

Abstract
BACKGROUND AND PURPOSE
Low doses of acetyl salicylic acid (ASA) and non-steroidal anti-inflammatory drugs (NSAIDs) cause gastrointestinal damage. The farnesoid X receptor (FXR) is a bile acid sensor essential for maintenance of intestinal homeostasis. Here, we have investigated whether FXR is required for mucosal protection in models of gastrointestinal injury caused by ASA and NSAIDs and if FXR activation has potential in the treatment or prevention of gastrointestinal injury caused by these agents.
EXPERIMENTAL APPROACH
FXR+/+ and FXR−/− mice were given ASA (10 to 100 mg·kg−1) or NSAIDs. Gastric and intestinal mucosal damage assessed by measuring lesion scores. FXR were activated by giving mice natural (chenodeoxycholic acid; CDCA) or synthetic (GW4064) FXR agonists.
KEY RESULTS
FXR, mRNA and protein, was detected in human and mouse stomach. FXR−/− mice were more prone to develop severe gastric and intestinal injury in response to ASA and NSAIDs and showed a severe reduction in the gastrointestinal expression of cystathionine-γ-lyase (CSE), an enzyme required for generation of hydrogen sulphide. CSE expression was reduced by ≈50% in wild-type mice challenged with ASA. Treating wild-type mice but not FXR−/− mice with CDCA or GW4064 protected against gastric injury caused by ASA and NSAIDs, by a CSE-dependent and cycloxygenase- and NO-independent, mechanism. FXR activation by GW4064 rescued mice from intestinal injury caused by naproxen.
CONCLUSIONS AND IMPLICATIONS
FXR was essential to maintain gastric and intestinal mucosal barriers. FXR agonists protected against gastric injury caused by ASA and NSAIDs by a CSE-mediated mechanism.
Keywords: Acetyl salicylic acid, bile acids, cystathionine-γ- lyase, farnesoid X receptor, hydrogen sulphide, nitric oxide, non-steroidal anti-inflammatory drugs, nuclear receptors
Introduction
Acetyl salicylic acid (ASA) and other non-steroidal anti-inflammatory drugs (NSAIDs) are widely used medications because of their demonstrated efficacy in preventing platelet aggregation and reducing pain and inflammation (Wolfe et al., 1999; Pusztaszeri et al., 2007; Fiorucci, 2009). However, long-term use of low doses of ASA and NSAIDs causes adverse gastrointestinal effects with life-threatening complications, gastrointestinal bleeding and perforation, occurring at a rate of ≈3% (Abraham et al., 2010). The basic mode of action of ASA and non-selective NSAIDs lies in the inhibition of cyclooxygenases (COX), with COX-2 generating prostaglandins at site of inflammation and COX-1 being responsible for generation of prostaglandins involved in protecting the gastrointestinal mucosa (Catella-Lawson et al., 2001; Fiorucci et al., 2007). While inhibition of COX-1 has provided an important explanation, the pathogenesis of gastrointestinal damage caused by ASA and NSAIDs is only partially understood. Indeed, COX-1-derived prostaglandins are not essential for maintaining gastric mucosal integrity while locally generated gaseous neurotransmitters, nitric oxide (NO) and hydrogen sulphide (H2S), are able to maintain mucosal integrity in a prostaglandin-depleted environment (Wallace, 2007; 2008;, Fiorucci and Santucci, 2011). We have reported that, in addition to COX inhibition, exposure to NSAIDs reduced the expression and activity of cystathionine-γ-lyase (CSE), a key enzyme involved in the generation of H2S (Fiorucci et al., 2005).
Bile acids, the end product of cholesterol metabolism, play a crucial role in integrating nutrient absorption and lipid metabolism, with liver and intestinal homeostasis (Makishima et al., 1999; Parks et al., 1999; Wang et al., 1999; Fiorucci et al., 2010). This homeostatic function has been linked to the activation of a family of receptors that includes the farnesoid X receptor (FXR), a nuclear receptor expressed in entero-hepatic tissues (Fiorucci et al., 2010; receptor nomenclature follows Alexander et al., 2009). In the small intestine, FXR regulates the expression of a number of homeostatic pathways including release of the growth factor fibroblast growth factor 19 (Inagaki et al., 2005), and lack of FXR predisposes to dysregulated immune homeostasis and intestinal damage in rodent models of colitis (Vavassori et al., 2009). Whether FXR is expressed in the gastric mucosa and exerts homeostatic functions in this tissue, however, is unknown. In the present study, by using FXR−/− mice and natural and synthetic FXR agonists, we have shown that FXR is essential in protecting the gastrointestinal tract against injury caused by ASA and NSAIDs. These data suggest that exploitation of FXR-regulated pathways might represent a novel mechanism for protecting gastrointestinal mucosa from the detrimental effects of ASA and NSAIDs.
Methods
Animals
All animal care and experimental protocols were approved by the local Animal Care and Ethics Committees of the University of Perugia. C57BL6 mice were from Harlan Nossan (Udine, Italy) and maintained on standard laboratory mouse chow on a 12 h light/dark cycle. FXR−/− mice (C57BL/6BJ6 background) were originally provided by Dr F. Gonzales (NIH, Bethesda) (Sinal et al, 2000). All mice were housed in a temperature-controlled room and had free access to food and water.
Acute damage
Mice were deprived of food for 16 h before being given one of the following oral suspensions: ASA (10–100 mg·kg−1), indomethacin (10 mg·kg−1), diclofenac (100 mg·kg−1) or ketoprofen (30 mg·kg−1). Mice were killed 3 h later, and gastric mucosal damage was measured. In some experiments, GW4064 (30 mg·kg−1) and chenodeoxycholic acid (CDCA: 15 mg·kg−1) were given (i.p. or p.o.) for 5 days before the NSAIDs. Gastric mucosal damage was assessed as described previously (Fiorucci et al., 2005). These measurements were made without knowledge of the treatments the mice had received. In addition, samples of the body region of the stomach were excised and divided for histology, RNA extraction and for measurement of myeloperoxidase (MPO) activity. To gain insights on the role of CSE, mice were pretreated with DL-propargylglycine at a dose of 10 mg·kg−1·day−1 i.p. alone or in combination with GW4064 (30 mg·kg−1) for 5 days and then given ASA (100 mg·kg−1) after 16 h fasting. Mice were killed 3 h later, and gastric mucosal injury and MPO activity assessed. To study the role of NO in the context of the protective effects of GW4064 (30 mg·kg−1 i.p. for 5 days before ASA administration), the animals were treated with Nω-nitro-L-arginine methyl ester (L-NAME), 30 mg·kg−1 i.p., a non-selective NOS inhibitor, 1 h before ASA treatment and killed 3 h later.
