

Abstract
Studies have demonstrated that the anti-tumour effect of natural killer (NK) cells is successful for patients with several cancers. Although interleukin-32 (IL-32) is endogenously expressed in NK cells, cytolytic function of NK cells against cancer cells has not been fully demonstrated. In the present study, we found that the growth of cancer cells was suppressed when colon cancer cells or prostate cancer cells were co-cultured with NK-92 cells, an NK cell line. We also found that the expression of tumour necrosis factor receptor 2 and death receptor 3 (DR3) was increased in PC3 cells, and the expression of FAS and DR3 was increased in SW620 cells by co-culture with NK-92 cells. However, cancer cell growth inhibition and IL-32 expression were abolished when cancer cells were co-cultured with NK cells transfected with small interfering (si) RNA of IL-32. DR3 expression was also diminished by co-culture with IL-32-specific siRNA-transfected NK-92 cells. Expression of APO3L, a ligand of DR3, was elevated in NK cells that were co-cultured with cancer cells. It was also found that expression of apoptosis-related proteins such as cleaved caspase-3 and bax was increased in cancer cells co-cultured with NK-92 cells, but their expression was abolished by co-culture with IL-32 siRNA-transfected NK-92 cells. Moreover, knockdown of DR3 in co-culture of NK-92 cells with cancer cells by siRNA or antibodies of DR3 and APO3L reversed the growth inhibitory effect of NK-92 cells. In conclusion, our study showed that IL-32 enhanced the cytotoxic effect of NK-92 cells on the cancer cells through activation of DR3 and caspase-3.
Keywords: apoptosis, cancer cells, death receptor 3, interleukin-32, natural killer-92 cells
Introduction
Natural killer (NK) cells are white blood lymphocytes and have an important role in innate immune defence by eliminating tumour cells or pathogen-infected cells through cytokine production and cytotoxicity.1,2 The NK cells comprise approximately 15% of all circulating lymphocytes and are also found in peripheral tissues including the liver, placenta and peritoneal cavity.3 Whereas resting NK cells circulate in the blood, cytokine-activated NK cells are capable of extravasation and infiltration into most tissues invaded by pathogens and tumour cells.4 They can release several cytokines such as granulocyte–macrophage colony-stimulating factor and interferon-γ, which can induce a tumour-killing effect.2,5,6 Natural killer cells also respond to many cytokines such as interleukin-2 (IL-2), IL-12, IL-15, IL-18 and type I interferons (α and β), which increase their cytolytic and anti-tumour functions, but their significant toxicity was observed after systemic administration of high doses.7–11 Besides these cytokines, there are several tumour necrosis factor (TNF) family ligands such as FAS ligand (FASL), TNF and TNF-related apoptosis-inducing ligand (TRAIL), which are also released by NK cells and have been shown to induce cancer cell apoptosis.1–3,12,13 Moreover, NK cells kill cancer cells by the release of cytoplasmic granules that contain a number of proteins, such as perforin and granzymes, which lyse target cells. The NK cells also kill cancer cells through nitric oxide signalling.1–3
Death receptor 3 (DR3) is also a member of the TNF receptor (TNFR) superfamily such as TNFR1 (DR1), FAS (DR2), DR3, DR4, DR5 and DR6, and is known as a death domain (DD) -containing receptor.14,15 The DR3 binds to its own ligand, APO3L, which is a type II membrane protein of the TNF family closely related to three death-inducing ligands; TRAIL, FasL and TNF-α. The extracellular sequence of APO3L shows highest identity to that of TNF. Soluble APO3L induced apoptosis in human cell lines.16,17 Once APO3L has bound to DR3 in cancer cells, apoptosis is triggered through TNFR-associated DD protein (TRADD), Fas-associated DD protein (FADD) and caspase-8. The APO3L interacts directly with the TRADD, and then TRADD recruits downstream signalling molecules including procaspase-8. Recruitment of caspase-8 to the death-inducing signalling complex leads to the activation of caspase-8 and subsequently activates downstream effector caspases such as caspase-3, -7 and -9. As a result, this receptor-mediated pathway induces apoptosis.18,19 Several cytokines such as IL-2, IL-7, IL-12, IL-15 and IL-18 activate NK cells.7–11 Activated NK cells induce death ligand, and then affect cancer cell apoptosis through interaction with death receptors of cancer cells.2,3 Normally, in cancer cells, the death receptors are down-regulated or mutated,20,21 but NK cells cause cancer cells to up-regulate death receptor expression and then kill the cancer cells by inducing apoptosis.22–24
In many malignant tumour cells, the expression level of MHC-I and MHC-II is repressed, thereby immunogenicity is repressed.25,26 Although cytotoxic T cells and helper T cells require both MHC class I and II for stimulation and tumour killing effect, NK cells do not require MHC for antigen presentation, so participate in the innate immune response.8,27 Therefore, anti-tumour immune responses through NK-cell-mediated mechanisms would be of potential benefit to anti-tumour therapy. We used NK-92, which expresses high levels of APO3L as well as FasL, TNF-α and TRAIL,28–29 and IL-2, an inducer of IL-32, -dependent cell line for survival.30,31 It can therefore be expected that IL-32 activates NK-92 cells, and hence increased anti-tumour activity of NK cells.
Interleukin-32, previously known as natural killer cell transcript 4 (NK4) was originally identified as a transcript that over-expressed in a cDNA library derived from IL-2-activated NK cells.30 Interleukin-32 was produced by NK cells, blood monocytes, T lymphocytes and epithelial cells,32,33 and has six splice variants, IL-32α, IL-32β, IL-32γ, IL-32δ, IL-32ε and IL-32ζ; IL-32α is the most abundant transcript.34 Although IL-32 does not share sequence worthy to know; the IL-32 gene is induced by IL-12 and IL-18, which are the cytokines that trigger the anti-tumour activity of NK cells, and IL-32 induces TNF-α, which promotes apoptotic activity of NK cells.30,32,33,35 Interleukin-32 is localized in both the cytosol and the nucleus, but the amounts of IL-32 secreted from the cells are small compared with those in the cytoplasm.36 Although IL-32 is highly expressed in NK cells, its cytolytic function in NK cells has not been investigated.
In this study, we demonstrate the enhancing effect of IL-32 on the anti-tumour activity of NK-92 cells through induction of death receptor and activation of caspase-3 pathway in cancer cells.
