

Abstract
In the following review we discuss several types of nanoparticles (such as TiO2, quantum dots, and gold nanoparticles) and their impact on the ability to image biological components in fixed cells. The review also discusses factors influencing nanoparticle imaging and uptake in live cells in vitro. Due to their unique size-dependent properties nanoparticles offer numerous advantages over traditional dyes and proteins. For example, the photostability, narrow emission peak, and ability to rationally modify both the size and surface chemistry of Quantum Dots allow for simultaneous analyses of multiple targets within the same cell. On the other hand, the surface characteristics of nanometer sized TiO2allow efficient conjugation to nucleic acids which enables their retention in specific subcellular compartments. We discuss cellular uptake mechanisms for the internalization of nanoparticles and studies showing the influence of nanoparticle size and charge and the cell type targeted on nanoparticle uptake. The predominant nanoparticle uptake mechanisms include clathrin-dependent mechanisms, macropinocytosis, and phagocytosis.
Keywords: Nanoparticle, Cellular uptake, Quantum dots, Titanium dioxide
Introduction
Implementation of nanoparticle use in cell biology has been one of the most exciting developments in this field in the past 5 years. The number of articles describing the use of nanoparticles is increasing so rapidly that this review will be limited only to applications of nanoparticles on whole cells, fixed or alive, and not on the numerous strictly in vitro or in vivo uses. The focus on cells in this review is based on the fact that understanding of the interactions between nanoparticles and cells is the first step toward mechanistic understanding of the relationship between organisms and nanomaterials. Therefore, cellular studies provide a preliminary step for nanoparticle use in in vivo therapeutic or imaging purposes. Herein we are particularly interested in nanoparticles applied to cells, and used for imaging of subcellular components. Although cytotoxicity and the effects of cell loading by nanoparticles are of little consequence in fixed cells, nanoparticle biocompatibility and cellular uptake mechanisms are particularly relevant to live cell studies. Studies of the effects of nanoparticles on cellular proliferation and viability have shown that in most cases toxicity/biocompatibility of nanoparticles depends on their concentration [1-9]. Depending on the type of cell treated, the size, and the surface charge of the nanoparticle conjugate (nanoconjugate), different cellular uptake mechanisms are used by cells—most often clathrin-dependent mechanisms, macropinocytosis, and phagocytosis [10-17].
Surface modifications are often used to increase the functionality of nanoconjugates. In work with cells, surface modifiers serve to (i) increase cellular uptake of nanoconjugates, (ii) increase the specificity of cellular uptake, and (iii) increase the efficiency of intracellular targeting or retention of nanoconjugates. These nanoparticle modifiers/conjugants include various antibodies and peptides which improve cell type and subcellular compartment targeting, while nucleic acids (and their mimics) have been demonstrated to modify subcellular retention of nanoconjugates.
This review focuses on optically fluorescent semiconductor quantum dots and noble metal nanoparticles with size- and shape-dependent optical properties. In addition, particular attention is given to a different type of semiconductor material—TiO2 which is easily functionalized by both optically fluorescent agents and molecules for subcellular targeting. For detection of nanoparticles in cells some of the most powerful techniques are still optical microscopy and electron microscopy. However, complementary newly emerging imaging approaches such as four photon microscopy [18], near-infrared surface enhanced Raman scattering [19,20], X-ray fluorescence micro- and nano-probe imaging [21-25], and coherent X-ray diffraction imaging [26-28] will significantly improve imaging work with nanoparticles in cells. Some of the future developments with these techniques are expected to allow for 3D imaging with resolution as good as 5 nm3 voxel (coherent X-ray diffraction imaging), permitting imaging of whole frozen cells with the nanoparticles distributed at specific destinations in the cellular interior.
Nanoparticle Chemistry
Nanoparticles are mesostructures with some unique properties compared to bulk materials on one hand and atomic or molecular structures on the other. Compared to the bulk materials with constant physical and chemical properties regardless of their sizes (until it reaches the nano-regime), the nanoparticles have size-dependent properties: for example, quantum confinement in different semiconductor nanoparticles, an absorbance of surface plasmon resonance in metal (particularly noble metals) nanoparticles, superparamagnetism in magnetic nanoparticles etc. Three main types of nanoparticles used for cellular imaging described in this review are: polymer/biomacromolecule nanoparticles, semiconductor nanoparticles, and metal nanoparticles.
Polymer/biomacromolecule nanoparticles are made of biocompatible nanomaterials. Often, they are used in combination with other types of materials to improve their biocompatibility or functionality. On their own they are also used for the applications in cellular imaging. Nanoparticles of this group discussed in this review are:
· polymer nanoparticles such as Poly(d,l-lactic-co-glycolic acid) (PLGA) nanoparticles [29-32], polystyrene [11,33], polyethylene glycol (PEG) covered or PEGylated nanoparticles [15,34], poly(ethylene glycol)-block-poly(aspartic acid) (PEG-PAA)-coated calcium phosphate [35,36], poly-vinyl-chloride (PVC) [37];
· lipids and lipoproteins [17,38];
· proteins condensed nanoparticles made with albumin and oligonucleotides [39,40];
· nanoparticles containing DNA in addition to inorganic molecules or non-nucleic acid polymers: polyethylene glycol/DNA nanoparticles [41], poly(methyl methacrylate)/poly(ethyleneimine)–nanoparticle/pDNA complexes [41,42], poly-l-Lysine-DNA complexes [15,43];
· various fluorescent polymer nanoparticles [14,44];
Semiconductor nanoparticles mentioned in this review include quantum dots [18,45-59], and other semiconductor metal oxides: SiO2, ZnO, Al2O3, CrO, SnO2 and TiO2[10,12,14,33,37,60-69].
The elemental components of quantum dots are from groups II–VI, III–V, or IV–IV in the periodic table. They are considered inorganic salts or metal oxides. The size of quantum dots is usually less than 10 nm which is smaller than a bulk excitation Bohr radius. The scale of quantum dots results in their unique photoelectron emission. After excitation of a quantum dot, electrons in the valence band of the quantum dot hop to its conductive band. When the excited electrons with higher energy move back to the valence band, photons are emitted and provide a fluorescent signal. Due to this, quantum dots have advantages over “classical” organic fluorescent dyes including outstanding photostability and narrow emission peaks; at the same time, quantum dots collectively cover a wide range of fluorescence emission wavelengths from blue to infrared light depending on their physical size, shape, and chemical components. This is very useful for photoluminescent labels and simultaneous multiple targets.
Different from quantum dots, TiO2 is a wide-gap semiconductor nanoparticle with photocatalytic ability. Upon excitation, TiO2 nanoparticles can trap multiple electrons, producing at the same time positively charged holes in the conjugated molecules (if present) or leading to formation of reactive oxygen species in the nanoparticle vicinity by removal of electrons from the molecules of water in contact with the TiO2 surface [70]. These electropositive holes can lead to oxidation of nearby biomolecules which may be useful for therapeutic purposes. The surface chemistry of TiO2 nanoparticles smaller than 20 nm relies on formation of “corner defects” on the surface of the nanoparticle which are very reactive with bidentate ligands [71,72]. Therefore, any molecule that can be synthesized or modified to include, for example, dopamine can be easily attached to the surface of TiO2 nanoparticles. This approach was used for attachment of DNA oligonucleotides enabling subcellularly specific retention of TiO2-DNA oligonucleotide nanoconjugates [24,66,67].
