

Abstract
Innate immunity constitutes the first line of defence against both external and endogenous threats in the brain, and microglia cells are considered key mediators of this process. Recent studies have shown that microRNAs (miRNAs) may play a determinant role in the regulation of gene expression during innate immune responses. The major goal of this work was to investigate the contribution of a specific miRNA – miR-155 – to the modulation of the microglia-mediated immune response. For this purpose, in vitro studies were performed in N9 microglia cells to evaluate changes in the levels of this miRNA following microglia activation. A strong up-regulation of miR-155 expression was observed following microglia exposure to lipopolysaccharide, which was consistent with a decrease in the levels of the suppressor of cytokine signalling 1 (SOCS-1) protein, a key inhibitor of the inflammatory process and a predicted target of miR-155. The miR-155 knockdown by anti-miRNA oligonucleotides up-regulated SOCS-1 mRNA and protein levels and significantly decreased the production of nitric oxide and the expression of inflammatory cytokines and inducible nitric oxide synthase. Finally, treatment of neuronal primary cultures with conditioned medium obtained from microglia cells, in which miR-155 was inhibited before cell activation, decreased inflammatory-mediated neuronal cell death. Overall, our results show that miR-155 has a pro-inflammatory role in microglia and is necessary for the progression of the immune response through the modulation of SOCS-1, suggesting that, in a chronic inflammatory context, miR-155 inhibition can have a neuroprotective effect.
Keywords: inflammation, innate immune response, microglia, miR-155, neurodegeneration
Introduction
Inflammation is believed to play an important role in several central nervous system (CNS) diseases of both acute and chronic nature. Local inflammatory reactions are early events following neuronal death as a consequence of stroke, infection and traumatic brain injury,1 but can also be a response to the accumulation of misfolded or aggregated proteins in neurodegenerative disorders such as Alzheimer's disease, Parkinson's disease and multiple sclerosis.2 As resident immune cells of the CNS, microglia cells are responsible for monitoring the CNS environment and sensing potential threats, through pattern recognition receptors, such as Toll-like receptors (TLRs), capable of binding highly conserved structural motifs present in different families of pathogens.3 Upon recognition of a specific pathogen-associated pattern, microglia change to an activated state and initiate both innate and adaptive immune responses, by producing an array of pro-inflammatory cytokines, free radicals and nitric oxide, while simultaneously initiating the recruitment of other immune-related cells.
Although microglia-mediated immune responses have the major purpose of promoting pathogen clearance and tissue regeneration, the resulting inflammatory state, if left unchecked, can aggravate neuronal injury. It is now believed that neuroinflammation is an important contributor to neurodegeneration in various CNS diseases, such as Alzheimer's disease4 and multiple sclerosis.5 Neurons are particularly susceptible to oxidative damage and to certain inflammatory mediators, which are either themselves neurotoxic or attract leucocytes with cytotoxic properties.6,7 This hypothesis has been supported by several studies showing that inhibiting microglia activation or blocking cytokine expression, cytokine receptor activation and the production of oxidative species contributes to neuronal survival in different models of brain injury.8–10
Compelling evidence now links small endogenous RNA molecules, known as microRNAs (miRNAs), to the regulation of many biological processes such as development, cellular differentiation and disease. These small RNA molecules exert their function by modulating mRNA half-life or inhibiting its translation via co-operative binding to the 3′ untranslated region (UTR) of target genes. Recently, miRNAs were shown to be directly involved in the control of both innate and adaptive immune responses, by directly interfering with TLR-mediated signal transduction mechanisms11 and the ensuing cytokine response.12 Altered miRNA expression has also been observed in chronic inflammatory diseases, such as psoriasis, atopic eczema and rheumatoid arthritis,13 which suggests the involvement of these RNA molecules in immune-mediated pathologies.
Among various miRNA, miR-155 has been associated with the regulation of different immune-related processes, such as haematopoiesis,14 B-cell and T-cell differentiation,15 cancer16 and innate immunity.12 The miR-155 is processed from an exon of a non-coding RNA transcribed from the B-cell Integration Cluster located on chromosome 21, showing strong sequence homology among humans, mice and hens, and is highly expressed in cells of lymphoid and myeloid origin.17 Recently, miR-155 has been identified and characterized as a component of macrophage and monocyte response to different types of inflammatory mediators, such as bacterial lipopolysaccharide (LPS), interferon-β (IFN-β), tumour necrosis factor-α (TNF-α) and polyriboinosinic-polyribocytidylic acid [poly(I:C)].12,18,19
Many of the miR-155 target transcripts identified so far are pro-apoptotic and anti-inflammatory proteins, such as the Fas-associated death domain protein, IκB kinase ε, inositol 5-phosphatase 1 and the suppressor of cytokine signalling-1 (SOCS-1). SOCS-1 belongs to a family of proteins known to regulate the response of immune cells to cytokines and other inflammatory stimuli, such as LPS, through direct inhibition of the Janus tyrosine kinase (JAK) and consequent inhibition of signal transducer and activator of transcription factors (STAT), as a ‘classical’ negative feedback loop. In addition, the C-terminal SOCS box domain interacts with components of the ubiquitin ligase system and mediates proteasomal degradation of associated proteins, including key elements of other pro-inflammatory pathways, such as the nuclear factor-κB and Jun N-terminal kinase pathways. Experimental evidence suggests that miR-155 plays a pro-inflammatory role and may be implicated in chronic inflammatory processes, such as those contributing to cancer and to certain neurodegenerative diseases.
Given the similarities between microglia and other cells of the immune system, such as macrophages and dendritic cells, where miR-155 has been found to be up-regulated upon activation,20 in this work we investigated the contribution of miRNA-155 to microglia activation and microglia-mediated immune responses. To our knowledge, this is the first study providing evidence that miR-155 has a strong pro-inflammatory role during microglia activation and is required for SOCS-1 post-transcriptional regulation and progression of the immune response in these cells. Moreover, our results suggest that miR-155 inhibition induces neuronal protection from microglia-induced damage, and miR-155 may therefore constitute an interesting and promising target for the control of neuronal inflammation.
Materials and methods
Materials
The cationic lipids 1,2 dioleoyl-3(trimethylammonium)propane (DOTAP) and 1,2-dioleoyl-sn-glycero-3-succinate (DOGS), and the helper lipid 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) were purchased from Avanti Polar Lipids (Alabaster, AL). The anti-miR-155 and negative control oligonucleotides were obtained from Ambion (Austin, TX). The plasmid encoding miR-155, the control plasmid and the plasmid encoding luciferase and the 3′ UTR of SOCS-1 were obtained from Origene (Rockville, MD). The SOCS-1 and inducible nitric oxide synthase (iNOS) antibodies were purchased from Cell Signaling (Danvers, MA). The anti-miR-155 locked nucleic acid (LNA) in situ hybridization probe, as well as all quantitative reverse transcription (qRT-) PCR primers for miRNA detection were purchased from Exiqon (Vedbaek, Denmark). The α-tubulin and actin antibodies were obtained from Sigma (St Louis, MO). All other chemicals were obtained from Sigma, unless stated otherwise.
Microglia cell culture and activation
N9 cells (immortalized mouse microglia cells) were cultured at 37° in a humidified atmosphere containing 5% CO2 and maintained in RPMI-1640 medium (Gibco, Paisley, UK) supplemented with 5% heat inactivated fetal bovine serum (Gibco), 100 μg/ml streptomycin and 1 U/ml penicillin. N9 microglia cells were plated 24 hr before the beginning of each experiment at a density of 250 000 cells/cm2 in uncoated six-well multi-well plates or at a density of 100 000 cells/cm2 in 12-well multi-well plates.
Primary microglia cells were obtained from 3-day-old C57BL/6 newborn mice. After digestion and dissociation of the dissected mouse cortices in Hanks’ buffered salt solution (136·7 mm NaCl, 2·1 mm NaHCO3, 0·22 μm KH2PO4, 5·3 mm KCl, 2·7 mm glucose, 10 mm HEPES, pH 7·3) supplemented with trypsin (1 mg/ml), mixed glial cultures were prepared by re-suspending the cell suspension in Dulbecco's modified Eagles’ medium : F12 Glutamax (Gibco), supplemented with 10% heat inactivated fetal bovine serum (Gibco) and 10 μg/ml gentamicin. Cells were plated at 20 × 106 cells/flask density onto 75 cm2 cell culture flasks, previously coated with poly-L lysine and maintained in culture at 37° in a humidified atmosphere containing 5% CO2 for 2 weeks. The cell medium was replaced each 5 days and, after the first medium change, M-CSF 0·25 ng/ml (macrophage colony-stimulating factor; PeproTech, Rocky Hill, NJ) was added to the flasks to promote microglia proliferation. After achieving 90% confluence, mixed glial cultures were subjected to shaking at 37° and 220 g for 2 hr, to promote microglia detachment from the flasks. The cell medium, containing the released microglia cells, was collected from each flask and centrifuged at 112 g for 5 min to promote cell sedimentation. Microglia cells were ressuspended in Dulbecco's modified Eagles’ medium:F12 Glutamax, supplemented with 10% fetal bovine serum and 10 μg/ml gentamicin, and plated onto 12-well multi-well plates at a density of 100 000 cells/well for qRT-PCR experiments or onto eight-well chamber slides at a density of 25 000 cells/well for in situ hybridization experiments. Regular characterization of primary microglia cultures indicated the presence of over 90% microglia cells, as determined by CD11b and GFAP immunostaining, so confirming the purity of these cultures.
