could not be determined at 9:1 and 7:3 lipid/surfactant ratios. Similarly, the particle size in Table III could not be determined at the 9:1 lipid/surfactant ratio. This effect at low surfactant content was due to phase separation of lipid as shown earlier in Figs. 5c and 4. It appears that there was not enough surfactant available to emulsify lipids at high dilution with aqueous media since much of the surfactant was apparently below the critical micelle concentration.
Fig. 5.
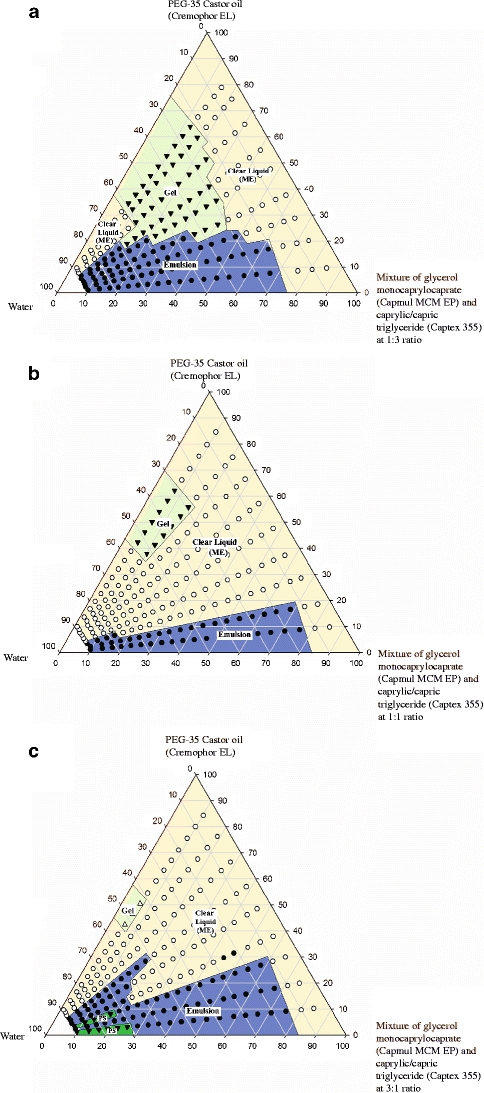
Phase diagram of mixtures of (a) glycerol monocaprylocaprate and caprylic/capric triglycerides (1:3), (b) glycerol monocaprylocaprate and caprylic/capric triglyceride (1:1), and (c) glycerol monocaprylocaprate and caprylic/capric triglycerides (3:1), in combination with water and PEG-35 castor oil. PS and ME in the figure indicate phase separation and microemulsion, respectively.
Table IV.
Particle Size (nm) with Dilution with Water for Mixture of Lipids: Surfactant at Different Ratios. (n = 2)
| % Water | Lipid : Surfactant ratio | |||||
|---|---|---|---|---|---|---|
| 9:1 | 7:3 | 1:1 | 3:7 | 2:8 | 1:9 | |
| Glycerol monocaprylocaprate : Caprylic/capric triglyceride (1:3) | ||||||
| 70 | 2532 | 9993 | 699 | 86 | 93 | 91 |
| 80 | 987 | 2688 | 344 | 31 | 17 | 20 |
| 90 | 1083 | 1063 | 261 | 20 | 18 | 16 |
| 99 | 163 | 769 | 119 | 22 | 19 | 17 |
| Glycerol monocaprylocaprate: Caprylic/capric triglyceride (1:1) | ||||||
| 70 | 3185 | 113 | 47 | 27 | 18 | 24 |
| 80 | 5303 | 349 | 21 | 18 | 14 | 33 |
| 90 | 867 | 533 | 21 | 16 | 18 | 16 |
| 99 | 434 | 40 | 24 | 19 | 18 | 17 |
| Glycerol monocaprylocaprate: Caprylic/capric triglyceride (3:1) | ||||||
| 70 | ND | ND | 2110 | 50 | 32 | 15 |
| 80 | ND | ND | 2314 | 20 | 14 | 13 |
| 90 | ND | ND | 880 | 16 | 14 | 12 |
| 99 | ND | ND | 164 | 18 | 18 | 15 |
ND Not determined due to phase separation of lipid
Effect of Low pH and Presence of Drug on Particle Size
Tables II, III and IV give the particle size data for drug-free lipid/surfactant mixtures up to the maximum 1:99 dilution with water (99% w/w). It is, however, recognized that after oral administration, a lipid-based delivery system would be diluted with an acidic gastric fluid rather than pure water. The gastrointestinal (GI) fluid volume according to the biopharmaceutical classification system is considered to be 250 mL, and the temperature in the GI environment is 37°C.Therefore, particle sizes of selected lipid/surfactant systems, with and without drug (2% w/w danazol), were determined after dilution with water (1:250) or dilute acid (0.01 M HCl) at 37°C. The results are presented in Table V. No significant effects from dilute acid (0.01 M HCl) or the presence of drug (danazol, 2%) were observed.
Table V.
Particle size (nm) of Emulsion/microemulsion System in 1:250 Dilutionith Water and 0.01N HCL at 37°C with Different Lipid: Surfactant Ratios (n = 2)
| Lipid/Mixture of lipids a | Surfactant b | Weight ratio | 1:250 dilution without drug in water | 1:250 dilution with drug (19 mg/g) in water | 1:250 dilution without drug in 0.01 N HCL | 1:250 dilution with drug (19 mg/g) in 0.01 N HCL |
|---|---|---|---|---|---|---|
| Capmul MCM | Cremophor EL | 1:1 | 297 | 290 | 293 | 296 |
| 3:7 | 236 | 236 | 231 | 233 | ||
| Glycerol Dicaprylate | Cremophor EL | 1:1 | 47 | 47 | 46 | 45 |
| 3:7 | 22 | 26 | 22 | 22 | ||
| Captex 8000 | Cremophor EL | 1:1 | 75 | 76 | 77 | 75 |
| 3:7 | 27 | 26 | 26 | 29 | ||
| Captex 355 | Cremophor EL | 1:1 | 80 | 83 | 77 | 75 |
| 3:7 | 30 | 30 | 28 | 28 | ||
| Captex 355-Capmul MCM (3:1) | Cremophor EL | 1:1 | 32 | 31 | 32 | 31 |
| 3:7 | 22 | 21 | 21 | 22 | ||
| Captex 355-Capmul MCM (1:1) | Cremophor EL | 1:1 | 23 | 25 | 26 | 23 |
| 3:7 | 20 | 19 | 20 | 21 | ||
| Captex 355-Capmul MCM (1:3) | Cremophor EL | 1:1 | 90 | 90 | 92 | 93 |
| 3:7 | 19 | 19 | 18 | 19 |
aCapmul MCM - Glycerol monocaprylocaprate , Captex 8000 - Glycerol tricaprylate and Captex 355 - caprylic/capric triglyceride
bCremophor EL – PEG-35 castor oil
Viscosity of Gels
The formation of a gel phase was observed in some of the phase diagrams for certain lipid/surfactant ratios with water concentrations from ca. 20–60% (glycerol dicaprylate, glycerol tricaprylate (Captex 8000) and caprylic/capric triglycerides (Captex 355)). The viscosities of various gel phases were measured and the results given in Table VI at 25°C. The gels were soft with low viscosity and, in general, the viscosity increased with increasing water concentration and then decreased upon further addition of water until the gel transitioned into a microemulsion or emulsion.
