

Abstract
Activation of Toll-like receptors (TLRs) triggers rapid inflammatory cytokine production in various cell types. The exogenous product of growth-arrest-specific gene 6 (Gas6) and Protein S (ProS) inhibit the TLR-triggered inflammatory responses through the activation of Tyro3, Axl and Mer (TAM) receptors. However, regulation of the Gas6/ProS-TAM system remains largely unknown. In the current study, mouse macrophages are shown to constitutively express Gas6 and ProS, which synergistically suppress the basal and TLR-triggered production of inflammatory cytokines, including those of tumour necrosis factor-α, interleukin-6 and interleukin-1β, by the macrophages in an autocrine manner. Notably, TLR signalling markedly decreases Gas6 and ProS expression in macrophages through the activation of the nuclear factor-κB. Further, the down-regulation of Gas6 and ProS by TLR signalling facilitates the TLR-mediated inflammatory cytokine production in mouse macrophages. These results describe a self-regulatory mechanism of TLR signalling through the suppression of Gas6 and ProS expression.
Keywords: Gas6, inflammation, protein S, TAM receptors, toll-like receptor
Introduction
Toll-like receptors (TLRs) are crucial triggers of innate immunity through the recognition of pathogen-associated molecular patterns.1 To date, 11 distinct TLRs have been found in humans, and 13 in mice.2 The ligands of most TLRs have been identified.3 For example, TLR4 recognizes the lipopolysaccharides (LPS) of Gram-negative bacteria;4 TLR3 recognizes the double-stranded RNA (dsRNA) produced by many viruses during replication, and is also activated by a synthetic dsRNA analogue, polyinosinic-polycytidylic acid [poly(I:C)],5 and TLR9 can be activated by CpG DNA motifs of both bacteria and viruses.6 Activation of TLR triggers two signalling pathways:3 the MyD88-dependent (D) pathway, which uses the adaptor molecule MyD88, leading to the activation of the nuclear factor-κB (NF-κB) and mitogen-activated protein kinases (MAPKs); and the MyD88-independent (I) pathway through the recruitment of Toll/interleukin-1 receptor domain-containing adaptor inducing interferon-β, resulting in the activation of NF-κB and interferon-regulating factor-3 (IRF3). With the exception of TLR3 and TLR4, all other TLRs trigger immune response exclusively through the D pathway. TLR3 signals exclusively through the I pathway,7 whereas TLR4 initiates both the D and I pathways.8 The TLR pathway-mediated activation of NF-κB and MAPKs induces the production of numerous pro-inflammatory cytokines including interleukin-1β (IL-1β), IL-6 and tumour necrosis factor-α (TNF-α). The I pathway-mediated IRF3 activation leads to the induction of type 1 interferons (IFN-α and IFN-β). These cytokines are involved in the host defence of pathogens through the regulation of immune responses or the direct killing of invading pathogens.
Although TLR-mediated inflammation is essential for host defence against pathogens, TLR signalling must be tightly controlled because unrestrained TLR activation generates a chronic inflammatory milieu that can result in various chronic inflammatory disorders.9 Several TLR signalling suppressors have been described in immune cells.10 Recent studies revealed that Tyro3, Axl and Mer (TAM) receptors play a pivotal role in negatively regulating innate immunity via the inhibition of the TLR-mediated inflammatory response and the promotion of phagocytic clearance of apoptotic cells.11–13 The TAM receptors belong to a subfamily of receptor tyrosine kinases. Of the 58 members of the receptor tyrosine kinase family,14 the TAM receptors are among the few that are specific to vertebrates. Analysis on TAM knockout mice revealed that TAM receptors play an essential role in the regulation of tissue homeostasis in the adult nervous, vascular and reproductive systems.15 Notably, TAM receptors have profound effects in the homeostatic regulation of innate immune responses.16,17 Two closely related proteins, the product of growth-arrest-specific gene 6 (Gas6) and Protein S (ProS), are common biological ligands of TAM receptors.18 Gas6 and ProS are two secreted soluble proteins that carry an N-terminal γ-carboxylated glutamic acid domain that confer the ability to bind phosphatidylserine on the surface of apoptotic cells,19 and a C-terminal sex hormone-binding globulin-like module that can bind and activate TAM receptors.20
Although the Gas6/ProS-TAM system has a pivotal role in regulating innate immunity, the regulation of this system remains largely unknown. In the current article, we provide evidence that TLR activation suppresses the expression of Gas6 and ProS, which facilitates the TLR-mediated inflammatory response in macrophages. The data provide insights into the regulation of Gas6 and ProS expression and function during the inflammatory response.
Materials and methods
Animals
C57BL/6 strain mice 8–10 weeks of age were obtained from the animal facility of Peking Union Medical College (Beijing, China). The mouse mutants for TAM receptors were provided by Dr Greg Lemke (Salk Institute for Biological Studies, La Jolla, CA). These mice were housed under specific pathogen-free conditions with a 12 : 12 hr light : dark cycle and had free access to food and water. The mice were handled in compliance with the Guideline for the Care and Use of Laboratory Animals established by the Chinese Council on Animal Care.