The N9 and primary microglia activation was achieved by exposure to LPS at 0·1, 0·5 or 1 μg/ml, for different periods of time, ranging from 30 min to 18 hr.
Liposome and lipoplex preparation
The delivery liposomal system (DLS) cationic liposomes were prepared by mixing 1 mg DOGS with 1 mg DOPE in 40 μl 90% ethanol, followed by the addition of 360 μl H2O, as described previously.21 After homogenization, the mixture was incubated for at least 30 min to allow liposome formation. The final lipid concentration was 5 mg/ml (2·5 mg DOGS and 2·5 mg DOPE). The DLS lipoplexes were prepared by gently mixing 10 μg anti-miRNA oligonucleotides with 190 μg total lipid in HEPES-buffered saline solution (HBS: 20 mm HEPES, 100 mm NaCl, pH 7·4) at a final volume of 1300 μl, followed by incubation for 30 min at room temperature.
Cationic liposomes composed of DOTAP : DOPE (1 : 1 molar ratio) were prepared as previously described by Campbell.22 Briefly, a mixture of 1 ml DOTAP and 1 ml DOPE in chloroform (from stock solutions of 25 mg/ml DOTAP and 26·6 mg/ml DOPE) was dried under nitrogen to obtain a thin lipid film. The film was dissolved in 100 μl ultrapure ethanol and the resulting ethanol solution was injected with a Hamilton syringe into 900 μl pre-heated (40°) HBS buffer, maintained continuously under vortex. The resulting multi-lamellar vesicles were briefly sonicated to obtain small uni-lamellar vesicles and diluted with HBS to a final DOTAP concentration of 1 mg/ml. Folate-associated lipoplexes (FA-lipoplexes) were prepared by incubating 41·9 μg DOTAP with 320 μg folate (32 μg/μg pDNA) for 15 min, followed by addition of 10 μg pDNA at a final volume of 1 ml in HBS. The mixture was further incubated for 30 min at room temperature. Both liposome formulations were stored at 4° until use and the lipoplexes were used immediately after preparation.
Transfection
Inhibition or over-expression of miR-155 was achieved by delivery of anti-miR-155 oligonucleotides or plasmid DNA encoding miR-155, respectively, to N9 cells. Immediately before transfection, cells were washed and the medium was replaced with Optimem (900 μl/well), free of serum and antibiotics. For inhibition of miR-155, 100 μl DLS lipoplexes containing 14·6 μg lipid and 0·1 nmol (0·772 μg) anti-miR-155 oligonucleotides were delivered to N9 cells, to obtain a final oligonucleotide concentration of 100 nm/well. Parallel experiments were performed using a negative control oligonucleotide sequence to ensure that the modulation of miR-155 targets could be attributed only to the specific anti-miR-155 oligonucleotide and not to the transfection process per se. Delivery of plasmid DNA to N9 cells was achieved through the use of FA-lipoplexes. One hundred microlitres of FA-lipoplexes, containing pmiR-155 were delivered to N9 cells to obtain a final plasmid concentration of 1 μg/well. In parallel experiments, a reporter plasmid, encoding the green fluorescent protein (GFP) was used as a control. Co-transfection experiments designed to validate the miR-155 binding site present in the 3′UTR of SOCS-1 were also performed using FA-associated liposomes. Two hundred microlitres of FA-lipoplexes, containing pmiR-155 and a plasmid encoding the reporter gene luciferase and the 3′UTR of SOCS-1 (pSOCS-1 3′UTR) were delivered to N9 cells to obtain a final plasmid concentration of 1 μg/well for each plasmid. In parallel experiments, plasmid (p) GFP was used in addition to pSOCS-1 3′UTR to serve as a control. In all transfection protocols, after 4 hr of incubation, the medium was replaced by new RPMI-1640 medium and N9 microglia cells were incubated for different periods of time, before further analysis.
Luciferase assay
Luciferase expression following co-transfection of pSOCS-1 3′UTR and pmiR-155 or pGFP was evaluated by assessing luciferase activity. Briefly, 48 hr after transfection, cells were washed twice with PBS and 100 μl lysis buffer (1 mm dithiothreitol, 1 mm EDTA, 25 mm Tris–phosphate, 8 mm MgCl2, 15% glycerol, 1% [volume/volume (v/v)] Triton X-100, pH 7·8) were added to each well. After cell lysis at −80°, 50 μl of each lysate were incubated with luciferin and ATP and light production was determined in a luminometer (Lmax II384; Molecular Devices, San Jose, CA). The protein content of the lysates was evaluated through the DC Protein Assay reagent (Bio-Rad, Hercules, CA), using BSA as the standard. Data were expressed as relative light units of luciferase per mg of total cell protein and presented as fold change with respect to control (untransfected cells).
Extraction of total RNA and cDNA synthesis
Total RNA, including small RNA species, was extracted from N9 microglia cells or primary microglia cultures using the miRCURY™ Isolation Kit – Cells (Exiqon), according to the manufacturer's recommendations for cultured cells. Briefly, after cell lysis, the total RNA was adsorbed to a silica matrix, washed with the recommended buffers and eluted with 35 μl RNase-free water by centrifugation. After RNA quantification, cDNA conversion for miRNA quantification was performed using the Universal cDNA Synthesis Kit (Exiqon). For each sample, cDNA for miRNA detection was produced from 20 ng total RNA according to the following protocol: 60 min at 42° followed by heat-inactivation of the reverse transcriptase for 5 min at 95°. The cDNA was diluted 80 × with RNase-free water before quantification by qRT-PCR. Synthesis of cDNA for mRNA quantification was performed using the iScript cDNA Synthesis Kit (Bio-Rad) and employing 1 μg total RNA for each reaction, by applying the following protocol: 5 min at 25°, 30 min at 42° and 5 min at 85°. Finally, the cDNA was diluted 1 : 4 with RNase free water.
Quantitative real-time PCR
Quantitative PCR was performed in an iQ5 thermocycler (Bio-Rad) using 96-well microtitre plates. For miRNA quantification the miRCURY LNA™ Universal RT microRNA PCR system (Exiqon) was used in combination with pre-designed primers (Exiqon) for miR-155 and sRNA U6 (reference gene). A master mix was designed for each primer set, according to the recommendations for the real-time PCR setup of individual assays suggested in this kit. For each reaction, 12 μl master mix was added to 8 μl template cDNA. All reactions were performed in duplicate (two cDNA reactions per RNA sample) at a final volume of 20 μl per well, using the iQ5 Optical System Software (Bio-Rad). The reaction conditions consisted of polymerase activation/denaturation and well-factor determination at 95° for 10 min, followed by 40 amplification cycles at 95° for 10 s and 65° for 1 min (ramp-rate 1·6°/s).
For mRNA quantification, the iQ SYBR Green Supermix Kit (Bio-Rad) was used. The primers for the target genes [SOCS-1, IFN-γ, interleukin-1β (IL-1β), IL-6, TNF-α and iNOS] and for the reference gene (HPRT) were pre-designed by Qiagen (QuantiTect Primer, Qiagen, Hilden, Germany). A master mix was prepared for each primer set, containing a fixed volume of SYBR Green Supermix and the appropriate amount of each primer to yield a final concentration of 150 nm. For each reaction, 20 μl master mix was added to 5 μl template cDNA. All reactions were performed in duplicate (two cDNA reactions per RNA sample) at a final volume of 25 μl per well, using the iQ5 Optical System software (Bio-Rad). The reaction conditions consisted of enzyme activation and well-factor determination at 95° for 1 min and 30 s, followed by 40 cycles at 95° for 10 s (denaturation), 30 s at 55° (annealing), and 30 s at 72° (elongation).
For both miRNA and mRNA quantification, a melting curve protocol was started immediately after amplification and consisted of 1 min heating at 55° followed by 80 steps of 10 s, with a 0·5° increase at each step. Threshold values for threshold cycle determination (Ct) were generated automatically by the iQ5 Optical System software. The miRNA and mRNA fold increase or fold decrease with respect to control samples was determined by the Pfaffl method, taking into consideration different amplification efficiencies of all genes and miRNAs in all experiments. The amplification efficiency for each target or reference RNA was determined according to the formula: E = 10(−1/S)−1, where S is the slope of the obtained standard curve.