Gastric adaptation study
Mice weighing 20–25 g were given a daily dose of ASA, 100 mg·kg−1, alone or in combination with GW 4064 (30 mg·kg−1) for one or 14 days. On day 14, the mice were fasted overnight. On day 15, mice were treated orally with an additional dose of ASA and killed 3 h later.
Intestinal damage, gastric and intestinal mucosal prostanoids, MPO and TNFα levels
Groups of six mice (not fasted) were treated daily for 5 days with naproxen (100 mg·kg−1 p.o.), in combination with GW4064 (30 mg·kg−1 i.p.) or vehicle. Six hours after the final administration of drugs, the mice were anesthetized with sodium pentobarbital (100 mg·kg−1) and blood samples were drawn, by intracardiac puncture, for determination of haematocrit (Reuter et al., 1997). The small intestine was excised, and the extent of haemorrhagic damage to the small intestine quantified, under a microscope, by measuring the lengths of each lesion in mm and then summing these to obtain a damage score for each mice (Reuter et al., 1997). These measurements were made without knowledge of the treatments the mice had received. Generation of PGE2 and 6-keto-PGF1α by gastric and intestinal mucosa was measured according to previously published methods (Fiorucci et al., 2005), using a specific elisa kit (Cayman Chemical Company, Ann Arbor, MI). Gastric myeloperoxidase (MPO) activity was measured using a spectrophotometric assay with tri-methylbenzidine (TMB) as a substrate (Vavassori et al., 2009). Activity is expressed as mU per mg protein. Stomach TNFα levels were quantified by elisa (Biosource).
Immunohistochemistry analysis of FXR expression
Human gastric tissue were obtained, with informed written consent, from patients during surgery for gastric cancer. The patients (4 males 1 female; 47-65 years old) had no chemotherapy before surgery. The samples used here were macroscopically normal. Human and mouse gastric tissue were cryoprotected in 0.1 M phosphate-buffered saline pH 7.4 (PBS), containing 30% sucrose for 24 h, then included in OCT and stored at −80°C. Cryostat sections (7 µm thickness) were processed for immunohistochemistry. Briefly, sections were washed in PBS, soaked in 3% H2O2 for 8 min, and incubated with 5% bovine serum albumin in PBS with Triton X-100 (0.3%) for 30 min. Sections were incubated with (as primary antibody) anti-FXR (Ab 28676, Abcam) or anti-CSE (sc-131905 Santa Cruz) in PBS with 0.3% Triton X-100 and 1% bovine serum albumin, at room temperature for 2 h. The sections were incubated with biotinylated anti-rabbit or anti-goat IgG (Vector) and then processed by the avidin-biotin-peroxidase method with Vectastain ABC kit (Vector, UK) and diaminobenzidine was used as chromogen.
Measurement of CSE activity
The CSE activity was assessed as described elsewhere (Ogasawara et al., 2002; Fiorucci et al., 2005).
RT-PCRs
The method and conditions for RT-PCR are the same described elsewhere (Fiorucci et al., 2005). Primers used were as follows (all for the murine genes): CSE: (s)tgctgccaccattacgatta and (as)gatgccaccctcctgaagta; intracellular adhesion molecule-1 (ICAM-1): (s)gggaatgtcaccaggaatgt and (as) tgagttttatggcctcctcct; TNFα: (s)acggcatggatctcaaagac and (as) gtgggtgaggagcacgtagt; COX-1: (s)tgccctctgtacccaaagac and (as) tgtgcaaagaaggcaaacag; COX-2: (s)agaaggaaatggctgcagaa and (as) gctcggcttccagtattgag; endothelial NO synthase (eNOS): (s)agaagagtccagcgaacagc and (as) tgggtgctgaactgacagag; inducible NOS (iNOS): (s)acgagacggataggcagaga and cacatgcaaggaagggaact; small heterodimer partner (SHP): tctcttcttccgccctatca and aagggcttgctggacagtta; and FXR: tgtgagggctgcaaaggttt and acatccccatctctctgcac.
Statistical analysis
All data are presented as the mean ± SEM. Comparisons of groups of data were performed using a one-way analysis of variance followed by the Student–Newman–Keuls post hoc test. An associated probability (P-value) of less than 5% was considered significant.
Materials
ASA; indomethacin; ketoprofen; diclofenac; DL-propargylglycine; Nω-nitro-L-arginine methyl ester hydrochloride (L-NAME), chenodeoxycholic acid (CDCA) and all other reagents were from Sigma Chemical Co. (Milan, Italy).
Results
Detection of FXR expression in the gastric mucosa and its functional characterization
As illustrated in Figure 1, FXR gene and protein, was detected in the gastric mucosa. The expression of the gene by quantitative RT-PCR was ≈30% of that expressed in adult mouse liver, the tissue that expresses the highest content of the gene in the adult animal. Using immunohistochemistry, expression of FXR was detected in the mucosa, periglandular areas, in the submucosa, with muscle layers of micro vessels expressing the highest levels of immunoreactivity (Figure S1). No staining was detected in gastric muscle layers (Figure S1). This pattern of expression was confirmed in samples of human stomach. The immunohistochemical analysis was carried out on normal human gastric mucosa, obtained from intact margins of surgically resected gastric cancers (Figure 1C) and showed nuclear staining for FXR in gastric epithelial cells.