Reagents
Ultra-pure S. Minnesota LPS, poly(I:C), CpG oligonucleotides, antagonists of TLR4 (tlrl-rslps) and TLR9 (tlrl-2088) were purchased from InvivoGen (San Diego, CA). Neutralizing anti-TLR3 antibody (TLR3.7) was purchased from Apotech (Geneva, Switzerland). Recombinant mouse Gas6 (986-GS-025), rat anti-Axl (MAB854), anti-Mer (MAB591) and anti-Tyro3 (MAB759) antibodies were purchased from R&D Systems (Minneapolis, MN). BAY 11-7082, SP600125, SB202190 and monoclonal antibodies against β-actin (A5316) were purchased from Sigma-Aldrich (St Louis, MO). Rabbit antibodies against NF-κBp65 (sc-372), p38 (sc-7149), Gas6 (sc-1935) and ProS (sc-27027) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit anti-phospho-p65 (No. 5970), anti-phospho-p38 (No. 4631) and anti-phospho-IRF3 (No. 3661) antibodies were purchased from Cell Signaling Technology (Beverly, MA). Rabbit anti-F4/80 (ab6640) antibodies were purchased from Abcam (Cambridge, UK). Fluorescein isothiocyanate-conjugated and horseradish peroxidase (HRP)-conjuated secondary antibodies were purchased from Zhongshan Biotechnology, Inc. (Beijing, China). Phycoerythrin (PE)-conjugated antibodies against F4/80 and FITC-conjugated annexin V were purchased from Biolegend (San Diego, CA).
Isolation and culture of peritoneal macrophages
Peritoneal macrophages were isolated based on a previous approach.21 Briefly, mice were anaesthetized with CO2 and then killed by cervical dislocation. The peritoneal cavities were lavaged with 5 ml ice-cold PBS to collect peritoneal cells. The cells were cultured in RPMI-1640 (Gibco-BRL, Grand Island, NY) supplemented with 10% fetal bovine serum (Gibco-BRL) in a humidified atmosphere containing 5% CO2 at 37°. After 2 hr, non-adherent cells were removed by vigorously washing with PBS, and the macrophages adhering to the dishes were identified by immunostaining for F4/80 (a marker for macrophages) and used for subsequent experiments.
Immunofluorescence staining
Mouse macrophages cultured on Lab-Tek chamber slides (Nunc, Naperville, IL) were fixed with cold methanol at −20° for 3 min, and permeabilized with 0·2% Triton X-100 in PBS for 15 min. The cells were blocked by incubation with 10% normal goat serum in PBS at room temperature for 30 min, and then incubated with rabbit anti-F4/80 antibodies in a humid chamber at 37° for 1 hr. After washing thrice with PBS, the cells were incubated with the FITC-conjugated goat anti-rabbit IgG for 30 min. Negative controls were incubated with pre-immune rabbit serum instead of the anti-F4/80 antibodies. The cells were washed thrice with PBS and subjected to a counterstaining for nuclei using 4′,6-diamidino-2-phenylindole (DAPI; Zhongshan Biotechnology, Inc.). The slides were mounted for examination under a fluorescence microscope (IX-71; Olympus, Tokyo, Japan).
Flow cytometry
Macrophages were detached by treatment with 5 mm EDTA for 5 min. After washing with cold PBS, the cells were stained with PE-conjugated antibodies against F4/80, FITC-conjugated annexin V following the manufacturer's instructions. The cells were analysed using a BD FACSSanto flow cytometer (BD Biosciences).
Quantitative real-time PCR
Total RNA was isolated from macrophages using TRIzol reagent (Invitrogen, Carlsbad, CA) in accordance with the manufacturer's instructions. RNA (1 μg) was reverse transcribed into cDNA in a 20-μl reaction mixture containing 2·5 μm random hexamers, 2 μm deoxynucleotide triphosphates and 200 U Moloney murine leukaemia virus reverse transcriptase (Promega, Madison, WI). Quantitative PCR was performed in a 20-μl reaction mixture containing 0·2 μl cDNA, 0·5 μm forward and reverse primers and 2× Power SYBR Green PCR Master Mix (Applied Biosystems, Foster city, CA) using an ABI PRISM 7300 real-time cycler (Applied Biosystems). The transcript levels of target genes were normalized to β-actin. The primers used for quantitative PCR are listed in Table 1.
Table 1.
Primers used for real-time PCRs
| Primer pairs (5′–3′) | ||
|---|---|---|
| Target genes | Forward | Reverse |
| Axl | TCATGTGAAGCCCACAATGC | GGAGCACTGTGATGGTGGCT |
| Mer | GCATGTTGCGGGATGACA | TGGCGGTAATAATCACCACTGTA |
| Tyro3 | GTGAAGCCCGCAACATAAAAG | GGTGCTTGAAGGCGAACAAT |
| Gas6 | CGAGTCTTCTCACACTGCTGTT | GCACTCTTGATATCGTGGATAGAAATAC |
| ProS | TTCCGTGTTGGCTCATTCC | TCTGAGGTCAGGATAAGCATTAGCT |
| TNF-α | CATCTTCTCAAAATTCGAGTGACA A | TGGGAGTAGACAAGGTACAACCC |
| IL-1β | CAACCAACAAGTGATATTCTCCATG | GATCCACACTCTCCAGCTGCA |
| IL-6 | GAGGATACCACTCCCAACAGACC | AAGTGCATCATCGTTGTTCATACA |
| β-actin | GAAATCGTGCGTGACATCAAAG | TGTAGTTTCATGGATGCCACAG |
Western blot analysis
Macrophages were lysed using RIPA lysis buffer (Applygen Technologies Inc., Beijing, China). Equal amounts of proteins were separated on 10% SDS–PAGE gel and subsequently electrotransferred onto PVDF membranes (Millipore, Bedford, MA). The membranes were blocked for 1 hr in Tris-buffered saline (TBS) containing 5% non-fat dried milk and incubated overnight with the primary antibodies at 4°. The membranes were then washed with TBS containing 0·1% Tween-20 (TBST) and incubated with the appropriate horseradish peroxidase-conjugated secondary antibodies (Zhongshan, Beijing, China) at room temperature for 1 hr. Peroxidase colour visualization was achieved using an enhanced chemiluminescence detection kit (Pierce Biotechnology, Rockford, IL).