In situ hybridization
Fluorescence in situ hybridization was performed in cultured adherent cells as described by Lu and Tsourkas,23 with some modifications. Briefly, microglia primary cells were seeded onto multi-chambered coverglass slides (Lab-Tek; Nalge Nunc, Rochester, NY) appropriate for confocal microscopy imaging. Following treatment with LPS, the cells were washed with PBS, fixed with 4% paraformaldehyde for 30 min at room temperature and permeabilized at 4° in 70% ethanol for 4 hr. Cells were then incubated with fresh acetylation solution [0·1 m triethanolamine and 0·5% (v/v) acetic anhydride] for 30 min at room temperature, rinsed twice in Tris-buffered saline (TBS) and pre-hybridized in the absence of the LNA probe in hybridization buffer [50% formamide, 5 × SSC, 5 × Denhardt's solution, 250 μg/ml yeast tRNA, 500 μg/ml salmon sperm DNA, 2% (w/v) blocking reagent, 0·1% CHAPs, 0·5% Tween) for 2 hr at a temperature 22–25° below the melting temperature (TM) of the probe. The hybridization step was carried out using the DIG-labelled (digoxigenin-labelled) LNA probes for miR-155 at the same temperature overnight. A scrambled probe (negative control) and U6snRNA (positive control) were also used in this experiment (data not shown). Three stringency washes were performed at the same temperature as probe hybridization to completely remove the non-hybridized probe. Endogenous peroxidase activity was inactivated by incubation in 3% hydrogen peroxide in TBS with 0·1% Tween-20 (TBS-T) for 30 min, followed by three washes with TBS-T. The slides were then placed in blocking solution (TBS-T, 10% heat-inactivated goat serum, 0·5% blocking agent) for 1 h at room temperature and incubated for the same period of time with an anti-DIG antibody (Roche, Amadora, Portugal) conjugated with the hydrogen peroxidase. To amplify the antibody signal, slides were further incubated with a TSA plus Cy3 (PerkinElmer, Waltham, MA) solution for 10 min in the dark, in accordance with the manufacturer's protocol. The cells were finally stained with the fluorescent DNA-binding dye Hoechst 33342 (Invitrogen Life Technologies, Paisley, UK) (1 μg/ml) for 5 min in the dark, washed with cold PBS, and mounted in Mowiol (Fluka; Sigma). Confocal images were acquired in a point scanning confocal microscope Zeiss LSM 510 Meta (Zeiss, Göttingen, Germany), with a 60 × oil objective. Digital images were acquired using the LSM 510 Meta software. All instrumental parameters pertaining to fluorescence detection and image analyses were held constant to allow sample comparison.
ELISA
The secretion of TLR-induced cytokines to the cell medium was determined using a Multi-Analyte ELISArray Kit (SA Biosciences Corporation, Frederik, MD). Briefly, 50 μl cell medium, collected from each well, was added to the ELISArray plate and incubated for 2 hr before the addition of the detection antibody. Following 1 hr of incubation, the samples were exposed to an avidin–horseradish peroxidase conjugate and to the development solution. After 15 min of incubation in the dark, the development reaction was stopped with the Stop solution and the optical density was measured at 450 nm in a microplate reader. Cytokine production was determined by comparison with both negative and positive controls present in the Multi-Analyte ELISArray.
Western blot analysis
Total protein extracts were obtained from N9 cells homogenized at 4° in lysis buffer (50 mm NaCl, 50 mm EDTA, 1% Triton X-100) supplemented with a protease inhibitor cocktail (Roche), 10 μg/ml dithiothreitol and 1 mm PMSF. Protein content was determined using the Bio-Rad Dc protein assay (Bio-Rad). Twenty micrograms total protein was resuspended in loading buffer (20% glycerol, 10% SDS and 0·1% bromophenol blue), incubated for 2 min at 95° and loaded onto a 10% polyacrylamide gel. After electrophoresis, the proteins were blotted onto a PVDF membrane according to standard protocols. After blocking in 5% non-fat milk, the membrane was incubated with the appropriate primary antibody (anti-iNOS, 1 : 500 or anti-SOCS-1 1 : 1000) overnight at 4°, and with the appropriate secondary antibody (1 : 10 000) (GE Healthcare, Waukesha, WI) for 2 hr at room temperature. Equal protein loading was shown by re-probing the membrane with an anti-actin antibody (1 : 10 000) (Sigma) and with the appropriate secondary antibody. After this incubation period, the blots were washed several times with saline buffer (TBS/T – 25 mm Tris–HCl, 150 mm NaCl, 0·1% Tween) and incubated with ECF substrate (enhanced chemifluorescence substrate) (alkaline phosphatase substrate; 20 μl ECF/cm2 of membrane) for 5 min at room temperature and then submitted to fluorescence detection at 570 nm using a Molecular Imager Versa Doc MP 4000 System (Bio-Rad). For each membrane, the analysis of band intensity was performed using the Quantity One software (Bio-Rad).
Nitrite quantification
Nitric oxide production was assessed by the Griess Reagent System (Promega Corporation, Madison, WI), a colorimetric assay that detects the presence of nitrite (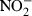 ), a stable reaction product of nitric oxide (NO) and molecular oxygen. Briefly, 50 μl cell medium, collected from each well, was incubated for 5 min with 50 μl sulfanilamide, followed by a further incubation of 5 min with 50 μl of N-1-napthylethylenediamide. The optical density of the samples was measured at 540 nm in a microplate reader and the nitrite concentration was determined by comparison with a standard curve obtained for a solution of sodium nitrite prepared in RPMI-1640.
), a stable reaction product of nitric oxide (NO) and molecular oxygen. Briefly, 50 μl cell medium, collected from each well, was incubated for 5 min with 50 μl sulfanilamide, followed by a further incubation of 5 min with 50 μl of N-1-napthylethylenediamide. The optical density of the samples was measured at 540 nm in a microplate reader and the nitrite concentration was determined by comparison with a standard curve obtained for a solution of sodium nitrite prepared in RPMI-1640.
Immunocytochemistry
Immunocytochemistry studies were performed in N9 microglia cells according to established protocols. Briefly, following transfection and LPS exposure, cells were washed twice with PBS and fixed with 4% paraformaldehyde in PBS for 20 min at room temperature. The cells were then permeabilized for 2 min with 0·2% Triton X-100 and non-specific binding epitopes were blocked by incubating the cells for 30 min with a 5% BSA solution prepared in PBS. Cells were incubated overnight at 4° with primary antibodies against the CD11b integrin (1 : 500) and α-tubulin (1 : 1000) prepared in PBS containing 1% BSA. Following two washing steps with PBS, cells were incubated for 2 hr at room temperature with the respective secondary antibodies (anti-rat Alexa Fluor-594 conjugate and anti-rabbit Alexa Fluor-488 conjugate; Molecular Probes, Leiden, the Netherlands) diluted 1 : 500 in PBS containing 1% BSA. Finally, all coverslips containing the samples were rinsed twice in PBS and incubated in the dark with DAPI (1 μg/ml) for 5 min, before being mounted on glass slides using Moviol (Sigma). The samples were then observed by epifluorescence microscopy under a Zeiss Axiovert microscope, equipped with a 20 × objective and the rhodamine, DAPI and FITC filters. Representative images were taken for each condition, using the same exposure time for each filter, to allow comparison of fluorescence intensity between different fields and conditions.
Neuronal primary cultures and evaluation of cell viability
Primary cultures of cortical neurons were obtained from C57BL/6 mice, at day 16 of gestation. After dissociation and centrifugation of the dissected cortices, the tissue was resuspended in Neurobasal medium (Invitrogen, San Diego, CA), supplemented with 2% (v/v) B27 supplement (Invitrogen) and 100 U/ml penicillin/streptomycin (Invitrogen). Cells were plated at a density of 500 000 cells/well in six-well multi-well plates previously coated with poly-l-lysine. Characterization of the embryonic neuronal cultures confirmed the presence of 95% neurons, as determined by GFAP and NeuN-immunostaining. Primary cultures were kept at 37° in a humidified atmosphere containing 5% CO2. After 8 days in culture, neurons were incubated for 24 hr with a 1 : 1 mixture of Neurobasal medium (500 μl) and conditioned medium (500 μl). The conditioned medium was obtained from untreated N9 cells, N9 cells exposed to LPS (0·1 μg/ml) for 24 hr, and N9 cells transfected with anti-miR-155 or control oligonucleotides 24 hr before exposure to LPS. In parallel experiments neurons were incubated with LPS (0·1 μg/ml) for 24 hr.
Cell viability of primary neuronal cultures was determined by a modified Alamar Blue assay. This assay measures the redox capacity of neurons and allows the determination of cell viability without the detachment of the cells, so cell integrity is maintained. Briefly, 1 ml Neurobasal medium supplemented with 10% (v/v) of Alamar Blue dye was added to each well following the 24-hr incubation period with conditioned medium or LPS. After 3 hr of incubation at 37°, 150 μl supernatant was collected from each well and transferred to 96-well plates. The optical density of the supernatant was measured at 570 and 600 nm in a microplate reader and cell viability was calculated as a percentage of control cells, using the formula: (A570–A600) of treated cells × 100/(A570–A600) of control cells.
Statistical analysis
All data are presented as mean ± standard deviation (SD) and are the result of three independent experiments, each performed at least in triplicate. One-way analysis of variance combined with Tukey post-hoc test was used for multiple comparisons in cell culture experiments. Statistical differences are presented at probability levels of P < 0·05 (*), P < 0·01 (**) and P < 0·001 (***). Calculations were performed with standard statistical software (GraphPad Prism 5, GraphPad Software, La Jolla, USA).
Results
LPS-mediated induction of miR-155 in microglia cells
Since miR-155 has been described as being up-regulated in various cells of myeloid origin upon their activation and as contributing to the modulation of the immune response mediated by these cells, we first investigated the expression of this miRNA in mouse N9 microglia cells and primary microglia cultures employing qRT-PCR. To activate and compare miR-155 levels in both resting and active microglia, N9 cells and primary microglia cultures were incubated with LPS, a specific ligand of TLR4, for 18 hr. Treatment of N9 cells with increasing concentrations of LPS (0·1, 0·5 and 1 μg/ml) showed a significant dose-dependent induction of miR-155 expression, which reached a 25-fold increase in miR-155 levels for the highest LPS concentration tested (Fig. 1a). A similar result was obtained in primary microglia cultures, where it was possible to observe a 12-fold or 21-fold increase in the expression of miR-155 following incubation with 0·1 or 1 μg/ml LPS, respectively (Fig. 1b).