Materials and methods
Regents and cell culture
RPMI-1640, minimum essential medium α, penicillin, streptomycin and fetal bovine serum were purchased from Gibco Life Technologies (Grand Island, NY). Small interfering (si) RNA species for IL-32 was purchased from Dhamacon (Lafayette, CO) and non-targeting control siRNA were purchased from Bioneer (Daejeon, Korea). The NK-92 natural killer cell, PC3 prostate cancer cell and SW620 colon cancer cell lines were obtained from the American Type Culture Collection (Manassas, VA). NK-92 cells were grown in the same conditions in minimum essential medium α that contained 20% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin and 10 ng/ml IL-2. PC3 and SW620 cells were grown at 37° in 5% CO2 humidified air in RPMI-1640 medium that contained 10% fetal bovine serum, 100 U/ml penicillin and 100 μg/ml streptomycin. In co-cultures the cells were grown in a mixed medium (1 : 1) of PC3 or SW620 and NK-92 medium in a trans-well system where the cells were separated by a porous polycarbonate membrane (pore size 0·4 μm; distance 6·5 mm). The PC3 or SW620 cells were first seeded at 5 × 104 cells/well plate, and then cultured overnight, thereafter the inserts containing NK-92 cells (5 × 104 cells/well) were added to the plate and cultured with the cancer cells.
Cell viability
To determine viable cell numbers, the SW620 colon cancer cells or PC3 prostate cancer cells were seeded onto 24-well plates (5 × 104 cells/well). The cells were trypsinized, pelleted by centrifugation for 5 min at 250 g, resuspended in 10 ml PBS, and 0·1 ml of 0·2% Trypan blue was added to the cancer cell suspension in each solution (0·9 ml each). Subsequently, a drop of suspension was placed in a Neubauer chamber, and the live cancer cells were counted. Cells that showed signs of Trypan blue uptake were considered to be dead, whereas those that excluded Trypan blue were considered to be viable. Each assay was carried out in triplicate.
Western blotting
Western blot analysis was performed as described previously.26 The membranes were immunoblotted with the following primary antibodies: mouse monoclonal antibodies directed against FAS and bax (1 : 500 dilutions; Santa Cruz Biotechnology, Santa Cruz, CA), and rabbit polyclonal antibodies directed against cleaved caspase-3 (1 : 1000 dilutions; Cell Signaling Technology, Beverly, MA), DR3, TNFR2, APO3L (1 : 500 dilutions; Santa Cruz Biotechnology). Interleukin-32 was detected using a monoclonal antibody, KU32-52, as described previously.36
Electroporation
NK-92 cells were counted, cell density was determined, and the required numbers of cells (1 × 106 cells per sample) were centrifuged at 160 g for 10 min at room temperature, and rinsed with PBS. The cell pellet was resuspended carefully in 100 μl human cell nucleofector solution (82 μl nucleofector solution + 18 μl supplement), according to the manufacturer's specification (Lonza, Walkersville, MD). One hundred microlitres of cell suspension was combined with 50 nm siRNA, and transferred into a certified cuvette, the selected program was applied electroporation program U-001 (Lonza), and 500 μl culture medium was added to the cuvette. The mixture was gently transferred into the 24-well plate, and analysed after 48 hr.
Transfection
PC3 cells or SW620 cells (5 × 104 cells/well) were plated in 24-well plates and transiently transfected with 0·4 μg of the empty vector or the constitutively activated 100 nm of negative siRNA or DR3 siRNA per well, using a mixture of plasmid and the WelFect-EX PLUS reagent in OPTI-MEN, according to the manufacturer's specification (WelGENE, Seoul, Korea).
Reverse transcription-PCR
Total RNA was extracted by RNeasy (Qiagen, Valencia, CA). The reverse transcription reaction was performed using an RNA to cDNA Kit (Applied Biosystems/Life Technologies Corporation, Carlsbad, CA). The PCR was performed with cDNA as a template using the primers below after an initial 1-min denaturation at 96°, followed by the indicated cycles of 96° for 1 min, 60 or 63° for 1 min and 72° for 1 min. The PCR primers used were 5′-ATGTGCTTCCCGAAGGTCCTC-3′ and 5′-TCATTTTGAAGGATTGGGGTTC-3′ for the primer of IL-32; 5′-ACCAAGTGCCACAAA GGAAC-3′ and 5′-CTGCAATTGAAGCACTGGAA-3′ for the human TNFR1; 5′-CTCAGGAGCATGGGGATAAA-3′ and 5′-AGCCAGCCAGTCTGACATCT-3′ for the human TNFR2; 5′-ATGGCGATGGCTGCGTGTCCTG-3′ and 5′-AGCGCCTCCTGGGTCTCGGGGTAG-3′ for human DR3; 5′-ACTTTGGTTGTTCCGTTGCTGTTG-3′ and 5′-GGCTTTCCATTTGCTGCTCA-3′ for human DR4; 5′-TGGAACAACGGGGACAGAACG-3′ and 5′-GCAGCGCAAGCAGAAAAGGAG-3′ for human DR5; 5′-AAGCCGGGGACCAAGGAGACAGACAAC-3′ and 5′-TGCCGGGGCCCCTTTTTCAGAGT-3′ for human DR6; 5′-CAAAGCCCATTTTTCTTCCA-3′ and 5′-GACAAAGCCACCCCAAGTTA-3′ for human FAS; 5′-TCG CAG AAG TGC ACC TAA AG-3′ and 5′-AGCCTTCCCCTCATCAAAGT-3′ for human APO3L; and 5′-GAAGGTGAAGGTCGGAGT-3′ and 5′-CTTCTACCACTACCCTAAAG-3′ for glyceraldehyde-3-phosphate dehydrogenase.
Flow cytometry
Anti-DR3 and FITC-labelled anti-rabbit antibodies were obtained from Santa Cruz Biotechnology. For flow cytometry, cultured cancer cells (2 × 105 cells/ml) were washed with PBS, and then incubated with 3% BSA/PBS solution (pH 7·0) containing primary antibody (DR3, 1 μg/ml) for 30 min at 4°, and then washed out. The cells were then incubated again for 30 min with the rabbit-FITC antibody (1 μg/ml). The cells expressing DR3 were analysed by flow cytometry on a FACScalibur® (Becton Dickinson, Franklin Lakes, NJ).
Nitric oxide determination
The nitrite accumulation in the supernatant was assessed by Griess reaction. Each 50 μl of culture supernatant was mixed with an equal volume of Griess reagent [0·1%N-(1-naphthyl)-ethylenediamine, 1% sulfanilamide in 5% phosphoric acid] and incubated at room temperature for 10 min. The absorbance at 540 nm was measured in a microplate absorbance reader, and a series of known concentrations of sodium nitrite was used as a standard.
