

Abstract
In the twentieth century vaccine development has moved from the use of attenuated or killed micro-organisms to protein sub-unit vaccines, with vaccine immunogenicity assessed by measuring antibodies induced by vaccination. However, for many infectious diseases T cells are an important part of naturally acquired protective immune responses, and inducing these by vaccination has been the aim of much research. The progress that has been made in developing effective T-cell-inducing vaccines against viral and parasitic diseases such as HIV and malaria is discussed, along with recent developments in therapeutic vaccine development for chronic viral infections and cancer. Although many ways of inducing T cells by vaccination have been assessed, the majority result in low level, non-protective responses. Sufficient clinical research has now been conducted to establish that replication-deficient viral vectored vaccines lead the field in inducing strong and broad responses, and efficacy studies of T-cell-inducing vaccines against a number of diseases are finally demonstrating that this is a valid approach to filling the gaps in our defence against not only infectious disease, but some forms of cancer.
Keywords: adjuvants, clinical trials, T cell, vaccine, viral vectored vaccines
Introduction
Although variolation to protect against death from smallpox infection was practised for many centuries before Edward Jenner tested the use of cowpox as a safer alternative to using small doses of variola virus, Jenner's work marks the beginning of modern vaccinology; the use of attenuated, killed pathogens or a component of the whole pathogen to induce adaptive immunity and protect against illness and death following a future exposure to that pathogen. During the nineteenth century Pasteur created the first attenuated bacterial vaccine against cholera in chickens, and rabies, cholera, typhoid plague vaccines were used in humans. The twentieth century saw a huge expansion in the number of vaccines licensed for use against bacterial and viral diseases, and the second decade of the twenty-first century has been designated the ‘Decade of Vaccines (http://www.gatesfoundation.org/vaccines/Pages/decade-of-vaccines.aspx), with calls for more research and improved access to existing vaccines. One aspect of vaccinology that is rapidly expanding is the development of so-called T-cell-inducing vaccines; vaccines designed to induce CD4+ and/or CD8+ T cells of sufficient magnitude and necessary phenotype or effector function that directly contribute to pathogen clearance via cell-mediated effector mechanisms, rather than only CD4+ T-cell help for B cells leading to protective antibody responses. Although some highly effective antibody-inducing vaccines against viral and bacterial diseases are available, to protect against more complex pathogens it will be necessary to engage the other arm of the adaptive immune system; T cells. In this review I will present an overview of the current research in this area with a particular emphasis on clinical research, highlighting the challenges and successes, and look towards the future to see where and how these vaccines might be deployed.
T-cell vaccines in the past
The first vaccines to be used consisted of attenuated or killed pathogens. Live organisms or inactivated virus particles are likely to induce T cells as well as antibody responses, so were these early vaccines really examples of T-cell-inducing vaccines? Although Jenner famously used cowpox taken from a pustule on the hand of a milkmaid, vaccinia virus, used in the twentieth century as the smallpox vaccine, is genetically distinct from cowpox.1 The precise origins of vaccinia virus will never be known, but it is possible that it was derived from a horsepox infection of a cow, followed by multiple passages of the virus from human to human, because material from the site of a recent inoculation was commonly used to vaccinate others. The mechanism of protection induced by vaccinia was also unknown at the time of smallpox eradication, but recent studies in primates indicate that whereas B cells are required for protection against smallpox, T cells are required to control the spread of the vaccinia virus itself following vaccination.2 A T-cell response to the vaccine is clearly induced and persists for many years, but is more important for the safety of the vaccine than for the desired protection against variola.
The rabies vaccine first used by Pasteur contained live virus, but was replaced in the early twentieth century by phenol-inactivated preparations, and inactivated vaccines are still in use today.3 The primary correlate of vaccine-induced protection is the presence of neutralizing antibody, and there does not appear to be a role for a cytotoxic T-cell response in protection.4 Hence the induction of T-cell responses by these two early vaccines was not an important protective mechanism. However, for the vaccine against tuberculosis first used in the 1920s and still in use today, a T-cell response is essential for protection. Bacille Calmette–Guérin (BCG) is a live attenuated strain of Mycobacterium bovis, the causative agent of bovine tuberculosis and a close relative of Mycobacterium tuberculosis. Both M. bovis and M. tuberculosis are intracellular bacteria, and are therefore shielded from attack by antibodies. Both CD4+ and CD8+ T cells are involved in protection against disease,5 although BCG is less efficient at priming CD8+ T-cell responses.6 BCG may therefore be considered the first T-cell-inducing vaccine, and is still the only licensed vaccine thought to work primarily through T-cell responses, but with its highly variable efficacy,7 does not represent a good model to follow for the future. However, much progress has been made in vaccination to induce protective T-cell responses.
Malaria
Although malaria transmission is declining in some parts of Africa, other countries are experiencing increases in the number of cases.8 No vaccine is available and control of malaria is almost entirely dependent on treatment of individual clinical episodes, which has become less effective as resistance to the anti-malarial drug chloroquine has spread across Africa.9 However, novel approaches that may also have benefits against other diseases are being evaluated in malaria vaccine development.10 The first effective vaccination of humans against malaria was reported in 1973, demonstrating that prophylactic vaccination against a protozoan parasite that employs many approaches to evading the human immune response could be achieved.11 However, the vaccination consisted of the bites of thousands of irradiated malaria-infected mosquitoes and was not considered a method suitable for mass deployment.
The complex life cycle of Plasmodium falciparum offers numerous opportunities for attack by the host's immune system, reviewed in ref. 10. The most advanced malaria vaccine in development, RTS,S, contains the repeat (R) and T-cell epitope (T) regions of the immunodominant CircumSporozoite Protein (CSP), which covers the exterior of the parasite when it first enters the body following the bite of an infected mosquito. The R and T regions are fused to the hepatitis B surface (S) antigen to form protein particles in the presence of additional S antigen, and administered with an adjuvant. Antibodies to R, if present at high titre,12 can neutralize the sporozoite before infection of hepatocytes occurs. Vaccine efficacy has been tested in a number of field trials in children in Africa, and in a trial of 2022 children in Mozambique vaccine was found to be 35% effective at preventing infection and 49% effective against severe malaria.13 Although proliferative T-cell responses to the vaccine can be detected, CD8+ T-cell responses to the vaccine antigen are not induced.14 A multi-centre phase III trial with this vaccine is now underway,15 but other research attempting to achieve protection via CD8+ T cells recognizing antigens expressed during the intra-hepatic stage of the parasite's life cycle is also progressing.