Table VI.
Viscosity (cP) of Gel with Dilution with Water at Different Lipid: Surfactant Ratios
| % water | Lipid: Surfactant ratio | ||
|---|---|---|---|
| 1:1 | 4:6 | 2:8 | |
| Glycerol dicaprylate | |||
| 20 | ND | ND | ND |
| 30 | 77 | 79 | 91 |
| 40 | 89 | 102 | 105 |
| 50 | 62 | 99 | 64 |
| 60 | ND | ND | ND |
| Glycerol tricaprylate | |||
| 20 | 116 | 116 | 184 |
| 30 | 86 | 179 | 122 |
| 40 | 169 | 125 | 195 |
| 50 | 109 | 79 | 106 |
| 60 | ND | ND | 91 |
| Caprylic/ Capric triglyceride | |||
| 20 | 120 | 77 | 130 |
| 30 | 110 | 114 | 194 |
| 40 | 99 | 107 | 175 |
| 50 | ND | 93 | 140 |
| 60 | ND | ND | 106 |
ND Not determined as gel was not formed
The addition of monoglyceride to di- and triglycerides not only decreased the gel phase, as previously mentioned, it also decreased the viscosity of any gel formed. The effect of the addition of monoglyceride (Capmul MCM EP) to a triglyceride (Captex 355) at a 1:3 ratio is shown graphically in Fig. 6. The effect of the addition of monoglyceride to triglyceride appeared to be a reduction in the viscosity of the gel formed until the gel almost or completely disappeared.
Fig. 6.
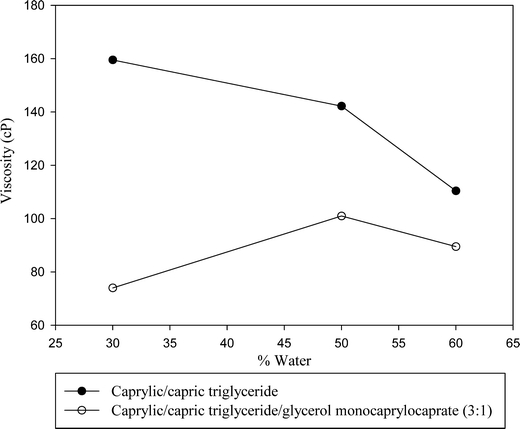
Comparison of viscosities (25°C) vs. percent water for caprylate/ capric triglyceride (Captex 355) and the mixture of caprylate/ capric triglyceride and glycerol monocaprylocaprate at 2:8 lipid to surfactant (Cremophor EL) ratio.
Solubility
The solubilities of danazol in different lipids individually and in mixtures with each other or with the surfactant (Cremophor EL) are given in Table VII. Danazol is a neutral compound with no ionizable functional group. Considering that its solubility is only 0.59 μg/mL in aqueous media, a major increase in solubility with lipids and the surfactant was observed. Among the lipids, both monoglyceride and diglyceride provided greater solubility (19 to 21 mg/g) than the triglycerides (7 mg/g). These results indicate the advantage of using the partially esterified medium chain glycerides (mono- and diglycerides) over the fully esterified glyceride (triglyceride). The solubility of danazol in Cremophor EL is higher than those in any of the lipids used (32 mg/g). Therefore, in addition to hydrophobic fatty acid chains, the presence of polar hydroxyl (mono- and diglycerides) and ethylene oxide (Cremophor EL) groups are also important in solubilizing a poorly soluble drug like danazol.
Table VII.
Solubility of Danazol at Different Ratios of Lipid/mixture of Lipids and Surfactant. (n = 2)
| Lipid: Surfactant a | Solubility (mg/g) (Experimental) | Solubility (mg/g) (Calculated) |
|---|---|---|
| Capmul MCM: Cremophor EL | ||
| 1:0 | 21 | – |
| 7:3 | 26 | 25 |
| 1:1 | 26 | 27 |
| 4:6 | 30 | 28 |
| 3:7 | 29 | 29 |
| 0:1 | 32 | – |
| Glycerol Dicaprylate : Cremophor EL | ||
| 1:0 | 19 | – |
| 7:3 | 22 | 23 |
| 1:1 | 24 | 26 |
| 4:6 | 28 | 27 |
| 3:7 | 29 | 28 |
| Captex 8000:Cremophor EL | ||
| 1:0 | 7 | – |
| 7:3 | 16 | 15 |
| 1:1 | 19 | 20 |
| 3:7 | 25 | 25 |
| Captex 355:Cremophor EL | ||
| 1:0 | 7 | – |
| 7:3 | 19 | 15 |
| 1:1 | 20 | 20 |
| 4:6 | 19 | 22 |
| 3:7 | 21 | 25 |
| Captex 355(3)-Capmul MCM(1): Cremophor EL | ||
| 1:0 | 10 | – |
| 7:3 | 18 | 17 |
| 1:1 | 27 | 21 |
| 3:7 | 27 | 25 |
| Captex 355(1)-Capmul MCM(1):Cremophor EL | ||
| 1:0 | 16 | – |
| 7:3 | 21 | 21 |
| 6:4 | 21 | 22 |
| 1:1 | 23 | 24 |
| 4:6 | 23 | 26 |
| 3:7 | 34 | 27 |
| Captex 355(1)-Capmul MCM(3) : Cremophor EL | ||
| 1:0 | 21 | – |
| 1:1 | 29 | 27 |
| 3:7 | 34 | 26 |
aCapmul MCM - Glycerol monocaprylocaprate , Captex 8000 - Glycerol tricaprylate, Captex 355 - caprylic/capric triglyceride and Cremophor EL – PEG-35 castor oil
When the lipids were mixed with each other or with the surfactants, the solubility changed in proportion to the solubility in the individual components. That is, the solubility behaved linearly. For example, the solubility of danazol in Capmul MCM and Cremophor EL are 21 and 32 mg/mL, respectively. When these two components were mixed (1:1), the solubility of danzaol was 26 mg/mL, which is essentially the average of its solubility in the individual components. The theoretical solubilty of danazol in different mixtures is given in Table VII. These data indicate that the experimentally determined solubility values for lipid/surfactant mixtures are similar to those calculated theoretically. When two components were mixed at different ratios, no significant synergistic increase (or decrease) in solubility was observed. Similar effects were noted previously by other investigators (38).
