

Abstract
BACKGROUND AND PURPOSE
Little information exists on the mechanisms that precipitate brain stem death, the legal definition of death in many developed countries. We investigated the role of tropomyocin receptor kinase B (TrkB) and its downstream signalling pathways in the rostral ventrolateral medulla (RVLM) during experimental brain stem death.
EXPERIMENTAL APPROACH
An experimental model of brain stem death that employed microinjection of the organophosphate insecticide mevinphos bilaterally into the RVLM of Sprague–Dawley rats was used, in conjunction with cardiovascular, pharmacological and biochemical evaluations.
KEY RESULTS
A significant increase in TrkB protein, phosphorylation of TrkB at Tyr516 (pTrkBY516), Shc at Tyr317 (pShcY317) or ERK at Thr202/Tyr204, or Ras activity in RVLM occurred preferentially during the pro-life phase of experimental brain stem death. Microinjection bilaterally into RVLM of a specific TrkB inhibitor, K252a, antagonized those increases. Pretreatment with anti-pShcY317 antiserum, Src homology 3 binding peptide (Grb2/SOS inhibitor), farnesylthioacetic acid (Ras inhibitor), manumycin A (Ras inhibitor) or GW5074 (Raf-1 inhibitor) blunted the preferential augmentation of Ras activity or ERK phosphorylation in RVLM and blocked the up-regulated NOS I/protein kinase G (PKG) signalling, the pro-life cascade that sustains central cardiovascular regulation during experimental brain stem death.
CONCLUSIONS AND IMPLICATIONS
Activation of TrkB, followed by recruitment of Shc/Grb2/SOS adaptor proteins, leading to activation of Ras/Raf-1/ERK signalling pathway plays a crucial role in ameliorating central cardiovascular regulatory dysfunction via up-regulation of NOS I/PKG signalling cascade in the RVLM in brain stem death. These findings provide novel information for developing therapeutic strategies against this fatal eventuality.
Keywords: tropomyocin receptor kinase B, rostral ventrolateral medulla, experimental brain stem death, mevinphos, central cardiovascular regulatory dysfunction, Shc/Grb2/SOS complex, Ras/Raf-1/ERK signalling, NOS I/protein kinase G cascade
Introduction
Whereas brain stem death is the legal definition of death stipulated in professional or statutory documents from the UK (Anonymous, 1976; 1979), USA (Anonymous, 1981), EU (Haupt and Rudolf, 1999) and Taiwan (Hung and Chen, 1995) and is of great clinical importance, a dearth of information exists in its mechanistic underpinning. The invariable prognosis that asystole occurs within hours or days after the diagnosis of brain stem death (Pallis, 1983) strongly suggests that permanent impairment of the brain stem circulatory control precedes death. It follows that better understanding of the cellular mechanisms that account for the progressive failure of brain stem cardiovascular regulation during the advancement towards brain stem death should offer new leads for the development of a therapeutic strategy against this fatal eventuality. Previous clinical work from our laboratory on critically ill patients (Kuo et al., 1997b; Yien et al., 1997; Yen et al., 2000) demonstrated that a dramatic reduction in or loss of the power density of the low-frequency (LF) component (0.04–0.15 Hz in human) in the power spectrum of systemic arterial pressure (SAP) signals consistently takes place before significant hypotension occurs in patients with confirmed brain stem death. We also showed (Kuo et al., 1997a) that the origin of this life-and-death signal resides in the rostral ventrolateral medulla (RVLM), which has been known for a long time to be responsible for the maintenance of sympathetic vasomotor tone and stable SAP (Spyer, 1994). As such, RVLM presents itself as a logical neural substrate for the mechanistic delineation of brain stem death (Chan et al., 2005b). It follows that evaluation of biochemical changes in the RVLM, whose neuronal activity is reflected in the waxing and waning of the life-and-death signal, should shed light on the cellular and molecular mechanisms of brain stem death.
Mevinphos (3-(dimethoxyphosphinyl-oxyl)-2-butenoic acid methyl ester; Mev), a US Environmental Protection Agency Toxicity Category I organophosphate pesticide, acts directly on the RVLM to elicit cardiovascular toxicity (Yen et al., 2001). Furthermore, the consecutive phases of augmentation followed by reduction of the LF component of the SAP spectrum manifested during Mev intoxication, which mirror those exhibited by patients who succumb to organophosphate poisoning (Yen et al., 2000), can be designated the pro-life and pro-death phase in this animal model of brain stem death (Chan et al., 2005b). Based on this Mev intoxication model (Chan et al., 2007a,b;), we have found that NO generated by NOS I in RVLM, followed by activation of the soluble guanylyl cyclase/cGMP/ PKG cascade, is responsible for the pro-life phase; peroxynitrite formed by a reaction between NOS II-derived NO and superoxide anion underlies the pro-death phase via progressive induction of apoptotic cell death in RVLM.