T cells that recognize and kill infected hepatocytes within the first week of infection have been the focus of much malaria vaccine development, as they provide the opportunity to stop the infection when a small number of parasites are present, and before any disease symptoms occur, so preventing illness in the immunized individual and blocking onward transmission. However, to achieve this, the vaccine must have an extremely high level of efficacy, as any parasites that are not destroyed within the first week will develop into blood-stage parasites and migrate out of the liver to infect erythrocytes. Partial efficacy is determined by the delay in detection of blood-stage parasites following malaria challenge.
The first attempt to compare multiple approaches to inducing protective T-cell responses against malaria antigens was described by Allsopp et al.16 employing a single immunodominant epitope from the rodent malaria Plasmodium berghei CSP antigen and testing the magnitude of CD8+ responses following immunization of mice with a number of different delivery systems suitable for human use. The strongest responses were induced following immunization with either a virus-like particle carrying the minimal epitope, or a lipopeptide, although the disadvantage of delivering a single epitope is that a large number of defined epitopes will be required to achieve vaccination coverage of an entire human population. Subsequent research substantially increased the number of epitopes carried on the virus-like particle using a multiple epitope (ME) string17 but immunization with first the protein particle and then a recombinant replication-deficient poxvirus (Modified Vaccinia virus Ankara; MVA) expressing the P. berghei CSP was required to achieve protection against challenge of the mice with P. berghei sporozoites.18 One advantage of the recombinant MVA is that the complete coding sequence of the malaria antigen could be expressed rather than a single epitope or string of epitopes linked together, obviating the requirement to define epitopes for multiple HLA types. The same is true of DNA vaccines, which were also tested in this mouse malaria model and found to be immunogenic, although again to achieve protection in this model, it was necessary to prime the immune response with the DNA vaccine and subsequently boost with the recombinant MVA, resulting in a greatly increased level of antigen-specific CD8+ T cells that correlated with protection.19
Following the demonstration of vaccine efficacy in the mouse malaria model, clinical trials of the DNA and MVA-vectored vaccines began, employing the ME string of T-cell epitopes fused to the complete coding sequence of the Thrombospondin-Related Adhesion Protein (TRAP) of P. falciparum. These trials were ground-breaking studies; the first trials of a recombinant MVA vaccine in healthy volunteers, the first use of a polyepitope vaccine in humans and the first test of DNA prime/recombinant virus boost in humans. Vaccine safety was therefore carefully assessed, and the vaccines were found to be well tolerated,20 immunogenic,21 and partially protective against malaria challenge when the ME-TRAP antigen was employed, but not when CSP was used instead.22 One out of eight malaria-naive volunteers immunized with DNA/DNA/MVA ME-TRAP was protected against P. falciparum sporozoite challenge, with the group as a whole experiencing a delay in the time to merozoites appearing in the blood, indicating that some of the intra-hepatic parasites had been killed. There was a significant positive correlation with peak ex vivo interferon-γ ELISpot response to ME-TRAP and time to malaria diagnosis. From these early trials it appeared possible to induce the required T-cell responses by vaccination and achieve at least partial protection against liver-stage infection using the stringent challenge model in which malaria-naive vaccinated or control (unvaccinated) volunteers receive the bites of five highly infectious mosquitoes. However, in a subsequent trial of semi-immune adults in The Gambia, although the vaccine was again safe and immunogenic, there was no significant vaccine efficacy against natural exposure to malaria infection.23
Further work employing the mouse malaria model demonstrated enhanced CD8+ responses and protective efficacy when the DNA vaccine was replaced by a recombinant avipoxvirus, fowlpox FP9,24 and this regimen was then tested in further clinical studies, again with ME-TRAP as the antigen. Sterile protection against challenge was achieved in some subjects, lasting as long as 20 months after vaccination,25 but again, although the vaccines were safe 26 and immunogenic in a malaria-exposed population27 there was no significant vaccine efficacy.28 Replacement of the avipoxvirus prime with a recombinant replication-deficient human adenovirus serotype 5 (Ad5) resulted in enhanced immunogenicity and protective efficacy in pre-clinical studies,29 but in humans, particularly in Africa, seroprevalence for Ad5 can reach 90%,30,31 and pre-existing immunity has been shown to reduce the immunogenicity of Ad5-vectored vaccines in animal models.32 To avoid this limitation on immunogenicity, less common serotypes such as Ad26 or Ad35,33 or simian adenoviruses34 have been employed as vaccine vectors, because the seroprevalence of these vectors is considerably lower than for Ad5.35–37
Clinical studies with Ad26-vectored and Ad35-vectored malaria vaccines have not been reported, but a number of clinical trials have been completed with the simian adenovirus vector ChAd63 and the first of these has now been published.38 Using a blood-stage malaria antigen, merozoite surface protein 1 (MSP1), priming with ChAd63 and boosting with MVA induced exceptionally strong T-cell responses, with a mixed CD4+/CD8+ phenotype, and also high antibody titres to the vaccine antigen. The use of this vector combination marks a new stage in T-cell-inducing vaccine development, resulting in the highest T-cell responses yet reported in clinical studies as well as very strong antibody responses using a two-dose regimen of replication-deficient viral vectors with no adjuvant. The incremental increases in T-cell immunogenicity obtained by use of different vaccination regimens are depicted in Table 1 and Fig. 1.
Table 1.