ELISA
Macrophages were cultured in 24-well plates at 37° at a density of 1 × 106 cells/well, and stimulated with TLR ligands. The concentration of cytokines in the culture medium was measured using ELISA kits: those for IL-1β (MLB00B), IL-6 (M6000B), TNF-α (MTA00B) and Gas6 (DY986) were purchased from R&D Systems (Minneapolis, MN); and the kit for ProS (E0735h) was purchased from Wuhan EIAab Science Co. Ltd (Wuhan, China). ELISAs were performed according to the manufacturer's instructions.
Statistical analysis
Data are presented as mean ± standard error of mean (SEM). These data were analysed using the Student's t-test or analysis of variance test. All calculations were performed with spss version 11.0 statistical software package (SPSS, Chicago, IL). Values of P < 0·05 and < 0·01 were considered significant and very significant, respectively.
Results
Expression of TAM receptors and their ligands in mouse macrophages
Peritoneal macrophages from 10-week-old C57BL/6 mice were used for Gas6/ProS-TAM expression analysis. Cell purity and viability were higher than 95%, based on immunofluorescence staining for F4/80 (Fig. 1a) and flow cytometry after double staining with PE-conjugated antibodies against F4/80 and FITC-conjugated annexin V (Fig. 1b). Axl and Mer were clearly detected, as well as very weak Tyro3 in wild-type (WT) macrophages, using quantitative PCR (Fig. 1c). In contrast, the mRNA of all three TAM receptors were absent in TAM knock-out (TAM−/−) macrophages. Gas6 and ProS mRNA were expressed in both WT and TAM−/− macrophages, with significantly high levels of ProS compared with Gas6 mRNA. Axl and Mer proteins, but not Tyro3, were detected in the WT cells by Western blotting (Fig. 1d), which is consistent with mRNAs. The TAM proteins were not detected in the TAM−/− macrophages. However, secreted Gas6 and ProS were detected in the culture media of both WT and TAM−/− macrophages. Based on the ELISA, the ProS level was significantly high in the medium compared with Gas6 24 hr after culture (Fig. 1e). The results indicate that mouse peritoneal macrophages constitutively express Axl and Mer, and synthesize their ligands Gas6 and ProS.
Figure 1.
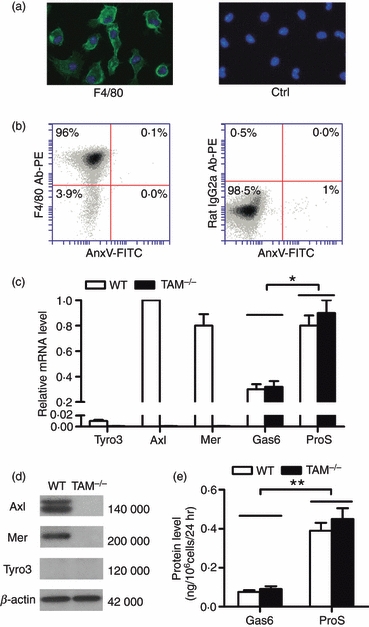
Gas6/ProS-TAM system expression in mouse macrophages. (a) Identification of mouse primary peritoneal macrophages. These cells were identified by immunofluorescence staining for F4/80 using anti-F4/80 antibodies and counter-stained with DAPI for the nuclei (left panel). Negative controls were incubated with pre-immune rabbit serum instead of the anti-F4/80 antibodies and counterstained with DAPI (right panel). (b) The purity and viability of macrophages were quantitatively assessed by flow cytometry after double staining with phycoerythrin (PE) -conjugated antibodies against F4/80 (F4/80 Ab-PE) and fluorescein isothiocyanate (FITC) -conjugated annexin V (AnxV-FITC). The living macrophages were determined to be an F4/80+/AnxV– population (left panel). An isotype control was obtained by labelling cells with FITC-conjugated rat IgG2a (FITC-IgG2a) (right panel). (c) Expression of Gas6, ProS and TAM receptors in the macrophages. Total RNA was extracted from the macrophages and the relative mRNA level was analysed using quantitative PCR. (d) Western blotting for detection of TAM receptors. β-Actin was used as the loading control. (e) Gas6 and ProS production by the macrophages. The macrophages were cultured in a serum-free medium for 24 hr. Gas6 and ProS concentration in the medium was measured by ELISA. These images are representatives of at least three independent experiments. Data represent the mean ± SEM of three experimental values (n = 1 mouse per experiment). *P < 0·05; **P < 0·01.
Gas6 and ProS inhibit the inflammatory cytokine expression by macrophages in an autocrine manner
Given that recombinant Gas6 and ProS inhibit TLR-mediated inflammatory cytokine production via the activation of TAM receptors in different types of cell,17,22 exogenous Gas6 and ProS significantly inhibit in a dose-dependent manner the expression of TNF-α, IL-6 and IL-1β by WT macrophages after stimulation with LPS (Fig. 2a). These effect were not observed in macrophages lacking TAM receptors (TAM−/−). Gas6 and ProS function were neutralized with antibodies to examine whether or not autocrine Gas6 and ProS regulate expression of the inflammatory cytokines in macrophages. The mRNA levels of TNF-α, IL-6 and IL-1β were significantly increased in WT macrophages 5 hr after treatment with the rabbit antibodies against Gas6 and ProS (Fig. 2b). The antibodies neutralizing Gas6 and ProS synergistically up-regulated the inflammatory cytokine expression in WT macrophages. The rabbit antibodies against p38 had no effect on expression of the cytokines, suggesting that the rabbit antibodies have no other components to induce the cytokine expression. In controls, an identical treatment on TAM−/− macrophages did not alter the cytokine expression. Further, similar effects of the antibodies against Gas6 and ProS on the LPS-induced inflammatory cytokine expression were observed (Fig. 2c). Notably, the basal and LPS-induced cytokine mRNA levels in TAM−/− macrophages were about fourfold higher than those in WT cells. These results suggest that Gas6 and ProS secreted by macrophages inhibit the basal and LPS-induced expression of inflammatory cytokines in an autocrine manner through TAM receptors.