Of the metal nanoparticles discussed in this review, the greatest emphasis will be given to gold nanoparticles [14,19,54,69,73-82] and then silver, cobalt and nickel nanoparticles [37]. Compared to other types of nanoparticles, metal nanoparticles, particularly the noble metal nanoparticles, easily form various stable nanostructures, are non-toxic and able to bind different targeting molecules. In particular, gold nanoparticles are easily modified with alkanethiols forming a chemical bond between gold and sulfur, while silver nanoparticles react with amino-compounds due to the formation of silver–nitrogen bond. This surface chemistry provides diverse ways for functionalizing through conjugation of nucleic acids (DNA, RNA, and synthetic nucleic acids such as locked nucleic acids [LNAs], peptide nucleic acids [PNAs] etc.), (poly)peptides or cellular ligands; e.g. thiocitic acid–polyethylene glycol–folate gold conjugates developed for targeting of cells with folate receptors [74]. The noble metal nanoparticles such as gold and silver have strong, size-dependent and shape-dependent optical properties with an absorbance of surface plasmon resonance. Thus different colors of the nanoparticles can be prepared from the same bulk metal by making nanoparticles of different sizes or shapes.
In summary, many nanoparticles have unique properties when compared to bulk materials of the same chemical composition. These unique chemical properties can be exploited for use in a variety of different applications including cellular imaging and delivery. The major types of nanoparticles that have been used for cellular imaging include polymer/biomacromolecular nanoparticles, semiconductor nanoparticles, and metal nanoparticles. Each of these types of nanoparticles has different properties that permit binding of proteins and nucleic acids that can be used for cellular and intracellular targeting.
Nanoparticles for Imaging in Fixed Cells
New developments in nanoparticle technology in recent years have offered numerous improvements to the study of fixed cells. Compared to traditional fluorescent dyes and proteins, modified quantum dots and gold nanoparticles possess alternative properties that enhance their imaging capabilities in cells that are fixed before imaging. Additionally, these nanoparticles enable multi-functional analyses of single samples using different forms of detection. Disadvantages of using nanoparticles are relatively minor and are increasingly being circumvented as technologies improve. For example, when conjugated to an antibody and used as a fluorescent tag, a quantum dot may be transformed into a non-fluorescent state upon initial illumination (termed blinking). This blinking may result in a false negative fluorescence reading during shorter periods of illumination. Li-Shishido et al. have demonstrated that this problem may be avoided by increasing the length of time that quantum dots are illuminated prior to recording of fluorescence intensity [52]. In that study, the majority of single dots were in the non-fluorescent state at the beginning of the illumination period, however, a 2- to 3-fold increase in fluorescence was observed after 10 min of illumination. Blinking (as well as bleaching) of the quantum dots was further suppressed in this study adding β-mercaptoethanol and glutathione to the sample [52]. Future nanoparticles will likely have an increased number and diversity of properties which will widen the scope of their use as cellular biomarkers [49].
Nanoparticle Based Biosensors have Enhanced Imaging Capabilities
Quantum dot nanocrystals functionalized by biomolecules are excellent fluorescent biosensors [55], proven to be active in many of the main cellular regions. Wu et al. (2003) used quantum dots to image cell surface markers (Her2), cytoplasmic proteins (actin and microtubules), and nuclear antigens [59]. Quantum dots can be designed to interact with a biological sample through electrostatic or hydrogen bonding [83] and are modifiable to suit their target. When coated with trimethoxysilylpropyl urea and acetate groups, quantum dots have shown the ability to bind to the nuclear membrane [45]. It is also possible for quantum dot nanocrystals to interact through ligand receptor interaction. CdSe–CdS core-shell nanocrystals with biotin covalently linked to the nanocrystal surface, have served as the secondary antibody, binding F-actin filaments in 3T3 mouse fibroblasts that had previously been labeled with phalloidin–biotin and streptavidin [45].
Relative to traditional fluorescent dyes and fluorescent proteins, the smaller size and increased photostability of quantum dots allow for prolonged and enhanced visualization of cellular detail. Wang et al. (2004) used quantum dots with maximum emission wavelength 605 nm (QD605) to detect the ovarian carcinoma marker CA125 in fixed cells. Antibody-conjugated quantum dots have demonstrated brighter and more specific signals as well as superior photostability compared to traditional organic FITC dyes. In one study, continuous illumination by an Argon laser 100 mW at 488 nm caused FITC signals to become undetectable after 24 min, while quantum dot-based probes maintained a bright signal after an hour [58]. The photostability of quantum dots also enables repeated imaging [50], which is valuable for higher resolution three-dimensional confocal imaging of fixed cells.
The broad excitation, narrow emission peak wavelengths of individual quantum dots, and availability of a wide range of quantum dots with different emission peaks allow for simultaneous imaging of multiple targets with multiple quantum nanoparticles. Taking advantage of spectral properties of quantum dots, flow cytometry has been used to resolve as many as 17 different fluorescent emissions, providing insight into complex phenotypic variations of numerous antigen-specific T-cell populations that would have previously eluded study [46]. There is an ever-increasing demand for multi-level analyses on single cell samples, where quantum dots and nanogold nanoparticles may be able to satisfy that need. Mittag et al. (2006) have demonstrated that a hyperchromatic cytometry approach, using quantum dots, allows for quantification and analysis of numerous areas of interest in a single cell [53]. Using a laser scanning cytometer and quantum dots, they were able to stain a sample with eight or more fluorochromes simultaneously through iterative restaining. The ability to relocate immobilized cells on a microscope slide enables extraction of many layers of information from a single cell after numerous rounds of treatments. The only factors that can limit the information gained by this multi-faceted approach are steric hindrance, the number of available antibodies [53], and resolution of optical microscopy (200 nm). Since quantum dots have broad excitation wavelengths and narrow emission wavelengths which can be varied through manipulation of nanoparticle size, they are well-suited for concurrent tracking. Using such multiplex assays and CdSe–ZnS core-shell quantum dots, Kriete et al. were able to quantify rapid changes in epidermal growth factor receptor internalization over time [84].
Additionally, quantum dots provide flexibility in that their particle size and surface chemistry can be varied to manipulate their chemical, optical, and electronic properties [85]. Quantum dots can even be used in molecular sensing as the optical properties of ZnS-capped CdSe quantum dots are affected by changes in pH and the presence of divalent cations [47]. This characteristic may even be used to monitor and optimize conditions during staining.
The relatively small size of quantum dots and gold nanoparticles provides an alternative manner to study fine cellular detail. Immunochemically functional quantum dots have been used for high magnification, three-dimensional erythrocyte reconstruction [57]. The quantum dot nanocrystals used in this study consisted of a < 10 nm CdSe semiconductor core surrounded by an inorganic ZnS shell. Conjugation of a monoclonal antibody to this quantum dot allowed for the detection of raft-like distribution of band 3 proteins in the erythrocyte membrane. Small differences in mtHSP70 and HSP60 between cancer and normal cells were visualized with the aid of quantum dots for simultaneous imaging [51].
Nanoparticles Used for Multi-modal Analyses
Nanoparticles enable multiple new approaches for imaging cellular samples. Since quantum dots are capable of narrow-spectrum emission when excited with light and readily absorb electrobeams, they can serve as imaging agents for both light microscopy and transmission electron microscopy [56]. Quantum dots have been used to label numerous endogenous proteins in fixed cells, permitting the visualization of these proteins by both light confocal and electron microscopy [50]. Quantum dots and immunogold nanoparticles have been used simultaneously, facilitating high resolution study of the potential interactions of multiple proteins. Streptavidin-conjugated Quantum Dot 605 and immunogold were used to detect primary rabbit anti-NH2-terminal CBP and mouse monoclonal anti-PML antibody 5E10, respectively [56]. Tang et al. (2007) have reported the ability of 60 nm colloidal gold nanoparticles to provide high spatial resolution data within individual fixed or live osteosarcoma cells using near-infrared surface-enhanced Raman scattering (SERS) [19,20]. Fahrni’s group used gold nanoparticles to do both optical imaging and X-ray fluorescence microscopy on the same cells [23,25]. To surmount the limitations of fluorescent microscopy and conventional multi-photon microscopy, Medda et al. (2006) have used quantum dots in conjunction with four photon microscopy to visualize the three dimensional co-localization of microtubule and mitochondrial networks with great detail [18]. This demonstrates the proof of principle of the manner in which quantum dots can be combined with currently “less common” imaging techniques to provide high resolution of detail that was not possible before.