Immunogenicity of T-cell-inducing vaccines in clinical trials, assessed by ELISpot assay
| Position in Fig. 1 | Disease | Description | ELISpot response SFU/106 PBMC | Reference |
|---|---|---|---|---|
| 1 | Malaria | DNA METRAP 3 doses of 1 mg | 55 | 21 |
| 2 | MVA METRAP 2 doses 6 × 107 PFU | 195 | 21 | |
| 3 | DNA/MVA METRAP 3 doses DNA1 μg, 1 dose MVA 1·5 × 108 PFU | 1430 | 21 | |
| 4 | FP9/MVA METRAP 2 doses FP9 1·0 × 108 PFU, 1 dose MVA1·5 × 108 PFU | 600 | 25 | |
| 5 | DNA/DNA/FP9/MVA METRAP 2 doses DNA 2 mg 1 dose FP9 1·0 × 108 PFU, 1 dose MVA1·5 × 108 PFU | 400 | 25 | |
| 6 | ChAd63 MSP1 1 dose 5 × 1010 vp | 2785 | 38 | |
| 7 | ChAd63/MVA MSP1 1 dose ChAD63 5 × 1010 vp, 1 dose MVA 5 × 108 PFU | 5090 | 38 | |
| 8 | HIV | Ad5 gag/pol/nef Summed response in subjects with no pre-existing responses to Ad5, 3 doses 3 × 1010 vp Cases | 1765 | 75 |
| 9 | Non-cases ALVAC-HIV Values not reported. Scored as +ve if more than 55 and above background, up to 11% were +ve | 1117 | 76 | |
| 10 | Influenza | MVA-NP+M1 influenza 1 dose 2·5 × 108 PFU | 1443 | 49 |
| 11 | Tuberculosis | BCG 100 μl BCG Glaxo | 38 | 52 |
| 12 | MVA-85A 1 dose 5 × 107 PFU | 1365 | 52 | |
| 13 | BCG/MVA 100 μl BCG Glaxo, 1 dose 5 × 107 PFU | 3248 | 52 | |
| 14 | Prostate cancer | Sipuleucel-T autologous PBMC therapy; mean of two patients | 183 | 77 |
PBMC, peripheral blood mononuclear cells; PFU, plaque-forming units; SFU, spot-forming units; vp, virus particles.
Figure 1.
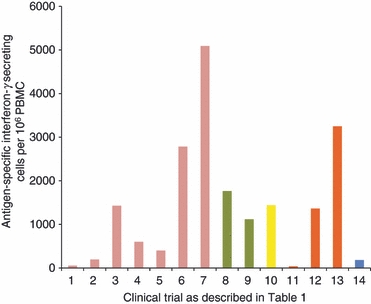
Immunogenicity of T-cell-inducing vaccines in clinical trials, assessed by ELISpot assay. PBMC, peripheral blood mononuclear cells.
HIV
After three decades of intensive research into HIV, there is much that is not understood about immune control of the virus and only one vaccine trial has demonstrated any efficacy in humans. Anti-viral drugs have increased the lifespan of those infected and reduced vertical transmission in Africa, which currently has the highest prevalence of seropositive adults, but in Eastern Europe the number of new cases is increasing. Although other factors may be involved, there is strong evidence for the role of CD8+ T cells specific for viral antigens in preventing and controlling infection, and much vaccine research has focused on inducing these responses.
The Step trial was a phase II study of Merck's Ad5-vectored gag/pol/nef vaccine in a three-dose regimen in 3000 volunteers with varying levels of pre-existing immunity to Ad5. The trial was stopped after a planned interim analysis demonstrated no likelihood of detecting vaccine efficacy if the trial continued. Unexpectedly, HIV infections were found to have increased in a subpopulation of the vaccinated cohort; uncircumcised men with pre-existing neutralizing antibodies to Ad5.39 No explanation has yet been found for this observation, which was a non-significant trend discovered during post hoc analysis and not repeated in a second study.40 The vaccine was clearly not effective, although it was deemed immunogenic, with 77% of vaccinees showing positive responses in ELISpot assays, and the majority of those recognizing two or three HIV antigens, as had previously been found in a phase I study of the same vaccine. However, compared with heterologous prime/boost regimens used to induce T-cell responses in malaria vaccine trials, the magnitude of the response was low (Fig. 1).
The only HIV vaccine trial to demonstrate efficacy in humans was the much criticized Rv144 trial involving > 16 400 volunteers at risk of HIV infection in Thailand.41 The vaccination schedule employed four doses of a recombinant canarypoxvirus (ALVAC) expressing gag, pro and env, along with recombinant gp120 protein with alum adjuvant at the last two doses. Earlier studies had shown that the ALVAC-vectored vaccine was poorly immunogenic and no efficacy had been demonstrated in a trial of the gp120 alone. Despite widespread scepticism, vaccine efficacy was 61% after the first year (after a retrospective analysis) and 31% after 3·5 years. The immune correlates of protection are not understood, and although the importance of the study is clear, the way forward is not.
Therapeutic HIV vaccine development has been explored, with good results in boosting both CD4+ and CD8+ responses to HIV antigens in seropositive individuals. Responses appear to broaden to recognize more epitopes following vaccination although it is not yet known whether this is simply a result of increasing many low-level pre-existing responses above the threshold of detection as opposed to priming new responses.42 Vaccination of seropositive individuals following viral load reduction with highly active anti-retroviral therapy (HAART) may provide an opportunity to safely interrupt HAART while maintaining viral containment.
Hepatitis C virus
Both therapeutic and prophylactic vaccination regimens against hepatitis C virus, in which T cells for non-structural proteins are primed or boosted are also showing promise, with adenovirus-vectored vaccines highly immunogenic in healthy volunteers43 and an MVA-vectored vaccine resulting in viral load reductions associated with T-cell response in a small clinical study.44
Influenza
The naturally acquired immune response to influenza includes CD8+ T cells recognizing conserved internal viral antigens as well as antibodies to the highly variable surface proteins, in particular haemagglutinin. Only haemagglutinin is induced by vaccination with inactivated influenza vaccines against seasonal influenza, and whereas live attenuated vaccines are able to prime T-cell responses in influenza-naive children, they do not boost pre-existing responses in adults. However, the continual antigenic drift in haemagglutinin requires annual vaccine reformulation and revaccination, and on average, 1 year in 20 the vaccine is not a good match for the circulating virus, resulting in low vaccine efficacy, which is at best around 80% and generally only around 40% in older adults.45,46
To provide useful protection against an acute infection, T cells specific for influenza must be present as effector or effector memory cells in the upper respiratory tract, ready to recognize and kill virus-infected cells. A central memory response can be maintained for many years following recovery from viral infection, but although it may result in a more rapid recovery from a severe infection, it is not likely to affect the course of a mild infection or prevent virus shedding and onward transmission. The half-life of circulating T cells specific to influenza has been calculated at 2 or 3 years,47 and without re-exposure to the virus, people therefore become susceptible to infection and illness over time. Boosting this T-cell response by vaccination could provide continued protection against subsequent infection, even following exposure to a virus of a different subtype to the initial infection.48
Using a recombinant replication-deficient MVA poxvirus to boost T-cell responses to conserved internal influenza antigens (nucleoprotein and matrix protein 1) has now been tested in a clinical trial, with the result that a large expansion in circulating influenza-specific T cells was observed after a single vaccination (Fig. 1).49 An influenza challenge efficacy study has also been completed in a phase IIa study (Lillie et al., manuscript submitted), and further development of the vaccine is planned.