Dispersion in Aqueous Media
The primary components of lipid-based pharmaceutical dosage forms are lipid, surfactant and drug. Such a mixture or preconcentrate is expected to disperse after oral ingestion as an emulsion or microemulsion in the gastrointestinal tract. It is also expected that the dissolved drug would not precipitate upon dilution. A dispersion test was performed for various lipid/surfactant mixtures, with and without dissolved danazol. This was to determine 1) how easily the preconcentrates emulsify in aqueous media, 2) what are the particle sizes of any emulsions formed, 3) what is the effect of the presence of drug on emulsification or particle size, and 4) if any precipitation of drug is observed (39). The test was conducted in 250 mL of 0.01 N HCL (pH 2) at 37°C by using the relatively low agitation of 50 rpm to mimic the physiological condition in the human stomach.
Figure 7a and b show the results of dispersion tests with individual lipids (lipid/surfactant 1:1 and 3:7). In these figures, the concentration of drug dispersed in water is plotted as a function of time. Although the capsule shell quickly disintegrated (< 5 min), an initial, slow dissolution rate (up to 15 min) was observed, except in the case of the monoglyceride (Capmul MCM). It has been shown in Fig. 1 that gels formed when water was added to lipid/surfactant mixtures of diglyceride and triglyceride. Thus, the lag time in the dispersions given in Fig. 7a and b may have been due to gel formation. The gel disappeared upon further stirring and complete dispersion (>80%) was observed within 30 to 60 min. Drug concentration data shown in Fig. 7a and b are for filtered samples (0.45 μm). The concentrations were about 5% higher when unfiltered samples were used (data not shown). It appeared that the lipids formed a thin layer at the surface of the dispersion medium and adsorbed slightly at the inner surface of the dispersion vessels, which could lower drug concentration in the dispersion medium. For these reasons, we considered a dispersion to be complete if a drug concentration greater than 80% was observed. Thus, it may be concluded from Fig. 7a and b that, although there may be an initial lag time, a complete dispersion of drug and lipid was obtained from lipid/surfactant mixtures of di- and triglycerides. No lag time for the dispersion of the monoglyceride was observed in Fig. 7a and b. This may be explained by the lack of gel formation in the phase diagram. However, the dispersion of drug from the monoglyceride was incomplete, which could be related to the formation of an emulsion rather than a microemulsion; and in the case of the monoglyceride/surfactant ratio of 3:7 in Fig. 7b, due to the precipitation of drug at ≥ 45 min. In a separate study, danazol was dissolved in the surfactant (Cremophor EL) and the dispersion test performed; danazol started to precipitate within 45 min. It is thus apparent that Cremophor EL alone, or in mixtures with monoglyceride (3:7), cannot keep the drug dispersed or solubilized in aqueous media. No such dispersion issue was observed for diglyceride and triglyceride. The mechanism of the precipitation of drug from the monoglyceride at 3:7 lipid/surfactant ratio, and not from diglyceride and triglyceride, was not investigated.
Fig. 7.
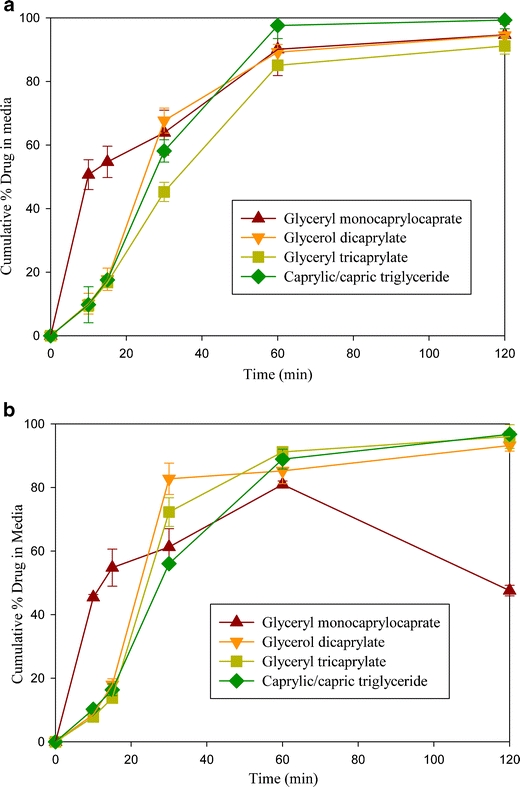
Cumulative % danazol present in dispersion media at (a) 1:1 ratio of lipid to surfactant (b) 3:7 ratio of lipid to surfactant (Cremophor EL).
Figure 8a and b show the dramatic effect of the dispersion of danazol when two lipids (mono- plus diglycerides or mono- plus triglycerides) were combined with the lipid/surfactant ratios of 1:1 and 3:7. It was demonstrated earlier that gel formation in the phase diagrams may be eliminated by using lipid mixtures (Fig. 5). In these cases, greater than 80% danazol dispersed within 10–15 min with no observable lag time, with the exception of the mixture of glycerol monocaprylocaprate (Capmul MCM) and caprylic/capric triglyceride (Captex 355) at ratio at 1:3 that exhibited gel formation at 1:1 lipid/surfactant mixture (Fig. 5a). Although Fig. 5a shows that there was also gel formation at the 3:7 lipid-surfactant ratios, no lag time was observed for the dispersion of the 1:3 mixture of glycerol monocaprylocaprate (Capmul MCM) and caprylic/capric triglyceride (Captex 355) in Fig. 8b. This could be related to the viscosity of the two systems; the 1:1 lipid/surfactant ratio gave higher viscosity than the 3:7 lipid/surfactant ratio (data not shown).
Fig. 8.
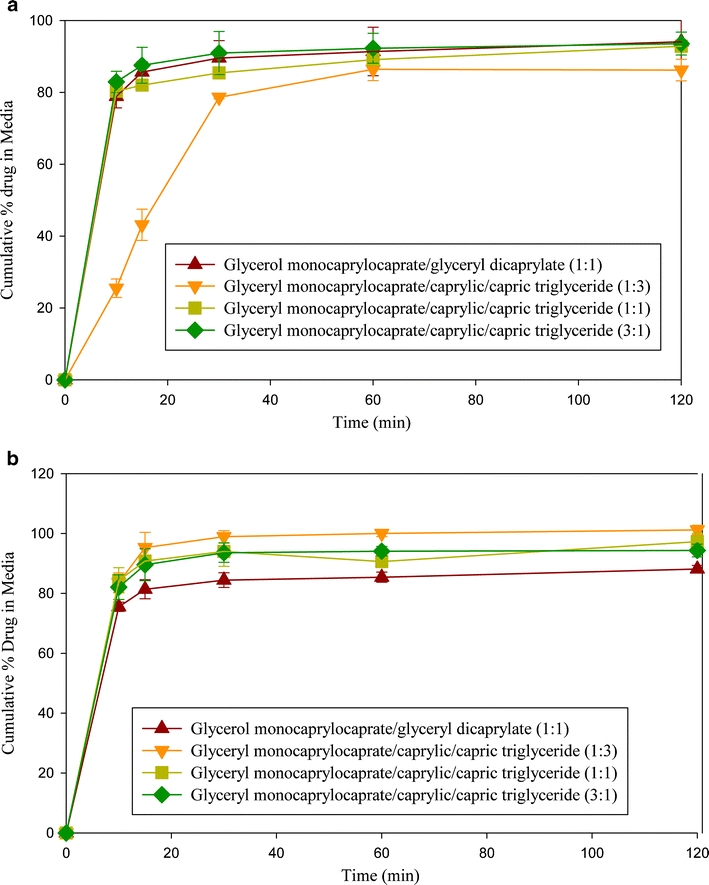
Cumulative % danazol present in dispersion media at (a) 1:1 ratio of mixture of lipids: surfactant (b) 3:7 ratio of mixture of lipids/surfactant (Cremophor EL).