In summary, innovations in nanoparticle technology over the last several years have provided many benefits to the imaging of fixed cells. The unique physical properties of nanoparticles make them highly photostable, convey a narrow emission spectra, and enable reiterative, high resolution imaging of samples using multiple forms of detection. Continued developments in the field of nanotechnology are likely to further enhance the benefits obtained from using nanoparticles for fixed cell imaging.
Nanoparticle Imaging in Live Cells
The use of nanoparticles in live cell imaging is already showing great promise. The ability to both rationally modify the surface chemistry of nanoparticles and conjugate them to biologically relevant molecules allows for an enhanced means to overcome some of the current limitations in live cell imaging, namely the rapid and efficient uptake of reagents. Moreover, nanoconjugates can be designed in such a way to take advantage of the physiological/molecular processes ongoing in cells in order to image subcellular compartments or illuminate certain aspects of cellular processes. A greater understanding of the effects nanoparticles have on living cells and the mechanisms the cell uses to take them up will have a direct impact on the ability to image with nanoparticles. This section describes some of the pertinent factors to consider when imaging live cells such as nanoparticle concentration, charge, size, and surface modifications.
Interactions of Nanoparticles and Living Cells
The growing selection and understanding of nanoparticles is opening new doors for cellular and medical imaging. They are also providing new insight into approaches such as antisense research [39,79], gene therapy [15,36,42], and drug delivery [30,86]. Despite this revolutionary potential, admittedly relatively little is known about the effects that nanoparticles have on living cells which could greatly impact live cell studies. There is currently a valid debate as to the possible deleterious effects that nanotechnology, if unchecked, will have on the environment and those exposed to it [87,88]. Several reviews on nanoparticle toxicity are available [1-6,8,9], and in this article we will not delve much into biocompatibility of nanoparticles.
TiO2 nanoparticles might be one of the best studied nanoparticles over the past several decades due to their potential uses in disinfection of polluted water and air (as reviewed in [89]). Although there are conflicting reports as to the extent of cytotoxicity that TiO2 nanoparticles exert on living cells [9,37,62,89-91], most studies to date show there is little effect on cell viability, even at high concentrations [90,92]. A study looking into the pathways activated by exposure to TiO2 nanoparticles, show that macrophage-like brain microglia BV2 cells have an increase in intracellular reactive oxygen species (ROS) due to oxidative burst and abnormal mitochondrial function [64]. A proteomics approach by Cha et al. showed that there are 20 proteins in bronchial epithelial BEAS-2B cells whose expression changed at least 2-fold upon exposure to TiO2 particles (0.29 μm) [60]. One of these proteins is macrophage migratory inhibitory factor (MIF) which has been shown to sustain a pro-inflammatory response by inhibiting p53 [93]. Another study comparing the effects of 5 different nanoparticles (TiO2, Co, Ni, Poly-Vinyl Chloride, and SiO2) on human dermal microvascular endothelial cells (HDMEC) showed that only Co and SiO2 nanoparticles had a significant effect on proliferation, viability, and pro-inflammatory potential [37]. TiO2 caused a minor, but detectable, increase in pro-inflammatory interleukin 8 (IL-8) release as detected by ELISA. The effects were slight compared to the response induced by Co and SiO2[37]. A separate study comparing the effects of several metal oxide nanoparticles (TiO2, ZnO, Fe3O4, Al2O3, CrO3) on mouse neuroblastoma Neuro-2A cells found that only ZnO nanoparticles were extremely toxic [62]. TiO2 and Fe3O4 nanoparticles had a slight effect on mitochondrial function at concentrations of 100 μg/ml, but cells treated with TiO2 or Al2O3 nanoparticles at the same concentration induced apoptosis in only 2% of cells. At high concentrations of 200 μg/ml, however, there was a noticeable effect on lactate dehydrogenase leakage, an indicator of cytotoxicity [62].
Quantum dots represent another prominent type of nanoparticle used in imaging whose cytotoxicity remains uncertain. Quantum dots commonly consist of a cadmium-selenide or cadmium-telluride core (CdSe or CdTe) enclosed within a zinc–sulfur shell [2,94]. The cadmium-based core is toxic to cells, but by coating it in a ZnS shell the core is sufficiently separated from the cell [95] to be non-toxic under functionally useful concentrations. Protection is also provided by coating the quantum dots with peptides, polyethylene glycol (PEG) or other biocompatible polymers [96-98]. A gene array experiment showed that human skin fibroblast (HSF-42) cells treated with PEG coated CdSe quantum dot had only 50 genes out of nearly 22,000 examined (∼0.2%) whose expression was altered significantly at concentrations of 8 or 80 nM [99]. Surprisingly, these did not include immune or inflammatory-related genes. The protection of PEG was further verified in human epidermal keratinocytes where carboxylic acid and PEG-amine coated quantum dots induced the release of pro-inflammatory cytokines IL-1β, IL-6, and IL-8, but PEG coated CdSe quantum dots did not [98]. Mercaptopropionic acid coated CdTe quantum dots, on the other hand, at 10 μg/ml were shown to cause a significant increase in intracellular reactive oxygen species (ROS) levels in MCF-7 cells and induced caspase-independent cell death [97]. Choi et al. showed that neuroblastoma SH-SY5Y cells treated with cysteamine-capped and N-acetylcysteine conjugated CdTe quantum dots have an increase in surface Fas expression, which is a known downstream target of ROS [100]. TEM showed the presence of autophagosomes in human mesenchymal stem cells treated with 5 nM of Q525, but not with larger Q605 having identical chemical composition [7]. This was confirmed with fluorescent confocal microscopy which showed elevated levels of LC3 expression in cells treated with Q525, but not Q605 [7].
Taken together, these studies clearly show some of the potential drawbacks of using nanoparticles for live cell experiments. By taking into consideration the type, size, surface chemistry, and the concentration of the nanoparticle being used to treat the cells, minimal cytotoxic effects can be achieved.
Cellular Uptake Mechanisms of Nanoparticles In vitro
Advancing the use of nanoparticles in cellular imaging and as potential drug delivery devices can only occur with a fundamental understanding of the cellular mechanisms involved in their uptake. Nanoparticle internalization in most cells occurs primarily through an active endocytic or phagocytic mechanism that is temperature and energy dependent (Table 1). For many cells, the key mechanisms of nanoparticle uptake include clathrin-mediated endocytosis, caveolin-dependent endocytosis, macropinocytosis, phagocytosis, and/or new uncharacterized mechanisms [10-15]. Clathrin-mediated endocytosis is the predominant mechanisms involved in non-macrophage cell nanoparticle uptake (reviewed elsewhere [17]); it results in the accumulation of extracellular macromolecules into clathrin coated vesicles which fuse to early endosomal vesicles eventually becoming degradative lysosomes. The function of many nanoparticles requires escape from the endosomes. Endosomal escape of fluorescent Poly(d,l-lactic-co-glycolic acid) nanoparticles was observed as the decreasing pH in maturing endosomes was believed to change the surface characteristics of the nanoparticles from anionic to cationic [101]. This reversal of surface charge was believed to cause association of the nanoparticles with the membrane of late endosomes, resulting in their rapid escape into the cytoplasm [101]. Bypassing endosomes can also occur by directly conjugating targeting molecules to the surface of the nanoparticle such as protein transduction domains [73].
Table 1.