As death represents the end of existence for an individual, we proposed previously (Chan et al., 2005b) that multiple pro-life and pro-death programmes must be activated in the RVLM during the advancement towards brain stem death. In this regard, the tropomyocin receptor kinase B (TrkB) and its downstream signalling pathways present themselves as another reasonable candidate for the pro-life programme. TrkB activation mediates neuroprotection against focal cerebral ischaemia (Krikov et al., 2008) and raises blood pressure transiently (Xu et al., 2010). Likewise, Ras/MAPK kinase (MEK)/ERK in the RVLM participates in sympatho-excitation (Kishi et al., 2010). Shc is an important adaptor protein (Nakamura et al., 1996) that links TrkB to the docking growth factor receptor-bound protein 2 (Grb2)/Son of Sevenless (SOS) complex (Egan et al., 1993; You et al., 2010). The Shc/Grb2/SOS complex determines Ras activity (Li et al., 1995; Rao, 1996), and the most common upstream regulator of MEK/ERK is Ras/Raf-1 (Marais and Marshall, 1996; Zebisch et al., 2007; McCubrey et al., 2008). We very recently found (Chan et al., 2010) that activation of MEK/ERK/MAPK signal-interacting kinase (MNK) in the RVLM plays a preferential pro-life role by sustaining central cardiovascular regulatory functions during brain stem death via up-regulation of NOS I/PKG signalling in the RVLM. Interestingly, ERK activation is induced by an overexpression of TrkB but is depressed by mutation of the Shc activation domain (Hollis et al., 2009).
In the present study we evaluated the hypothesis that TrkB in the RVLM, by activating the NOS I/PKG cascade, with the participation of Shc/Grb2/SOS adaptor protein complex and Ras/Raf-1 cascade as interposing signals between TrkB and MEK/ERK, sustains the central cardiovascular regulatory machinery and has a pro-life role in experimental brain stem death. Using our Mev intoxication model of brain stem death, we demonstrated that activation of TrkB, followed by recruitment of Shc/Grb2/SOS adaptor proteins, which leads to activation of the Ras/Raf-1/ERK signalling pathway, plays a crucial role in ameliorating central cardiovascular regulatory dysfunction via upregulation of NOS I/PKG signalling cascade in the RVLM.
Methods
All animal care and experimental procedures complied with the guidelines of the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) International and were approved by the Laboratory Animal Committee of Chang Gung Memorial Hospital-Kaohsiung Medical Center.
Animals
Adult male Sprague–Dawley rats (280–345 g, n = 252) purchased from the Experimental Animal Center of the National Science Council, Taiwan, Republic of China were used. Animals were housed in groups of two to three in individually ventilated cages, in a temperature-controlled room (22 ± 2°C) with 12 h light/12 h dark cycles (lights on at 07:00 h) and with free access to rat chow and water. All efforts were made to minimize animal suffering and to reduce the number of animal used.
General preparation
Preparatory surgery, which included cannulation of a femoral artery and a femoral vein, together with tracheal intubation, was carried out under pentobarbital sodium (50 mg·kg−1, i.p.)-induced anaesthesia. SAP signals were recorded from the femoral artery, and heart rate (HR) was derived instantaneously from SAP signals. During the recording session, which routinely commenced 60 min after the administration of pentobarbital sodium, anaesthesia was maintained by i.v. infusion of propofol (Zeneca, Macclesfield, UK) at 20–25 mg·kg−1·h−1. We have demonstrated previously (Yang et al., 1995) that this scheme provided satisfactory anaesthetic maintenance while preserving the capacity of central cardiovascular regulation. Thus, stable mean SAP (MSAP; 119.5 ± 3.8 mmHg), HR (340.5 ± 13.6 beats min−1) and power density of LF component (14.1 ± 2.5 mmHg2) was achieved across all experimental groups (mean ± SEM, n = 252). Body temperature of the animals was maintained at 37°C with a heating pad, and rats were allowed to breathe spontaneously with room air during the entire recording session.