Figure 2.
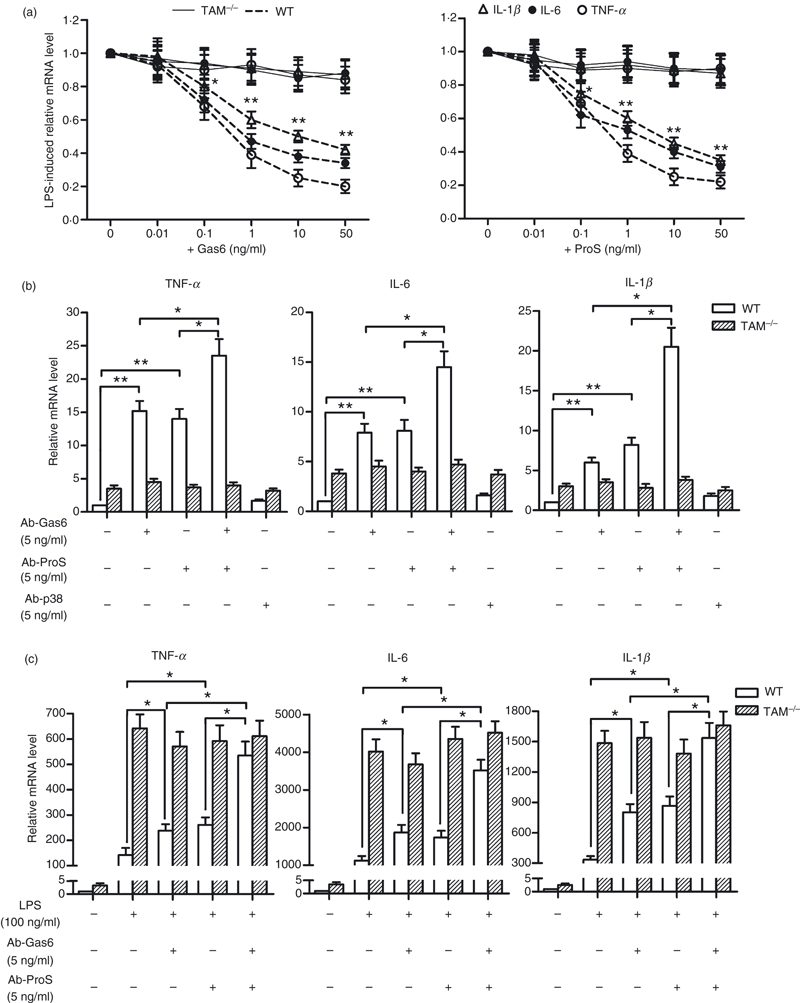
Gas6 and ProS inhibit inflammatory cytokine expression in an autocrine manner. Mouse peritoneal macrophages were cultured in serum-free medium for 6 hr before treatment. (a) Effects of recombinant Gas6 and ProS on the LPS-induced inflammatory cytokine expression. The wild-type (WT) and TAM triple mutant (TAM−/−) macrophages were treated with lipopolysaccharide (LPS) after a 2-hr pre-incubation with the indicated doses of Gas6 and/or ProS for 4 hr. The relative mRNA levels of the cytokines were analysed using quantitative PCR. (b) Effects of the neutralizing antibodies against Gas6 and ProS on the expression of inflammatory cytokines in the basal condition. The WT and TAM−/− macrophages were incubated with the rabbit antibodies against Gas6 (Ab-Gas6) and/or ProS (Ab-ProS), and p38 (Ab-p38) for 5 hr. The relative mRNA levels of the cytokines were analysed using quantitative PCR. (c) Effects of the antibodies on the LPS-induced inflammatory cytokine expression. The WT and TAM−/− macrophages were treated with LPS alone or with LPS after a 3-hr pre-incubation with Ab-Gas6 and/or Ab-ProS for 4 hr. The relative mRNA levels of the cytokines were analysed using quantitative PCR. Data are the mean ± SEM of three experiments (n = 3 mice per experiment). *P < 0·05; **P < 0·01.
TLR ligands suppress Gas6 and ProS expression
The expression of Gas6, ProS and TAM receptors in macrophages after treatment with TLR ligands was investigated to determine whether or not TLR activation regulates the Gas6/ProS-TAM system. LPS (a TLR4 ligand) markedly inhibited the expression of both Gas6 and ProS at the mRNA levels in a time-dependent manner (Fig. 3a). A significant reduction in mRNA was first observed 4 hr after cell stimulation with 100 ng/ml LPS, and the expression was completely aborted at 12 hr. Further, poly(I:C) (a TLR3 ligand) and CpG (a TLR9 ligand) significantly inhibited both Gas6 and ProS expression in the macrophages (Fig. 3b,c). Consistent with the reduction of mRNAs, Gas6 and ProS proteins in medium were dramatically decreased 24 hr after cell stimulation with the TLR ligands (Fig. 3d). The inhibitory effects of the TLR ligands on Gas6 and ProS production were significantly reduced by the TLR inhibitors, which implies that the TLR ligands inhibit Gas6 and ProS production via activation of their respective TLRs. In contrast, the TLR ligands did not affect TAM receptor expression (data not shown).
Figure 3.