Variables affecting nanoparticle uptake and subcellular localization
| Nanoparticle | Cell type | Localization | Uptake mechanism | References |
|---|---|---|---|---|
|
1. NP size | ||||
| 50 nm silica magnetic NP |
A549 lung cancer |
Endosomal |
CME |
Kim et al. [12] |
| 24 and 43 nm Polystyrene(PST) |
HeLa |
24 nm = perinuclear, 43 nm = lysosome |
24 nm = CME independent, 43 nm = CME |
Lai et al. [13] |
| 100 nm PLGA |
Primary RHEC |
Membrane bound, intracellular |
Clathrin?, Caveolin independent |
Qaddoumi et al. [32] |
| 78 nm–1 μm Microspheres |
RBC |
Cytoplasm |
Passive uptake? |
Rothen-Rutishauser et al. [14] |
| 40 nm–4.5 μm Microshperes |
Dendritic |
Cytoplasm and membrane bound |
No experimental evidence |
Foged et al. [11] |
|
2. NP charge | ||||
| PEG-PLA NP(+) and (−) charge |
HeLa |
Both types perinuclear |
(+) NP = CME/macropinocytosis, (−) NP = CME/caveolin independent |
Harush-Frenkel et al. [30] |
| 100 nm MSN (uncoated, weak, moderate, and strong (+) charge) |
hMSC and 3T3-L1 |
No experimental evidence |
hMSC: uncoated, weak, mod. (+) = CME, strong (+) unknown. 3T3-L1 = All CME |
Chung et al. [10] |
|
3. Cell type | ||||
| 78 nm–1 μm PST Microsphere |
Macrophage vs. RBC |
Intracellular, not membrane bound |
Macrophage: 1 μm = phagocytosis, .078–0.2 μm = actin-independent; RBC: all actin-independent |
Geiser et al. [33] |
| MSN strongly (+) |
hMSC vs. 3T3-L1 |
No experimental evidence |
hMSC = CME-independent, 3T3-L1 = CME |
Chung et al. [10] |
|
4. Surface modifications | ||||
| Folic acid-LDL NP |
KB Cells (FR+) |
Cytoplasm, not in nucleus |
Receptor mediated endocytosis |
Zheng et al. [38] |
| PVA and vitamin E TPGS coated PLGA NPs |
Caco-2 |
Cytoplasm and nucleus |
No experimental evidence |
Win and Feng [108] |
| Trastuzumab—HSA NP |
BT-474 and SK-BR-3 |
No experimental evidence |
Receptor mediated endocytosis |
Steinhauser et al. [40] |
| Tat peptide conjugated Gold NP | hTERT-BJ1 fibroblast | Nucleus | No experimental evidence | de la Fuente and Berry [73] |
NP = nanoparticle, PEG = poly(ethylene glycol), CME = clathrin-mediated endocytosis, PLGA = Poly(d,l-lactic-co-glycolic acid), (+) = positively charged, (−) = negatively charged, MSN = mesoporous silica nanoparticle, hMSC = human mesenchymal stem cell, RBC = red blood cell, FR+ = folate receptor positive, LDL = low density lipoprotein, PVA = polyvinyl alcohol, TPGS = d-alpha-tocopheryl polyethylene glycol 1000 succinate, HSA = human serum albumin
Clathrin-mediated endocytosis has been involved, at some level, in the uptake of a majority of the nanoparticles in non-macrophage cells. In osteosarcoma MNNG/HOS cells, fluorescently labeled FITC-layered double hydroxide nanoparticles were observed to co-localize with several proteins significant for clathrin-mediated endocytosis, but not with caveolin-1 [102]. Immunofluorescent confocal microscopy revealed that the FITC-LDH nanoparticle distribution matched that of fluorescent anti-clathrin, anti-eps15, and anti-dynamin antibodies [102]. This was further validated by treatment of the cells with the clathrin-mediated endocytosis inhibitor, chlorpromazine [102]. In human cervical epithelial carcinoma (HeLa) and primary human umbilical vein endothelial cells (HUVEC), 43 nm carboxyl-modified fluorescent polystyrene nanoparticles were also found to enter the cell via clathrin-dependent endocytosis [13]. Treatment with chlorpromazine inhibited uptake by as much as 43%, while caveolin-dependent uptake inhibitors (filipin and genistein) had no effect [13]. This was confirmed by the co-localization of the nanoparticles with the lysosomal stain Lysotracker (Molecular Probes). In A549 lung cancer cells, hyperosmotic sucrose was used to suppress coated pit function, resulting in decreased silica coated magnetic nanoparticle uptake [16]. Transmission Electron Microscopy (TEM) analysis also showed the presence of the magnetic nanoparticles localized within endosomes, all pointing to the fact that clathrin-mediated endocytosis is responsible for uptake [16].
The considerable absence of evidence implicating caveolin-dependent endocytosis in nanoparticle uptake in vitro may be due to particle size. A thorough study by Rejman et al. showed fluorescent latex microspheres were taken up primarily by clathrin-mediated endocytosis at sizes ranging from 50 to 200 nm, while particles 500 nm and above were taken up in a caveolin-dependent fashion by murine melanoma B16-F10 cells [103]. The lack of absolute specificity for some of the inhibitors used in many of the experiments might also contribute to some confusion discerning the exact mechanism involved in nanoparticle uptake [104]. Finally, the discrepancy in caveolin expression among different cell lines may also affect results. When NIH/3T3 cells were transformed by oncogene expression, caveolin expression was dramatically decreased at both the mRNA and protein levels [105].
Macropinocytosis is expected to be responsible for uptake of pegylated poly-lysine (C1K30-polyethylene glycol)-compacted DNA nanoparticles in Cos-7 cells [15]. Rhodamine labeled DNA was complexed with C1K30-polyethylene glycol and only slightly co-localized with early endosomal antigen-1 (EEA1). The distribution of the nanoparticle-DNA complex did not overlap with that of receptor-mediated endocytosis (a subset of clathrin-mediated endocytosis) marker transferrin or with late endosomal-marker lysobisphosphatidic acid (LBPA) [15]. Additionally, treatment with chlorpromazine or filipin had no effect on the amount of C1K30-DNA uptake. When cells were incubated with amiloride, an inhibitor of macropinocytosis [106], intracellular fluorescent rhodamine was significantly reduced [15].
In primary rabbit conjunctival epithelial cells (RCEC) uptake of fluorescent Poly(d,l-lactic-co-glycolic acid) nanoparticles was inhibited upon potassium depletion (clathrin-mediated endocytosis inhibitor) but not by filipin and nystatin (caveolin inhibitors) [32]. However, when clathrin was specifically knocked-down using antisense oligonucleotides targeting the rabbit clathrin HC gene, there was no effect on Poly(d,l-lactic-co-glycolic acid) nanoparticle uptake. Fluorescent transferrin internalization was decreased upon treatment with the antisense oligonucleotides, suggesting clathrin-mediated endocytosis was specifically targeted [32]. The authors concluded that uptake was clathrin- and caveolin-independent, and they hypothesized that it may occur via macropinocytosis or adsorptive endocytosis.
The study of nanoparticles has also brought to light and helped characterize some potentially new uptake mechanisms. A study by Chung et al. found that in human mesenchymal stem cells (hMSC) uptake of strongly positive mesoporous silica nanoparticles was not affected by any of the inhibitors used targeting clathrin-mediated endocytosis, caveolin-dependent endocytosis, actin polymerization, or microtubule polymerization [10]. Similarly, a study looking at ultrafine particles (78 nm–1 μm) observed that non-phagocytic red blood cells were able to internalize particles in the presence of cytochalasin D, which inhibits actin polymerization [33]. The authors concluded that internalization must occur through “adhesive interaction” or diffusion.
Variables Affecting In vitro Uptake of Nanoparticles in Living Cells
The ability to rationally design nanoparticles allows for the manipulation of their size, surface chemistry, and charge, invariably affecting their mechanism of uptake. Since the mode of internalization has a direct consequence on the subcellular localization and stability of the nanoparticle, it is imperative to consider these factors in live cell studies. The key variables elucidated thus far, appearing to be the most critical for the efficiency and mechanism of nanoparticle uptake include the size of the nanoparticle, the charge of the nanoparticle surface (ignoring targeting molecule conjugations), and the cell type being used [10,22,29,107] (Fig. 1 and Table 1). Several studies have shown that by simply altering one of these three variables the type and efficiency of uptake can be considerably changed.