Tuberculosis
Although BCG administered to adolescents in tropical regions may have extremely low efficacy against pulmonary tuberculosis (TB), vaccinating within the first few months of life has a higher degree of efficacy against disseminated TB in childhood, and is widely used.7,50 Development of a TB vaccine has concentrated on improving the level of efficacy of BCG rather than throwing the baby out with the bathwater. This has led to more immunogenic versions of BCG, or boosting vaccines that increase the T-cell response to mycobacterial antigens after an initial BCG vaccination early in life.51
The most advanced of these improved vaccines is MVA-85A, using the replication-deficient poxvirus to deliver the immunodominant antigen 85A as a booster following BCG vaccination. Although T-cell responses to BCG antigens are low, there are many of them to antigens shared with M. tuberculosis, and it is likely to be this broad response that provides protection. However, using MVA-85A, the T-cell response to that single antigen increases dramatically (Fig. 1).52 The first efficacy study of this vaccine is now underway in South Africa, with results expected in 2012. However, using the bovine/M. bovis model, vaccine efficacy has already been demonstrated.53 Boosting BCG with MVA-85A or Ad5-8A resulted in improved efficacy over the use of BCG alone. This bodes well for the future deployment of a more effective TB vaccine.
Cancer
A prophylactic anti-cancer vaccine has now been licensed, for use in adolescent girls, to induce antibodies that prevent human papillomavirus infection, which may lead to cervical cancer. However, therapeutic vaccines are also in development, with the aim of recruiting T cells to destroy cancerous cells within the body. It is likely that these would be used in conjunction with other established treatments, for example surgery to remove a solid tumour along with vaccination to prevent metastases and recurrence of the cancer. The first of these to demonstrate efficacy in a phase III trial is quite unlike any prophylactic vaccine in current use. Sipuleucel-T (also known as Provenge) is an autologous cellular therapy produced from the patient's white blood cells collected by leukapheresis and cultured with a recombinant fusion protein of human prostatic acid phosphatase with granulocyte–macrophage colony-stimulating factor.54 Despite inducing a T-cell response that was barely measureable by ELISpot, (Fig. 1) median survival time of the treatment group was increased by 4 months.
Colorectal adenocarcinoma, which has a lifetime risk of 5–6%, is commonly treated by surgery, but in around a third of cases surgery alone does not provide a cure, and metastases are common.55 Viral vectored vaccines have been tested for this form of cancer. Using ALVAC with p53 as the antigen, immune responses were directed against the viral vector rather than p53 in the majority of patients. In a different trial using MVA expressing the oncofetal antigen 5T4 (TroVax), strong proliferative responses recognizing the antigen were obtained and in a phase II study this was associated with longer survival.56,57 TroVax has also been assessed for hormone-refractory prostate cancer and renal cell carcinoma.58
Antigen delivery for T-cell induction
What conclusions can we draw from the above to aid us in choosing the type of vaccine to use for induction of protective T-cell responses? DNA vaccines were at one time thought to be the ideal way to induce T-cell responses. After injection they express the encoded antigen inside the host cells resulting in both cellular and humoral immunity. They can be manipulated to express cytokines or other molecules intended to enhance the immune response, and are simple to produce.59 Unfortunately, the early successes in pre-clinical studies did not translate into clinical trials, and whereas DNA vaccines are safe to use and do induce T-cell responses in humans, they are of a very low magnitude.21 Efforts to increase immunogenicity by use of a ‘gene-gun’ resulted in more efficient delivery such that the dose could be considerably reduced, but the response was not increased, and although efforts to find an adjuvant to increase the immunogenicity of DNA vaccines in humans, success has so far been modest.60 The same is true of peptide vaccines.61,62
Other research has concentrated on developing adjuvants to increase the immunogenicity of protein vaccines, reviewed in ref. 63, but again although responses can be induced in pre-clinical studies, they are not of high magnitude and in many cases have not yet been tested in clinical studies. Of all the replication-deficient viral vectors available, adenovirus is the most potent in priming T-cell responses to the recombinant antigen, and recent studies employing simian adenoviruses or rare human serotypes appear to hold the most promise. As extremely high T-cell responses can be primed with a single dose,38 the problem of anti-vector immunity is largely avoided.
Recombinant replication-deficient poxviruses have also been used with considerable success, and when used to boost an existing response rather than prime a novel one, the response becomes focused on the recombinant antigen rather than inducing significant anti-vector immunity.64 Replication-deficient viral vectors can be manufactured at large scale, thermostable formulations are available65 and the number of clinical trials now completed or underway is testament to the broad utility of these vectors.66
Complex therapeutic regimens requiring ex vivo treatment of patient's lymphocytes have been trialled for use against cancer, but there is no plausible reason why the most immunogenic vaccination regimens tested for infectious diseases should not also be highly effective at inducing T-cell responses against cancer antigens. Pre-clinical studies demonstrating that tolerance to self-antigens can be broken have been performed.67
Antigens and epitopes
Both BCG and irradiated sporozoite vaccines induce T-cell responses to a large number of antigens, but for most diseases vaccine development must rely on the use of a small number of antigens. The choice of antigen(s) is determined by whether T-cell responses against the antigen have been found to be protective, either in humans or animal models, and the degree of polymorphism found between different isolates, with highly conserved antigens clearly favoured, although it is possible to combine conserved regions from more than one protein to produce a synthetic vaccine antigen.68,69 It is preferable to include more than one antigen in the vaccine to reduce the likelihood of immune escape.69 T-cell epitopes within the antigens may be identified either by bioinformatics prediction and experimental confirmation, or by taking an empirical approach using a library of peptides spanning the complete antigen sequence.70 However, although knowledge of epitopes presented by common HLA types may be helpful in conducting detailed phenotypic studies of T-cell responses in clinical research and vaccine development, a complete knowledge of the possible epitopes contained within the antigen(s) is not necessary, and only becomes important when using delivery systems with severe restrictions on the length of peptide sequence that can be encoded.
Looking to the future
Over the last decade we have learned that vaccines tested in animal models are generally more immunogenic and more protective than the same vaccines in clinical studies, but the hierarchy of immunogenicity is maintained, and although small clinical studies should be viewed as an essential part of vaccine development, there is still a role for animal models in prioritizing the candidates for clinical trials.