Statistical analysis
The data were analysed using the GraphPad Prism 4 ver. 4.03 software (GraphPad Software, La Jolla, CA). Data are presented as mean ± SD. The differences in all data were assessed by one-way analysis of variance. When the P-value in the analysis of variance test indicated statistical significance, the differences were assessed by the Dunnett's test. A value of P < 0·05 was considered to be statistically significant.
Results
Effect of NK-92 cells on colon and prostate cancer cell growth and expression of death receptors
First, we investigated the anti-cancer effect of NK cells to cancer cells. When the PC3 prostate cancer cell or SW620 colon cancer cells were co-cultured with NK-92 cell, the growth of cancer cells was significantly inhibited (Fig. 1a). The TNF family ligands such as FASL, TNF, TRAIL and APO3L are released by NK-92 cells and have been shown to induce cancer cell death.12,13,28,29 These ligands bind with different death receptors such as TNFR1 (for TNF-α), TNFR2 (for TNF-β), DR2 (for FASL), DR3 (for APO3L) and DR4 or DR5 (for TRAIL) to cause cancer cell death through expression of death receptor in cancer cells.3,6,18 For this reason, we determined the mRNA expression of the death receptors in cancer cells after co-culture with NK cells, and we found that the TNFR2 and DR3 are increased in PC3 cells, and FAS and DR3 are increased in SW620 cells by co-culture with NK-92 cells (see Supplementary material, Fig. S1). We also confirmed the increased expression of these death receptors by Western blot (Fig. 1b). Because the DR3 was increased in both PC3 and SW620 cells, we showed the DR3 expression on the cell surface by flow cytometry (Fig. 1c). We showed that the DR3-positive cells are increased from 2·17% to 72·56% in PC3 cells and from 1·75% to 85·93% in SW620 cells by co-culture with NK-92 cells. After increase of DR3 expression, apoptosis is induced through activation of caspase-3 and bax,18,19 so we investigated the role of NK-92 cells on the expression of these apoptosis-related proteins in cancer cells. The expression of apoptosis-related proteins such as cleaved caspase-3 and bax was increased in PC3 and SW620 cells co-cultured with NK-92 cells (Fig. 1d).
Figure 1.
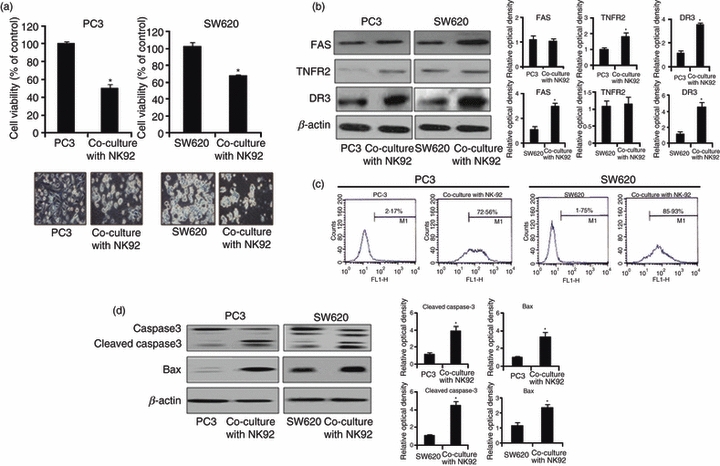
Effect of natural killer (NK) cells on the cytotoxicity to prostate and colon cancer cells. (a) Colon cancer cells (SW620) and prostate cancer cells (PC3) were cultured in 24-well plates (5 × 104 cells/well), and then co-cultured with NK-92 cells (5 × 104) for 48 hr. Thereafter, cell growth was measured by direct counting after Trypan blue staining. (b) NK-92 cells (5 × 104) were co-cultured with PC3 or SW620 cells for 48 hr, and then harvested. Death receptor [FAS, tumour necrosis factor receptor 2 (TNFR2) and death receptor 3 (DR3)] expression was detected by Western blotting in PC3 and SW620 cells. (c) NK-92 cells (5 × 104) were co-cultured with PC3 or SW620 cells for 48 hr, and then harvested. DR3 receptor expression was investigated by flow cytometry using anti-DR3 in PC3 and SW620 cells as described in Materials and methods. (d) SW620 and PC3 were cultured into 24-well plates (5 × 104 cells/well), and thereafter co-cultured with NK-92 cells (5 × 104) for 48 hr, and then harvested. Apoptotic protein expression such as cleaved caspase-3 and bax was determined by Western blotting. Relative optical density of specific bands is expressed versus actin amount. The figures are representatives of three experiments with replicates. Values are mean ± SD of three experiments with replicates. *P<0·05 indicates significantly different from the control group.
IL-32 expression in NK cells and enhancing cytotoxic effect of NK-92 cells on cancer cells
We then investigated whether IL-32 expression in NK-92 cells could affect the cytotoxic effect of NK-92 cells against cancer cells. Interleukin-32 was endogenously expressed in NK-92 cells (Fig. 2a left), and enforced expression by transfection of IL-32 over-expressed plasmid did not increase the endogenous expression of IL-32 in NK-92 cells. However, IL-32 siRNA reduced its expression (Fig. 2a right), suggesting that IL-32 is endogenously expressed in NK-92 cells. We then investigated whether the anti-cancer effect of NK cells is mediated by IL-32. When these cancer cells were co-cultured with NK-92 cells transfected with IL-32-specific siRNA, the NK cell-induced inhibition of cancer cell growth was abolished in both prostate and colon cancer cells (Fig. 2b). As shown in Fig. 1, the DR3 was commonly over-expressed in both cancer cell lines after co-culture with NK cells. Hence, to further demonstrate whether the increased expression of DR3 is related to IL-32 expression by NK cells, DR3 expression was determined in PC3 and SW620 cancer cells co-cultured with NK cells transfected with siRNA of IL-32. The increased DR3 in PC3 and SW620 cells was diminished by co-culture with IL-32-specific siRNA-transfected NK-92 cells (Fig. 2c). These data again indicated that the DR3 is important in the anti-cancer effect on prostate cancer or colon cancer cells by NK cells, and IL-32 may have a critical role in the expression of NK cell-induced DR3 in these cancers. The increased expression of apoptosis-related proteins such as cleaved caspase-3 and bax in PC3 and SW620 cells co-cultured with NK-92 cell was also abolished by IL-32-specific siRNA (Fig. 2d). These data indicated that the IL-32 has a critical role in the induction of NK cell-induced prostate cancer and colon cancer cell death.