Figure 1.
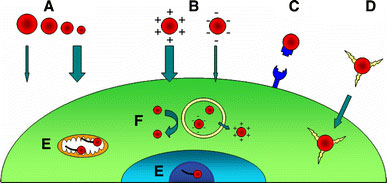
Factors affecting nanoparticle uptake. (A) Generally, smaller nanoparticles are internalized more efficiently than larger ones with similar surface characteristics. (B) Due to the negative charge of the cellular membrane, positively charged particles are preferentially taken up by living cells. (C) Cell-specific targeting by conjugating ligands for surface receptors to nanoparticles. (D) Rapid uptake and endosome bypassing can be achieved by conjugating protein transduction domains to the surface of the nanoparticle. (E) Conjugation of ODN was found to aid in specific subcellular localization based on the presence of complimentary cellular DNA. (F) Endosome escape has been reported to occur for nanoparticles whose surface is positively charged inside the low pH of late endosomes. Small nanoparticles have been reported to bypass degradation pathways better than larger particles of same chemical composition (see text for details)
The relevance of size was dramatically exemplified by looking at the variation in internalization of polystyrene nanoparticles whose only difference was geometric size. Lai et al. compared the mechanism of uptake and subcellular localization of 24 and 43 nm nanoparticles in HeLa cells [13]. Although uptake of both nanoparticles was temperature-dependent and caveolin-independent, the larger nanoparticles appeared to enter the cell through a clathrin/degradative pathway while the smaller nanoparticles did not [13]. In fact, the smaller 24 nm particles appeared in a perinuclear localization that did not significantly overlap with early endosome markers or Lysotracker. Therefore, it appears that the polystyrene nanoparticles entered the cell via an entirely different mechanism based purely on size, with the smaller nanoparticles able to avoid endosomal/lysosomal entrapment [13]. Dendritic cells, treated with fluorescent polystyrene nano- and micro-particles (40 nm–15 μm) of similar charge, showed a preferential uptake of smaller rather than larger particles [11]. In fact nanoparticles ranging from 40 to 500 nm had increased cell association compared to particles ranging from 1 to 4.5 μm as determined by flow cytometry [11]. The authors hypothesized that the 40–100 nm particles were taken up by macropinocytosis while larger particles up to 15 μm were taken up by phagocytosis [11]. In human colon adenocarcinoma Caco-2 cells, however, it was noted that the smaller polystyrene nanoparticles did not necessarily have the highest uptake efficiency [108]. In fact, 50 nm nanoparticles appeared to be taken up about half as well as 100 nm particles after 1 h of treatment [108]. Also, PEG coated quantum dots of different sizes (Q565 and Q655) but with similar charges did not co-localize within human epidermal keratinocyte cells, further demonstrating the effect of nanoparticle size on subcellular localization [98].
The effect of surface charge also has a profound effect on internalization capability. This is partly due to the fact that the cell membrane is negatively charged and will have a higher affinity for positively charged molecules. In dendritic cells, coating large 1 μm fluorescent polystyrene particles with positively charged poly-l-lysine increased uptake almost 10-fold compared to uncoated and negatively charged tetanus toxoid coated particles [11]. In smaller 100 nm nanoparticles, although uptake of poly-l-lysine coated nanoparticles was significantly higher than that of uncoated nanoparticles, there was no difference compared to uptake of the negatively charged nanoparticles [11]. In a separate study, confocal microscopy revealed a higher fluorescence intensity for cells treated with fluorescent positively charged polyethylene glycol-d,l-polylactide nanoparticles compared to negatively charged nanoparticles in HeLa cells [30]. Flow cytometry analysis also showed the rate of uptake was significantly higher for the positively charged nanoparticles than for their negative counterparts. In order to determine if the mechanism was also affected by changing the nanoparticle surface charge, cells were also infected with adenoviruses expressing dominant negative alleles of proteins involved in endocytosis [30]. From this the authors deduced that the inferior rate of uptake of negative nanoparticles occurs through a clathrin- and caveolin-independent mechanism while the more rapid uptake of positively charged nanoparticles occurs through a clathrin-dependent mechanism [30]. Interestingly, when a dominant negative form of Dynamin I was expressed (inhibiting both clathrin-mediated endocytosis and caveolin-dependent endocytosis) there was a significant increase in cellular fluorescence of cells treated with positively charged fluorescent nanoparticles [30]. This suggests that if the predominant mechanisms involving clathrin and caveolin are interrupted, a more efficient compensatory mechanism takes over. The authors speculated that this compensatory mechanism may in fact be macropinocytosis, although further study is required [30].
Different cell types obviously have unique efficiencies of nanoparticle uptake and respond differently to various kinds of nanoparticles. For example, when treating hMSC and 3T3-L1 cells with mesoporous silica nanoparticles of different surface charges, it was observed that uptake differed between the cell types [10]. Regardless of the extent of charge, 3T3-L1 cells took up the nanoparticles via clathrin-mediated endocytosis, but uptake of strong positively charged nanoparticles in hMSC cells occurs by an alternate and undefined mechanism [10].
Effect of Surface Modifications on In vitro Nanoparticle Uptake
In an attempt to target nanoparticles to specific cell types, to increase uptake efficiency, and to bypass intracellular obstacles (e.g. endosomes) there is an increasing amount of work being done to conjugate targeting molecules to the surface of nanoparticles. Among the most effective and interesting conjugants are protein transduction domains [73,109]. These are short amphipathic peptide sequences that translocate across cell membranes in a rapid manner [110-112]. Although there is much debate as to the mechanism they use to cross the cell membrane, there is little doubt that it occurs rapidly and efficiently. When the protein transduction domain of HIV-Tat (GRKKRRQRRR) was conjugated to 2.8 nm gold nanoparticles, TEM showed that it translocated across the cell membrane and was localized within the nucleus of human fibroblast cells [73]. Gold nanoparticles lacking the Tat peptide, on the other hand, were found surrounding the mitochondria or in cytoplasmic vacuoles [73]. In another study, when 20 nm gold nanoparticles were conjugated to the HIV-Tat peptide, the nanoparticle-peptide nanoconjugate was found to be localized mainly in the cytoplasm and not in the nucleus [80], once again substantiating the fact that nanoparticle size is one of the key factors affecting the uptake. In HeLa cells, when iron oxide CLIO nanoparticles were labeled with Cy3.5 and conjugated to a fluorescent Tat peptide-FITC conjugate, there was a rapid and sustained internalization of the nanoparticles as determined by flow cytometry [109]. Fluorescent confocal microscopy revealed that at 24 h post-treatment there was extensive co-localization of FITC and Cy3.5 in the nucleus and cytoplasm of cells treated with Tat peptide-FITC-Cy3.5-CLIO nanoparticles [109]. The nuclear localization was lost by 72 h.
An alternative approach is to specifically target tumor cells that overexpress surface receptors such as Her2/Neu or folate receptor. Therefore, by conjugating the ligands of these receptors to nanoparticles it is possible to achieve cell-specific internalization for potential drug delivery or imaging. Human Serum Albumin nanoparticles complexed with Trastuzumab (antibody against Her2) showed specific uptake of nanoparticles only in Her2 overexpressing cell lines [40]. Likewise, conjugation of nanoparticles to folate has been successful in targeting folate receptor overexpressing prostate and nasopharyngeal cancer cells [38,74,113].