Figure 1.
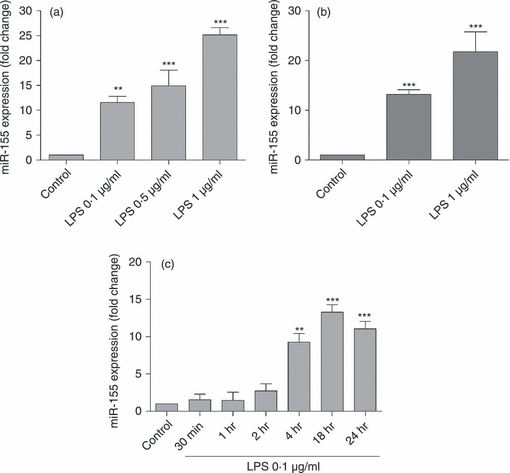
Quantification of microRNA-155 (miR-155) expression following microglia activation with lipopolysaccharide (LPS). N9 microglia cells (a) or primary microglia cultures (b) were incubated with LPS at 0.1, 0.5 or 1 μg/ml for 18 hr. Alternatively, N9 cells were incubated with LPS at 0.1 μg/ml for different periods of time (30 min, 1 hr, 2 hr, 4 hr, 18 hr and 24 hr) (c). Following cell incubation with LPS, total RNA was extracted and miR-155 levels were determined by quantitative real-time RT-PCR using specific LNA probes for the mature form of this miRNA. Results are presented as miR-155 fold change with respect to control (untreated cells). **P< 0.01 and ***P< 0.001 compared with control. Results are representative of three independent experiments performed in triplicates.
To establish a time–course for this event, changes in miR-155 levels were monitored by qRT-PCR at different time-points (30 min, 1, 2, 4, 18 and 24 hr), following stimulation of N9 cells with the lowest concentration of LPS (0·1 μg/ml). The levels of miR-155 remained constant until 4 hr after the beginning of the stimulus, when a significant increase was observed with respect to control levels (Fig. 1c). Levels of miR-155 continued to increase, reaching a maximum at 18 hr, but showed a tendency to decrease after an incubation period of 24 hr.
To confirm the results obtained by qRT-PCR, in situ hybridization studies were performed in primary microglia cultures exposed to 0·1 or 1 μg/ml LPS, using an LNA probe specific for the mature form of miR-155 (Fig. 2). The miR-155 labelling was significantly more intense in the cytoplasm of microglia cells incubated with LPS than in control cells. Since the probe only recognizes the mature form of this miRNA, these results further validate the qRT-PCR data presented in Fig. 1(b) and confirm that, under inflammatory conditions, miR-155 expression increases not only in N9 microglia cells but also in microglia primary cells.
Figure 2.
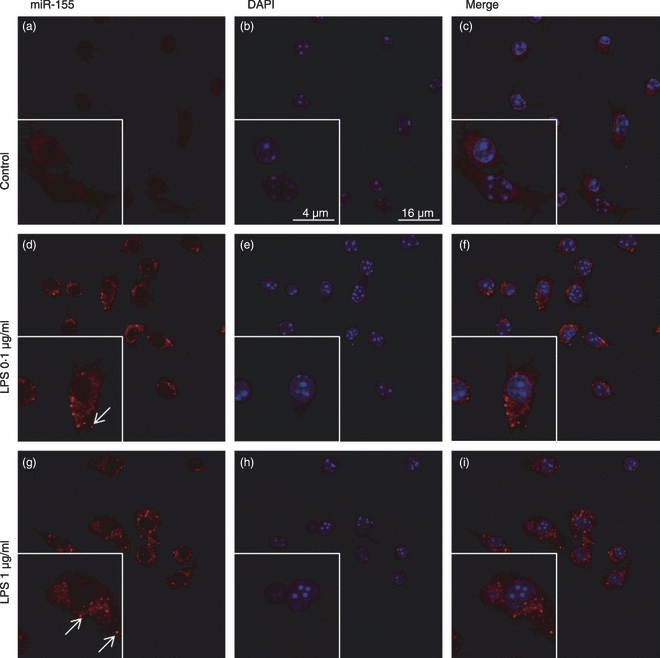
Evaluation of microRNA-155 (miR-155) expression in primary microglia cultures using in situ hybridization. Primary microglia cultures were incubated with lipopolysaccharide (LPS) at 0.1 or 1 μg/ml for 18 hr before miR-155 labelling using a DIG-conjugated LNA-based probe, specific for the mature form of this miRNA. Expression of miR-155 (red) was detected using an anti-DIG antibody and the TSA Cy3 signal amplification system (a), (d) and (g). In parallel studies, the levels of miR-155 were also analysed in untreated microglia cells. The cell nuclei were labelled with DAPI (blue) (b), (e) and (h). Representative confocal microscopy images of all experimental conditions are presented at a 600 × magnification. (c), (f) and (i) show merged images of all three channels.
Primary microglia cells are not easily obtained with high yield, are extremely difficult to transfect and are easily activated by cell culture procedures, also, the responses of N9 cells and primary microglia cultures to LPS treatment are similar, so the subsequent studies were performed in N9 cells. This cell line, which comprises immortalized mouse-derived microglia cells, has been described as mimicking the behaviour of primary microglia regarding TLR expression, cytokine release and NO production, and has been employed in several studies as an in vitro model to study microglia activation.24–26
SOCS-1 is a target of miR-155 in microglia cells
The miRNAs exert their regulatory effects mainly at the post-transcriptional level, by targeting complementary or partly complementary mRNAs and inducing mRNA cleavage or translation repression. To identify potential targets of miR-155 that might be relevant in the microglia immune response, we screened the mouse and human miR-155 sequences using the miRBase and PicTar miRNA target identification programmes. Among other important targets, such as inositol 5-phosphatase 1, IκB kinase ε, c/ebp β and receptor-interacting protein kinase, this analysis allowed the identification of the SOCS-1 protein as a potential target of miR-155 in both human and mouse cells (Fig. 3a).
Figure 3.
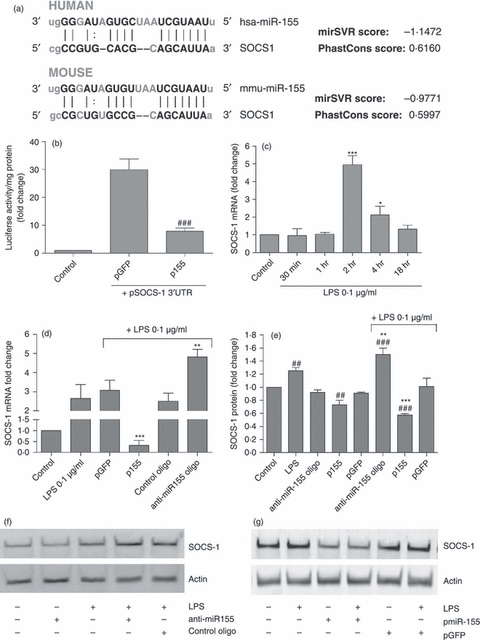
Modulation of suppressor of cytokine signalling 1 (SOCS-1) mRNA and protein levels by microRNA-155 (miR-155). (a) SOCS-1 was found to be a predicted target of miR-155 in both humans and mice and (b) validation of miR-155 binding to the 3′UTR of SOCS-1 was achieved using a luciferase reporter assay. N9 cells were transfected with pmiR-155 and pSOCS-1 3′UTR for 4 hr. Forty-eight hours after transfection, luciferase activity and protein levels were determined in all samples. Cell co-transfection with pGFP and pSOCS-1 3′UTR was used as a positive control in this experiment. Results are presented as the fold increase in luciferase activity/mg protein with respect to control (non-transfected cells). ###P< 0.001 compared with cells transfected with pGFP and pSOCS-1 3′UTR (c) The time–course of SOCS-1 expression was determined by quantitative real-time (q) RT-PCR. N9 cells were incubated with lipopolysaccharide (LPS) at 0.1 μg/ml for different periods of time (30 min, 1 hr, 2 hr, 4 hr and 18 hr). Results are presented as SOCS-1 mRNA fold change with respect to control (untreated cells). *P < 0.05 and ***P < 0.001 compared with control. SOCS-1 (d) mRNA and (e) protein levels were determined by qRT-PCR and Western blot, respectively. N9 cells were transfected with anti-miR-155 oligonucleotides (anti-miR155 oligo) or miR-155-encoding plasmid (p155) for 4 hr. Alternatively, N9 cells were transfected with non-targeting oligonucleotides (control oligo) or a plasmid encoding GFP (pGFP). Twenty-four hours after transfection, cells were incubated with LPS at 0.1 μg/ml for 4 hr and RNA and protein extractions were performed. Representative Western blot images illustrate the reduction or increase in SOCS-1 protein levels, following miR-155 (f) down-regulation or (g) up-regulation, respectively. Results are expressed as SOCS-1 mRNA or protein fold change with respect to control (non-transfected and untreated cells). ##P < 0.01 and ###P < 0.001 compared with control (untreated and non-transfected cells) and **P < 0.01 and ***P < 0.001 compared with LPS-treated cells in the absence of transfection. All results are representative of three independent experiments performed in triplicate.