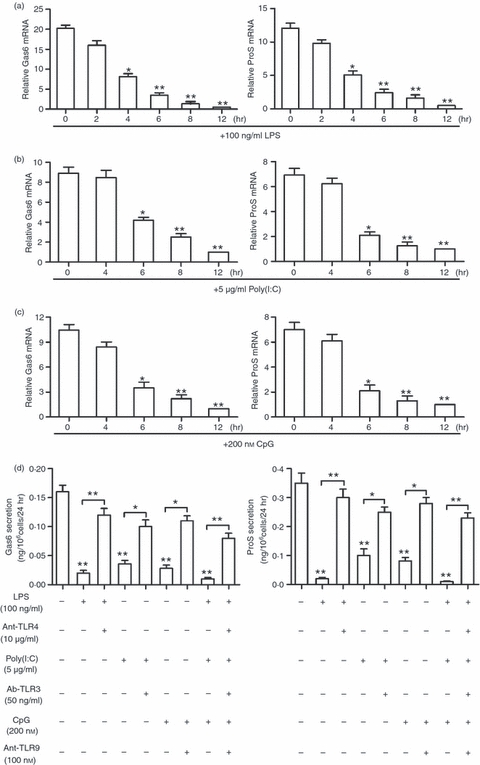
Toll-like receptor (TLR) ligand-mediated inhibition of Gas6 and ProS expression. (a–c), Time-dependent suppression of Gas6 and ProS expression by TLR ligands. The macrophages in the serum-free medium were treated with 100 ng/ml lipopolysaccharide (LPS) (a), 5 μg/ml poly(I:C) (b) and 200 nm CpG (c) for the indicated time. The relative mRNA levels of Gas6 and ProS were determined using quantitative PCR. (d) Suppression of Gas6 and ProS secretion by TLR ligands. The macrophages were treated with the TLR ligands individually or with the TLR ligands following 2-hr pre-incubation with respective TLR inhibitors: antagonists of TLR4 (Ant-TLR4) and TLR9 (Ant-TLR9), neutralizing antibodies against TLR3 (Ab-TLR3). The concentrations of Gas6 (left panel) and ProS (right panel) in the medium were measured using ELISA 24 hr following the treatments. Data are the mean ± SEM of three experiments (n = 4 mice per experiment). *P < 0·05; **P < 0·01.
TLR-mediated inhibition of Gas6 and ProS expression is dependent on NF-κB activation
The involvement of the TLR signalling pathways in the inhibition of Gas6 and ProS expression was investigated to further understand the mechanism underlying the TLR-mediated inhibition. LPS activated NF-κB in the macrophages through the time-dependent phosphorylation of subunit p65 (see Supplementary material, Fig. S1). All three TLR ligands evidently phosphorylated NF-κBp65 2 hr after treatment (see Supplementary material, Fig. S2). The IRF3 was phosphorylated by LPS and poly(I:C), but not by CpG (see Supplementary material, Fig. S3). In contrast, LPS and CpG induced phosphorylation of MAPK p38 (see Supplementary material, Fig. S4); poly(I:C) did not exhibit any effect. Inhibitors of NF-κB, IRF3 and p38 activation efficiently decreased the LPS-induced phosphorylation of the target proteins (see Supplementary material, Fig. S5). Notably, LPS inhibition of Gas6 and ProS expression was significantly reversed by BAY 11-7082, a NF-κB activation inhibitor (Fig. 4a). However, blockage of IRF3 and p38 phosphorylation by their respective inhibitors (SP 600125 for IRF3, SB202190 for p38) did not change the inhibitory effect of LPS on Gas6 and ProS expression. Similarly, the inhibition of Gas6 and ProS expression by poly(I:C) and CpG was attributed to NF-κB activation (Fig. 4b,c).
Figure 4.
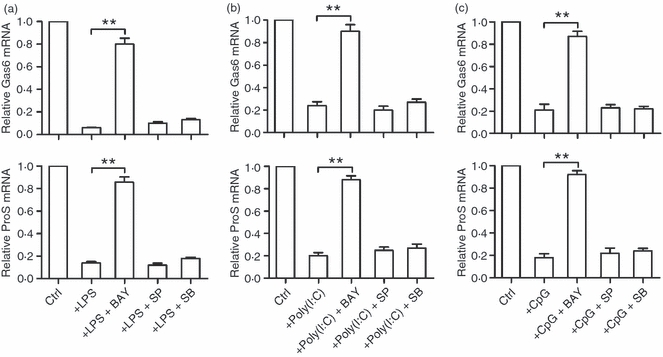
Toll-like receptor (TLR) signalling involved in the inhibition of Gas6 and ProS expression. (a) Macrophages were treated with lipopolysaccharide (LPS) alone or with LPS and the respective inhibitors [10 μm BAY11-7082 (BAY) for p65, 50 μm SP600125 (SP) for interferon-regulating factor-3 (IRF3), 10 μm SB202190 (SB) for p38] for 8 hr. (b) Macrophages were treated with poly(I:C) alone or with poly(I:C) and the respective inhibitors for 8 hr. (c) Macrophages were treated with CpG alone or with CpG and the respective inhibitors for 8 hr. The relative mRNA levels were determined using quantitative PCR. Data represent the mean ± SEM of three experiments (n = 3 mice per experiment). **P < 0·01.