Viewed from outside the field, HIV vaccine research appears to have been trying to run before it can walk. Extremely large, long and expensive field efficacy trials should follow small, short and less expensive trials that indicate strong immunogenicity and a plausible mechanism for protection. Diverse strategies should be tested and compared, with detailed immunological investigation to hunt for immune responses associated with protection. The scientific strategic plan (2010) of the Global HIV Vaccine Enterprise (GHAVE) now states that ‘clinical efficacy trials should not be perceived as the culmination of a series of basic science experiments but rather as an integral part of the discovery process’.71 Following the approaches recommended by GHAVE, we may now expect to see steady progress in HIV vaccine assessment, but patience on the part of researchers and funders will be required before any major breakthrough in prophylactic HIV vaccine development is achieved.
If early successes in small-scale trials are to be built on, it will be necessary to understand the reasons for success and optimize them. Makedonas and Betts72 argue for a broader range of assays to be used in vaccine trials, in particular assessing anti-viral activity, and this fits well with small clinical studies in which a wide range of exploratory assays may be used before refining the approach for larger trials. However, this should be coupled with the use of highly immunogenic vaccine regimens, because a detailed assessment of a low to moderate immune response is considerably less informative than when the response is strong. Multi-parameter flow cytometry is a powerful tool, but only when the number of cells available for analysis is sufficient to provide meaningful numbers in multiple subgroups. Global gene expression studies are becoming an important means of assessing responses to vaccination and have the potential to help define immune ‘signatures of protection’ rather than relying on a single assay readout to identify what is likely to be a complex series of immune system interactions that lead to protection against difficult pathogens.73
For therapeutic cancer vaccines, the challenge is now to find a non-patient-specific, more immunogenic method of inducing the required immune response, which requires breaking immunological tolerance to a self-antigen. Again, highly immunogenic regimens should be assessed. Therapeutic vaccination is likely to be a growth area for treatment of chronic viral infections.
Recent clinical trials have now demonstrated efficacy of T-cell-inducing vaccines against a number of diseases, and although many approaches to assessing protective T-cell responses may be taken, the ELISpot assay has become established as the most suitable means of determining vaccine immunogenicity.74 The magnitude of the ELISpot response required for protection will need to be determined for each vaccine and disease, but it is now possible to envisage licensing vaccines based on T-cell immunogenicity and clinical efficacy, without any requirement for humoral immunity.
Finally, it must be recognized that the most effective way to immunize against many infectious diseases is likely to be to employ both cellular and humoral immune responses against the pathogen. Having recognized that the induction of protective T-cell-mediated responses is essential in some areas of vaccine development, we should not neglect antibody induction. The ‘Decade of Vaccines’ will see important developments in this area.
Acknowledgments
SCG is a Jenner Investigator and receives financial support from the Medical Research Council, UK, the Wellcome Trust, and the Oxford Biomedical Research Centre. I am grateful to Simon Draper and Teresa Lambe for their helpful comments on the manuscript.
Disclosures
SCG holds a number of patents relating to means of inducing T-cell responses by vaccination.
References
- 1.Gubser C, Hue S, Kellam P, Smith GL. Poxvirus genomes: a phylogenetic analysis. J Gen Virol. 2004;85(Pt 1):105–17. doi: 10.1099/vir.0.19565-0. [DOI] [PubMed] [Google Scholar]
- 2.Gordon SN, Cecchinato V, Andresen V, et al. Smallpox vaccine safety is dependent on T cells and not B cells. J Infect Dis. 2011;203:1043–53. doi: 10.1093/infdis/jiq162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kammer AR, Ertl HC. Rabies vaccines: from the past to the 21st century. Hybrid Hybridomics. 2002;21:123–7. doi: 10.1089/153685902317401726. [DOI] [PubMed] [Google Scholar]
- 4.Johnson N, Cunningham AF, Fooks AR. The immune response to rabies virus infection and vaccination. Vaccine. 2010;28:3896–901. doi: 10.1016/j.vaccine.2010.03.039. [DOI] [PubMed] [Google Scholar]
- 5.Thaiss CA, Kaufmann SH. Toward novel vaccines against tuberculosis: current hopes and obstacles. Yale J Biol Med. 2010;83:209–15. [PMC free article] [PubMed] [Google Scholar]
- 6.Ryan AA, Nambiar JK, Wozniak TM, Roediger B, Shklovskaya E, Britton WJ, Fazekas de St Groth B, Triccas JA. Antigen load governs the differential priming of CD8 T cells in response to the bacille Calmette–Guérin vaccine or Mycobacterium tuberculosis infection. J Immunol. 2009;182:7172–7. doi: 10.4049/jimmunol.0801694. [DOI] [PubMed] [Google Scholar]
- 7.Colditz GA, Brewer TF, Berkey CS, Wilson ME, Burdick E, Fineberg HV, Mosteller F. Efficacy of BCG vaccine in the prevention of tuberculosis. Meta-analysis of the published literature. JAMA. 1994;271:698–702. [PubMed] [Google Scholar]
- 8.Okiro EA, Bitira D, Mbabazi G, Mpimbaza A, Alegana VA, Talisuna AO, Snow RW. Increasing malaria hospital admissions in Uganda between 1999 and 2009. BMC Med. 2011;9:37. doi: 10.1186/1741-7015-9-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Snow RW, Trape JF, Marsh K. The past, present and future of childhood malaria mortality in Africa. Trends Parasitol. 2001;17:593–7. doi: 10.1016/s1471-4922(01)02031-1. [DOI] [PubMed] [Google Scholar]