Because SOCS-1 is expressed in microglia, acting as a negative regulator of several inflammatory pathways triggered by cytokines and LPS, we investigated the contribution of miR-155 to the regulation of SOCS-1 expression in these cells. A recent study, using a luciferase reporter assay, has provided functional evidence that miR-155 is able to bind to the 3′UTR of SOCS-1 mRNA in HEK293T cells.27 Using a similar assay, which comprises the co-transfection of pmiR-155 and a plasmid encoding both the luciferase gene and the 3′UTR sequence of SOCS-1 (pSOCS-1 3′UTR), followed by the evaluation of luciferase activity 48 hr after transfection, we were also able to validate miR-155 binding to the untranslated repeat of this protein in N9 cells (Fig. 3b). With this experiment, it was possible to observe the expected increase in luciferase activity following the delivery of both pSOCS-1 3′UTR and the pGFP plasmids. However, delivery of pmiR-155 in addition to pSOCS-1 3′UTR resulted in reduced luciferase activity levels, which were significantly lower than those obtained following transfection with the control plasmid (pGFP) and pSOCS-1 3′UTR. These results indicate that, similar to what was reported in HEK293T cells, miR-155 expression in N9 cells is able to block luciferase expression through binding to the 3′UTR sequence of SOCS-1, which precedes the luciferase gene. The miR-155–mRNA pairing leads to post-transcription repression or mRNA degradation, decreasing luciferase expression and hence luciferase activity, so validating SOCS-1 as a target of miR-155.
Aiming at ascertaining a possible temporal relation between miR-155 and SOCS-1 expression levels, we performed a qRT-PCR time–course study to identify changes in SOCS-1 levels following microglia incubation with LPS (0·1 μg/ml). The results displayed in Fig. 3(c) show that following 2 hr of incubation with LPS, SOCS-1 mRNA levels present a sharp increase of fivefold, but decrease afterwards, approaching only a twofold increase after 4 hr of incubation and reaching basal levels at 18 hr. These results correlate temporally with those shown in Fig. 1(c) and support the hypothesis that miR-155 may contribute directly to the observed decrease in SOCS-1 levels by targeting SOCS-1 mRNA. To confirm this possibility we determined whether over-expression or inhibition of miR-155 would lead to significant changes in SOCS-1 mRNA and protein levels. For this purpose, N9 microglia cells were transfected with a plasmid encoding miR-155 (p155) or with anti-miR-155 oligonucleotides, which bind with high affinity to miR-155 and avoid miRNA–target mRNA interactions. N9 cells were exposed 24 hr later to LPS (0·1 μg/ml). A non-inhibitory oligonucleotide (control oligonucleotide) and a plasmid encoding GFP (pGFP) were used as negative controls, to detect possible transfection-related unspecific changes in SOCS-1. The SOCS-1 mRNA and protein levels were determined by qRT-PCR and Western blot, respectively, after 4 hr of incubation with LPS (Fig. 3d–g). The SOCS-1 mRNA and protein levels in N9 cells stimulated with LPS increased following miRNA inhibition and decreased upon miR-155 over-expression. Furthermore, under resting conditions, a decrease in SOCS-1 protein levels was observed following over-expression of miR-155 (Fig. 3e) and a similar result was observed in mRNA levels (data not shown). However, no increase in SOCS-1 mRNA or protein levels was observed following transfection with anti-miR-155 oligonucleotides, probably because of the low levels of miR-155 in resting cells. As no significant changes were observed in cells transfected with the control oligonucleotide or with pGFP, the results presented in Fig. 3 validate miR-155 as a specific modulator of SOCS-1 in microglia cells.
Expression of miR-155 is necessary for the production of inflammatory cytokines following microglia activation
To assess the effects of miR-155 and SOCS-1 modulation on microglia activation and on the production of inflammatory mediators, initial studies addressed the time-dependent expression of IFN-β, a classical target of SOCS-1 negative feedback regulation, following microglia activation with LPS (0·1 μg/ml). Results in Fig. 4(a) clearly show that although IFN-β levels start to increase quickly after LPS exposure, achieving a twofold increase after 1 hr of incubation, this effect becomes much more pronounced following a 4-hr incubation period. These results correlate with our previous observations of an increase in miR-155 levels (Fig. 1a) and a decrease in SOCS-1 expression levels (Fig. 3a) at this same time point, suggesting that the observed IFN-β response is dependent on both miR-155 and SOCS-1 expression. To confirm the relation among IFN-β, miR-155 and SOCS-1, we evaluated the functional consequences of miR-155 inhibition or over-expression in IFN-β mRNA levels following microglia activation. For this purpose, N9 microglia cells were transfected again with a plasmid encoding miR-155 or with anti-miR-155 oligonucleotides 24 hr before N9 exposure to LPS (0·1 μg/ml). Interferon-β mRNA levels were determined by qRT-PCR following an 18-hr incubation with LPS (Fig. 4b). A very strong increase in IFN-β mRNA levels was observed following over-expression of miR-155 and incubation with LPS, whereas an inhibition of this miRNA reduced IFN-β expression levels to basal levels even in the presence of LPS. These data indicate that changes in miR-155 levels are sufficient to modulate IFN-β production in activated microglia cells. No significant changes in IFN-β expression levels were observed in cells transfected with control oligonucleotides or with the control plasmid (pGFP), which further attests that the observed effect is specific for miR-155 modulation.
Figure 4.
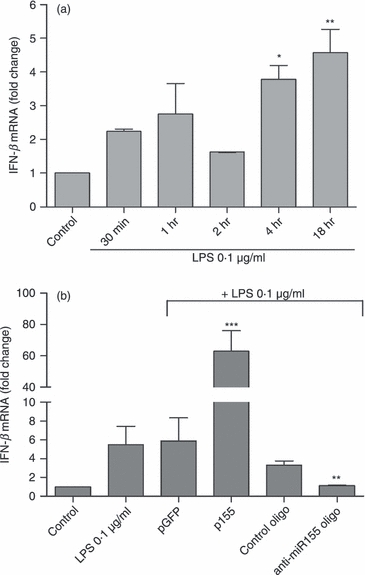
Modulation of interferon-β (IFN-β) expression by microRNA-155 (miR-155). (a) N9 microglia cells were incubated with lipopolysaccharide (LPS) at 0.1 μg/ml for different periods of time (30 min, 1 hr, 2 hr, 4 hr and 18 hr). Results are presented as IFN-β mRNA fold change with respect to control (untreated cells). *P < 0.05 and **P < 0.01 compared with control. (b) The IFN-β expression levels were determined by quantitative real-time RT-PCR. N9 cells were transfected with anti-miR-155 oligonucleotides (anti-miR155 oligo) or miR-155-encoding plasmid (p155) complexed with cationic liposomes for 4 hr. Alternatively, N9 cells were transfected with non-targeting oligonucleotide (control oligo) or a plasmid encoding GFP (pGFP). Twenty-four hours after transfection, cells were incubated with LPS at 0.1 μg/ml for 18 hr and RNA extraction was performed. Results are expressed as IFN-β mRNA fold change with respect to control (untranfected and untreated cells). **P < 0.01 and ***P < 0.001 compared with LPS-treated cells in the absence of transfection. Results are representative of three independent experiments performed in triplicate.
Following the demonstration of the effect of miR-155 and SOCS-1 manipulation on the IFN response, the mRNA levels of other pro-inflammatory cytokines, such as TNF-α, IL-1β and IL-6, were also determined in N9 cells after miR-155 inhibition and LPS exposure, using a similar experimental protocol (Fig. 5). As observed, TNF-α and IL-6 mRNA levels (Fig. 5a,b) were also significantly decreased following miR-155 inhibition. Although a decrease was observed for IL-1β (Fig. 5c), this effect was not statistically significant. As mRNA levels reflect cellular gene expression but not protein secretion, medium was collected from N9 cells following transfection with anti-miR-155 or control oligonucleotides and LPS treatment, and analysed by an ELISA to determine the levels of nine cytokines/chemokines expressed following microglia activation (Fig. 5d). This assay confirmed that miR-155 inhibition decreases the secretion of TNF-α and IL-6, but has no effect on IL-1β or any other of the tested cytokines, with the exception of TARC (thymus and activation regulated chemokine), whose levels although significantly lower compared with those of TNF-α and IL-6, were also found to be decreased. No significant differences were found between non-transfected N9 cells treated with LPS and cells transfected with control oligonucleotides before LPS exposure (data not shown), which further confirms the specificity of the effects observed with the anti-miR-155 oligonucleotides. Taken together, these results indicate that miR-155 can act as a strong inducer of cytokine production following microglia activation and that miR-155 inhibition decreases both the expression and the secretion of specific pro-inflammatory cytokines.
Figure 5.