Table VIII summarizes the particle size analyses vs. time during the dispersion tests. The presence of drug in the dispersed phase gave no observable difference in the particle size. The particle size analysis during dispersion testing confirms the earlier observations in the phase diagrams. Emulsions with relatively large particles (~400–500 μm) were formed when Capmul MCM alone was used with the surfactant (lipid/surfactant 1:1), while the particle size decreased by a factor of almost 10 (< 50 nm) when Capmul MCM was mixed with either a diglyceride or triglyceride (Captex 355) at 1:1 ratio. This was a major improvement over the performance of the individual lipids. The monoglyceride (Capmul MCM) gave particle sizes in the range of 400 to 500 nm, while the diglyceride and triglycerides (Captex 355 and Captex 8000) gave particle sizes in the range of 100 to 200 nm. When the lipid ratio of monoglyceride/triglyceride was increased from 1:1 to 3:1, that is the percentage of monoglyceride was increased, the particle size after dispersion increased about 10 fold (ca. 30 nm to 300 nm.) The particle size, however, remained small (40–70 nm) at higher surfactant concentration. These results are in agreement with the phase diagram in Fig. 5c, which showed that the mixture of monoglyceride/triglyceride (3:1) formed a microemulsion upon dilution with water only at a high surfactant level.
Table VIII.
Particle Size at Different Time Interval of Dispersion Test of Danazol at Different Ratios of Lipid: Surfactant and Mixture of Lipids: Surfactant (n = 3)
| Time (min) | Particle size (nm) ± s.d. | |||
|---|---|---|---|---|
| 1:1 (Lipid: Surfactant) | 3:7 (Lipid:Surafactant) | |||
| Control | With drug | Control | With drug | |
| Capmul MCM a | ||||
| 10 | 434 ± 19 | 424 ± 17 | 420 ± 19 | 430 ± 12 |
| 15 | 471 ± 17 | 440 ± 19 | 492 ± 12 | 500 ± 15 |
| 30 | 517 ± 20 | 486 ± 20 | 556 ± 13 | 557 ± 15 |
| 60 | 493 ± 12 | 487 ± 16 | 499 ± 12 | 563 ± 16 |
| 120 | 506 ± 13 | 522 ± 11 | 496 ± 14 | 565 ± 13 |
| Glycerol dicaprylate | ||||
| 10 | 94 ± 4 | 92 ± 5 | 80 ± 5 | 69 ± 3 |
| 15 | 104 ± 5 | 107 ± 3 | 80 ± 4 | 68 ± 2 |
| 30 | 113 ± 2 | 115 ± 2 | 67 ± 5 | 69 ± 1 |
| 60 | 114 ± 3 | 119 ± 3 | 69 ± 5 | 56 ± 1 |
| 120 | 113 ± 2 | 116 ± 1 | 61 ± 2 | 66 ± 3 |
| Captex 8000 b | ||||
| 10 | 123 ± 3 | 125 ± 3 | 30 ± 1 | 29 ± 3 |
| 15 | 161 ± 2 | 156 ± 5 | 29 ± 1 | 27 ± 3 |
| 30 | 179 ± 4 | 173 ± 4 | 29 ± 1 | 26 ± 2 |
| 60 | 183 ± 5 | 176 ± 2 | 28 ± 1 | 26 ± 1 |
| 120 | 175 ± 4 | 176 ± 3 | 27 ± 2 | 25 ± 1 |
| Captex 355 c | ||||
| 10 | 159 ± 3 | 127 ± 5 | 28 ± 2 | 29 ± 1 |
| 15 | 197 ± 5 | 168 ± 4 | 27 ± 3 | 28 ± 1 |
| 30 | 217 ± 4 | 196 ± 3 | 28 ± 1 | 23 ± 3 |
| 60 | 225 ± 3 | 199 ± 5 | 26 ± 3 | 28 ± 1 |
| 120 | 216 ± 2 | 200 ± 2 | 25 ± 3 | 28 ± 1 |
| Capmul MCM :Glyceryl dicaprylate (1:1) | ||||
| 10 | 62 ± 3 | 41 ± 1 | 325 ± 20 | 236 ± 24 |
| 15 | 59 ± 2 | 40 ± 3 | 372 ± 24 | 253 ± 18 |
| 30 | 57 ± 4 | 40 ± 2 | 363 ± 19 | 379 ± 20 |
| 60 | 50 ± 1 | 38 ± 2 | 377 ± 20 | 469 ± 19 |
| 120 | 67 ± 2 | 40 ± 3 | 404 ± 25 | 451 ± 21 |
| Capmul MCM : Captex 355 (1:3) | ||||
| 10 | 31 ± 3 | 31 ± 3 | 21 ± 1 | 21 ± 2 |
| 15 | 40 ± 1 | 32 ± 1 | 24 ± 2 | 21 ± 1 |
| 30 | 49 ± 3 | 39 ± 4 | 21 ± 3 | 21 ± 2 |
| 60 | 36 ± 2 | 36 ± 1 | 23 ± 1 | 21 ± 3 |
| 120 | 36 ± 1 | 34 ± 1 | 21 ± 1 | 22 ± 1 |
| Capmul MCM : Captex 355 (1:1) | ||||
| 10 | 28 ± 1 | 25 ± 2 | 19 ± 1 | 21 ± 1 |
| 15 | 27 ± 2 | 28 ± 1 | 20 ± 2 | 22 ± 1 |
| 30 | 32 ± 1 | 29 ± 2 | 20 ± 3 | 23 ± 2 |
| 60 | 29 ± 3 | 28 ± 2 | 19 ± 2 | 22 ± 2 |
| 120 | 29 ± 1 | 28 ± 1 | 20 ± 1 | 24 ± 1 |
| Capmul MCM : Captex 355 (3:1) | ||||
| 10 | 266 ± 10 | 325 ± 11 | 47 ± 1 | 49 ± 2 |
| 15 | 299 ± 9 | 324 ± 7 | 54 ± 2 | 51 ± 1 |
| 30 | 291 ± 8 | 426 ± 7 | 69 ± 1 | 67 ± 3 |
| 60 | 323 ± 7 | 421 ± 5 | 66 ± 2 | 44 ± 3 |
| 120 | 287 ± 5 | 320 ± 5 | 60 ± 2 | 40 ± 2 |
aCapmul MCM - Glycerol monocaprylocaprate
bCaptex 8000 - Glycerol tricaprylate
cCaptex 355 - Caprylic/capric triglyceride
DISCUSSION
Although it is desirable for the development of lipid-based pharmaceutical dosage forms that a drug has high solubility in the lipids used, there is no clear consensus in the pharmaceutical literature as to what determines the solubility of drugs in lipids. The selection of lipids is still made by trial and error. The medium chain lipids, however, have gained popularity in recent years as they have demonstrated higher drug solubilities compared to long chain lipids (40). Medium chain lipids are liquids at room temperature; whereas, many long chain lipids, especially partial glycerides, are viscous semisolids or solids at room temperature (11). The results of the present investigation showed that lipid/surfactant mixtures, which are generally used in self-emulsifying pharmaceutical dosage forms, can exist in different phases upon dilution with water. Since the different phases formed may have different utility and serve different purposes in formulations, it is essential that we discuss these phases in more detail.