Mev intoxication model of brain stem death
Since Mev induces comparable cardiovascular responses when given systemically or directly to the RVLM, in our animal model a microinjection of Mev was administered bilaterally into the RVLM to elicit site-specific effects (Yen et al., 2001). SAP signals were subjected to simultaneous power spectral analysis using a computer algorithm developed by our laboratory (Kuo and Chan, 1993). In brief, SAP signals were digitized at a rate of 2048 Hz, followed by a bunching (64-point average) algorithm to reduce the sampling rate to 32 Hz. The SAP data to be analysed were then truncated into successive 32 s (1024 points) time segments with 50% overlapping. For each time segment, our algorithm estimated the power density of the spectral components based on fast Fourier transform. The resultant two-dimensional spectrogramme and values of each spectral component were recorded either graphically or numerically. By repeating the procedures continuously, we were able to examine the spectral changes of SAP signals over time in an on-line and real-time manner. We were particularly interested in the LF (0.25–0.8 Hz) component of the SAP spectrum because it is the best experimental index for mechanistic evaluation of central cardiovascular regulatory dysfunction during brain stem death. There are three reasons for this. Firstly, the power density of the LF component represents the most crucial link between our animal model and clinical observations, and is a more sensitive prognostic index than SAP for brain stem death (Chan et al., 2005b). A dramatic reduction or loss of the LF power is associated with brain stem death (Kuo et al., 1997b), which consistently takes place before significant hypotension occurs in patients who succumb to organophosphate poisoning (Yen et al., 2000). However, whereas a patient proclaimed brain stem dead (legal death) invariably exhibits asystole (clinical death), this eventuality may occur within hours or days and there is no fixed time-interval between these two events. As such, the sequential increase and decrease in LF power exhibited in our Mev intoxication model can only be used to reflect lower (pro-life) or higher (pro-death) probability of exhibiting brain stem death and cannot be related to clinical death. Secondly, the LF component of the SAP spectrum originates from the RVLM (Kuo et al., 1997a). Thirdly, the power density of the LF component mirrors the prevalence of baroreflex-mediated sympathetic neurogenic vasomotor discharges that emanate from the RVLM (Li et al., 2001) and is, therefore, a reasonable indicator of brain stem cardiovascular regulation. Temporal changes in pulsatile SAP, MSAP, HR and power density of the LF component were routinely followed for 180 min after the administration of Mev, in an on-line and real-time manner (Yen et al., 2001; Chang et al., 2009a).
Microinjection of test agents
Microinjection bilaterally of test agents into the RVLM, each at a volume of 50 nL, was carried out stereotaxically and sequentially (Chan et al., 2007a,b;) via a glass micropipette connected to a 0.5 µL Hamilton (Reno, NV) microsyringe. The co-ordinates used were: 4.5–5 mm posterior to lambda, 1.8–2.1 mm lateral to midline and 8.1–8.4 mm below the dorsal surface of cerebellum. Test agents used included Mev (kindly provided by Huikwang Corporation, Tainan, Taiwan; 10 nmol); a TrkB inhibitor (Levine et al., 1995; Katsuki et al., 2009), K252a (Calbiochem, San Diego, CA; 1 pmol); a depletor of endogenous TrkB ligands (Sahenk et al., 2010), mouse recombinant TrkB-Fc fusion protein (R&D Systems, Minneapolis, MN; 5 nmol); a specific inhibitor of Grb2/SOS interaction (Su et al., 1996; Upadhyay et al., 2004), Src homology 3 binding peptide (SH3b-p; Calbiochem; 25 fmol); two specific inhibitors of Ras activity (Amos et al., 2006; Park et al., 2006; 40 pmol), farnesylthioacetic acid (FTA; Enzo Life Sciences, Farmingdale, NY; 40 pmol) or manumycin A (Sigma-Aldrich, Saint Louis, MO; 40 pmol); a specific inhibitor of c-Raf1 activity (Hughes and Brown, 2006; Park et al., 2006), GW5074 (Sigma-Aldrich; 40 pmol); goat polyclonal antiserum (Santa Cruz Biotechnology, Santa Cruz, CA) against phosphorylated Shc at tyrosine 239/240 (pShcY239/240; 1:20) or 317 (pShcY317; 1:20) (Ishihara et al., 1998) site or normal goat serum (NGS, Sigma-Aldrich). All test agents were dissolved in artificial CSF (aCSF), with the exception of K252a, FTA, manumycin A and GW5074, which was prepared with 0.2% DMSO. As in previous studies (Chan et al., 2007a,b;), we added 0.02% Triton X-100 (Sigma-Aldrich) to anti-pShcY239/240 or anti-pShcY317 antiserum or NGS to facilitate its transport across the cell membrane of RVLM neurons. All test agents used for pretreatments were given 30 min before the administration of Mev. The doses used were adopted from previous reports (Upadhyay et al., 2004; Amos et al., 2006; Hughes and Brown, 2006; Park et al., 2006; Katsuki et al., 2009) that used those test agents for the same purpose as in this study and were found to effectively inhibit Mev-induced cardiovascular responses in preliminary experiments. Application of the same amount of aCSF, 0.2% DMSO or NGS served as the vehicle control to account for possible volume effects of the microinjection. The composition of aCSF was (mM): NaCl 117, NaHCO3 25, glucose 11, KCl 4.7, CaCl2 2.5, MgCl2 1.2 and NaH2PO4 1.2. To avoid the confounding effects of drug interactions, each animal received only one pharmacological treatment.