Down-regulation of Gas6 and ProS facilitates the TLR-mediated inflammatory cytokine production
The TLR-mediated down-regulation of Gas6 and ProS is thought to facilitate the inflammatory cytokine production because Gas6 and ProS negatively regulate TLR-induced inflammatory cytokine expression by macrophages in an autocrine manner (Fig. 2c). For this reason, the correlation between the inflammatory cytokine and the Gas6/ProS levels in the medium after the LPS treatment of macrophages was analysed. The results of ELISA showed that IL-6, TNF-α and IL-1β reached high plateau levels in media of WT macrophages 8–12 hr after LPS treatment, and declined to low levels at 20–24 hr (Fig. 5a, left panel). The cytokines were again slightly up-regulated 28–32 hr after LPS treatment. About a twofold increase in the cytokine production by TAM−/− macrophages compared with WT cells was observed (Fig. 5a, right panel). However, the secondary up-regulation of cytokines 28–32 hr after LPS treatment was not observed in TAM−/− cells. In contrast, levels of Gas6 and ProS secreted by WT and TAM−/− macrophages reached similar peaks at 8 hr and declined to very low levels 24–32 hr after LPS treatment (Fig. 5b). In particular, a supply of exogenous Gas6 or ProS 24 hr after LPS treatment completely abolished the secondary up-regulation of cytokines in WT macrophages 28–32 hr after treatment (Fig. 5c, left panel). Exogenous Gas6 or ProS did not affect the cytokine production in TAM−/− cells (Fig. 5c, right panel). These results suggest that Gas6 and ProS down-regulation both contribute to increased cytokine production after 24 hr of LPS treatment.
Figure 5.
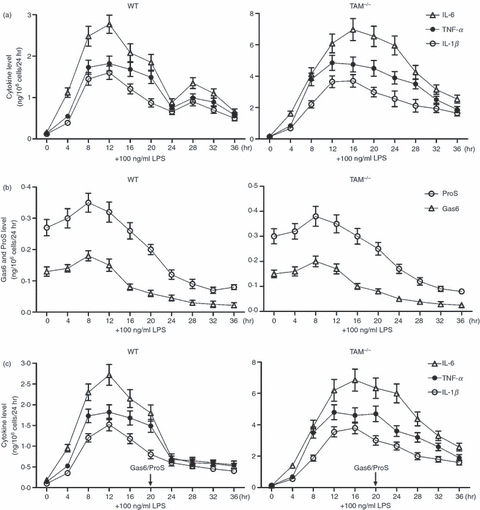
Dynamics of inflammatory cytokine and Gas6/ProS production. (a and b) Effects of lipopolysaccharide (LPS) on inflammatory cytokine, Gas6 and ProS production. At 6 hr after culture in serum-free medium, the macrophages were treated with 100 ng/ml LPS for the indicated duration. The inflammatory cytokine level (a), as well as Gas6 and ProS levels (b) in the medium were measured using ELISA. (c) The macrophages were treated with 100 ng/ml LPS for the indicated duration. Exogenous Gas6 or ProS (0·2 ng/ml) were added into the culture 20 hr after LPS treatment. The cytokine production was measured using ELISA. Data are the mean ± SEM of three experiments (n = 3 mice per experiment). *P < 0·05; **P < 0·01.
Discussion
Inflammatory responses are regulated by pro-inflammatory and anti-inflammatory factors in opposite manners. In the current study, Gas6 and ProS are expressed in the primary mouse macrophages and negatively regulate the basal and TLR-mediated pro-inflammatory cytokine expression in an autocrine manner. In this context, Gas6 and ProS can be considered as anti-inflammatory factors. Intriguingly, TLR signalling inhibits Gas6 and ProS expression in macrophages, which feeds forward the production of pro-inflammatory cytokines. These data describe a novel inter-regulatory system between pro-inflammatory and anti-inflammatory factors.
Gas6 and ProS belong to a family of vitamin K-dependent proteins, and have a high structural homology.23 In addition to a critical role for ProS in anti-coagulation,24 both Gas6 and ProS play various important roles in regulating cell survival, adhesion, migration, phagocytosis and immunity through the activation of TAM receptors.20 Inhibition of the Gas6/ProS-TAM system on TLR-driven inflammatory cytokine production was first demonstrated by Rothlin et al.17 in mouse dendritic cells (DCs). Our results in mouse macrophages correspond to those observations in DCs. Rothlin et al. provided evidence that the Gas6/ProS-TAM system represents a new pathway for the inhibition of inflammation through inhibiting TLR signalling, in which TLR-induced Axl is implicated. They did not investigate Gas6/ProS expression upon TLR activation in DCs. Up-regulation of Axl by TLR activation might negatively feed back inflammation. We describe in this study that TLR signalling would positively feed forward inflammation by reducing the Gas6/ProS levels. Our data provide an additional insight into the regulation of inflammation by the Gas6/ProS-TAM system. However, we did not find TAM receptor induction by TLR ligands in macrophages (data not shown). The discrepancy between our results and those of Rothlin et al. might be reconciled by the fact that different cell types were used in the two studies. In vivo, most migratory DCs will transit the inflammation cycle only once, before their apoptotic elimination. Axl induction might facilitate the resolution of inflammation through the inhibition of TLR signalling at the final stage of the inflammatory cycle. By contrast, macrophages transit the cycle reiteratively. Gas6 and ProS down-regulation may be required for a reiterative cycling macrophage to be fully responsive to subsequent pathogen encounter, which might facilitate the elimination of pathogens through the burst of cytokines.
Toll-like receptors are potent triggers of the inflammatory response against invading pathogens.25,26 However, TLR-initiated inflammation must be properly regulated because unrestrained TLR signalling generates a chronic inflammatory milieu that often leads to autoimmunity.27 Activation of TLR evidently drives the production of negative regulators that in turn inhibit TLR signaling.10 Suppressor of cytokine signalling (SOCS) proteins are critical in such TLR-driven inhibitors.28,29 The Gas6/ProS-TAM system is a negative regulator of innate immunity by inhibiting TLR signalling in DCs.17 TAM activation leads to SOCS1 and SOCS3 upon the type 1 interferon receptor (IFNAR)/transcription factor STAT1 signalling. This IFNAR/STAT1 signalling up-regulates Axl, which may feed forward SOCS protein production. SOCS1 promotes the degradation of the TLR4 adaptor protein MAL,28 and SOCS3 inhibits TRAF6 ubiquitylation.29 In the present study, we demonstrate that TLR ligands reduce Gas6/ProS expression via NF-κB activation. NF-κB activation results in the induction of various cytokines. Whether NF-κB activation-driven cytokines are involved in the reduction of Gas6/ProS should be investigated.