While peptides direct nanoparticle uptake, conjugation of nucleic acids have a marked effect on nanoparticle subcellular retention [66,67,79]. Our own laboratory has shown the effects of conjugating oligonucleotides to the surface of TiO2 nanoparticles that target different organelles in living cells. X-ray-fluorescence microscopy (reviewed in [24,114]) and TEM have shown that by altering the oligonucleotide sequence bound to the nanoparticle the subcellular localization of the TiO2-DNA nanoconjugate can change based on the location of available cellular complimentary DNA [66,67]. For example, when breast cancer MCF-7/WS8 cells were treated with TiO2 nanoconjugates complimentary to genomic DNA encoding 18S rRNA (of which 200–300 copies reside in the nucleolus [115]), nanoconjugates were detected by X-ray-fluorescence microscopy and TEM within the nucleus. On the other hand, when the oligonucleotide sequence bound to the TiO2 nanoparticle was complimentary to mitochondrial DNA, there was a more disperse Ti signal found throughout the cytoplasm as detected by X-ray fluorescence microscopy. Also, TEM showed the presence of electron dense nanoparticles in the mitochondria [67]. Furthermore, results in Fig. 2 show the combination of X-ray-fluorescence microscopy and fluorescent confocal microscopy for imaging the same cell treated with a TiO2-DNA nanoconjugate whose nucleic acid component is labeled with tetramethylrhodamine (TAMRA). Clearly there is both a titanium signal and fluorescent TAMRA signal in the nucleus as well as in the perinuclear region. This strongly suggests that TiO2-DNA nanoconjugates are stable in cells.
Figure 2.
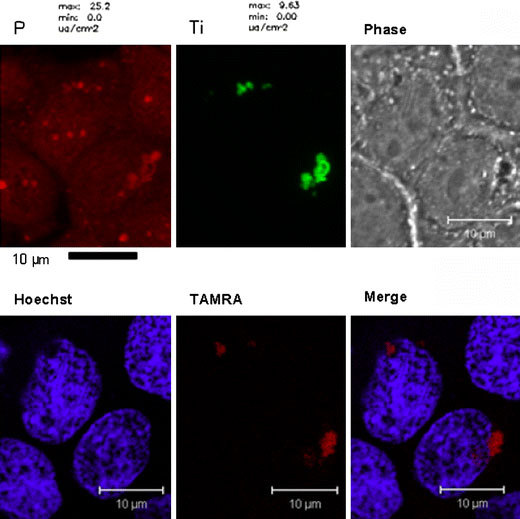
Combining X-ray fluorescence microscopy and fluorescent confocal microscopy for the imaging of intracellular nanoconjugates. MCF-7 cells were transfected with TiO2-DNA nanoconjugates complimentary to genomic DNA encoding r18S rRNA. The DNA was fluorescently labeled with TAMRA. After treatment, cells were washed, fixed, and stained with Hoechst dye. Then they were analyzed by fluorescent confocal microscopy for the localization of TAMRA. Next, the same cells were dehydrated in 100% ethanol and analyzed at the 2-ID-D Beamline at the Advanced Photon Source at Argonne National Laboratories for the presence of titanium. Black bar scale represents 10 μm for XFM (top left and middle), and the white bar 10 μm for fluorescent confocal microscopy (top right, bottom row)
In conclusion, imaging of live cells is important in assessing biological function, and nanotechnology offers many new approaches for such studies. In most cases, imaging of live cells is dependent upon the development of nanomaterials that can penetrate the cell membrane and not cause the subsequent death of the cell. Many groups are exploring the use of bionanoconjugates that can be designed to probe functional biological pathways in living cells, and the identification of pathways important in nanomaterial uptake into cells will facilitate this work.
Conclusions
In this review we focused on nanoparticles that have been used for cellular imaging; either in live or fixed cells. We chose this as a focus area because whole cell imaging and manipulation by nanoparticles are at this time gathering momentum. The cell is the best starting point for development of new therapeutics and new cellular molecular biology techniques, because the whole cell as a biological entity has always been the first target en route to mechanistic understanding of both intracellular and whole organism pathways and processes. New types of nanoparticles are developed daily and we can anticipate that new uses for them in the field of cell imaging and manipulation will be discovered with great rapidity as well. Most of the cellular manipulation with nanoparticles is, at this moment, devoted to improvements of new therapies and imaging tools. It is our opinion, however, that development of nanoparticles as tools for basic science may revolutionize cellular and molecular biology techniques as much as understanding and utilization of enzymatic reactions did, leading to creation of molecular biology we know today.
Acknowledgements
The authors would like to extend their gratitude to Benjamin Haley and David Paunesku for their input and advice. Work was supported by NIH Grants: CA107467, EB002100, P50 CA89018, U54CA119341. Use of the Advanced Photon Source was supported by the U.S. Department of Energy Basic Energy Sciences; under contract number DE-AC02-06CH11357.
References
- Gojova A, Guo B, Kota RS, Rutledge JC, Kennedy IM, Barakat AI. Environ. 2007. p. 3. [DOI] [PMC free article] [PubMed]
- Hardman R. Environ. 2006. p. 2. [DOI] [PMC free article] [PubMed]
- Lanone S, Boczkowski J. Curr. 2006. p. 6. [DOI] [PubMed]
- Medina C, Santos-Martinez MJ, Radomski A, Corrigan OI, Radomski MW. Br. 2007. p. 5. [DOI] [PMC free article] [PubMed]
- Nel A, Xia T, Madler L, Li N. Science. 2006. p. 5761. [DOI] [PubMed]
- Priestly BG, Harford AJ, Sim MR. Med. 2007. p. 4. [DOI] [PubMed]
- Seleverstov O, Zabirnyk O, Zscharnack M, Bulavina L, Nowicki M, Heinrich JM, Yezhelyev M, Emmrich F, O’Regan R, Bader A. Nano Lett. 2006. p. 12. [DOI] [PubMed]
- Tsuji JS, Maynard AD, Howard PC, James JT, Lam CW, Warheit DB, Santamaria AB. Toxicol. 2006. p. 1. [DOI] [PubMed]
- Warheit DB, Hoke RA, Finlay C, Donner EM, Reed KL, Sayes CM. Toxicol. 2007. p. 99. COI number [1:CAS:528:DC%2BD2sXotVahu7o%3D] [DOI] [PubMed]
- Chung TH, Wu SH, Yao M, Lu CW, Lin YS, Hung Y, Mou CY, Che YC, Huang DM. Biomaterials. 2007. p. 2959. COI number [1:CAS:528:DC%2BD2sXktFOktL4%3D] [DOI] [PubMed]
- Foged C, Brodin B, Frokjaer S, Sundblad A. Int. 2005. p. 2. [DOI] [PubMed]
- Kim JS, Yoon TJ, Yu KN, Noh MS, Woo M, Kim BG, Lee KH, Sohn BH, Park SB, Lee JK, Cho MH. J. 2006. p. 4. [DOI] [PMC free article] [PubMed]
- Lai SK, Hida K, Man ST, Chen C, Machamer C, Schroer TA, Hanes J. Biomaterials. 2007. p. 18. [DOI] [PubMed]
- Rothen-Rutishauser BM, Schurch S, Haenni B, Kapp N, Gehr P. Environ. 2006. p. 14. [DOI] [PubMed]
- Walsh M, Tangney M, O’Neill MJ, Larkin JO, Soden DM, McKenna SL, Darcy R, O’Sullivan GC, O’Driscoll CM. Mol. 2006. p. 6. [DOI] [PubMed]
- Huang M, Ma Z, Khor E, Lim LY. Pharm. 2002. p. 10. COI number [1:CAS:528:DC%2BD38XosVymtw%3D%3D] [DOI] [PubMed]
- Mousavi SA, Malerod L, Berg T, Kjeken R. Biochem. 2004. p. 1. COI number [1:CAS:528:DC%2BD2cXis1Sqtw%3D%3D] [DOI] [PMC free article] [PubMed]
- Medda R, Jakobs S, Hell SW, Bewersdorf J. J. 2006. p. 3. [DOI] [PubMed]
- Tang HW, Yang XB, Kirkham J, Smith DA. Anal. 2007. p. 3646. COI number [1:CAS:528:DC%2BD2sXkt1Ors7g%3D] [DOI] [PubMed]
- Yang PH, Sun X, Chiu JF, Sun H, He QY. Bioconjug. 2005. p. 3. [DOI] [PubMed]
- Lai B, Maser J, Paunesku T, Woloschak GE. Int. 2002. p. 8. [DOI] [PubMed]
- Lai B, Maser J, Vogt S, Paunesku T, Woloschak GE. Int. 2004. p. 6. COI number [1:CAS:528:DC%2BD2cXms1Gnsbs%3D] [DOI] [PubMed]
- McRae R, Lai B, Vogt S, Fahrni CJ. J. 2006. p. 1. [DOI] [PubMed]
- Paunesku T, Vogt S, Maser J, Lai B, Woloschak G. J. 2006. p. 6. [DOI] [PubMed]
- Yang L, McRae R, Henary MM, Patel R, Lai B, Vogt S, Fahrni CJ. Proc. Natl. Acad. Sci. USA. 2005. p. 32. [DOI] [PMC free article] [PubMed]
- Pfeifer MA, Williams GJ, Vartanyants IA, Harder R, Robinson IK. Nature. 2006. p. 7098. [DOI] [PubMed]
- Robinson IK, Vartanyants IA, Williams GJ, Pfeifer MA, Pitney JA. Phys. 2001. p. 19. [DOI] [PubMed]
- Miao J, Charalambous P, Kirz J, Sayre D. Nature. 1999. p. 6742.