Figure 1.
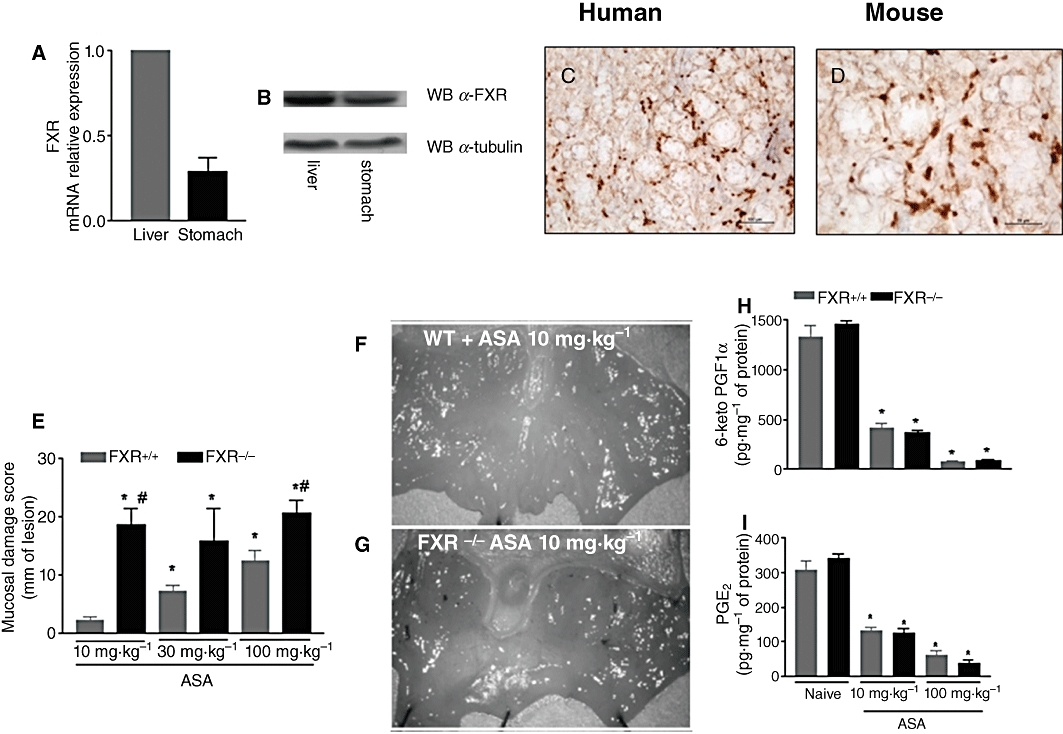
Effcts of FXR gene ablation on gastric responses to ASA and NSAIDS. (A,B) qRT-PCR and Western blot analysis of FXR expression in the liver and stomach of wild-type (FXR+/+) mice. (C,D) Immunohistochemical analysis of FXR expression in the human and mouse mucosa. Nuclear staining of gastric glandular epithelial cells is shown. Magnification 40×. (E) Gastric injury caused by ASA was exacerbated in FXR−/− mice. *P < 0.05 significantly different from naïve; #P < 0.05 significantly different from FXR+/+ treated with ASA (n = 6). (F,G) Representative example of macroscopic appearance of gastric injury caused by 10 mg·kg−1 ASA to FXR+/+ (WT) and FXR−/− mice. (H,I) Ex vivo generation of gastric prostaglandins after in vivo exposure to ASA. *P < 0.05 significantly different from naïve (n = 6).
To gain insights on the functional role of FXR in the gastric mucosa, we have investigated whether the gene for this receptor was essential to maintain intestinal homeostasis in a setting of mucosal injury caused by ASA and NSAIDs. Because a putative homeostatic activity of FXR would manifest itself by an increased susceptibility of FXR−/− mice to injury caused by ASA and NSAIDs, we challenged these mice with increasing doses of ASA. The severity of the damage was significantly exacerbated in FXR−/− mice, as the mucosal damage following 10 mg·kg−1 ASA was sixfold higher in FXR−/− mice than in wild-type mice and close the the level of damage caused by treating FXR wild-type mice with 100 mg·kg−1 ASA. An example of the gastric injury caused by ASA to FXR wild-type and FXR−/− mice is shown in Figure 1F and G. The differential susceptibility of FXR−/− mice, compared with wild-type mice, was not due to a different inhibition of gastric prostanoids, because as shown in Figure 1H and I, ASA caused a comparable suppression of 6-keto-PGF1α and PGE2 formation by the gastric mucosa. We therefore assessed the expression of a number of mediators that have been shown to be mechanistically linked to development of gastric injury in this model. As shown in Figure 2, FXR−/− mice had higher mucosal levels of MPO and TNFα (P < 0.05; n = 6) than wild-type mice, while expression of ICAM-1, an adhesion molecule required for neutrophil margination into the gastric mucosa, was similar in the two strains. We found no difference in the level of regulation of COX-1 and COX-2 in response to ASA, further confirming that the differential susceptibility of FXR−/− mice to ASA was COX-independent.
Figure 2.