Figure 2.
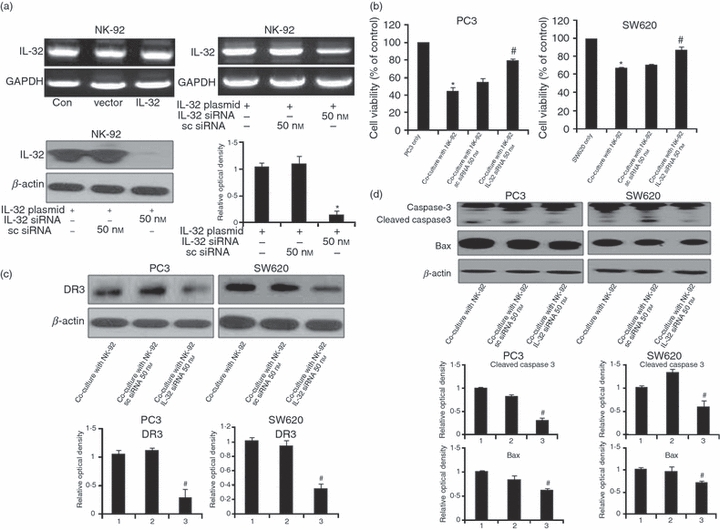
Expression of interleukin-32 (IL-32), and effects of IL-32 knockdown in NK-92 cells on the expression of death receptor 3 and apoptosis-related proteins of cancer cells. (a) IL-32 is endogenously expressed in NK-92 cells, and its expression by IL-32 plasmid transfection. NK-92 cells (1 × 106) were transfected with the IL-32 plasmid (1·5 μg) by electroporation and then incubated for 48 hr. The IL-32 expression was detected by reverse transcription-PCR (upper panel). NK-92 cells transfected with non-targeting control small interfering (si) RNA or IL-32 siRNA (50 nm/well) by electroporation were incubated for 48 hr. IL-32 expression was detected by Western blotting (lower panel). (b) Effect of silencing endogenous IL-32 expression on the cytotoxic effect of NK-92 cells. SW620 and PC3 were cultured into 24-well plates (5 × 104 cells/well), and then co-cultured with IL-32 siRNA-transfected NK-92 cells (5 × 104 cells) for 48 hr. Thereafter, cell growth was measured by direct counting after Trypan blue staining. Sc-siRNA, IL-32 non-targeting control siRNA. The figures are representatives of three experiments with replicates. (c) Effects of IL-32 knockdown in NK-92 cells on the expression of DR3 of cancer cells. NK-92 cells transfected with non-targeting control siRNA or IL-32 siRNA (50 nm/well) by electroporation for 24 hr were co-cultured with colon cancer cells (SW620) or prostate cancer cells (PC3) for 48 hr. The expression of DR3 was determined by Western blotting. (d) Effects of IL-32 knockdown in NK-92 cells on the expression of apoptosis-related proteins of cancer cells. NK-92 cells transfected with non-targeting control siRNA or with IL-32 siRNA (50 nm/well) by electroporation were co-cultured with SW620 or PC3 cells for 48 hr. The expression of cleaved caspase-3 and bax was determined by Western blotting. Quantitative analysis of immunoblot was performed by Western blot density in PC3 and SW620 cells. Relative optical density of specific bands is expressed versus actin amount. The figures are representatives of three experiments with replicates. Values are mean ± SD of three experiments with replicates. *P<0·05 indicates significantly different from the control group. #P<0·05 indicates significantly different from the co-cultured cell group.
Effect of IL-32 on the expression of APO3L in NK cells, and effect of APO3L and DR3 knockdown on the growth of cancer cells co-cultured with NK cells
To confirm that the DR3 has a critical role in the death of prostate cancer or colon cancer cells by the enhancing effect of IL-32 on NK-cell-induced cytotoxicity, we investigated the expression of APO3L, a ligand of DR3, in NK cells. Consistent with DR3 expression in cancer cells, the expression of APO3L was also increased in NK-92 cells after co-culture with cancer cells (Fig. 3a), The expression of APO3L in NK-92 cells was blocked by IL-32 siRNA treatment (Fig. 3b). When the PC3 or SW620 cells were transfected with DR3 siRNA, and then co-cultured with NK cells, the growth inhibition of cancer cells by NK-92 cells was diminished (Fig. 3c). When the anti-DR3 pre-treated PC3 or SW620 cells were co-cultured with NK-92 cells, the growth inhibitory effect was also diminished, and the PC3 or SW620 cells were co-cultured with APO3L pre-treated NK-92 cells, the growth inhibitory effect was also diminished (Fig. 3d). These data indicated that the cytotoxic effect of NK-92 cells to the cancer cells is mediated through DR3.
Figure 3.