Possible Structural Changes Following Addition of Water to Lipid/Surfactant Mixtures
The fate of different lipid/surfactant mixtures upon dilution with water may be explained with the schematic phase diagram in Fig. 9a. The line A to B depicts the dilution of a lipid/surfactant (1:1) mixture with water and the line C to D depicts the dilution of a lipid/surfactant (7:3) mixture with water. If one follows along the line A to B, a clear liquid is observed at low water content, which converts to a gel upon addition of more water and then reconverts to a clear or transluscent liquid phase upon further addition of water. There are many reports in the literature that analyzed microstructures of such phase transitions (41–46).It is generally recognized that upon addition of water the lipid/surfactant mixture successively forms a water-in-oil (w/o) microemulsion, a bicontinuous gel phase, followed by an oil-in-water (o/w) microemulsion. The possible structures of theses phases and the change in curvature of the surfactant layer along the change from w/o to o/w microemulsion are shown schematically in Fig. 9b. The gel is an intermediate phase occurring at the phase inversion from a w/o to o/w microemulsion. It may be comprised of different structures beginning with a lamellar bilayer as shown in Fig. 9b that may evolve into hexagonal phases in which surfactant molecules aggregate into circular cylindrical micelles and/or bicontinuous cubic phases. In all of these structures, the bilayer divides the space into two interwoven continuous networks of water and lipid/surfactant combinations (41). Sagalowicz et al. (45) reported that there may be as many as six different self-assembled structures of hexagonal, lamellar and cubic types in the transition from the w/o reversed micellar or microemulsion system to an o/w micellar or microemulsion system. Since a detailed investigation of the microstructure of the gel phase has not been conducted in the present study, we depicted the gel phase only with a lamellar structure for the purpose of illustrating a change in the curvature of the surfactant layer during phase transition from w/o and o/w microemulsions (Fig. 9b).
Fig. 9.
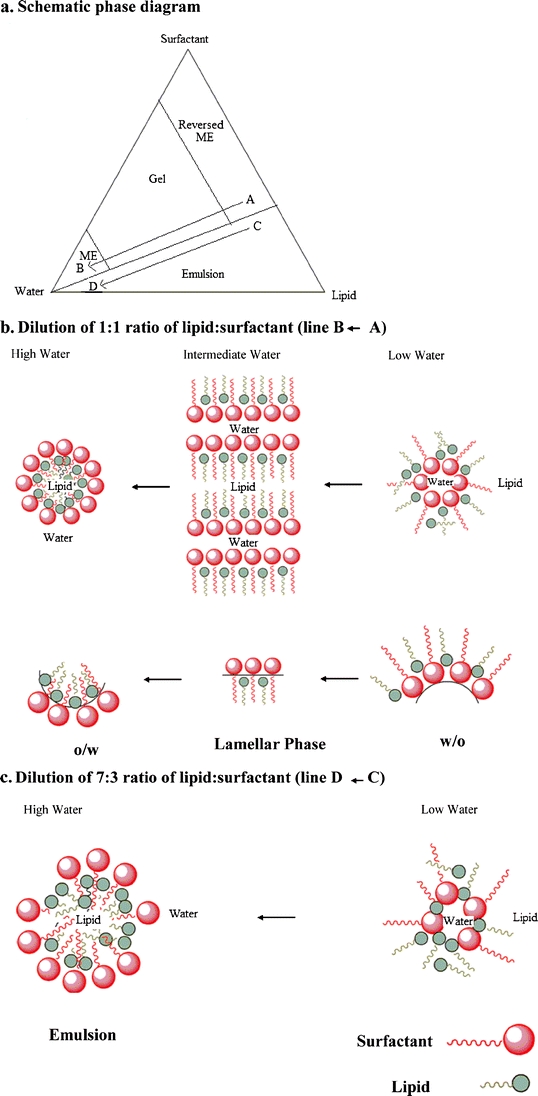
Schematic representation of (a) phase diagram with dilution curve, (b) conversion from w/o into o/w microemulsion passing through the gel phase as shown by line A to B in the phase diagram, and (c) conversion from w/o into o/w emulsion as shown by line C to D in the phase diagram.
Some of the different lipids (mono- and diglycerides) used in the present study are surface active in nature as they have polar functional groups as well as nonpolar hydrocarbon chains. However, the glycerides are less hydrophilic and have lower HLB values than Cremophor EL whose long polyethylene oxide chain makes it more hydrophilic. As shown in Fig. 9b, when the lipid is mixed with the surfactant at a ratio of around 1:1 (or lower), the two species may participate in forming the bilayer and then the o/w microemulsion. This is not the case when the lipid to surfactant ratio is higher, as shown by line C to D in the schematic phase diagram of Fig. 9a. The lipid/surfactant mixture converts to an emulsion instead of a gel when the lipid/surfactant ratio is high. The possible reason for this phenomenon is that the concentration of the hydrophilic surfactant is not sufficient enough to enable the formation of bilayer along with lipid. Instead, Fig. 9c illustrates that the water is initially absorbed into the lipid/surfactant mixture. Upon dilution, this is followed by the formation of a biphasic emulsion system where the lipid aggregates at the core of the emulsion droplet and the hydrophilic surfactant forms the outer layer.
The mechanism of the dramatic effect of combining monoglyceride with di-or triglycerides in eliminating the gel phase, expanding the microemulsion region, and reducing the particle size was not elucidated in the present study. It appears that the monoglyceride is acting as a cosurfactant with Cremophor EL as it has an intermediate HLB value. Further work in this area is continuing.
Pharmaceutical Application of Lipid/Surfactant/Water Phases
A few of the possible pharmaceutically important phases of lipid/surfactant/water systems depicted in Figs. 1, 4 and 5 are described below:
Lipid/Surfactant Mixture and Water-in-Oil Microemulsion
The clear region of either neat lipid/surfactant mixture with no water present or the lipid/surfactant mixture with a relatively low water content (<15%) in the phase diagrams often serves as the carrier for lipid-based drug delivery systems. Since some of these mixtures form o/w microemulsion upon dilution with water, they are sometimes referred to as microemulsion preconcentrates. Depending on drug solubility, a drug formulation in such mixtures results in a soft or hard gelatin capsule or a liquid solution dosage form.