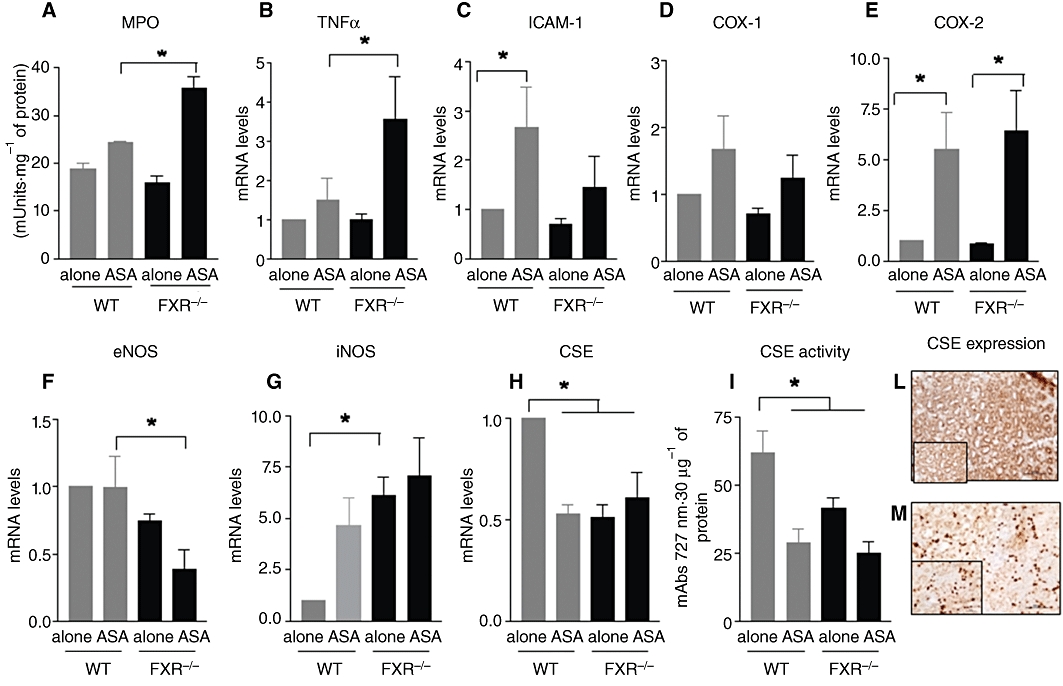
(A-I) Assessment of biochemical markers of gastric damage in FXR+/+ (WT) and FXR−/− mice exposed to ASA. *P < 0.05 significantly different from wild-type naïve. (L,M) Expression of CSE in the mouse (upper) and human (below) gastric mucosa. Magnification 20×. Insets. Magnification 40×.
Figure 2F–G demonstrates that FXR−/− mice were characterized by a reduced expression of eNOS but had fivefold higher levels of iNOS. However, there was no difference in the level of expression of iNOS induced by ASA, a known mechanism of adaptation to injury caused by this agent. A striking difference between wild-type and naïve FXR−/− mice was the reduced gastric expression of CSE mRNA in FXR−/− mice (P < 0.05 vs. FXR+/+; n = 6). CSE mRNA was reduced by 50% by treating FXR+/+ mice with ASA. Levels of CSE mRNA in FXR−/− were ≈50% of that of wild-type mice. No further decrease was measured in these mice after exposure to ASA. Changes in the expression of the CSE gene were mirrored by a similar decrease in CSE activity (Figure 2I). As shown in Figure 2L and M, CSE was abundantly expressed in the gastric mucosa, from mice or humans, with staining of glandular areas with a predominantly cytosolic localization.
Adaptation to ASA is a phenomenon characterized by a reduction of the extent of gastric hemorrhagic and erosive lesions, that occurs in rodents and humans, despite continuous treatment with ASA or an NSAID. The mechanism is COX-independent. To investigate whether FXR is involved in gastric adaptation to ASA, wild-type and FXR−/− mice were challenged with ASA, 100 mg·kg−1, for 14 days and gastric injury measured on day one and on day 14. As illustrated in Figure S2, both wild-type and FXR−/− mice adapted to ASA, indicating that this phenomenon was independent of the presence of FXR.
Activation of FXR protects against gastrointestinal injury caused by ASA and NSAIDs
Because these data highlight a functional role for FXR in the gastric mucosa that is protective, we have then investigated whether activation of FXR by natural and synthetic ligands will protect against gastric injury caused by ASA. Data shown in Figure 3A demonstrate that activation of FXR with GW4064, a synthetic FXR agonist, administered orally or i.p. at adose of 30 mg·kg−1, and by CDCA, a natural FXR agonist, administered orally at a dose of 15 mg·kg−1, protected against acute gastric damage caused by 100 mg·kg−1 ASA (n = 6, P < 0.05). Protection exerted by FXR agonists was confirmed by a reduced macroscopic score, attenuation of histopathological changes, reduced release of TNFα and attenuated influx of neutrophils (Figure 3A–G). In addition, treating mice with GW4064 resulted in a strong induction of CSE, mRNA and activity (Figure 3H and I). Finally, to ascertain whether protection exerted by FXR agonists extended to NSAIDs, mice left untreated or pretreated with GW4064 (30 mg·kg−1) were given indomethacin, ketoprofen or naproxen. As indicated in Figure 3L, damage caused by these non-selective NSAIDs was significantly attenuated by pretreatment with GW4064 (n = 6; P < 0.05).
Figure 3.