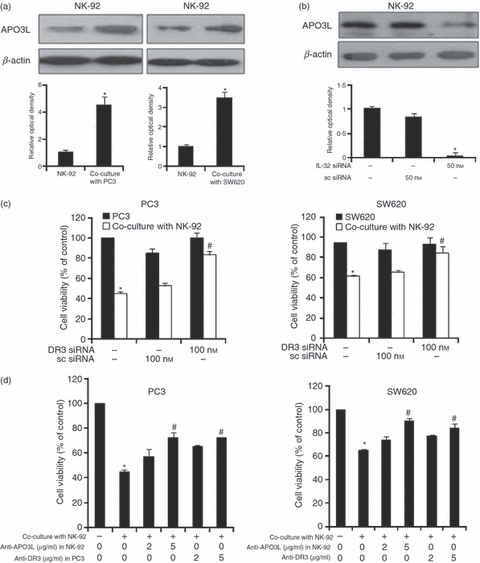
Effect of interleukin-32 (IL-32) on the expression of APO3L in natural killer (NK) cells, and effect of APO3L and death regulator 3 (DR3) knockdown on the growth of cancer cells co-cultured with NK cells. (a) Effect of co-culture on the expression of APO3L. NK-92 cells (5 × 104) were co-cultured with PC3 or SW620 cells for 48 hr. APO3L expression was detected by Western blotting in NK-92 cells. (b) Effect of silencing endogenous IL-32 expression on the expression of APO3L in NK-92 cells. NK-92 cells were transfected with non-targeting control small interfering (si) RNA or IL-32 siRNA (50 nm/well) by electroporation and then incubated for 48 hr. APO3L expression was detected by Western blotting. (c) Effects of DR3 knockdown on the cancer cell growth by co-culture with NK cells. SW620 and PC3 (5 × 104 cells/well) were transfected with non-targeting control siRNA or DR3 siRNA (100 nm/well) for 24 hr. Thereafter co-cultured with NK-92 cells for 48 hr, and cancer cell growth was measured by direct counting after Trypan blue staining. (d) Effects of anti-APO3L in NK-32 cells and anti-DR3 in cancer cells on the cancer cell growth by co-culture with NK cells. Before initiation of co-culture, anti-APO3L (2 or 5 μg/ml) was incubated in NK-92 cells for 2 hr. Pre-incubated NK-92 cells (5 × 104) were co-cultured with PC3 or SW620 cells for 48 hr, and then cancer cell growth was measured by direct counting after trypan blue staining. Alternatively, cancer cells were pre-incubated with anti-DR3 (2 or 5 μg/ml) for 2 hr. Pre-incubated cancer cells (5 × 104) were co-cultured with PC3 or SW620 cells for 48 hr, and then cancer cell growth was measured by direct counting after Trypan blue staining. Quantitative analysis of immunoblot was performed by Western blot density in PC3 and SW620 cells. Relative optical density of specific bands is expressed versus actin amount. The figures are representatives of three experiments with replicates. Values are mean ± SD of three experiments with replicates. *P<0·05 indicates significantly different from the control group. #P<0·05 indicates significantly different from the co-cultured cell group.
Effect of IL-32 on the level of nitric oxide and the expression of perforin/granzyme expression in NK cells
In addition to the death receptor pathway, NK cells can use the perforin/granzyme-containing granule exocytosis pathway and the nitric oxide pathway in the tumour killing effect.3,37–39 We studied the expression of perforin and granzyme B, and nitric oxide release in NK cells and NK cells cultured with cancer cells. We found that perforin and granzyme B expression in NK-92 cells was not changed after co-culture with cancer cells (Fig. 4a). We also found that the expression of perforin and granzyme B was not changed after knockdown of IL-32 with IL-32 siRNA in NK-92 cells (Fig. 4b). The release of nitric oxide into the medium was not changed after co-culture with cancer cells, either (Fig. 4c). After knockdown of IL-32 with IL-32 siRNA in NK-92 cells, nitric oxide level was not changed (Fig. 4d). These results indicate that the nitric oxide pathway and the perforin/granzyme pathway were not affected by IL-32 in the cytotoxic processing of NK cells and by co-culture with cancer cells.
Figure 4.
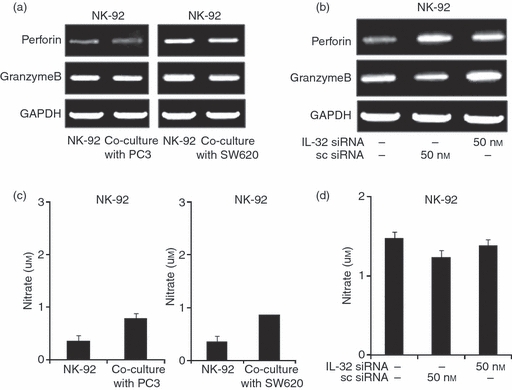
Effects of interleukin-32 (IL-32) on the expression of perforin/granzyme B and the level of nitric oxide (NO) of cancer cells co-cultured with natural killer (NK) cells. (a) Effect of co-culture on the expression of perforin and granzyme B. SW620 and PC3 cells were cultured into 24-well plates (5 × 104 cells/well) and then co-cultured with NK-92 cells (5 × 104) for 48 hr. Thereafter, expression of perforin and granzyme B was determined by reverse transcription-PCR in NK-92 cells. (b) Effects of IL-32 knockdown in NK-92 cells on the expression of perforin and granzyme B. NK-92 cells were transfected with non-targeting control small interfering (si) RNA or IL-32 siRNA (50 nm/well) by electroporation and then incubated for 48 hr, thereafter co-cultured with colon cancer cells (SW620) or prostate cancer cells (PC3) for 48 hr. The expression of perforin and granzyme B was determined by reverse transcription-PCR in NK-92 cells. The figures are representatives of three experiments with replicates. (c) Effect of co-culture on the level of nitric oxide. SW620 and PC3 were cultured into 24-well plates (5 × 104 cells/well) and then co-cultured with NK-92 cells (5 × 104) for 48 hr. Thereafter, the nitrite release in the supernatant was assessed by Griess reaction as described in Materials and methods. (d) Effects of IL-32 knockdown in NK-92 cells on the level of nitric oxide. NK-92 cells transfected with non-targeting control siRNA or IL-32 siRNA (50 nm/well) by electroporation were co-cultured with SW620 or PC3 for 48 hr. The nitrite release in the supernatant was assessed by Griess reaction as described in Materials and methods. The figures are representatives of three experiments with replicates. Values are mean ± SD of three experiments with replicates.
Discussion
In this study, we demonstrated that IL-32 plays a role in the cancer-cell-killing ability of NK cells through activation of the DR-3 and caspase-3 pathways in cancer cells. We showed that the IL-32 is endogenously, highly expressed in NK-92 cells, and the cancer cell growth was suppressed when cancer cells were co-cultured with NK-92 cells. Further study showed that the cancer-cell-killing ability of NK cells was abolished when the cancer cells were co-cultured with NK-92 cells transfected with IL-32 siRNA. These data indicated that the cytotoxic effect of NK-92 cells on the cancer cells could be in part mediated by IL-32 expression. Several studies demonstrated that the anti-tumour effect of NK cells is successful in patients with lung cancer, hepatic cancer, renal cell carcinoma and malignant melanoma.40–42 A recent study described the use of cytokine-enriched NK cells to treat patients with breast cancer.43 In these studies, the effective cytokine that has an effect on NK cell function is closely related to IL-2. The administration of IL-2 to widely metastasized cancer in humans effectively inhibited tumour growth.44,45In vivo administration of IL-18 elevates NK activity in mice, and in vitro treatment with IL-18 augments NK activity of murine splenocytes and human peripheral blood mononuclear cells.46–48 However, IL-18-deficient mice showed impaired NK cell activity against cancer cell growth.49 Interleukin-12 also stimulates NK cells and enhances cytolytic activity by inducing interferon-γ production,50 but IL-12-deficient mice showed impaired NK cell activity.51 In mice lacking IL-12 and IL-18 cytokines, the cytolytic activity of the NK cells is further impaired, indicating that the cytolytic activity of NK cells is synergized by IL-12 and IL-18 in vivo. It was known that IL-12 and IL-18 induce IL-32 to increase the survival of NK-92 cells so our data indicated that IL-32 could be involved in NK cell killing ability against cancer cells. However, its action mechanism is not clear.