- Davda J, Labhasetwar V. Int. 2002. p. 51. COI number [1:CAS:528:DC%2BD38XhslymsLg%3D] [DOI] [PubMed]
- Harush-Frenkel O, Debotton N, Benita S, Altschuler Y. Biochem. 2007. p. 1. [DOI] [PubMed]
- Kim SH, Jeong JH, Chun KW, Park TG. Langmuir. 2005. p. 19. [DOI] [PubMed]
- Qaddoumi MG, Gukasyan HJ, Davda J, Labhasetwar V, Kim KJ, Lee VH. Mol. 2003. p. 559. COI number [1:CAS:528:DC%2BD3sXpt1aisbk%3D] [PubMed]
- Geiser M, Rothen-Rutishauser B, Kapp N, Schurch S, Kreyling W, Schulz H, Semmler M, Im Hof V, Heyder J, Gehr P. Environ. 2005. p. 11. [DOI] [PMC free article] [PubMed]
- Reddy GR, Bhojani MS, McConville P, Moody J, Moffat BA, Hall DE, Kim G, Koo YE, Woolliscroft MJ, Sugai JV, Johnson TD, Philbert MA, Kopelman R, Rehemtulla A, Ross BD. Clin. 2006. p. 22. [DOI] [PubMed]
- Kakizawa Y, Furukawa S, Kataoka K. J. Control Release. 2004. p. 2. [DOI] [PubMed]
- Sokolova V, Kovtun A, Heumann R, Epple M. J. 2007. p. 2. [DOI] [PubMed]
- Peters K, Unger RE, Kirkpatrick CJ, Gatti AM, Monari E. J. 2004. p. 4. [DOI] [PubMed]
- Zheng G, Chen J, Li H, Glickson JD. Proc. Natl. Acad. Sci. USA. 2005. p. 49. [DOI] [PMC free article] [PubMed]
- Arnedo A, Irache JM, Merodio M, Espuelas Millan MS. J. Control Release. 2004. p. 1. [DOI] [PubMed]
- Steinhauser I, Spankuch B, Strebhardt K, Langer K. Biomaterials. 2006. p. 28. [DOI] [PubMed]
- Brigger I, Dubernet C, Couvreur P. Adv. 2002. p. 5. [DOI] [PubMed]
- Feng M, Lee D, Li P. Int. 2006. p. 209. COI number [1:CAS:528:DC%2BD28XhvF2mu7Y%3D] [DOI] [PubMed]
- Lucas B, Remaut K, Sanders NN, Braeckmans K, De Smedt SC, Demeester J. Biochemistry (Mosc.) 2005. p. 29. [DOI] [PubMed]
- Vogt A, Combadiere B, Hadam S, Stieler KM, Lademann J, Schaefer H, Autran B, Sterry W, Blume-Peytavi U. J. 2006. p. 6. [DOI] [PubMed]
- Bruchez M, Moronne M, Gin P, Weiss S, Alivisatos AP. Science. 1998. p. 5385. [DOI] [PubMed]
- Chattopadhyay PK, Price DA, Harper TF, Betts MR, Yu J, Gostick E, Perfetto SP, Goepfert P, Koup RA, De Rosa SC, Bruchez MP, Roederer M. Nat. 2006. p. 8. [DOI] [PubMed]
- Gao X, Chan WC, Nie S. J. 2002. p. 4. [DOI] [PubMed]
- Gao X, Cui Y, Levenson RM, Chung LW, Nie S. Nat. 2004. p. 8. [DOI] [PubMed]
- Giepmans BN, Adams SR, Ellisman MH, Tsien RY. Science. 2006. p. 5771. [DOI] [PubMed]
- Giepmans BN, Deerinck TJ, Smarr BL, Jones YZ, Ellisman MH. Nat. Methods. 2005. p. 10. [DOI] [PubMed]
- Kaul Z, Yaguchi T, Kaul SC, Wadhwa R. Ann. 2006. p. 469. COI number [1:CAS:528:DC%2BD28Xms1OktL0%3D] [DOI] [PubMed]
- Li-Shishido S, Watanabe TM, Tada H, Higuchi H, Ohuchi N. Biochem. 2006. p. 1. [DOI] [PubMed]
- Mittag A, Lenz D, Gerstner AOH, Tarnok A. Cytom Part A. 2006. p. 7. [DOI] [PubMed]
- Prow T, Smith JN, Grebe R, Salazar JH, Wang N, Kotov N, Lutty G, Leary J. Mol. 2006. p. 606. COI number [1:CAS:528:DC%2BD28XntVels74%3D] [PubMed]
- Sapsford K, Pons T, Medintz I, Mattoussi H. Sensors. 2006. p. 925. COI number [1:CAS:528:DC%2BD28XhtFamu7nF] [DOI]
- Nisman R, Dellaire G, Ren Y, Li R, Bazett-Jones DP. J. 2004. p. 1. [DOI] [PubMed]
- Tokumasu F, Dvorak J. J. 2003. p. 256. COI number [1:CAS:528:DC%2BD3sXotVeltbY%3D] [DOI] [PubMed]
- Wang HZ, Wang HY, Liang RQ, Ruan KC. Acta Biochim. Biophys. Sin. (Shanghai) 2004. p. 10. COI number [1:CAS:528:DC%2BD2MXktFOru7c%3D] [DOI] [PubMed]
- Wu X, Liu H, Liu J, Haley KN, Treadway JA, Larson JP, Ge N, Peale F, Bruchez MP. Nat. 2003. p. 1. [DOI] [PubMed]
- Cha MH, Rhim T, Kim KH, Jang AS, Paik YK, Park CS. Mol. Cell. Proteomics. 2007. p. 1. [DOI] [PubMed]
- Herr JK, Smith JE, Medley CD, Shangguan D, Tan W. Anal. 2006. p. 9. [DOI] [PubMed]
- Jeng HA, Swanson J. J. 2006. p. 12. [DOI] [PubMed]
- Liu J, Saponijc Z, Dimitrijevic NM, Luo S, Preuss D, Rajh T. Proc. SPIE. 2006. p. 48.