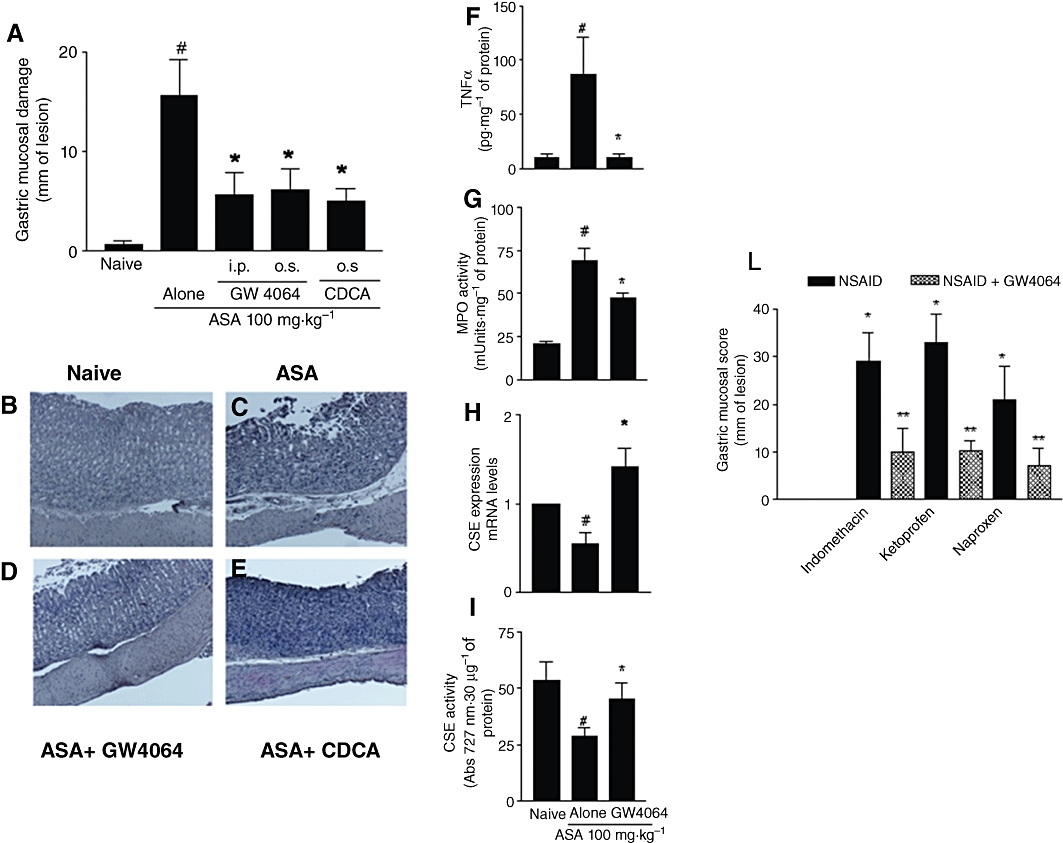
FXR activation by natural and synthetic ligands protects against development of gastric injury caused by ASA and NSAIDs. (A) Administration of GW4064 (30 mg·kg−1) either i.p. or p.o.. and CDCA (10 mg·kg−1) p.o.. attenuates gastric injury caused by dosing wild-type rats with ASA. #P < 0.05 significantly different from naïve; P < 0.05 significantly different from ASA alone (n = 5–7). (B–E) H&E staining of gastric mucosa of wild-type mice exposed to ASA alone or in combination with GW4064 or CDCA. Magnification 4×. (F–I) Assessment of biochemical markers of gastric damage in wild-type mice exposed to ASA alone or in combination with GW4064. #P < 0.05 significantly different from naïve; *P < 0.05 significantly different from ASA alone (n = 5–7). (L) Activation of FXR by GW4064 protects against gastric damage caused by NSAIDs. *P < 0.05 significantly different from naïve; **P < 0.05 significantly different from NSAID alone (n = 6).
Protection exerted by FXR agonists is NO-independent but requires activity of CSE
Because previous studies have demonstrated that FXR activation might regulate the expression and activity of eNOS and CSE, two enzymes involved in the formation of NO and H2S, we then investigated whether inhibition of these pathways affected the ability of FXR agonists to attenuate damage caused by ASA. L-NAME is a non-selective inhibitor of eNOS, known to exacerbate gastric injury caused by ASA and NSAIDs. As illustrated in Figure 4A, exposure to L-NAME, 30 mg·kg−1 i.p. administered 1 h before ASA, exacerbated the severity of gastric injury caused by ASA in wild-type mice, although it failed to do so in FXR−/− mice. Additionally, protection exerted by GW4064, in FXR wild-type mice exposed to 100 mg·kg−1 ASA was maintained, indicating that protection exerted by GW4064 in this setting was NO-independent.
Figure 4.
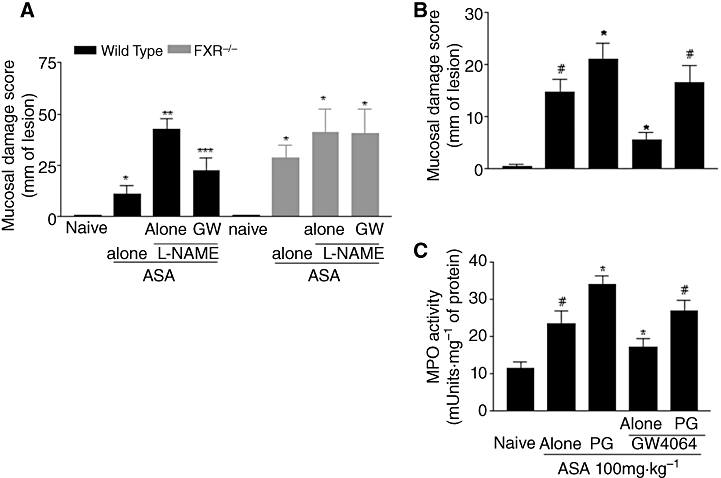
Protection exerted by FXR activation is independent of eNOS but prevented by inhibition of CSE. (A) Administration of L-NAME exacerbates gastric injury caused by 100 mg·kg−1 ASA in wild-type but not in FXR−/− mice. Treating mice with GW4064 protects against injury caused by ASA + L-NAME in wild-type mice but not in FXR−/− mice. #P < 0.05 significantly different from naïve; *P < 0.05 significantly different from ASA alone; **P < 0.05 significantly different from ASA + L-NAME (n = 6). (B,C) Inhibition of CSE activity by DL-propargylglycine (PG) prevents GW4064 protecting against damage caused by ASA in wild-type mice. #P < 0.05 significantly different from naïve; *P < 0.05 significantly different from ASA alone (n = 6).
DL-propargylglycine is a non-specific CSE inhibitor that reduces H2S formation in the stomach (Fiorucci et al., 2005). As illustrated in Figure 4B and C, treatment with DL-propargylglycine increased significantly the extent of gastric injury caused by ASA and almost completely reversed protection exerted by GW4064 in this setting. Similar data were obtained by measuring MPO activity in the stomach (n = 4–6; P < 0.05 vs. ASA alone or ASA plus GW4064.)