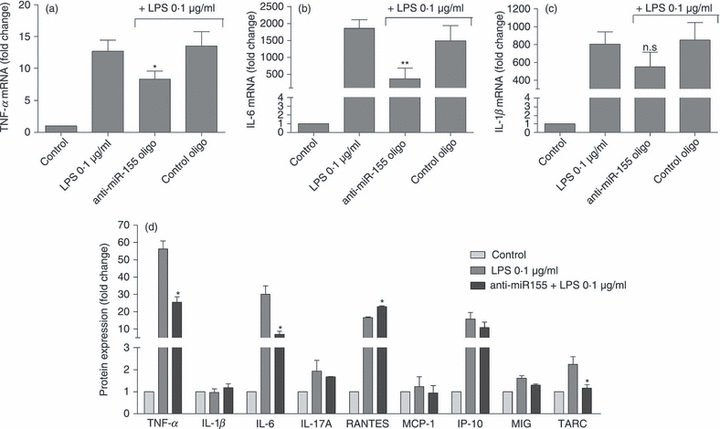
Modulation of cytokine expression and secretion by microRNA-155 (miR-155). The mRNA of (a) tumour necrosis factor-α (TNF-α), (b) interleukin-6 (IL-6) and (c) IL-1β mRNA were quantified by quantitative RT-PCR. (d) The secretion of several inflammatory cytokines in response to relation with Toll-like receptor (TLR) activation was determined by ELISA. N9 cells were transfected with anti-miR-155 oligonucleotides (anti-miR155 oligo) or with a non-targeting oligonucleotide (control oligo) complexed with cationic liposomes for 4 hr. Twenty-four hours after transfection, cells were incubated with lipopolysaccharide (LPS) at 0.1 μg/ml for 18 hr. The cell culture medium was then collected to determine cytokine protein levels by ELISA and the RNA was extracted from the cells. Results are expressed as fold change in cytokine mRNA or protein levels with respect to control (untranfected and untreated cells). *P < 0.05 and **P < 0.01 compared with LPS-treated cells in the absence of transfection. Results in (a), (b) and (c) are representative of three independent experiments performed in triplicate. Results in (d) are representative of two independent experiments performed in triplicate.
Inhibition of miR-155 reduces NO production and iNOS expression following microglia activation
Nitric oxide is an inflammatory mediator whose production by iNOS is a well-described hallmark of microglia activation. Although NO is a volatile gas, it is possible to monitor its release to the cell culture medium by measuring the levels of nitrites, the sub-products of NO oxidation, through the Griess reaction. Aiming at assessing the contribution of miR-155 for NO production, N9 microglia cells were transfected with anti-miR155 oligonucleotides or a plasmid encoding miR-155, before LPS treatment (0·1 μg/ml for 18 hr). As expected, cells exposed to LPS presented a strong increase in nitrite production (Fig. 5a). However, miR-155 inhibition before LPS treatment led to a significant decrease in nitrite release to the medium (40%), with respect to LPS-treated untransfected cells, whereas miR-155 over-expression had the opposite effect, increasing nitrite levels. These results could not be reproduced using a control oligonucleotide or a control plasmid, which indicates that the changes in NO and nitrite production are a specific response to miR-155 modulation. Moreover, a decrease in iNOS mRNA, as assessed by qRT-PCR (Fig. 6b), and in protein levels, as assessed by Western blot (Fig. 5c,d), was observed following miR-155 inhibition, but not following transfection with the control oligonucleotides. Western blot analysis also showed an increase in iNOS levels after miR-155 over-expression, which further confirms the contribution of miR-155 to the regulation of NO synthesis by modulating iNOS expression.
Figure 6.
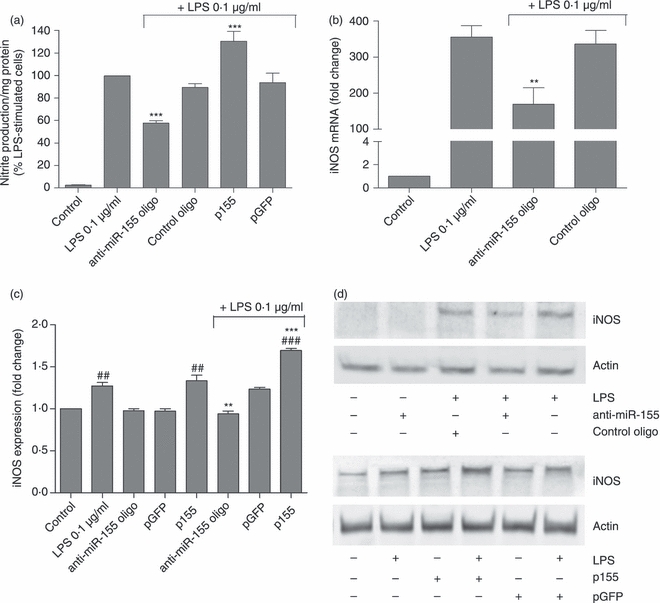
Effect of microRNA-155 (miR-155) modulation on nitrite production and inducible nitric oxide synthase (iNOS) expression. (a) Nitrite production was quantified by the Griess Assay. The effect of miR-155 modulation on (b) iNOS mRNA was determined by quantitative RT-PCR and on (c) protein expression levels was determined by Western blot. N9 cells were transfected with anti-miR-155 oligonucleotides (anti-miR155 oligo) or miR-155-encoding plasmid (p155) complexed with cationic liposomes for 4 hr. Alternatively, N9 cells were transfected with non-targeting oligonucleotides (control oligo) or a plasmid encoding GFP (pGFP). Twenty-four hours after transfection, cells were incubated with lipopolysaccharide (LPS) at 0.1 μg/ml for 18 hr. The extracellular cell medium was then collected to quantify nitrite secretion through the Griess reaction and RNA and protein were extracted from the cells. (d) Representative Western blot images illustrate the increase or reduction in iNOS protein levels following miR-155 up-regulation or down-regulation, respectively. Results in (a) are expressed as % of nitrite production/mg protein in LPS-treated cells, in the absence of transfection. Results in (b) and (c) are expressed as mRNA or protein fold change with respect to control (untransfected and untreated cells). ##P < 0.01 and ###P < 0.001 compared with control (untreated and untransfected cells) and **P < 0.01 and ***P < 0.001 compared with LPS-treated cells in the absence of transfection. Results are representative of three independent experiments performed in triplicate.
Both LPS-mediated TLR activation and NO production have been related to an increase in the expression and activation of the CD11b integrin β, a surface marker whose expression is increased in activated microglia cells.28,29 To assess the consequences of miR-155 inhibition, and the resulting decrease in NO production, on CD11b expression, we performed immunocytochemistry to evaluate CD11b labelling in N9 cells. For this purpose, N9 microglia cells were transfected with anti-miR-155 or control oligonucleotides 24 hr before exposure to LPS (0·1 μg/ml). Following 18 hr of incubation with LPS, cells were fixed and labelled with the nuclear dye DAPI, with a specific anti-CD11b antibody and an antibody against the structural protein tubulin (Fig. 7). Results in Fig. 7 clearly show that exposure to LPS increases CD11b labelling in N9 cells (Fig. 7e), with respect to control cells (Fig. 7a). In this regard, it was also possible to observe striking differences in cell morphology, because LPS-treated cells lose the characteristic star shape of resting N9 cells and become round and amoeboid, a common feature of activated microglia cells. Similar results were observed in N9 cells transfected with control oligonucleotides followed by LPS exposure (Fig. 7m). These cells present the same intense CD11b labelling and round shape of untransfected, LPS-treated cells. However, cells transfected with the anti-miR-155 oligonucleotides before LPS treatment showed less intense CD11b labelling and a morphology closer to that of control cells (Fig. 7i), indicating lower levels of CD11b.
Figure 7.
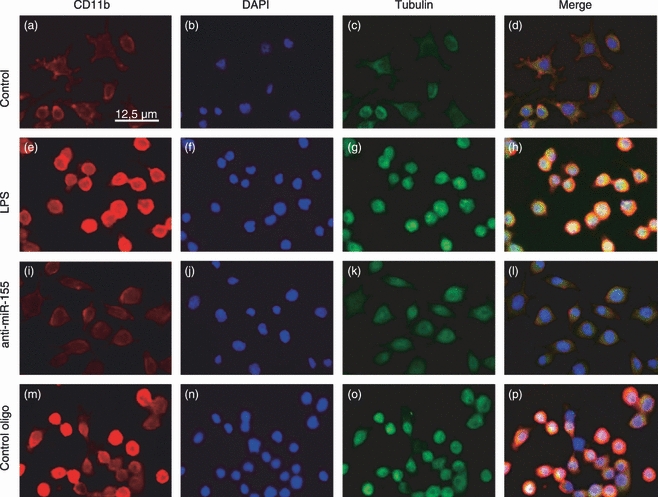
Effect of microRNA-155 (miR-155) modulation on CD11b protein levels and cell morphology. The expression levels of CD11b and morphology of N9 cells were assessed by immunocytochemistry. N9 cells were transfected with anti-miR-155 oligonucleotides (anti-miR155 oligo) or non-targeting oligonucleotides (control oligo) for 4 hr. Twenty-four hours after transfection, cells were incubated with lipopolysaccharide (LPS) at 0.1 μg/ml for 18 hr before fixation and immunocytochemistry. N9 cells were labelled with anti-CD11b antibody (red) (a), (e), (i) and (m) or anti-tubulin antibody (green) (c), (g), (k) and (o). The nuclei were labelled with DAPI (blue) (b), (f), (j) and (n). Representative fluorescence microscopy images of all experimental conditions are presented at a 200 × magnification. (d), (h), (l) and (p) show merged images of all three channels.