The NK cells kill cancer cells through at least three mechanisms. NK cells can use the perforin/granzyme-containing granule exocytosis pathway, the nitric oxide pathway and the death receptor-ligand pathway.3,37–39 The perforin/granzyme pathway is the principle pathway by which NK cells kills the cancer cells or virus-infected cells by lysis, and this pathway occurs primarily in the spleen and lung. Nitric oxide is one of the most powerful effector molecules in the cytotoxic function of NK cells against tumour cells.39 We found that the expression of perforin and granzyme B by reverse transcription-PCR was not changed when NK-92 cells were co-cultured with cancer cells. The release of nitric oxide from NK-92 cells into the culture medium was hardly changed. These results indicated that the perforin/granzyme-containing granule exocytosis pathway and nitric oxide pathway may not be significant in our study conditions. So, we questioned that the reason for the enhanced cytotoxic effect of NK cells caused by IL-32 is related to the death receptor/ligand pathway. We found that the expression of APO3L (DR3 ligand) in NK-92 cells was elevated when NK cells were co-cultured with cancer cells, and knockdown of IL-32 by siRNA decreased the expression of APO3L in NK-92 cells. We also demonstrated that the mRNA expression of the death receptors was increased after co-culture of cancer cells with NK cells. We showed that the TNFR2 and DR3 are increased in PC3 cells, and FAS and DR3 were increased in SW620 cells by co-culture with NK-92 cells transfected with IL-32. It has been demonstrated that, depending on the type of cytokines and cancer cells, differential DRs and DR ligands are involved. Similar to our findings, other studies have also shown that IL-15 activates NK cells, so significantly increasing the cytolytic ability of NK cells against most solid tumour cells (cervical carcinoma, larynx cancer, human melanoma, lung cancer, liver hepatoma, breast cancer and gastric cancer) and haematological tumour cells. They showed that IL-15 can improve the function of NK cells through an NKG2D-dependent mechanism, and also augment the expression of cytotoxic effector molecules such as TRAIL and FasL. Interleukin-2 FasL, TNFα, granulocyte–macrophage colony-stimulating factor, interferon-γ, MIP (Macrophage inflammatory protein)-1α and MIP-1β dose dependently increased the expression of death receptors in cancer cells, and death receptor ligands in NK cells resulting in increased cytotoxicity against tumour cells.52 Moreover, it was also found that the increased DR3 in PC3 and SW620 cells by co-culture with NK-92 cell was diminished by co-culture with IL-32-specific siRNA-transfected NK-92 cells. Knockdown of DR3 in the co-culture of cancer cells with NK-92 cells abolished cancer cell growth inhibition. In many malignant tumour cells, the expression level of MHC-I and MHC-II is repressed, thereby immunogenicity is repressed.25,26 Although, cytotoxic T cell and helper T cells require both MHC class I and II for stimulation and tumour killing effect, NK cells do not require MHC for antigen presentation, even though they participate in the innate immune response.8,27 These results indicate that increase of APO3L in NK-92 cells by IL-32 could cause DR3 in cancer cells leading to anti-cancer activity of NK-92 cells.
Activation of DR3 leads to activation of apoptosis through TRADD, FADD and caspase-8 pathways, and subsequently activates apoptosis-related proteins such as caspase-3 and bax.15,18,19 The increased apoptosis-related proteins such as cleaved caspase-3 and bax in cancer cells were induced by NK-92 cells, and their expression was abolished by IL-32 knockdown. These results showed that IL-32 may be critical in the NK-cell-induced cell death in prostate and colon cancer cells. In our previous study, we found that the infiltration of NK cells into tumour tissue was elevated in nude mice xenografted with IL-32γ-transfected colon cancer cells as well as IL-32γ-over-expressed mice inoculated with melanoma cells.53 Overall, our study is the first to identify that IL-32 has anti-tumour effects on colon cancer cells and prostate cancer cells, and these effects may be related to the enhancement of the cytotoxic effect of NK cells via activation of DR3 and caspase 3 pathways in colon and prostate cancer cells as well as APO3L in NK cells. Altogether, our findings are important in understanding the role of IL-32 as anti-tumour cytokine, and it may be of benefit to cancer therapy.
Acknowledgments
This work was supported by Korea Research Foundation Grant (MRC 9440, R13-2010-002948).