FXR activation protects against intestinal injury caused by NSAIDs
Because FXR has a role in maintaining the integrity of intestinal mucosa, we then investigated whether intestinal damage caused by treatment with naproxen, 100 mg·kg−1 day for 5 days, would be exacerbated in FXR−/− mice and whether FXR activation by GW4064 would rescue wild-type mice from intestinal damage in this model. Figure 5A demonstrates that naproxen-induced injury of small intestine was attenuated by activation of FXR in wild-type mice (P < 0.05 vs. naproxen alone; n = 6). In contrast, FXR gene ablation resulted in a striking exacerbation of the injury caused by naproxen and the protective effect exerted by GW4064 was lost. These data were supported by the analysis of MPO activity and blood haematocrit. A striking reduction of ≈10% in haematocrit levels, a measure of intestinal bleeding, was observed in wild-type mice-given naproxen and this fall was significantly attenuated by GW4064 (P < 0.05; n = 6). A similar pattern was observed in FXR−/− mice, but, in this case, GW4064 failed to protect against gastrointestinal bleeding (n = 6; P < 0.05 vs. naïve and P < 0.05 GW4064 vs. naproxen alone). As shown in Figure 5D, protection exerted by GW4064 was COX-independent (n = 6; P < 0.05 vs. naïve; not significant GW4064 vs. naproxen alone).
Figure 5.
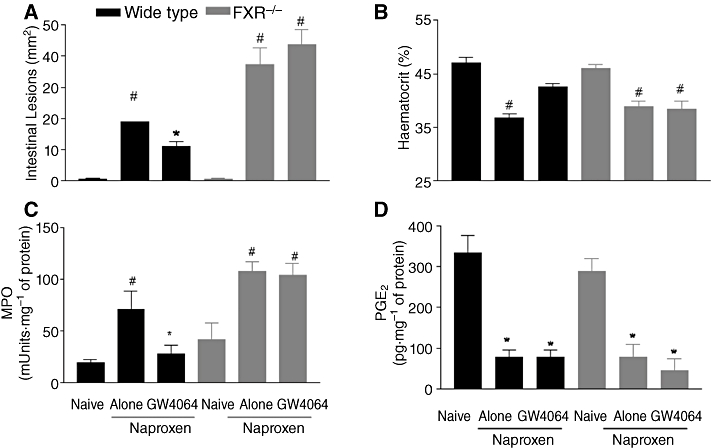
FXR is essential to maintain intestinal integrity in a model of intestinal injury caused by naproxen. (A) Intestinal injury score, (B) haematocrit, (C) MPO activity and (D) PGE2 generation in wild-type and FXR−/− mice challenged with naproxen, 100 mg·kg−1·day−1, for 5 days. #P < 0.05 significantly different from naïve; *P > 0.05 significantly different from naproxen alone (n = 6).
Discussion
In the present study, we have provided evidence that the bile acid sensor FXR is expressed in the human and rodent stomach and that this nuclear receptor provides signals that are essential for maintenance of mucosal integrity. This previously unrecognized role of FXR was revealed by challenging mice with ASA and NSAIDs. Gastrointestinal injury, ranging from acute erosions to ulcer bleeding and perforation, represents a well recognized complication of the use of ASA and NSAIDs. The risk of gastrointestinal bleeding in patients taking low doses of ASA (<100 mg·day−1) or medium doses of any NSAID increases approximately fourfold in comparison with untreated populations (Wolfe et al., 1999). The pathogenesis of injury caused by ASA and NSAIDs is largely related to the suppression of COX-1-derived prostanoids in the gastrointestinal tract; however, additional mechanisms seem to be involved. This contention is well exemplified by the demonstration that COX-1 gene ablation results in mutant mice that survive well, have no gastric pathology, and show less indomethacin-induced gastric ulceration than wild-type mice, even though their gastric PGE2 levels are only ≈1% of wild-type mice (Langenbach et al., 1995).
In the present study, we have provided evidence that the bile acid sensor FXR is required for maintenance of gastrointestinal mucosal integrity in a model of ASA and NSAIDs-induced injury and that this protective effect is prostaglandin-independent. This contention emerges from the results of studies carried out in FXR−/− mice. Although these mice show normal gastric morphology, they develop severe gastric and intestinal lesions when challenged with ASA or NSAIDs. This enhanced susceptibility is COX-independent because expression of COX-1 and COX-2 mRNAs in the gastric mucosa was regulated in a similar manner and ASA caused a comparable suppression of the formation of 6-keto-PGF1α and PGE2. This enhanced susceptibility to COX-inhibiting agents extends beyond the stomach, because FXR deficiency predisposes to the development of more severe intestinal injury in response to naproxen, a validated model of intestinal damage. Again, the mechanisms underlying this enhanced susceptibility were COX-independent because exposure of FXR+/+ and FXR−/− mice to naproxen caused a similar suppression of PGE2 formation in the intestine.
In the search for a mechanism that could support the enhanced susceptibility of FXR−/− to gastrointestinal damage caused by ASA and NSAIDs, we have investigated the role of NO and H2S, two gaseous mediators that exert homeostatic functions in the gastric mucosa. A previous study has demonstrated that the eNOS promoter contains an imperfect inverted repeat DNA motif, -628AGCTCAgtGGACCT-641, that might function as a putative FXR-responsive element. The activation of this non-canonical response element triggered eNOS transcription and exposing vascular endothelial cells to an FXR ligand up-regulated the expression of eNOS mRNA and protein and increased the production of nitrite/nitrate (Li et al., 2008). One important finding of the present study is that FXR−/− mice had reduced levels of eNOS in their gastric mucosa, highlighting the functional relevance of FXR in regulating eNOS gene transcription. However, we believe that these reduced levels of eNOS did not represent the main cause for the enhanced susceptibility of FXR−/− mice to ASA and NSAIDs for at least two reasons. Firstly, eNOS deficiency seems to be compensated by a robust induction of iNOS (≈fivefold in comparison to naïve FXR+/+ mice) and secondly, activation of FXR by CDCA and GW4064 attenuated gastric injury in wild-type mice treated with L-NAME.