Evidence that autocrine Gas6 and ProS synergistically inhibit inflammatory cytokine expression by macrophages in baseline conditions suggests that these two cytokines play important roles in the maintenance of immune homeostasis in normal physiological conditions. Several observations are consistent with this speculation. Regarding autoimmunity, patients with systemic lupus erythematosus have low circulating levels of ProS.30,31 Because ProS is a TAM ligand, a low ProS level may lead to reduced TAM signalling, which consequently leads to immune hyperactivation. At the same time, patients with systemic lupus erythematosus are prone to thrombosis,32 which corresponds to a role of ProS as a blood anticoagulant.24 On the other hand, increased TAM signalling may also result in diseases. A clinical report showed that circulating Gas6 levels were elevated in patients with severe sepsis, and that Gas6 elevation was correlated with a patient's clinical score and the occurrence of septic shock,33 suggesting that hyperactive TAM signalling might play a role in sepsis.
The treatment of macrophages with TLR ligands rapidly up-regulated the production of IL-6, TNF-α and IL-1β, which declined to low levels 24 hr after the treatment. Thereafter a secondary mild up-regulation of the inflammatory cytokines was observed, the mechanism of which has yet to be determined. Evidence that reduced Gas6 and ProS levels are responsible for the secondary up-regulation of inflammatory cytokines after 24 hr of LPS stimulation is provided. The results provide novel insights into inflammatory regulations.
In recent years, increasing research on the Gas6/ProS-TAM system function has been observed, which has provided essential clues on the biological implications of this system. Gas6 and ProS have exhibited crucial roles in the clearance of apoptotic cells and autoimmune diseases.12,13 Interestingly, links between the two phenomena have been known for many years.34 However, the regulation of Gas6 and ProS expression remains largely unknown. In the current study, evidence that Gas6 and ProS can be down-regulated by TLR activation in macrophages is provided, which may feed forward the inflammatory responses against infectious pathogens. Appropriate TAM signalling is critical in the homeostatic regulation of the immune system and resolution of inflammation. Therefore, understanding the regulatory mechanism of the Gas6/ProS-TAM system will have important implications for human immune disorder interventions.
Acknowledgments
We thank Dr Qingxian Lu and Dr Greg Lemke for proving TAM mutant mice. This work was supported by the National Natural Science Foundation of China (Grant No. 30971459) and the Special Funds for Major State Basic Research Project of China (Grant No. 2007CB947504).
Disclosures
The authors indicated no potential conflicts of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. The macrophages in serum-free medium were stimulated with 100 ng/ml lipopolysaccharide (LPS) for the indicated time.
Figures S2, S3 and S4. The cell lysates were prepared from macrophages 2 hr after treatment with Toll-like receptor ligands [5 μg/ml poly(I:C), 100 ng/ml lipopolysaccharide (LPS) and 200 nM CpG].
Figure S5. Inhibition of p65, interferon-regulating factor-3 (IRF-3) and p38 phosphorylation by their respective inhibitors.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than about missing material) should be directed to the corresponding author for the article.
References
- 1.Zhu J, Mohan C. Toll-like receptor signaling pathways – therapeutic opportunities. Mediators Inflamm. 2010;2010:781235. doi: 10.1155/2010/781235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li M, Zhou Y, Feng G, Su SB. The critical role of Toll-like receptor signaling pathways in the induction and progression of autoimmune diseases. Curr Mol Med. 2009;9:365–74. doi: 10.2174/156652409787847137. [DOI] [PubMed] [Google Scholar]
- 3.Kawai T, Akira S. TLR signaling. Cell Death Differ. 2006;13:816–25. doi: 10.1038/sj.cdd.4401850. [DOI] [PubMed] [Google Scholar]
- 4.Nagai Y, Akashi S, Nagafuku M, et al. Essential role of MD-2 in LPS responsiveness and TLR4 distribution. Nat Immunol. 2002;3:667–72. doi: 10.1038/ni809. [DOI] [PubMed] [Google Scholar]
- 5.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–8. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 6.Hemmi H, Takeuchi O, Kawai T, et al. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–5. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 7.Yamamoto M, Sato S, Hemmi H, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–3. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 8.Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine. 2008;42:145–51. doi: 10.1016/j.cyto.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 9.Wang J, Hu Y, Deng WW, Sun B. Negative regulation of Toll-like receptor signaling pathway. Microbes Infect. 2009;11:321–7. doi: 10.1016/j.micinf.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 10.Liew FY, Xu D, Brint EK, O'Neill LA. Negative regulation of toll-like receptor-mediated immune responses. Nat Rev Immunol. 2005;5:446–58. doi: 10.1038/nri1630. [DOI] [PubMed] [Google Scholar]