- 10.Moorthy VS, Good MF, Hill AV. Malaria vaccine developments. Lancet. 2004;363:150–6. doi: 10.1016/S0140-6736(03)15267-1. [DOI] [PubMed] [Google Scholar]
- 11.Clyde DF, Most H, McCarthy VC, Vanderberg JP. Immunization of man against sporozoite-induced falciparum malaria. Am J Med Sci. 1973;266:169–77. doi: 10.1097/00000441-197309000-00002. [DOI] [PubMed] [Google Scholar]
- 12.Olotu A, Lusingu J, Leach A, et al. Efficacy of RTS,S/AS01E malaria vaccine and exploratory analysis on anti-circumsporozoite antibody titres and protection in children aged 5-17 months in Kenya and Tanzania: a randomised controlled trial. Lancet Infect Dis. 2011;11:102–9. doi: 10.1016/S1473-3099(10)70262-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alonso PL, Sacarlal J, Aponte JJ, et al. Duration of protection with RTS,S/AS02A malaria vaccine in prevention of Plasmodium falciparum disease in Mozambican children: single-blind extended follow-up of a randomised controlled trial. Lancet. 2005;366:2012–8. doi: 10.1016/S0140-6736(05)67669-6. [DOI] [PubMed] [Google Scholar]
- 14.Lalvani A, Moris P, Voss G, et al. Potent induction of focused Th1-type cellular and humoral immune responses by RTS,S/SBAS2, a recombinant Plasmodium falciparum malaria vaccine. J Infect Dis. 1999;180:1656–64. doi: 10.1086/315074. [DOI] [PubMed] [Google Scholar]
- 15.Regules JA, Cummings JF, Ockenhouse CF. The RTS,S vaccine candidate for malaria. Expert Rev Vaccines. 2011;10:589–99. doi: 10.1586/erv.11.57. [DOI] [PubMed] [Google Scholar]
- 16.Allsopp CE, Plebanski M, Gilbert S, et al. Comparison of numerous delivery systems for the induction of cytotoxic T lymphocytes by immunization. Eur J Immunol. 1996;26:1951–9. doi: 10.1002/eji.1830260841. [DOI] [PubMed] [Google Scholar]
- 17.Gilbert SC, Hill AV. Protein particle vaccines against malaria. Parasitol Today. 1997;13:302–6. doi: 10.1016/s0169-4758(97)01091-0. [DOI] [PubMed] [Google Scholar]
- 18.Plebanski M, Gilbert SC, Schneider J, et al. Protection from Plasmodium berghei infection by priming and boosting T cells to a single class I-restricted epitope with recombinant carriers suitable for human use. Eur J Immunol. 1998;28:4345–55. doi: 10.1002/(SICI)1521-4141(199812)28:12<4345::AID-IMMU4345>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 19.Schneider J, Gilbert SC, Hannan CM, Degano P, Prieur E, Sheu EG, Plebanski M, Hill AV. Induction of CD8+ T cells using heterologous prime-boost immunisation strategies. Immunol Rev. 1999;170:29–38. doi: 10.1111/j.1600-065x.1999.tb01326.x. [DOI] [PubMed] [Google Scholar]
- 20.Moorthy VS, McConkey S, Roberts M, et al. Safety of DNA and modified vaccinia virus Ankara vaccines against liver-stage P. falciparum malaria in non-immune volunteers. Vaccine. 2003;21:2004–11. doi: 10.1016/s0264-410x(02)00771-5. [DOI] [PubMed] [Google Scholar]
- 21.McConkey SJ, Reece WH, Moorthy VS, et al. Enhanced T-cell immunogenicity of plasmid DNA vaccines boosted by recombinant modified vaccinia virus Ankara in humans. Nat Med. 2003;9:729–35. doi: 10.1038/nm881. [DOI] [PubMed] [Google Scholar]
- 22.Dunachie SJ, Walther M, Epstein JE, et al. A DNA prime-modified vaccinia virus ankara boost vaccine encoding thrombospondin-related adhesion protein but not circumsporozoite protein partially protects healthy malaria-naive adults against Plasmodium falciparum sporozoite challenge. Infect Immun. 2006;74:5933–42. doi: 10.1128/IAI.00590-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moorthy VS, Imoukhuede EB, Milligan P, et al. A randomised, double-blind, controlled vaccine efficacy trial of DNA/MVA ME-TRAP against malaria infection in Gambian adults. Plos Med. 2004;1:e33. doi: 10.1371/journal.pmed.0010033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anderson RJ, Hannan CM, Gilbert SC, et al. Enhanced CD8+ T cell immune responses and protection elicited against Plasmodium berghei malaria by prime boost immunization regimens using a novel attenuated fowlpox virus. J Immunol. 2004;172:3094–100. doi: 10.4049/jimmunol.172.5.3094. [DOI] [PubMed] [Google Scholar]
- 25.Webster DP, Dunachie S, Vuola JM, et al. Enhanced T cell-mediated protection against malaria in human challenges by using the recombinant poxviruses FP9 and modified vaccinia virus Ankara. Proc Natl Acad Sci USA. 2005;102:4836–41. doi: 10.1073/pnas.0406381102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bejon P, Peshu N, Gilbert SC, et al. Safety profile of the viral vectors of attenuated fowlpox strain FP9 and modified vaccinia virus Ankara recombinant for either of 2 preerythrocytic malaria antigens, ME-TRAP or the circumsporozoite protein, in children and adults in Kenya. Clin Infect Dis. 2006;42:1102–10. doi: 10.1086/501459. [DOI] [PubMed] [Google Scholar]
- 27.Bejon P, Mwacharo J, Kai OK, et al. Immunogenicity of the candidate malaria vaccines FP9 and modified vaccinia virus Ankara encoding the pre-erythrocytic antigen ME-TRAP in 1-6 year old children in a malaria endemic area. Vaccine. 2006;24:4709–15. doi: 10.1016/j.vaccine.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 28.Bejon P, Mwacharo J, Kai O, et al. A phase 2b randomised trial of the candidate malaria vaccines FP9 ME-TRAP and MVA ME-TRAP among children in Kenya. PLoS Clin Trials. 2006;1:e29. doi: 10.1371/journal.pctr.0010029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gilbert SC, Schneider J, Hannan CM, Hu JT, Plebanski M, Sinden R, Hill AV. Enhanced CD8 T cell immunogenicity and protective efficacy in a mouse malaria model using a recombinant adenoviral vaccine in heterologous prime-boost immunisation regimes. Vaccine. 2002;20:1039–45. doi: 10.1016/s0264-410x(01)00450-9. [DOI] [PubMed] [Google Scholar]