In the past few years, we and others have shown that CSE, an enzyme involved in H2S generation from homocysteine and L-cysteine, is selectively targeted by NSAIDs in the gastric mucosa (Fiorucci et al., 2005; Fiorucci and Santucci, 2011). The mechanistic readout of this interaction is that the stomach of rodent exposed to NSAIDs generates less H2S than control animals. Because exogenous H2S attenuates gastric injury caused by NSAIDs despite a continuous inhibition of COXs and NOSs, it has been proposed that H2S alone is able to protect the gastric mucosa (Fiorucci et al., 2005). We have previously shown that the CSE promoter contains a canonical FXR-responsive element in its 5′ flanking region and that this regulatory element has a functional relevance in controlling the level of expression of CSE, mRNA and protein, in the liver (Renga et al., 2009). We have now confirmed these findings by demonstrating FXR−/− mice express ≈50% of CSE mRNA in the stomach and that the activity of this enzyme was profoundly impaired by FXR deficiency. Considering that administration of ASA reduced the gastric expression and activity of CSE in wild-type mice; that CSE was localized in the gastric mucosa in human and rodents, that protection by GW4064 against damage caused by ASA was accompanied with a robust induction of CSE expression and activity, and that inhibition of CSE with DL-propargylglycine attenuated protection afforded by FXR agonists, all our results support the view that a dysregulated expression of CSE in the gastrointestinal mucosa makes an important contribution to the enhanced susceptibility of FXR−/− mice to damage caused by ASA and NSAIDs. However, due to the lack of selectivity of DL-propargylglycine for CSE, more specific inhibitors are required to fully establish the specificity of this interaction.
Taken together with previous findings showing an enhanced susceptibility of FXR−/− mice to develop unchecked inflammation in the intestine and colon (Inagaki et al., 2005; Vavassori et al., 2009) as well as an enhanced tendency to develop intestinal cancer (Maran et al., 2009), our present results seem to support the notion that FXR plays an important homeostatic function in these tissues. Loss of FXR translates in a variety of functional deficiencies and subtle alterations in the balance of protective mechanisms including an altered expression of COXs, eNOS and iNOS and CSE. Because FXR orchestrates the integration of nutrient metabolism with intestinal homeostasis, it might be that alterations of this gene could have a role in the development of disorders of the gastrointestinal tract in humans.
These findings might have important translational readouts (Wallace, 2007; Wallace et al., 2010). Indeed, the FXR-CSE-H2S pathways could be exploited therapeutically by designing novel COX inhibitor compounds that also release H2S or activate FXR-CSE, to spare the gastrointestinal mucosa. Indeed, while a number of approaches are presently available to protect the gastric mucosa against damage caused by ASA and NSAIDs, the same is not true for intestinal injury caused by these agents (Goldstein et al., 2007). Intestinal complications caused by COX-inhibiting agents are not efficiently prevented by co-treatment with proton pump inhibitors (Fiorucci, 2009) and, while selective COX inhibitors spare the intestinal mucosa (Laine et al., 2008), their use is associated with an increased risk of cardiovascular ischaemic events (Fitzgerald, 2004). In this context development of FXR agonists to protect the intestinal mucosa might have relevance in the treatment of patients taking ASA or NSAIDs who are at high risk of developing gastrointestinal and cardiovascular complications and for whom selective COX-2 inhibitors are not recommended.
In summary, we have shown that FXR, a bile acid receptor, plays an essential role in maintaining gastric and intestinal barrier integrity. Exploitation of FXR-regulated pathways might represent a novel mechanism for protecting gastric and intestinal mucosa from damage induced by ASA and NSAIDs.
Acknowledgments
There were no external funds used in these studies.
Glossary
Abbreviations
- ASA
acetyl- salicylic acid
- CDCA
chenodeoxycholic acid
- CSE
cystathionine-γ- lyase
- FXR
farnesoid X receptor
- L-NAME
Nω-nitro-L-arginine methyl ester hydrochloride
- MPO
myeloperoxidase
Conflict of interest
None of the authors has any conflict of interest to be disclosed.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Immunostaining for FXR in mouse gastric mucosa. FXR immunoreactive cells were detected in the mucosa (B) and in the vessel (A) of submucosa.
Figure S2 Gastric adaptation to ASA is not impaired by FXR gene deletion. Mice were given ASA 100 mg·kg−1 for 1 or 12 days and mucosal injury assessed 24 h after the last administration. #P < 0.05 versus naïve; *P < 0.05 versus acute (1 day).