- 11.Lemke G, Rothlin CV. Immunobiology of the TAM receptors. Nat Rev Immunol. 2008;8:327–36. doi: 10.1038/nri2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lemke G, Burstyn-Cohen T. TAM receptors and the clearance of apoptotic cells. Ann N Y Acad Sci. 2010;1209:23–9. doi: 10.1111/j.1749-6632.2010.05744.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rothlin CV, Lemke G. TAM receptor signaling and autoimmune disease. Curr Opin Immunol. 2010;22:740–6. doi: 10.1016/j.coi.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–34. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 15.Lu Q, Gore M, Zhang Q, et al. Tyro-3 family receptors are essential regulators of mammalian spermatogenesis. Nature. 1999;398:723–8. doi: 10.1038/19554. [DOI] [PubMed] [Google Scholar]
- 16.Lu Q, Lemke G. Homeostatic regulation of the immune system by receptor tyrosine kinases of the Tyro 3 family. Science. 2001;293:306–11. doi: 10.1126/science.1061663. [DOI] [PubMed] [Google Scholar]
- 17.Rothlin CV, Ghosh S, Zuniga EI, Oldstone MB, Lemke G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell. 2007;131:1124–36. doi: 10.1016/j.cell.2007.10.034. [DOI] [PubMed] [Google Scholar]
- 18.Stitt TN, Conn G, Gore M, et al. The anticoagulation factor protein S and its relative, Gas6, are ligands for the Tyro 3/Axl family of receptor tyrosine kinases. Cell. 1995;80:661–70. doi: 10.1016/0092-8674(95)90520-0. [DOI] [PubMed] [Google Scholar]
- 19.Nakano T, Ishimoto Y, Kishino J, et al. Cell adhesion to phosphatidylserine mediated by a product of growth arrest-specific gene 6. J Biol Chem. 1997;272:29411–4. doi: 10.1074/jbc.272.47.29411. [DOI] [PubMed] [Google Scholar]
- 20.Nagata K, Ohashi K, Nakano T, Arita H, Zong C, Hanafusa H, Mizuno K. Identification of the product of growth arrest-specific gene 6 as a common ligand for Axl, Sky, and Mer receptor tyrosine kinases. J Biol Chem. 1996;271:30022–7. doi: 10.1074/jbc.271.47.30022. [DOI] [PubMed] [Google Scholar]
- 21.McPhillips KA, Erwig LP. Assessment of apoptotic cell phagocytosis by macrophages. Methods Mol Biol. 2009;559:247–56. doi: 10.1007/978-1-60327-017-5_17. [DOI] [PubMed] [Google Scholar]
- 22.Sun B, Qi N, Shang T, Wu H, Deng T, Han D. Sertoli cell-initiated testicular innate immune response through toll-like receptor-3 activation is negatively regulated by Tyro3, Axl, and mer receptors. Endocrinology. 2010;151:2886–97. doi: 10.1210/en.2009-1498. [DOI] [PubMed] [Google Scholar]
- 23.Manfioletti G, Brancolini C, Avanzi G, Schneider C. The protein encoded by a growth arrest-specific gene (gas6) is a new member of the vitamin K-dependent proteins related to protein S, a negative coregulator in the blood coagulation cascade. Mol Cell Biol. 1993;13:4976–85. doi: 10.1128/mcb.13.8.4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.ten Kate MK, van der Meer J. Protein S deficiency: a clinical perspective. Haemophilia. 2008;14:1222–8. doi: 10.1111/j.1365-2516.2008.01775.x. [DOI] [PubMed] [Google Scholar]
- 25.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 26.Beutler B, Jiang Z, Georgel P, Crozat K, Croker B, Rutschmann S, Du X, Hoebe K. Genetic analysis of host resistance: Toll-like receptor signaling and immunity at large. Annu Rev Immunol. 2006;24:353–89. doi: 10.1146/annurev.immunol.24.021605.090552. [DOI] [PubMed] [Google Scholar]
- 27.Banchereau J, Pascual V. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity. 2006;25:383–92. doi: 10.1016/j.immuni.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 28.Mansell A, Smith R, Doyle SL, et al. Suppressor of cytokine signaling 1 negatively regulates Toll-like receptor signaling by mediating Mal degradation. Nat Immunol. 2006;7:148–55. doi: 10.1038/ni1299. [DOI] [PubMed] [Google Scholar]
- 29.Frobose H, Ronn SG, Heding PE, Mendoza H, Cohen P, Mandrup-Poulsen T, Billestrup N. Suppressor of cytokine signaling-3 inhibits interleukin-1 signaling by targeting the TRAF-6/TAK1 complex. Mol Endocrinol. 2006;20:1587–96. doi: 10.1210/me.2005-0301. [DOI] [PubMed] [Google Scholar]
- 30.Meesters EW, Hansen H, Spronk HM, Hamulyak K, Rosing J, Rowshani AT, ten Berge IJ, ten Cate H. The inflammation and coagulation cross-talk in patients with systemic lupus erythematosus. Blood Coagul Fibrinolysis. 2007;18:21–8. doi: 10.1097/01.mbc.0000256022.01900.c2. [DOI] [PubMed] [Google Scholar]
- 31.Suh CH, Hilliard B, Li S, Merrill JT, Cohen PL. TAM receptor ligands in lupus: protein S but not Gas6 levels reflect disease activity in systemic lupus erythematosus. Arthritis Res Ther. 2010;12:R146. doi: 10.1186/ar3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ruiz-Irastorza G, Khamashta MA, Castellino G, Hughes GR. Systemic lupus erythematosus. Lancet. 2001;357:1027–32. doi: 10.1016/S0140-6736(00)04239-2. [DOI] [PubMed] [Google Scholar]
- 33.Borgel D, Clauser S, Bornstain C, et al. Elevated growth-arrest-specific protein 6 plasma levels in patients with severe sepsis. Crit Care Med. 2006;34:219–22. doi: 10.1097/01.ccm.0000195014.56254.8a. [DOI] [PubMed] [Google Scholar]
- 34.Nagata S, Hanayama R, Kawane K. Autoimmunity and the clearance of dead cells. Cell. 2010;140:619–30. doi: 10.1016/j.cell.2010.02.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.