- Long TC, Saleh N, Tilton RD, Lowry GV, Veronesi B. Environ. 2006. p. 14. [DOI] [PubMed]
- Mechery SJ, Zhao XJJ, Wang L, Hilliard LR, Munteanu A, Tan WH. Chem-Asian J. 2006. p. 3. [DOI] [PubMed]
- Paunesku T, Rajh T, Wiederrecht G, Maser J, Vogt S, Stojicevic N, Protic M, Lai B, Oryhon J, Thurnauer M, Woloschak G. Nat. 2003. p. 5. [DOI] [PubMed]
- Paunesku T, Vogt S, Lai B, Maser J, Stojicevic N, Thurn KT, Osipo C, Liu H, Legnini D, Wang Z, Lee C, Woloschak GE. Nano Lett. 2007. p. 3. [DOI] [PMC free article] [PubMed]
- Peters R. Traffic. 2005. p. 5. [DOI] [PubMed]
- Zhang HD, Williams PS, Zborowski M, Chalmers JJ. Biotechnol. 2006. p. 5. [DOI] [PubMed]
- Fujishima A, Honda K. Nature. 1972. p. 5358. [DOI] [PubMed]
- Michelmore A, Gong W, Jenkins P, Ralston J. Phys. 2000. p. 2985. COI number [1:CAS:528:DC%2BD3cXktlCntL8%3D]
- Rajh T, Chen LX, Lukas K, Liu T, Thurnauer M, Tiede DM. J. Phys. Chem. B. 2002. p. 41. [DOI]
- de la Fuente JM, Berry CC. Bioconjug. 2005. p. 5. [DOI] [PubMed]
- Dixit V, Van den Bossche J, Sherman DM, Thompson DH, Andres RP. Bioconjug. 2006. p. 3. [DOI] [PubMed]
- Herdt AR, Drawz SM, Kang Y, Taton TA. Colloids Surf. 2006. p. 2. [DOI] [PubMed]
- Kirpotin DB, Drummond DC, Shao Y, Shalaby MR, Hong K, Nielsen UB, Marks JD, Benz CC, Park JW. Cancer Res. 2006. p. 13. [DOI] [PubMed]
- Park SY, Lee JS, Georganopoulou D, Mirkin CA, Schatz GC. J. 2006. p. 25. [DOI] [PubMed]
- Qin WJ, Yung LY. Biomacromolecules. 2006. p. 11. [DOI] [PubMed]
- Rosi NL, Giljohann DA, Thaxton CS, Lytton-Jean AK, Han MS, Mirkin CA. Science. 2006. p. 5776. [DOI] [PubMed]
- Tkachenko AG, Xie H, Coleman D, Glomm W, Ryan J, Anderson MF, Franzen S, Feldheim DL. J. 2003. p. 16. [DOI] [PubMed]
- Tkachenko AG, Xie H, Liu Y, Coleman D, Ryan J, Glomm WR, Shipton MK, Franzen S, Feldheim DL. Bioconjug. 2004. p. 3. [DOI] [PubMed]
- Salnikov V, Lukyanenko YO, Frederick CA, Lederer WJ, Lukyanenko V. Biophys. 2007. p. 3. [DOI] [PMC free article] [PubMed]
- Wilchek M, Bayer EA. Trends Biochem. 1989. p. 10. [DOI] [PubMed]
- Kriete A, Papazoglou E, Edrissi B, Pais H, Pourrezaei K. J. 2006. p. 22. COI number [1:STN:280:DC%2BD28zgsl2rtQ%3D%3D] [DOI] [PubMed]
- Jaiswal JK, Mattoussi H, Mauro JM, Simon SM. Nat. 2003. p. 1. [DOI] [PubMed]
- Ferrari M. Nat. Rev. Cancer. 2005. p. 3. [DOI] [PubMed]
- Kagan VE, Bayir H, Shvedova AA. Nanomedicine. 2005. p. 4. [DOI] [PubMed]
- Oberdorster G, Oberdorster E, Oberdorster J. Environ. 2005. p. 7. COI number [1:CAS:528:DC%2BD2MXntVyls7Y%3D] [DOI] [PMC free article] [PubMed]
- Blake DM, Maness PC, Huang Z, Wolfrum EJ, Huang J, Jacoby WA. Separ. 1999. p. 1. COI number [1:CAS:528:DyaK1MXjsVamsrc%3D] [DOI] [PMC free article] [PubMed]
- Zhang AP, Sun YP. World J. 2004. p. 21. COI number [1:CAS:528:DC%2BD2cXntFKjsL8%3D] [DOI] [PMC free article] [PubMed]
- Garabrant DH, Fine LJ, Oliver C, Bernstein L, Peters JM. Scand. J. Work. Environ. Health. 1987. p. 1. [DOI] [PubMed]
- Seo JW, Chung H, Kim MY, Lee J, Choi IH, Cheon J. Small. 2007. p. 5. [DOI] [PubMed]
- Mitchell RA, Liao H, Chesney J, Fingerle-Rowson G, Baugh J, David J, Bucala R. Proc. Natl. Acad. Sci. USA. 2002. p. 1. [DOI] [PMC free article] [PubMed]
- Medintz IL, Uyeda HT, Goldman ER, Mattoussi H. Nat. 2005. p. 6. [DOI] [PubMed]
- Derfus AM, Chan WC, Bhatia SN. Nano Lett. 2004. p. 1. [DOI] [PMC free article] [PubMed]
- Hoshino A, Fujioka K, Oku T, Suga M, Sasaki YF, Ohta T, Yasuhara M, Suzuki K, Yamamoto K. Nano Lett. 2004. p. 11. [DOI]
- Lovric J, Cho SJ, Winnik FM, Maysinger D. Chem. 2005. p. 11. [DOI] [PubMed]
- Ryman-Rasmussen JP, Riviere JE, Monteiro-Riviere NA. J. 2007. p. 1. [DOI] [PubMed]
- Zhang T, Stilwell JL, Gerion D, Ding L, Elboudwarej O, Cooke PA, Gray JW, Alivisatos AP, Chen FF. Nano Lett. 2006. p. 4. COI number [1:CAS:528:DC%2BD2sXhsVOiur8%3D] [DOI] [PMC free article] [PubMed]
- Choi AO, Cho SJ, Desbarats J, Lovric J, Maysinger D. J. 2007. p. 1. [DOI] [PMC free article] [PubMed]
- Panyam J, Zhou WZ, Prabha S, Sahoo SK, Labhasetwar V. FASEB J. 2002. p. 10. [DOI] [PubMed]
- Oh JM, Choi SJ, Kim ST, Choy JH. Bioconjug. 2006. p. 6. [DOI] [PubMed]
- Rejman J, Oberle V, Zuhorn IS, Hoekstra D. Biochem. 2004. p. 159. COI number [1:CAS:528:DC%2BD2cXis1Wnsg%3D%3D] [DOI] [PMC free article] [PubMed]
- Khalil IA, Kogure K, Akita H, Harashima H. Pharmacol. 2006. p. 1. [DOI] [PubMed]
- Koleske AJ, Baltimore D, Lisanti MP. Proc. Natl. Acad. Sci. USA. 1995. p. 5. [DOI] [PMC free article] [PubMed]
- Hewlett LJ, Prescott AR, Watts C. J. 1994. p. 5. [DOI] [PMC free article] [PubMed]
- Chavanpatil MD, Khdair A, Panyam J. J. 2006. p. 2651. COI number [1:CAS:528:DC%2BD28XhtVKgu7nN] [DOI] [PubMed]
- Win KY, Feng SS. Biomaterials. 2005. p. 15. [DOI] [PubMed]
- Koch AM, Reynolds F, Kircher MF, Merkle HP, Weissleder R, Josephson L. Bioconjug. 2003. p. 6. [DOI] [PubMed]
- Gupta B, Levchenko TS, Torchilin VP. Adv. 2005. p. 4. [DOI] [PubMed]
- Vives E, Brodin P, Lebleu B. J. 1997. p. 25. [DOI] [PubMed]
- Zorko M, Langel U. Adv. 2005. p. 4. [DOI] [PubMed]
- Hattori Y, Maitani Y. Cancer Gene Ther. 2005. p. 10. [DOI] [PubMed]
- Fahrni CJ. Curr. 2007. p. 2. [DOI] [PubMed]
- Makalowski W. Acta Biochim. 2001. p. 3. [PubMed]