Inhibition of miR-155 prevents neuronal death following microglia activation
In view of the pro-inflammatory role of miR-155 in activated microglia, as evidenced by our results on N9 cells, we evaluated the potential of miR-155 modulation as an anti-inflammatory and neuroprotective strategy. For this purpose, N9 microglia cells were transfected with anti-miR-155 or control oligonucleotides 24 hr before exposure to LPS (0·1 μg/ml). Following 18 hr of incubation with LPS, the medium of N9 cells was collected and mixed with Neurobasal medium at a ratio of 1 : 1 (v/v). Primary cultures of cortical neurons were incubated with this mixture (conditioned medium) for 24 hr before assessment of cell viability using the Alamar Blue assay (Fig. 8). In parallel, cortical cultures were exposed directly to the same concentration of LPS (0·1 μg/ml). Figure 8 shows that neurons exposed to conditioned medium collected from N9 cells, previously incubated with LPS in the absence of transfection, presented a reduction in viability of 40%. Similar results were observed in neurons incubated with conditioned medium collected from cells transfected with control oligonucleotides. However, neurons treated with medium conditioned by N9 cells, in which miR-155 had been inhibited before LPS treatment, presented only a slight decrease in viability (10%) with respect to control neuronal cells. Moreover, neurons exposed directly to LPS or to medium collected from untransfected and untreated N9 cells did not show significant changes in viability, suggesting that the observed neuronal death was caused by inflammatory mediators secreted by N9 cells rather than as a direct consequence of the presence of LPS.
Figure 8.
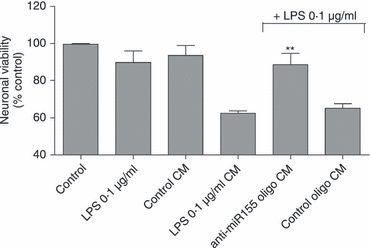
Neuronal viability in the presence of microglia conditioned medium. Neuronal viability was assessed by the Alamar Blue Assay after exposure to conditioned medium obtained from N9 cells. N9 cells were transfected with anti-microRNA-155 (miR-155) oligonucleotides (anti-miR155 oligo) or non-targeting oligonucleotides (control oligo) for 4 hr. Twenty-four hours after transfection, cells were incubated with lipopolysaccharide (LPS) at 0.1 μg/ml for 18 hr and the N9 conditioned medium (CM) was collected. CM was also collected from untransfected and untreated N9 cells (control CM), or from untransfected LPS-treated cells (LPS 0.1 μg/ml CM). Primary cortical neurons were obtained at day 16 of gestation and cultured in Neurobasal medium (NBM) for 10 days before incubation with a mixture of CM and NBM at a ratio of 1 : 1 (v/v) for 24 hr. Alternatively, neuronal cultures were incubated directly with LPS at 0.1 μg/ml for the same period of time. Results are presented as a % of neuronal viability with respect to control cells. **P < 0.01 compared with neurons incubated with LPS 0.1 μg/ml CM. Results are representative of three independent experiments performed in triplicate.
Taken together, the results obtained in this study clearly demonstrate the important role of miR-155 in the regulation of different aspects of the immune response mediated by microglia, such as cytokine expression, NO production and neurotoxicity, and reveal a new and promising therapeutic application of miRNA modulation strategies.
Discussion
Recent studies have shown a role for specific miRNAs in the control of adaptive and innate immune responses, and the deregulation of these miRNAs has been associated with several pathologies that present an inflammatory component, including cancer,27 rheumatoid arthritis13 and neurodegenerative disorders such as Alzheimer's disease. The miR-155 belongs to this group of miRNAs and has been found to be expressed in several cells of the immune system, such as macrophages, monocytes, dendritic cells and haematopoietic progenitors/stem cells.12 In the present work we provide evidence, for the first time, that miR-155 is also significantly up-regulated in both primary microglia cells and N9 microglia cells following cell activation upon exposure to the TLR4 ligand LPS (Figs 1 and 2). The observed time–course for miR-155 up-regulation was similar to what was previously described in other cells.27 Although it was initially detected at very low levels in N9 microglia cells, upon cell activation the levels of this miRNA increased rapidly, starting to rise 4 hr after LPS exposure.
While much has been discovered concerning miR-155 expression patterns and basic functions through the study of miR-155−/−mice, the molecular pathways and targets affected by this miRNA are poorly characterized, particularly in the CNS. To further clarify the role of this miRNA in CNS inflammatory processes, we searched for miR-155 candidate targets that could be involved in microglia activation and microglia-mediated innate immune responses in the brain. Using bioinformatic tools, and based on the information already available in the literature, we identified SOCS-1 as a possible target of miR-155 in human and mice cells and confirmed that miR-155 is able to bind to the 3′UTR of this protein (Fig. 3b). SOCS-1 has been described as having a short half-life (1–2 hr) and its expression levels are reported to increase rapidly following macrophage exposure to inflammatory cytokines and TLR ligands.30 The stability of this protein can be regulated by its association with other proteins, including PIM 1 (Proto-oncogene serine/threonine-protein kinase 1) and ubiquitin, although these mechanisms are not sufficient to explain the quick modulation of SOCS-1 protein levels upon cell activation.30 In this work, we were able to observe the expected rapid increase in SOCS-1 levels following microglia exposure to LPS. The SOCS-1 mRNA levels peaked 2 hr after the beginning of the stimulus and decreased rapidly thereafter, approaching basal levels at 4 hr (Fig. 3c). Interestingly, the time course of changes in SOCS-1 mRNA levels, following LPS exposure, correlated closely with the time–course observed for miR-155 increase, suggesting that this miRNA may be involved in the post-transcriptional regulation of SOCS-1. Using approaches involving the inhibition or over-expression of miR-155, we demonstrated that miR-155 is indeed able to repress SOCS-1 upon microglia activation, at both the mRNA and protein levels (Fig. 3). Our results are consistent with the hypothesis that miR-155 constitutes a new and important modulator of SOCS-1 in microglia and can, therefore, act as a key intervenient in the regulation of several inflammatory pathways triggered in these cells, by promoting the post-transcription repression of SOCS-1 mRNA. Our findings also correlate with recent studies showing increased miR-155 expression and a decrease in SOCS-1 levels in other cell types, such as mature dendritic cells,20 which further reflects the important role of miRNAs in the fine tuning of gene regulation in the context of innate immunity.
It is considered that miR-155 is a pro-inflammatory miRNA, because miR-155-deficient mice present defects in germinal centre formation and in antibody isotype class switching, being unable to produce significant levels of IL-2 and IFN-γ, following immunization.31 Moreover, up-regulation of miR-155 following exposure to LPS has been shown to enhance TNF-α production in macrophages, both in vitro and in vivo, and such over-expression has been reported in rheumatoid arthritis patients with respect to healthy controls.32 These results strongly suggest that miR-155 is involved in protective immunity when properly regulated, yet it can also contribute to malignant conditions upon its deregulated expression.
In microglia cells, miR-155 increase upon cell activation seems to be necessary for the progression of the immune response and the production of inflammatory mediators. A decrease in miR-155 levels, following transfection with anti-miR-155 oligonucleotides, led to a significant reduction in the expression of IL-6, IFN-β and TNF-α and in the secretion of both IL-6 and TNF-α (Figs 4 and 5). In addition, a decrease in the production of NO and in the expression of iNOS (Fig. 6) was observed following miR-155 inhibition. In contrast, the over-expression of miR-155 before exposure of microglia cells to LPS had the opposite effect, increasing the expression of IFN-β (Fig. 4) and iNOS (Fig. 6b–d) and the production of NO (Fig. 6a). Although iNOS is not a predicted target of miR-155, these results suggest a possible role for miR-155 in regulating the production of NO, an important mediator of microglia immune response, probably by directly interfering with proteins that act upstream of iNOS, such as SOCS-1 and elements of the nuclear factor-κB pathway. Our results also showed an LPS-induced up-regulation in the expression of the microglia surface marker CD11b, a molecule that binds the intracellular adhesion molecule-1 and complement C3b. A change in cell morphology was also observed under the same conditions, with N9 cells adopting a round shape characteristic of activated microglia. However, a strong decrease in CD11b immunolabelling was observed upon miR-155 inhibition, even following LPS stimulation (Fig. 7), as well as a small number of round cells. Our findings clearly show that inhibition of miR-155 recapitulated almost completely the resting phenotype of microglia cells, even in the presence of LPS, suggesting that miR-155 has a pro-inflammatory role in microglia cells, and further supporting what has been previously described in other cells of the immune system.
Although microglia activation, in the presence of an external or internal threat, can be beneficial in the CNS, it is now believed that the failure to terminate microglia-mediated immune responses at the appropriate moment can lead to the over-expression of inflammatory mediators and to the establishment of a chronic inflammatory state with deleterious consequences to the surrounding neurons. Our results establish a direct link among miR-155 expression, SOCS-1 inhibition and the production of inflammatory mediators, suggesting that the deregulation of miR-155 can constitute a contributing factor to inflammatory processes in the CNS, by disturbing the normal function of SOCS-1 and increasing cytokine and NO production. Of note, we found that this miRNA is up-regulated in the brain of mice transgenic for Alzheimer's disease, with respect to their wild-type littermates, in an age-dependent manner (manuscript in preparation), which further supports the hypothesis that miR-155 may play a role in neurodegenerative pathologies. If this is the case, miR-155 inhibition in microglia cells may constitute a new and promising anti-inflammatory approach to decrease microglia-mediated neuronal damage. Our findings suggest that miR-155 inhibition in N9 microglia cells before activation with LPS is sufficient to reduce neuronal damage induced upon cell exposure to microglia-conditioned medium (Fig. 8). The observed increase in neuronal viability is most probably the result of a decrease in the levels of inflammatory cytokines and NO present in the conditioned medium, because direct treatment of neuronal cultures with LPS did not decrease cell viability.