- 30.Nwanegbo E, Vardas E, Gao W, Whittle H, Sun H, Rowe D, Robbins PD, Gambotto A. Prevalence of neutralizing antibodies to adenoviral serotypes 5 and 35 in the adult populations of The Gambia, South Africa, and the United States. Clin Diagn Lab Immunol. 2004;11:351–7. doi: 10.1128/CDLI.11.2.351-357.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kostense S, Koudstaal W, Sprangers M, et al. Adenovirus types 5 and 35 seroprevalence in AIDS risk groups supports type 35 as a vaccine vector. AIDS. 2004;18:1213–6. doi: 10.1097/00002030-200405210-00019. [DOI] [PubMed] [Google Scholar]
- 32.Yang ZY, Wyatt LS, Kong WP, Moodie Z, Moss B, Nabel GJ. Overcoming immunity to a viral vaccine by DNA priming before vector boosting. J Virol. 2003;77:799–803. doi: 10.1128/JVI.77.1.799-803.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Radosevic K, Rodriguez A, Lemckert AA, et al. The Th1 immune response to Plasmodium falciparum circumsporozoite protein is boosted by adenovirus vectors 35 and 26 with a homologous insert. Clin Vaccine Immunol. 2010;17:1687–94. doi: 10.1128/CVI.00311-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hill AV, Reyes-Sandoval A, O'Hara G, et al. Prime-boost vectored malaria vaccines: progress and prospects. Hum Vaccin. 2010;6:78–83. doi: 10.4161/hv.6.1.10116. [DOI] [PubMed] [Google Scholar]
- 35.Ersching J, Hernandez MI, Cezarotto FS, et al. Neutralizing antibodies to human and simian adenoviruses in humans and New-World monkeys. Virology. 2010;407:1–6. doi: 10.1016/j.virol.2010.07.043. [DOI] [PubMed] [Google Scholar]
- 36.Barouch DH, Kik SV, Weverling GJ, et al. International seroepidemiology of adenovirus serotypes 5, 26, 35, and 48 in pediatric and adult populations. Vaccine. 2011;29:5203–9. doi: 10.1016/j.vaccine.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dudareva M, Andrews L, Gilbert SC, et al. Prevalence of serum neutralizing antibodies against chimpanzee adenovirus 63 and human adenovirus 5 in Kenyan children, in the context of vaccine vector efficacy. Vaccine. 2009;27:3501–4. doi: 10.1016/j.vaccine.2009.03.080. [DOI] [PubMed] [Google Scholar]
- 38.Sheehy SH, Duncan CJA, Elias SC, et al. Phase Ia clinical evaluation of the Plasmodium falciparum blood-stage antigen MSP1 in ChAd63 and MVA vaccine vectors. Mol Ther. 2011 doi: 10.1038/mt.2011.176. doi: 10.1038/mt.2011.176 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Buchbinder SP, Mehrotra DV, Duerr A, et al. Efficacy assessment of a cell-mediated immunity HIV-1 vaccine (the Step Study): a double-blind, randomised, placebo-controlled, test-of-concept trial. Lancet. 2008;372:1881–93. doi: 10.1016/S0140-6736(08)61591-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gray G, Buchbinder S, Duerr A. Overview of STEP and Phambili trial results: two phase IIb test-of-concept studies investigating the efficacy of MRK adenovirus type 5 gag/pol/nef subtype B HIV vaccine. Curr Opin HIV AIDS. 2010;5:357–61. doi: 10.1097/COH.0b013e32833d2d2b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rerks-Ngarm S, Pitisuttithum P, Nitayaphan S, et al. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N Eng J Med. 2009;361:2209–20. doi: 10.1056/NEJMoa0908492. [DOI] [PubMed] [Google Scholar]
- 42.Dorrell L, Yang H, Ondondo B, et al. Expansion and diversification of virus-specific T cells following immunization of human immunodeficiency virus type 1 (HIV-1)-infected individuals with a recombinant modified vaccinia virus Ankara/HIV-1 Gag vaccine. J Virol. 2006;80:4705–16. doi: 10.1128/JVI.80.10.4705-4716.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Halliday J, Klenerman P, Barnes E. Vaccination for hepatitis C virus: closing in on an evasive target. Expert Rev Vaccines. 2011;10:659–72. doi: 10.1586/erv.11.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Habersetzer F, Zarski J-P, Leroy V. A novel vectorized HCV therapeutic vaccine (TG4040): results of a phase I study in naive patients chronically infected by HCV. Denmark: 44th Annual Meeting of the European Association for the Study of the Liver; 2009. [Google Scholar]
- 45.Hannoun C, Megas F, Piercy J. Immunogenicity and protective efficacy of influenza vaccination. Virus Res. 2004;103:133–8. doi: 10.1016/j.virusres.2004.02.025. [DOI] [PubMed] [Google Scholar]
- 46.Monto AS, Ansaldi F, Aspinall R, et al. Influenza control in the 21st century: optimizing protection of older adults. Vaccine. 2009;27:5043–53. doi: 10.1016/j.vaccine.2009.06.032. [DOI] [PubMed] [Google Scholar]
- 47.McMichael AJ, Gotch FM, Dongworth DW, Clark A, Potter CW. Declining T-cell immunity to influenza, 1977–82. Lancet. 1983;2:762–4. doi: 10.1016/s0140-6736(83)92297-3. [DOI] [PubMed] [Google Scholar]
- 48.Thomas PG, Keating R, Hulse-Post DJ, Doherty PC. Cell-mediated protection in influenza infection. Emerg Infect Dis. 2006;12:48–54. doi: 10.3201/eid1201.051237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Berthoud TK, Hamill M, Lillie PJ, et al. Potent CD8+ T-cell immunogenicity in humans of a novel heterosubtypic influenza A vaccine, MVA-NP+M1. Clin Infect Dis. 2011;52:1–7. doi: 10.1093/cid/ciq015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fine PE. Variation in protection by BCG: implications of and for heterologous immunity. Lancet. 1995;346:1339–45. doi: 10.1016/s0140-6736(95)92348-9. [DOI] [PubMed] [Google Scholar]
- 51.Hatherill M. Prospects for elimination of childhood tuberculosis: the role of new vaccines. Arch Dis Child. 2011;96:851–6. doi: 10.1136/adc.2011.214494. [DOI] [PubMed] [Google Scholar]
- 52.McShane H, Pathan AA, Sander CR, Keating SM, Gilbert SC, Huygen K, Fletcher HA, Hill AV. Recombinant modified vaccinia virus Ankara expressing antigen 85A boosts BCG-primed and naturally acquired antimycobacterial immunity in humans. Nat Med. 2004;10:1240–4. doi: 10.1038/nm1128. [DOI] [PubMed] [Google Scholar]