Gel
The gel phase could be a transition phase in between w/o microemulsions and o/w microemulsions. The dispersion experiments in the present investigation showed that although the gel formation could initially retard dispersion of a formulation upon dilution with water, a complete dispersion with the formation of either a microemulsion or an emulsion occurred in all cases within 60 min. Thus, the gel formation may not have a major impact on the bioavailability of an oral dosage form.
Gels per se may also be used as pharmaceutical dosage forms because of their semisolid and viscous properties (47–51). Gels may prolong the release of drug after oral administration or topical application. In particular, gels may be applied to body cavities, such as eye, nose, vagina, etc., for increasing contact time and prolonging drug release. There is a recent report that a gel may also be used for periodontal anesthesia (47). Gels may be formed within cavities in situ when formulations with lipid/surfactant mixtures come in contact with the limited water present in body cavities, or they may be administered as finished products formulated by adding water. In addition to poorly soluble drugs, the gel may also serve as a reservoir for proteins and water-soluble drugs. There is also the potential of the lipid/surfactant mixtures, with or without a low level of water, administered intramuscularly for the formation of in situ gels such as the long-acting parenteral depot system.
Microemulsion
The clear or translucent phase (≥ 50% water) is often referred to as the microemulsion, or more precisely, the o/w microemulsion phase. The phase diagrams in the present investigation were initially constructed by visual observation of the different phases formed. Subsequent analysis indicated that the particle size of the microemulsion phase was < 200 nm. There has been a long controversy over what constitutes a microemulsion and whether it is an emulsion or a micelle (52,53). It is now generally recognized as the thermodynamically stable micellar or swollen micellar system of lipid, surfactant and water (54). It is formed spontaneously upon addition of water to lipid/surfactant or lipid/surfactant/cosurfactant mixtures.
The phase diagrams generated in the present investigation identified various microemulsion regions produced following dilution of lipid/surfactant mixtures with water. They also demonstrate how the microemulsion phase may be expanded with higher lipid content by using mixtures of two lipids instead of using them individually. These results should help formulators in selecting microemulsion preconcentrates (lipid/surfactant mixtures) for various dosage forms.
Emulsion
When the lipid/surfactant mixtures turned cloudy upon dilution with water, we considered them emulsions. In most cases where the preconcentrates were diluted with water at 1:99 or 1:250 ratios and the emulsions were formed, the particle sizes were still relatively low, usually in the submicron range (Tables II, III and IV). Depending on the properties of drugs, some formulators may find such a reduction in particle size suitable for bioavailability enhancement. The drug concentrated in emulsion globules may be released and available for absorption through lipolysis within the gastrointestinal tract (55,56). Therefore, it may not always be necessary to form a microemulsion for bioavailability enhancement as emulsions with particle sizes under one micron (or even higher) may be sufficient. If the surfactant concentration is low in the emulsion phase due to a low lipid/surfactant ratio, one should, however, be mindful of the potential phase separation of the lipid from aqueous media as indicated by the phase diagrams of Figs. 4 and 5c.
CONCLUSION
Mixtures of lipids and surfactants are often used as solvents or carriers for poorly water-soluble drugs. They form a microemulsion (particle size < 200 nm) or an emulsion (particle size > 200 nm) upon dilution with water or GI fluids, if taken orally. Although it is often expected that lipid/surfactant mixtures would produce a microemulsion upon dilution with water, we demonstrated using mono-, di- and triglycerides of medium chain fatty acids in combination with a common surfactant (PEG-35 castor oil) that the formation of microemulsion with these glycerides is often complex. For many lipid/surfactant mixtures, the formation of macroemulsions rather than microemulsions was observed, making the microemulsion phase rather small. In addition, gel or liquid crystalline phases were produced in the cases of diglycerides and triglycerides. The microemulsion phase could be expanded and the gel phase practically eliminated by combining monoglyceride with diglyceride or triglyceride at 1:1 ratios. However, it is also recognized that the gel formation may be desirable if the formulation is used topically or administered to body cavities (e.g., nose). There was no effect of the presence of a model drug, danazol, on the phase diagram or particle size after dilution with water. The dispersion rate of microemulsion preconcentrates (danazol dissolved in lipid/surfactant mixture) in aqueous media could be enhanced and the particle size of the microemulsion or emulsion formed reduced by using mixtures of lipids. It is hoped that this paper will provide practical guidance to formulation scientists in selecting appropriate medium chain glycerides for dosage form development based on lipid-surfactant ratio, particle size, drug solubility, dispersion of preconcentrate in aqueous media, absence or presence of gel phase and so forth.
ACKNOWLEDGMENTS & DISCLOSURES
This study was supported, in part, with a generous research grant from ABITEC Corporation, 501 W. 1st Avenue, Columbus, OH 43215. The authors thank Dr. Donald Kelemen, Dr. Jim Williams, Mr. Charles Nider III and Ms. Monna Manning of ABITEC Corporation for helpful discussions and valuable suggestions, and Mr. Darshil Patel of St. John’s University for technical assistance. The authors also acknowledge the assistance of Dr. Jim Williams in the preparation of glycerol dicaprylate used.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
REFERENCES
- 1.Lipinski CA. Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Meth. 2000;44(1):235–249. doi: 10.1016/S1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 2.Serajuddin ATM, Li P, Haefele T. Development of lipid-based drug delivery systems for poorly water-soluble drugs as viable oral dosage forms—Present status and future prospects. Am Pharm Rev. 2008;11:34–42. [Google Scholar]
- 3.Hauss DJ, Fogal SE, Ficorilli JV, Price CA, Roy T, Jayaraj AW, Keirns JJ. Lipid-based delivery systems for improving the bioavailability and lymphatic transport of a poorly water-soluble LTB4 inhibitor. J Pharm Sci. 1997;87(2):164–169. doi: 10.1021/js970300n. [DOI] [PubMed] [Google Scholar]
- 4.Humberstone AJ, Charman WN. Lipid-based vehicles for the oral delivery of poorly water soluble drugs. Adv Drug Del Rev. 1997;25(1):103–128. doi: 10.1016/S0169-409X(96)00494-2. [DOI] [Google Scholar]
- 5.Charman WN. Lipids, lipophilic drugs, and oral delivery—Some emerging concepts. J Pharm Sci. 2000;89(8):967–978. doi: 10.1002/1520-6017(200008)89:8<967::AID-JPS1>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 6.Kawakami K, Yoshikawa T, Moroto Y, Kanaoka E, Takahashi K, Nishihara Y, Masuda K. Microemulsion formulation for enhanced absorption of poorly soluble drugs. I. Prescription design. J Control Rel. 2002;81(1–2):64–74. doi: 10.1016/s0168-3659(02)00049-4. [DOI] [PubMed] [Google Scholar]
- 7.Vasanthavada M, Serajuddin ATM. Lipid-based self-emulsifying solid dispersions. In: Hauss D, editor. Lipid-based formulations for oral drug delivery: Enhancing bioavailability of poorly water-soluble drugs. New York: Informa Healthcare; 2007. pp. 149–184. [Google Scholar]
- 8.Porter CJH, Trevaskis NL, Charman WN. Lipids and lipid-based formulations: Optimizing the oral delivery of lipophilic drugs. Nature Rev. 2007;6:231–48. doi: 10.1038/nrd2197. [DOI] [PubMed] [Google Scholar]
- 9.Porter CJH, Pouton CW, Cuine JF, Charman WN. Enhancing intestinal drug solubilization using lipid-based drug delivery systems. Adv Drug Del Rev. 2008;60(6):673–691. doi: 10.1016/j.addr.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 10.Pouton CW, Porter JH. Formulation of lipid based delivery system for oral administration: Materials, methods and strategies. Adv Drug Del Rev. 2008;60(6):625–637. doi: 10.1016/j.addr.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 11.Hauss DJ. Oral lipid-based formulations. Adv Drug Del Rev. 2007;59(7):667–676. doi: 10.1016/j.addr.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 12.Chakraborty S, Shukla D, Mishra B, Singh S. Lipid—An emerging platform for oral delivery of drugs with poor bioavailability. Eur J Pharm Sci. 2009;73(1):1–15. doi: 10.1016/j.ejpb.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 13.O’Driscoll CM, Griffin BT. Biopharmaceutical challenges associated with drugs low aqueous solubility—the potential impacts of lipid-based formulations. Adv Drug Del Rev. 2008;60(6):617–624. doi: 10.1016/j.addr.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 14.Strickley RG. Currently marketed oral lipid-based dosage forms: Drug products and excipients. In: Hauss D, editor. Lipid-based formulations for oral delivery: Enhancing bioavailability of poorly water-soluble drugs. New York: Informa Healthcare; 2007. pp. 1–31. [Google Scholar]
- 15.Gursoy RN, Benita S. Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomed Pharmacother. 2004;58(3):173–182. doi: 10.1016/j.biopha.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 16.Hauss DJ. Oral lipid-based drug delivery—A case of implementation failing to keep up with innovation? Am Pharm Rev. 2006.