Overall, our results demonstrate that miR-155 silencing is able to decrease microglia-mediated neurotoxicity and may, therefore, represent a valuable therapeutic strategy in the context of chronic inflammation.
Acknowledgments
The authors would like to acknowledge Professor Carlos B. Duarte (Faculty of Science and Technology, University of Coimbra, Portugal) for his critical reading of this manuscript. Ana Cardoso is the recipient of a fellowship from the Portuguese Foundation for Science and Technology (SFRH/BPD/46228/2008). This work was partially financed by a grant from the Portuguese Foundation for Science and Technology (PTDC/BIO/65627/2006).
References
- 1.Amor S, Puentes F, Baker D, van der Valk P. Inflammation in neurodegenerative diseases. Immunology. 2010;129:154–69. doi: 10.1111/j.1365-2567.2009.03225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Czlonkowska A, Kurkowska-Jastrzebska I. Inflammation and gliosis in neurological diseases – clinical implications. J Neuroimmunol. 2010;231:78–85. doi: 10.1016/j.jneuroim.2010.09.020. [DOI] [PubMed] [Google Scholar]
- 3.Lehnardt S. Innate immunity and neuroinflammation in the CNS: the role of microglia in Toll-like receptor-mediated neuronal injury. Glia. 2010;58:253–63. doi: 10.1002/glia.20928. [DOI] [PubMed] [Google Scholar]
- 4.Qin L, Liu Y, Cooper C, Liu B, Wilson B, Hong JS. Microglia enhance β-amyloid peptide-induced toxicity in cortical and mesencephalic neurons by producing reactive oxygen species. J Neurochem. 2002;83:973–83. doi: 10.1046/j.1471-4159.2002.01210.x. [DOI] [PubMed] [Google Scholar]
- 5.Takeuchi H, Wang J, Kawanokuchi J, Mitsuma N, Mizuno T, Suzumura A. Interferon-gamma induces microglial-activation-induced cell death: a hypothetical mechanism of relapse and remission in multiple sclerosis. Neurobiol Dis. 2006;22:33–9. doi: 10.1016/j.nbd.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 6.Larbi A, Pawelec G, Witkowski JM, Schipper HM, Derhovanessian E, Goldeck D, Fulop T. Dramatic shifts in circulating CD4 but not CD8 T cell subsets in mild Alzheimer's disease. J Alzheimers Dis. 2009;17:91–103. doi: 10.3233/JAD-2009-1015. [DOI] [PubMed] [Google Scholar]
- 7.McDole J, Johnson AJ, Pirko I. The role of CD8+ T-cells in lesion formation and axonal dysfunction in multiple sclerosis. Neurol Res. 2006;28:256–61. doi: 10.1179/016164106X98125. [DOI] [PubMed] [Google Scholar]
- 8.Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 9.Purisai MG, McCormack AL, Cumine S, Li J, Isla MZ, Di Monte DA. Microglial activation as a priming event leading to paraquat-induced dopaminergic cell degeneration. Neurobiol Dis. 2007;25:392–400. doi: 10.1016/j.nbd.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schwartz M. Macrophages and microglia in central nervous system injury: are they helpful or harmful? J Cereb Blood Flow Metab. 2003;23:385–94. doi: 10.1097/01.WCB.0000061881.75234.5E. [DOI] [PubMed] [Google Scholar]
- 11.Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-κB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A. 2006;103:12481–6. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O'Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci U S A. 2007;104:1604–9. doi: 10.1073/pnas.0610731104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sonkoly E, Wei T, Janson PC, et al. MicroRNAs: novel regulators involved in the pathogenesis of psoriasis? PLoS ONE. 2007;2:e610. doi: 10.1371/journal.pone.0000610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Masaki S, Ohtsuka R, Abe Y, Muta K, Umemura T. Expression patterns of microRNAs 155 and 451 during normal human erythropoiesis. Biochem Biophys Res Commun. 2007;364:509–14. doi: 10.1016/j.bbrc.2007.10.077. [DOI] [PubMed] [Google Scholar]
- 15.Kluiver J, Poppema S, de Jong D, Blokzijl T, Harms G, Jacobs S, Kroesen BJ, van den Berg A. BIC and miR-155 are highly expressed in Hodgkin, primary mediastinal and diffuse large B cell lymphomas. J Pathol. 2005;207:243–9. doi: 10.1002/path.1825. [DOI] [PubMed] [Google Scholar]
- 16.Gironella M, Seux M, Xie MJ, et al. Tumor protein 53-induced nuclear protein 1 expression is repressed by miR-155, and its restoration inhibits pancreatic tumor development. Proc Natl Acad Sci U S A. 2007;104:16170–5. doi: 10.1073/pnas.0703942104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Faraoni I, Antonetti FR, Cardone J, Bonmassar E. miR-155 gene: a typical multifunctional microRNA. Biochim Biophys Acta. 2009;1792:497–505. doi: 10.1016/j.bbadis.2009.02.013. [DOI] [PubMed] [Google Scholar]
- 18.Kutty RK, Nagineni CN, Samuel W, Vijayasarathy C, Hooks JJ, Redmond TM. Inflammatory cytokines regulate microRNA-155 expression in human retinal pigment epithelial cells by activating JAK/STAT pathway. Biochem Biophys Res Commun. 2010;402:390–5. doi: 10.1016/j.bbrc.2010.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tili E, Michaille JJ, Cimino A, et al. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J Immunol. 2007;179:5082–9. doi: 10.4049/jimmunol.179.8.5082. [DOI] [PubMed] [Google Scholar]
- 20.Lu C, Huang X, Zhang X, et al. miR-221 and miR-155 regulate human dendritic cell development, apoptosis, and IL-12 production through targeting of p27kip1, KPC1, and SOCS-1. Blood. 2011;117:4293–303. doi: 10.1182/blood-2010-12-322503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lavigne C, Thierry AR. Enhanced antisense inhibition of human immunodeficiency virus type 1 in cell cultures by DLS delivery system. Biochem Biophys Res Commun. 1997;237:566–71. doi: 10.1006/bbrc.1997.7191. [DOI] [PubMed] [Google Scholar]
- 22.Campbell MJ. Lipofection reagents prepared by a simple ethanol injection technique. BioTechniques. 1995;18:1027–32. [PubMed] [Google Scholar]
- 23.Lu J, Tsourkas A. Imaging individual microRNAs in single mammalian cells in situ. Nucleic Acids Res. 2009;37:e100. doi: 10.1093/nar/gkp482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferreira R, Xapelli S, Santos T, Silva AP, Cristovao A, Cortes L, Malva JO. Neuropeptide Y modulation of interleukin-1β (IL-1β)-induced nitric oxide production in microglia. J Biol Chem. 2011;285:41921–34. doi: 10.1074/jbc.M110.164020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mayo L, Levy A, Jacob-Hirsch J, Amariglio N, Rechavi G, Stein R. Bid regulates the immunological profile of murine microglia and macrophages. Glia. 2011;59:397–412. doi: 10.1002/glia.21109. [DOI] [PubMed] [Google Scholar]
- 26.Soria JA, Arroyo DS, Gaviglio EA, Rodriguez-Galan MC, Wang JM, Iribarren P. Interleukin 4 induces the apoptosis of mouse microglial cells by a caspase-dependent mechanism. Neurobiol Dis. 2011;43:616–24. doi: 10.1016/j.nbd.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang S, Zhang HW, Lu MH, He XH, Li Y, Gu H, Liu MF, Wang ED. MicroRNA-155 functions as an OncomiR in breast cancer by targeting the suppressor of cytokine signaling 1 gene. Cancer Res. 2010;70:3119–27. doi: 10.1158/0008-5472.CAN-09-4250. [DOI] [PubMed] [Google Scholar]
- 28.Means TK, Luster AD. Integrins limit the Toll. Nat Immunol. 2010;11:691–3. doi: 10.1038/ni0810-691. [DOI] [PubMed] [Google Scholar]
- 29.Roy A, Fung YK, Liu X, Pahan K. Up-regulation of microglial CD11b expression by nitric oxide. J Biol Chem. 2006;281:14971–80. doi: 10.1074/jbc.M600236200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baker BJ, Akhtar LN, Benveniste EN. SOCS1 and SOCS3 in the control of CNS immunity. Trends Immunol. 2009;30:392–400. doi: 10.1016/j.it.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rodriguez A, Vigorito E, Clare S, et al. Requirement of bic/microRNA-155 for normal immune function. Science. 2007;316:608–11. doi: 10.1126/science.1139253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stanczyk J, Pedrioli DM, Brentano F, et al. Altered expression of MicroRNA in synovial fibroblasts and synovial tissue in rheumatoid arthritis. Arthritis Rheum. 2008;58:1001–9. doi: 10.1002/art.23386. [DOI] [PubMed] [Google Scholar]