Disclosures
The author declares no conflict of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Effect of natural killer (NK) cells on the expression of DRs in prostate cancer and colon cancer cells.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Cerwenka A, Lanier LL. Natural killer cells, viruses and cancer. Nat Rev Immunol. 2001;1:41–9. doi: 10.1038/35095564. [DOI] [PubMed] [Google Scholar]
- 2.Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol. 2001;22:633–40. doi: 10.1016/s1471-4906(01)02060-9. [DOI] [PubMed] [Google Scholar]
- 3.Smyth MJ, Hayakawa Y, Takeda K, Yagita H. New aspects of natural-killer-cell surveillance and therapy of cancer. Nat Rev Cancer. 2002;2:850–61. doi: 10.1038/nrc928. [DOI] [PubMed] [Google Scholar]
- 4.Biron CA. Activation and function of natural killer cell responses during viral infections. Curr Opin Immunol. 1997;9:24–34. doi: 10.1016/s0952-7915(97)80155-0. [DOI] [PubMed] [Google Scholar]
- 5.Glas R, Franksson L, Une C, et al. Recruitment and activation of natural killer (NK) cells in vivo determined by the target cell phenotype. An adaptive component of NK cell-mediated responses. J Exp Med. 2000;191:129–38. doi: 10.1084/jem.191.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brady J, Carotta S, Thong RP, et al. The interactions of multiple cytokines control NK cell maturation. J Immunol. 2010;185:6679–88. doi: 10.4049/jimmunol.0903354. [DOI] [PubMed] [Google Scholar]
- 7.Waldmann TA, Tagaya Y. The multifaceted regulation of interleukin-15 expression and the role of this cytokine in NK cell differentiation and host response to intracellular pathogen. Annu Rev Immunol. 1999;17:19–49. doi: 10.1146/annurev.immunol.17.1.19. [DOI] [PubMed] [Google Scholar]
- 8.Suzuki K, Nakazato H, Matsui H, et al. NK cell-mediated anti-tumor immune response to human prostate cancer cell, PC-3: immunogene therapy using a highly secretable form of interleukin-15 gene transfer. J Leukoc Biol. 2001;69:531–7. [PubMed] [Google Scholar]
- 9.Ferlazzo G, Pack M, Thomas D, et al. Distinct roles of IL-12 and IL-15 in human natural killer cell activation by dendritic cells from secondary lymphoid organs. Proc Natl Acad Sci USA. 2004;101:16606–11. doi: 10.1073/pnas.0407522101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Granucci F, Zanoni I, Pavelka N, et al. A contribution of mouse dendritic cell-derived IL-2 for NK cell activation. J Exp Med. 2004;200:287–95. doi: 10.1084/jem.20040370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zamai L, Ponti C, Mirandola P, et al. NK cells and cancer. J Immunol. 2007;178:4011–6. doi: 10.4049/jimmunol.178.7.4011. [DOI] [PubMed] [Google Scholar]
- 12.Screpanti V, Wallin RP, Grandien A, Ljunggren HG. Impact of FASL-induced apoptosis in the elimination of tumor cells by NK cells. Mol Immunol. 2005;42:495–9. doi: 10.1016/j.molimm.2004.07.033. [DOI] [PubMed] [Google Scholar]
- 13.Smyth MJ, Cretney E, Takeda K, et al. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) contributes to interferon γ-dependent natural killer cell protection from tumor metastasis. J Exp Med. 2001;193:661–70. doi: 10.1084/jem.193.6.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang S, El-Deiry WS. TRAIL and apoptosis induction by TNF-family death receptors. Oncogene. 2003;22:8628–33. doi: 10.1038/sj.onc.1207232. [DOI] [PubMed] [Google Scholar]
- 15.Osawa K, Takami N, Shiozawa K, Hashiramoto A, Shiozawa S. Death receptor 3 (DR3) gene duplication in a chromosome region 1p36.3: gene duplication is more prevalent in rheumatoid arthritis. Genes Immun. 2004;5:439–43. doi: 10.1038/sj.gene.6364097. [DOI] [PubMed] [Google Scholar]
- 16.Wiley SR, Winkles JA. TWEAK, a member of the TNF superfamily, is a multifunctional cytokine that binds the TweakR/Fn14 receptor. Cytokine Growth Factor Rev. 2003;14:241–9. doi: 10.1016/s1359-6101(03)00019-4. [DOI] [PubMed] [Google Scholar]
- 17.Marsters SA, Sheridan JP, Pitti RM, Brush J, Goddard A, Ashkenazi A. Identification of a ligand for the death-domain-containing receptor Apo3. Curr Biol. 1998;8:525–8. doi: 10.1016/s0960-9822(98)70204-0. [DOI] [PubMed] [Google Scholar]
- 18.Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281:1305–8. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- 19.Chinnaiyan AM, O'Rourke K, Yu GL, et al. Signal transduction by DR3, a death domain-containing receptor related to TNFR-1 and CD95. Science. 1996;274:990–2. doi: 10.1126/science.274.5289.990. [DOI] [PubMed] [Google Scholar]
- 20.Hahne M, Rimoldi D, Schroter M, et al. Melanoma cell expression of Fas (Apo-1/CD95) ligand: implications for tumor immune escape. Science. 1996;274:1363–6. doi: 10.1126/science.274.5291.1363. [DOI] [PubMed] [Google Scholar]
- 21.Ozören N, El-Deiry WS. Cell surface Death Receptor signaling in normal and cancer cells. Semin Cancer Biol. 2003;13:135–47. doi: 10.1016/s1044-579x(02)00131-1. [DOI] [PubMed] [Google Scholar]
- 22.Albertsson PA, Basse PH, Hokland M, et al. NK cells and the tumour microenvironment: implications for NK-cell function and anti-tumour activity. Trends Immunol. 2003;24:603–9. doi: 10.1016/j.it.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 23.Smyth MJ, Takeda K, Hayakawa Y, Peschon JJ, van den Brink MR, Yagita H. Nature's TRAIL – on a path to cancer immunotherapy. Immunity. 2003;18:1–6. doi: 10.1016/s1074-7613(02)00502-2. [DOI] [PubMed] [Google Scholar]
- 24.Screpanti V, Wallin RP, Ljunggren HG, Grandien A. A central role for death receptor-mediated apoptosis in the rejection of tumors by NK cells. J Immunol. 2001;167:2068–73. doi: 10.4049/jimmunol.167.4.2068. [DOI] [PubMed] [Google Scholar]
- 25.Sanda MG, Restifo NP, Walsh JC, Kawakami Y, Nelson WG, Pardoll DM, Simons JW. Molecular characterization of defective antigen processing in human prostate cancer. J Natl Cancer Inst. 1995;87:280–5. doi: 10.1093/jnci/87.4.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bander NH, Yao D, Liu H, et al. MHC class I and II expression in prostate carcinoma and modulation by interferon-α and -γ. Prostate. 1997;33:233–9. doi: 10.1002/(sici)1097-0045(19971201)33:4<233::aid-pros2>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 27.Robertson MJ, Ritz J. Biology and clinical relevance of human natural killer cells. Blood. 1990;76:2421–38. [PubMed] [Google Scholar]