- 17.Vonderscher J, Meinzer A. Rationale for the development of Sandimmune Neoral. Transplant Proc. 1994;26(5):2925–2927. [PubMed] [Google Scholar]
- 18.Meinzer A, Mueller E, Vondersher J. Microemulsion—A suitable galenical approach for the absorption enhancement of a low soluble compound? BT Gattefosse. 1995;88:21–26. [Google Scholar]
- 19.Kovarik JM, Mueller EA, van Bree JB, Tetzloff W, Kutz K. Reduced inter- and intraindividual variability in cyclosporine pharmacokinetics from a microemulsion formulation. J Pharm Sci. 1994;83(3):444–446. doi: 10.1002/jps.2600830336. [DOI] [PubMed] [Google Scholar]
- 20.Mueller EA, Kovarik JM, van Bree JB, Grevel J, Lucker PW, Kutz K. Influence of a fat-rich meal on the pharmacokinetics of a new oral formulation of cyclosporine in a crossover comparison with the market formulation. Pharm Res. 1994;11(1):151–155. doi: 10.1023/A:1018922517162. [DOI] [PubMed] [Google Scholar]
- 21.Mueller EA, Kovarik JM, Kutz K. Minor influence of a fat-rich meal on the pharmacokinetics of a new oral formulation of cyclosporine. Transplant Proc. 1994;26(5):2957–2958. [PubMed] [Google Scholar]
- 22.Sheen PC, Kim SI, Petillo JJ, Serajuddin ATM. Bioavailability of a poorly water-soluble drug from tablet and solid dispersion in man. J. Pharm. Sci. 1991;80(7):712–714. doi: 10.1002/jps.2600800722. [DOI] [PubMed] [Google Scholar]
- 23.Cannon JB, Long MA. Emulsions, microemulsions, and lipid-based drug delivery systems for drug solubilization and delivery—Part II: Oral applications. In: Liu R, editor. Water-insoluble drug formulation. Boca Raton: CRC; 2008. pp. 227–253. [Google Scholar]
- 24.Pouton CW. Lipid formulations for oral administration of drugs: non-emulsifying, self-emulsifying and ‘self-mcroemulsifying’ drug delivery systems. Eur J Pharm Sci. 2000;11(Suppl.2):S-93–S98. doi: 10.1016/s0928-0987(00)00167-6. [DOI] [PubMed] [Google Scholar]
- 25.Pouton CW. Formulation of poorly water-soluble drugs for oral administration: Physicochemical and physiological issues and the lipid formulation classification system. Eur J Pharm Sci. 2006;29(3–4):278–287. doi: 10.1016/j.ejps.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 26.Shah NH, Carvajal MT, Patel CI, Infeld MH, Malick AW. Self-emulsifying drug delivery systems (SEDDS) with polycolized glycerides for improving in vitro dissolution and oral absorption of lipophilic drugs. Int J Pharm. 1994;106(1):15–23. doi: 10.1016/0378-5173(94)90271-2. [DOI] [Google Scholar]
- 27.Nazzal S, Smalyukh II, Lavrentovich OD, Khan MA. Preparation and in vitro characterization of a euticetic based semisolid self-enoemulsified drug delivery system (SNEDDS) of utiquinone: Mechanism and progress of emulstion formation. Int J Pharm. 2002;235(1–2):247–265. doi: 10.1016/S0378-5173(02)00003-0. [DOI] [PubMed] [Google Scholar]
- 28.Rao SVR, Shao J. Self-nanoemulsifying drug delivery systems (SNEDDS) for oral delivery of protein drugs. I. Formulation development. Int J Pharm. 2008;362(1–2):2–9. doi: 10.1016/j.ijpharm.2008.05.018. [DOI] [PubMed] [Google Scholar]
- 29.Zhang P, Liu Y, Feng N, Xu J. Preparation and evaluation of self-microemulsifyig drug delivery system of oridin. Int J Pharm. 2008;355:269–276. doi: 10.1016/j.ijpharm.2007.12.026. [DOI] [PubMed] [Google Scholar]
- 30.Elnaggar YSR, El-Massik MA, Abdallah OY. Self-nanoemulsifying drug delivery systems of tamoxifen citrate: Design and optimization. Int J Pharm. 2009;380(1–2):133–144. doi: 10.1016/j.ijpharm.2009.07.015. [DOI] [PubMed] [Google Scholar]
- 31.Shafiq S, Shakeel F, Talegaonkar S, Ahmad FJ, Khar RK, Ali M. Development and bioavailability assessment of ramipril nanoemulsion formation. Eur J Pharm Biopharm. 2007;66(2):227–243. doi: 10.1016/j.ejpb.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 32.Kang BK, Lee JS, Chon SK, Jeong Sy, Yuk SH, Khang G, Lee HB, Cho SH. Development of self-microemulsifying drug delivery systems (SMEDDS) for oral bioavailability enhancement of simvastatin in beagle dogs. Int J Pharm. 2004;274(1–2):65–73. doi: 10.1016/j.ijpharm.2003.12.028. [DOI] [PubMed] [Google Scholar]
- 33.Constantinides PP, Scalart J. Formulation and physical characterization of water-in-oil microemulsions containing long- versus medium chain glycerides. Int J Pharm. 1997;158(1):57–68. doi: 10.1016/S0378-5173(97)00248-2. [DOI] [Google Scholar]
- 34.Li P, Ghosh A, Wagner RF, Holinej J, Krill S, Joshi YM, Serajuddin ATM. Effect of combined use of nonionic surfactant on oil-in-Water microemulsions. Int. J. Pharm. 2005;288(1):27–34. doi: 10.1016/j.ijpharm.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 35.Ekwall P, Mandell L, Fontell K. Ternary systems of potassium soap, alcohol and water. J Colloid Interface Sci. 1969;31(4):508–512. doi: 10.1016/0021-9797(69)90052-6. [DOI] [PubMed] [Google Scholar]