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Abraham NS, Hlatky MA, Antman EM, Bhatt DL, Bjorkman DJ, Clark CB, et al. ACCF/ACG/AHA ACCF/ACG/AHA 2010 expert consensus document on the concomitant use of proton pump inhibitors and thienopyridines: a focused update of the ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of antiplatelet therapy and NSAID use. Am J Gastroenterol. 2010;105:2533–2549. doi: 10.1038/ajg.2010.445. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catella-Lawson F, Reilly MP, Kapoor SC, Cucchiara AJ, DeMarco S, Tournier B, et al. Cyclooxygenase inhibitors and the antiplatelet effects of aspirin. N Engl J Med. 2001;345:1809–1817. doi: 10.1056/NEJMoa003199. [DOI] [PubMed] [Google Scholar]
- Fiorucci S. Prevention of nonsteroidal anti-inflammatory drug-induced ulcer: looking to the future. Gastroenterol Clin North Am. 2009;38:315–332. doi: 10.1016/j.gtc.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Fiorucci S, Santucci L. Hydrogen sulfide-based therapies: focus on H2S-releasing NSAIDs. Inflamm Allergy Drug Targets. 2011;10:133–140. doi: 10.2174/187152811794776213. [DOI] [PubMed] [Google Scholar]
- Fiorucci S, Antonelli E, Distrutti E, Rizzo G, Mencarelli A, Orlandi S, et al. Inhibition of hydrogen sulfide generation contributes to gastric injury caused by anti-inflammatory nonsteroidal drugs. Gastroenterology. 2005;129:1210–1224. doi: 10.1053/j.gastro.2005.07.060. [DOI] [PubMed] [Google Scholar]
- Fiorucci S, Santucci L, Distrutti E. NSAIDs, coxibs, CINOD and H2S-releasing NSAIDs: what lies beyond the horizon. Dig Liver Dis. 2007;39:1043–1051. doi: 10.1016/j.dld.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Fiorucci S, Cipriani S, Baldelli F, Mencarelli A. Bile acid-activated receptors in the treatment of dyslipidemia and related disorders. Prog Lipid Res. 2010;49:171–185. doi: 10.1016/j.plipres.2009.11.001. [DOI] [PubMed] [Google Scholar]
- Fitzgerald GA. Coxibs and cardiovascular disease. N Engl J Med. 2004;351:1709–1711. doi: 10.1056/NEJMp048288. [DOI] [PubMed] [Google Scholar]
- Goldstein JL, Eisen GM, Lewis B, Gralnek IM, Aisenberg J, Bhadra P, et al. Small bowel mucosal injury is reduced in healthy subjects treated with celecoxib compared with ibuprofen plus omeprazole, as assessed by video capsule endoscopy. Aliment Pharmacol Ther. 2007;25:1211–1222. doi: 10.1111/j.1365-2036.2007.03312.x. [DOI] [PubMed] [Google Scholar]
- Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2:217–225. doi: 10.1016/j.cmet.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Laine L, Curtis SP, Langman M, Jensen DM, Cryer B, Kaur A, et al. Lower gastrointestinal events in a double-blind trial of the cyclo-oxygenase-2 selective inhibitor etoricoxib and the traditional nonsteroidal anti-inflammatory drug diclofenac. Gastroenterology. 2008;135:1517–1525. doi: 10.1053/j.gastro.2008.07.067. [DOI] [PubMed] [Google Scholar]
- Langenbach R, Morham SG, Tiano HF, Loftin CD, Ghanayem BI, Chulada PC, et al. Prostaglandin synthase 1 gene disruption in mice reduces arachidonic acid-induced inflammation and indomethacin-induced gastric ulceration. Cell. 1995;83:483–492. doi: 10.1016/0092-8674(95)90126-4. [DOI] [PubMed] [Google Scholar]
- Li J, Wilson A, Kuruba R, Zhang Q, Gao X, He F, et al. FXR-mediated regulation of eNOS expression in vascular endothelial cells. Cardiovasc Res. 2008;77:169–177. doi: 10.1093/cvr/cvm016. [DOI] [PubMed] [Google Scholar]
- Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, et al. Identification of a nuclear receptor for bile acids. Science. 1999;284:1362–1365. doi: 10.1126/science.284.5418.1362. [DOI] [PubMed] [Google Scholar]
- Maran RR, Thomas A, Roth M, Sheng Z, Esterly N, Pinson D, et al. Farnesoid X receptor deficiency in mice leads to increased intestinal epithelial cell proliferation and tumor development. J Pharmacol Exp Ther. 2009;328:469–477. doi: 10.1124/jpet.108.145409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogasawara Y, Ishii K, Tanabe S. Enzymatic assay of gamma-cystathionase activity using pyruvate oxidase-peroxidase sequential reaction. J Biochem Biophys Methods. 2002;51:139–150. doi: 10.1016/s0165-022x(02)00010-6. [DOI] [PubMed] [Google Scholar]
- Parks DJ, Blanchard SG, Bledsoe RK, Chandra G, Consler TG, Kliewer SA, et al. Bile acids: natural ligands for an orphan nuclear receptor. Science. 1999;284:1365–1368. doi: 10.1126/science.284.5418.1365. [DOI] [PubMed] [Google Scholar]
- Pusztaszeri MP, Genta RM, Cryer BL. Drug-induced injury in the gastrointestinal tract: clinical and pathologic considerations. Nat Clin Pract Gastroenterol Hepatol. 2007;4:442–453. doi: 10.1038/ncpgasthep0896. [DOI] [PubMed] [Google Scholar]
- Renga B, Mencarelli A, Migliorati M, Cipriani S, Distrutti E, Fiorucci S. Bile-acid-activated farnesoid X receptor regulates hydrogen sulfide production and hepatic microcirculation. World J Gastroenterol. 2009;15:2097–2108. doi: 10.3748/wjg.15.2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter BK, Davies NM, Wallace JL. Nonsteroidal anti-inflammatory drug enteropathy in rats: role of permeability, bacteria, and enterohepatic circulation. Gastroenterology. 1997;112:109–117. doi: 10.1016/s0016-5085(97)70225-7. [DOI] [PubMed] [Google Scholar]
- Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102:731–744. doi: 10.1016/s0092-8674(00)00062-3. [DOI] [PubMed] [Google Scholar]
- Vavassori P, Mencarelli A, Renga B, Distrutti E, Fiorucci S. The bile acid receptor FXR is a modulator of intestinal innate immunity. J Immunol. 2009;183:6251–6261. doi: 10.4049/jimmunol.0803978. [DOI] [PubMed] [Google Scholar]
- Wallace JL. Hydrogen sulfide-releasing anti-inflammatory drugs. Trends Pharmacol Sci. 2007;28:501–550. doi: 10.1016/j.tips.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Wallace JL. Prostaglandins, NSAIDs, and gastric mucosal protection: why doesn't the stomach digest itself? Physiol Rev. 2008;88:1547–1565. doi: 10.1152/physrev.00004.2008. [DOI] [PubMed] [Google Scholar]
- Wallace JL, Caliendo G, Santagada V, Cirino G. Markedly reduced toxicity of a hydrogen sulphide-releasing derivative of naproxen (ATB-346) Br J Pharmacol. 2010;159:1236–1246. doi: 10.1111/j.1476-5381.2009.00611.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Chen J, Hollister K, Sowers LC, Forman BM. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol Cell. 1999;3:543–553. doi: 10.1016/s1097-2765(00)80348-2. [DOI] [PubMed] [Google Scholar]
- Wolfe MM, Lichtenstein DR, Singh G. Gastrointestinal toxicity of nonsteroidal antiinflammatory drugs. N Engl J Med. 1999;340:1888–1899. doi: 10.1056/NEJM199906173402407. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.