- 53.Vordermeier HM, Villarreal-Ramos B, Cockle PJ, et al. Viral booster vaccines improve Mycobacterium bovis BCG-induced protection against bovine tuberculosis. Infect Immun. 2009;77:3364–73. doi: 10.1128/IAI.00287-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sims RB. Sipuleucel-T: autologous cellular immunotherapy for men with asymptomatic or minimally symptomatic metastatic castrate resistant prostate cancer. J Cancer. 2011;2:357–9. doi: 10.7150/jca.2.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Speetjens FM, Zeestraten EC, Kuppen PJ, Melief CJ, van der Burg SH. Colorectal cancer vaccines in clinical trials. Expert Rev Vaccines. 2011;10:899–921. doi: 10.1586/erv.11.63. [DOI] [PubMed] [Google Scholar]
- 56.Elkord E, Dangoor A, Drury NL, et al. An MVA-based vaccine targeting the oncofetal antigen 5T4 in patients undergoing surgical resection of colorectal cancer liver metastases. J Immunother. 2008;31:820–9. doi: 10.1097/CJI.0b013e3181876ab3. [DOI] [PubMed] [Google Scholar]
- 57.Elkord E, Dangoor A, Burt DJ, et al. Immune evasion mechanisms in colorectal cancer liver metastasis patients vaccinated with TroVax (MVA-5T4) Cancer Immunol Immunother. 2009;58:1657–67. doi: 10.1007/s00262-009-0674-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tykodi SS, Thompson JA. Development of modified vaccinia Ankara-5T4 as specific immunotherapy for advanced human cancer. Expert Opin Biol Ther. 2008;8:1947–53. doi: 10.1517/14712590802567298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Webster RG, Robinson HL. DNA vaccines: a review of developments. BioDrugs. 1997;8:273–92. doi: 10.2165/00063030-199708040-00004. [DOI] [PubMed] [Google Scholar]
- 60.Baden LR, Blattner WA, Morgan C, et al. Timing of plasmid cytokine (IL-2/Ig) administration affects HIV-1 vaccine immunogenicity in HIV-seronegative subjects. J Infect Dis. 2011;204:1541–9. doi: 10.1093/infdis/jir615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Perez SA, von Hofe E, Kallinteris NL, Gritzapis AD, Peoples GE, Papamichail M, Baxevanis CN. A new era in anticancer peptide vaccines. Cancer. 2010;116:2071–80. doi: 10.1002/cncr.24988. [DOI] [PubMed] [Google Scholar]
- 62.Nardin E. The past decade in malaria synthetic peptide vaccine clinical trials. Hum Vaccin. 2010;6:27–38. doi: 10.4161/hv.6.1.9601. [DOI] [PubMed] [Google Scholar]
- 63.Foged C, Hansen J, Agger EM. License to kill: formulation requirements for optimal priming of CD8+ CTL responses with particulate vaccine delivery systems. Eur J Pharm Sci. 2011 doi: 10.1016/j.ejps.2011.08.016. doi: 10.1016/j.ejps.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 64.Woodland DL. Jump-starting the immune system: prime-boosting comes of age. Trends Immunol. 2004;25:98–104. doi: 10.1016/j.it.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 65.Alcock R, Cottingham MG, Rollier CS, et al. Long-term thermostabilization of live poxviral and adenoviral vaccine vectors at supraphysiological temperatures in carbohydrate glass. Sci Transl Med. 2010;2:19ra2. doi: 10.1126/scitranslmed.3000490. [DOI] [PubMed] [Google Scholar]
- 66.Rollier CS, Reyes-Sandoval A, Cottingham MG, Ewer K, Hill AV. Viral vectors as vaccine platforms: deployment in sight. Curr Opin Immunol. 2011;23:377–82. doi: 10.1016/j.coi.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 67.Peruzzi D, Dharmapuri S, Cirillo A, et al. A novel chimpanzee serotype-based adenoviral vector as delivery tool for cancer vaccines. Vaccine. 2009;27:1293–300. doi: 10.1016/j.vaccine.2008.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Goodman AL, Epp C, Moss D, et al. New candidate vaccines against blood-stage Plasmodium falciparum malaria: prime-boost immunization regimens incorporating human and simian adenoviral vectors and poxviral vectors expressing an optimized antigen based on merozoite surface protein 1. Infect Immun. 2010;78:4601–12. doi: 10.1128/IAI.00315-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Letourneau S, Im EJ, Mashishi T, et al. Design and pre-clinical evaluation of a universal HIV-1 vaccine. PLoS ONE. 2007;2:e984. doi: 10.1371/journal.pone.0000984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu J, Zhang S, Tan S, Zheng B, Gao GF. Revival of the identification of cytotoxic T-lymphocyte epitopes for immunological diagnosis, therapy and vaccine development. Exp Biol Med (Maywood) 2011;236:253–67. doi: 10.1258/ebm.2010.010278. [DOI] [PubMed] [Google Scholar]
- 71.Berkley S, Bertram K, Delfraissy J-F, et al. The 2010 scientific strategic plan of the Global HIV Vaccine Enterprise. Nat Med. 2010;16:981–9. doi: 10.1038/nm0910-981. [DOI] [PubMed] [Google Scholar]
- 72.Makedonas G, Betts MR. Living in a house of cards: re-evaluating CD8+ T-cell immune correlates against HIV. Immunol Rev. 2011;239:109–24. doi: 10.1111/j.1600-065X.2010.00968.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pulendran B, Li S, Nakaya HI. Systems vaccinology. Immunity. 2010;33:516–29. doi: 10.1016/j.immuni.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Slota M, Lim JB, Dang Y, Disis ML. ELISpot for measuring human immune responses to vaccines. Expert Rev Vaccines. 2011;10:299–306. doi: 10.1586/erv.10.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McElrath MJ, De Rosa SC, Moodie Z, et al. HIV-1 vaccine-induced immunity in the test-of-concept step study: a case-cohort analysis. Lancet. 2008;372:1894–905. doi: 10.1016/S0140-6736(08)61592-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nitayaphan S, Pitisuttithum P, Karnasuta C, et al. Safety and immunogenicity of an HIV subtype B and E prime-boost vaccine combination in HIV-negative Thai adults. J Infect Dis. 2004;190:702–6. doi: 10.1086/422258. [DOI] [PubMed] [Google Scholar]
- 77.Small EJ, Fratesi P, Reese DM, Strang G, Laus R, Peshwa MV, Valone FH. Immunotherapy of hormone-refractory prostate cancer with antigen-loaded dendritic cells. J Clin Oncol. 2000;18:3894–903. doi: 10.1200/JCO.2000.18.23.3894. [DOI] [PubMed] [Google Scholar]