- 36.Lo I, Madsen F, Florence AT, Treguier J-P, Seiller M, Puisieux F. The influence of surfactant HLB and the nature of the oil phase on the phase diagrams of nonionic surfactant-oil-water systems. J Colloid Interface Sci. 1977;59(2):319–325. doi: 10.1016/0021-9797(77)90014-5. [DOI] [Google Scholar]
- 37.Kaukonen AM, Boyd BJ, Porter CJH, Charman WN. Drug solubilization behavior during in vitro digestion of simple triglyceride lipid solution formulations. Pharm Res. 2003;21(2):245–253. doi: 10.1023/B:PHAM.0000016282.77887.1f. [DOI] [PubMed] [Google Scholar]
- 38.Kawakami K, Yoshikawa T, Moroto Y, Kanaoka E, Takahashi K, Nishihara Y, Masuda K. Microemulsion formation for enhanced absorption of poorly water soluble drugs I. Prescription design. J Control Rel. 2002;81(1–2):65–74. doi: 10.1016/S0168-3659(02)00049-4. [DOI] [PubMed] [Google Scholar]
- 39.Mohsin K, Long MA, Pouton CW. Design of lipid-based formulations for oral administration of poorly water-soluble drugs: Precipitation of drug after dispersion of formulations in aqueous solution. J Pharm Sci. 2009;98(10):3582–3595. doi: 10.1002/jps.21659. [DOI] [PubMed] [Google Scholar]
- 40.Li P, Pudipeddi M, Hynes SR, Royce AE, Serajuddin ATM. Development and characterization of a solid microemulsion preconcentrate system for oral delivery of poorly water soluble drugs. J. Pharm. Sci. 2009;98(4):1750–1763. doi: 10.1002/jps.21547. [DOI] [PubMed] [Google Scholar]
- 41.Ezrahi S, Aserin A, Garti N. Aggregation behavior in one-phase (Winsor IV) microemulsion systems. In: Kumar P, Mittal KL, editors. Handbook of microemulsion science and technology. New York: Informa Inc.; 1999. pp. 185–246. [Google Scholar]
- 42.Gradzielski M, Hoffmann H. Rheological properties of microemulsions. In: Kumar P, Mittal KL, editors. Handbook of microemulsion science and technology. New York: Informa In; 1999. pp. 357–386. [Google Scholar]
- 43.Lawrence MJ, Rees GD. Microemulsion-based media as novel drug delivery systems. Adv Drug Del Rev. 2000;45(1):89–121. doi: 10.1016/S0169-409X(00)00103-4. [DOI] [PubMed] [Google Scholar]
- 44.Salager J, Anton RE, Sabatini DA, Harwell JH, Acosta EJ, Tolosa LI. Enhancing solubilization in microemulsions—State of the art and current trends. J Surfactant Detergent. 2005;8(1):3–21. doi: 10.1007/s11743-005-0328-4. [DOI] [Google Scholar]
- 45.Sagalowicz L, Leser ME, Watzke HJ, Michel M. Monoglyceride self-assembly structures as delivery vehicles. Trends in Food Sci Technol. 2006;17(5):204–214. doi: 10.1016/j.tifs.2005.12.012. [DOI] [Google Scholar]
- 46.Malmsten M. Phase transformations in self-assembly systems for drug delivery applications. J Disp Sci Technol. 2007;28(1):63–72. doi: 10.1080/01932690600991755. [DOI] [Google Scholar]
- 47.Scherlund M, Malmsten M, Holmqvist P, Brodin A. Thermosetting microemulsions and mixed micellar solutions as drug delivery systems for periodontal anesthesia. Int J Pharm. 2000;194(1):103–116. doi: 10.1016/S0378-5173(99)00366-X. [DOI] [PubMed] [Google Scholar]
- 48.Scherlund M, Welin-Berger K, Brodin A, Malmsten M. Local anesthetic block copolymer system undergoing phase transition on dilution with water. Eur J Pharm Sci. 14:53–61. [DOI] [PubMed]
- 49.Shah JC, Sadhale Y, Chilukuri DM. Cubic phase gels as drug delivery systems. Adv Drug Del Rev. 2001;47(2–3):229–250. doi: 10.1016/S0169-409X(01)00108-9. [DOI] [PubMed] [Google Scholar]
- 50.Alam MM, Varade D, Aramaki K. Solubilization of triglycerides in liquid crystals of nonionic surfactant. J Colloid Interface Sci. 2008;325(1):243–249. doi: 10.1016/j.jcis.2008.05.066. [DOI] [PubMed] [Google Scholar]
- 51.Carvalho FC, Sarmento VHV, Chiavacci LA, Barbi MS, Gremiao MPD. Development and in vitro evaluation of surfactant systems for controlled release of zidovudine. J Pharm Sci. 2010;99(5):2367–2374. doi: 10.1002/jps.22005. [DOI] [PubMed] [Google Scholar]
- 52.Prince LM. Microemulsions versus micelles. J Colloid Interface Sci. 1975;52(1):182–188. doi: 10.1016/0021-9797(75)90315-X. [DOI] [Google Scholar]
- 53.Rosano HL. Introduction. In: Rosano HL, Clausse, editors. Microemulsion system. New York: Informa; 1987. pp. xv–xix. [Google Scholar]
- 54.Shah DO. Micelles, microemulsions and monolayers: Quarter Century Progress at the University of Florida. In: Shas DO, editor. Micelles, microemulsion and monolayers: Science and technology. New York: Informa; 1999. pp. 1–52. [Google Scholar]
- 55.Cuine JF, Charman WN, Pouton CW, Edwards GA, Porter CJH. Increasing the proportional content of surfactant (Cremophor EL) relative to lipid in self-emulsifying lipid-based formulations of danazol reduces oral bioavailability in beagle dogs. Pharm Res. 2007;24(4):248–757. doi: 10.1007/s11095-006-9194-z. [DOI] [PubMed] [Google Scholar]
- 56.Larsen A, Holm R, Pedersen ML, Mullertz A. Lipid-based formulation for danazol containing a digestible surfactant, labrafill M2125CS: In vitro bioavailability and dynamic in vitro analysis. Pharm Res. 2008;25(12):2769–2777. doi: 10.1007/s11095-008-9641-0. [DOI] [PubMed] [Google Scholar]