- 28.Migone TS, Zhang J, Luo X, et al. TL1A is a TNF-like ligand for DR3 and TR6/DcR3 and functions as a T cell costimulator. Immunity. 2002;16:479–92. doi: 10.1016/s1074-7613(02)00283-2. [DOI] [PubMed] [Google Scholar]
- 29.Maki G, Klingemann HG, Martinson JA, Tam YK. Factors regulating the cytotoxic activity of the human natural killer cell line, NK-92. J Hematother Stem Cell Res. 2001;10:369–83. doi: 10.1089/152581601750288975. [DOI] [PubMed] [Google Scholar]
- 30.Dahl CA, Schall RP, He HL, Cairns JS. Identification of a novel gene expressed in activated natural killer cells and T cells. J Immunol. 1992;148:597–603. [PubMed] [Google Scholar]
- 31.Tam YK, Maki G, Miyagawa B, Hennemann B, Tonn T, Klingemann HG. Characterization of genetically altered, interleukin 2-independent natural killer cell lines suitable for adoptive cellular immunotherapy. Hum Gene Ther. 1999;10:1359–73. doi: 10.1089/10430349950018030. [DOI] [PubMed] [Google Scholar]
- 32.Kim SH, Han SY, Azam T, Yoon DY, Dinarello CA. Interleukin-32: a cytokine and inducer of TNFα. Immunity. 2005;22:131–42. doi: 10.1016/j.immuni.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 33.Shoda H, Fujio K, Yamaguchi Y, et al. Interactions between IL-32 and tumor necrosis factor α contribute to the exacerbation of immune-inflammatory diseases. Arthritis Res Ther. 2006;8:166. doi: 10.1186/ar2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nold-Petry CA, Nold MF, Zepp JA, Kim SH, Voelkel NF, Dinarello CA. IL-32-dependent effects of IL-1β on endothelial cell functions. Proc Natl Acad Sci USA. 2009;106:3883–8. doi: 10.1073/pnas.0813334106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dinarello CA, Kim SH. IL-32, a novel cytokine with a possible role in disease. Ann Rheum Dis. 2006;65:61–4. doi: 10.1136/ard.2006.058511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kang JW, Choi SC, Cho MC, et al. A proinflammatory cytokine interleukin-32β promotes the production of an anti-inflammatory cytokine interleukin-10. Immunology. 2009;128:532–40. doi: 10.1111/j.1365-2567.2008.03025.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Henkart PA. Lymphocyte-mediated cytotoxicity: two pathways and multiple effector molecules. Immunity. 1994;1:343–6. doi: 10.1016/1074-7613(94)90063-9. [DOI] [PubMed] [Google Scholar]
- 38.Kojima H, Shinohara N, Hanaoka S, et al. Two distinct pathways of specific killing revealed by perforin mutant cytotoxic T lymphocytes. Immunity. 1994;1:357–64. doi: 10.1016/1074-7613(94)90066-3. [DOI] [PubMed] [Google Scholar]
- 39.Cifone MG, Ulisse S, Santoni A. Natural killer cells and nitric oxide. Int Immunopharmacol. 2001;1:1513–24. doi: 10.1016/s1567-5769(01)00095-9. [DOI] [PubMed] [Google Scholar]
- 40.Rosenberg SA, Lotze MT, Yang JC, et al. Prospective randomized trial of high-dose interleukin-2 alone or in conjunction with lymphokine-activated killer cells for the treatment of patients with advanced cancer. J Natl Cancer Inst. 1993;85:622–32. doi: 10.1093/jnci/85.8.622. [DOI] [PubMed] [Google Scholar]
- 41.Margolin KA. Interleukin-2 in the treatment of renal cancer. Semin Oncol. 2000;27:194–203. [PubMed] [Google Scholar]
- 42.Semino C, Martini L, Queirolo P, et al. Adoptive immunotherapy of advanced solid tumors: an eight year clinical experience. Anticancer Res. 1999;19:5645–9. [PubMed] [Google Scholar]
- 43.Burns LJ, Weisdorf DJ, DeFor TE, et al. Enhancement of the anti-tumor activity of a peripheral blood progenitor cell graft by mobilization with interleukin 2 plus granulocyte colony-stimulating factor in patients with advanced breast cancer. Exp Hematol. 2000;28:96–103. doi: 10.1016/s0301-472x(99)00129-0. [DOI] [PubMed] [Google Scholar]
- 44.Carson WE, Parihar R, Lindemann MJ, et al. Interleukin-2 enhances the natural killer cell response to Herceptin-coated Her2/neu-positive breast cancer cells. Eur J Immunol. 2001;31:3016–25. doi: 10.1002/1521-4141(2001010)31:10<3016::aid-immu3016>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 45.Kossman SE, Scheinberg DA, Jurcic JG, Jimenez J, Caron PC. A phase I trial of humanized monoclonal antibody HuM195 (anti-CD33) with low-dose interleukin 2 in acute myelogenous leukemia. Clin Cancer Res. 1999;5:2748–55. [PubMed] [Google Scholar]
- 46.Okamura H, Tsutsui H, Kashiwamura S, Yoshimoto T, Nakanishi K. Interleukin-18 (IL-18): a novel cytokine that augments both innate and acquired immunity. Adv Immunol. 1998;70:281. doi: 10.1016/s0065-2776(08)60389-2. [DOI] [PubMed] [Google Scholar]
- 47.Takeda K, Tsutsui H, Yoshimoto T, et al. Defective NK cell activity and Th1 response in IL-18-deficient mice. Immunity. 1998;8:383. doi: 10.1016/s1074-7613(00)80543-9. [DOI] [PubMed] [Google Scholar]
- 48.Hyodo Y, Matsui K, Hayashi N, et al. IL-18 up-regulates perforin-mediated NK activity without increasing perforin messenger RNA expression by binding to constitutively expressed IL-18 receptor. J Immunol. 1999;162:1662–8. [PubMed] [Google Scholar]
- 49.Shi FD, Takeda K, Akira S, Sarvetnick N, Ljunggren HG. IL-18 directs autoreactive T cells and promotes autodestruction in the central nervous system via induction of IFN-γ by NK cells. J Immunol. 2000;165:3099–104. doi: 10.4049/jimmunol.165.6.3099. [DOI] [PubMed] [Google Scholar]
- 50.Hunter CA, Timans J, Pisacane P, et al. Comparison of the effects of interleukin-1 α, interleukin-1β and interferon-γ-inducing factor on the production of interferon-γ by natural killer. Eur J Immunol. 1997;27:2787–92. doi: 10.1002/eji.1830271107. [DOI] [PubMed] [Google Scholar]
- 51.Ehl S, Bischoff R, Ostler T, et al. The role of Toll-like receptor 4 versus interleukin-12 in immunity to respiratory syncytial virus. Eur J Immunol. 2004;34:1146–53. doi: 10.1002/eji.200324449. [DOI] [PubMed] [Google Scholar]
- 52.Berg M, Lundqvist A, McCoy PJ, et al. Clinical-grade ex vivo-expanded human natural killer cells up-regulate activating receptors and death receptor ligands and have enhanced cytolytic activity against tumor cells. Cytotherapy. 2009;11:341–55. doi: 10.1080/14653240902807034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oh JH, Cho MC, Kim JH, et al. L-32γ inhibits cancer cell growth through inactivation of NF-κB and STAT3 signals. Oncogene. 2011;30:3345–59. doi: 10.1038/onc.2011.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.