Collection of tissue samples from ventrolateral medulla
We routinely collected tissue samples for subsequent biochemical evaluations (Chan et al., 2007a,b;) during the peak of phase I or II (Mev group), or 30 or 180 min after microinjection of aCSF, 0.2% DMSO or NGS into RVLM (vehicle control group). Animals were killed with an overdose of pentobarbital sodium and tissues from both sides of the ventrolateral medulla, at the level of RVLM (0.5–1.5 mm rostral to the obex), were collected by micropunches made with a 1 mm (i.d.) stainless steel bore to cover the anatomical boundaries of the RVLM, with a diameter of 625–650 µm (Supporting Figure S1). Medullary tissues collected from anaesthetized animals that had not received pharmacological treatments served as the sham controls. The total concentration of proteins extracted from those tissue samples was determined by the BCA protein assay (Pierce, Rockford, IL).
elisa
Commercial kits for elisa were used (Chan et al., 2010; Dai et al., 2010) to detect Ras activity (Millipore, Billerica, MA), PKG activity (CycLex, Nagano, Japan), or to determine the levels of total Trk protein (Cell Signaling, Danvers, MA), TrkB protein (Cell Signaling), phosphorylated TrkB at Tyr516 (pTrkBY516) (Cell Signaling), ERK1/2 (Cell Signaling), phosphorylated ERK1/2 at Thr202/Tyr204 (Cell Signaling), NOS I (Cell Signaling), NOS II (Cell Signaling) or 3-nitrotyrosine (Cell Biolabs, San Diego, CA) in cell lysate from RVLM. The final absorbance of reaction solution was determined by spectrophotometry using an elisa microtiter plate reader (Anthros Labtec, Salzburg, Austria) and expressed as fold changes against sham-controls, except 3-nitrotyrosine.
Western blot analysis
Western blot analysis, including gel electrophoresis and transfer to or blotting on PVDF membrane (Millipore) was carried out (Chan et al., 2007a,b; Chang et al., 2009a) using primary antisera that included a rabbit polyclonal antiserum against TrkC (Santa Cruz Biotechnology); a goat polyclonal antiserum against phosphorylated Shc at Tyr239 or Tyr240 (pShcY239/240) (Santa Cruz Biotechnology) or phosphorylated Shc at Tyr317 (pShcY317) (Santa Cruz Biotechnology), or a mouse monoclonal antiserum against β-actin (Chemicon, Temecula, CA). This was followed by incubation with horseradish peroxidase-conjugated donkey anti-rabbit IgG (Amersham Biosciences, Little Chalfont, Bucks, UK) for TrkC, rabbit anti-goat IgG (Amersham Biosciences) for pShcY239/240 or pShcY317, or sheep anti-mouse IgG (Amersham Biosciences) for β-actin. As a routine, 20–30 µg of protein per sample was loaded, and each antiserum was applied to a fresh blot. Specific antibody-antigen complex was detected by an enhanced chemiluminescence Western blot detection system (NEN, Boston, MA). The amount of protein product was quantified by the ImageMaster Video Documentation System (Amersham Pharmacia Biotech) and was expressed as the ratio to β-actin protein.
Histology
In some animals that were not used for biochemical analysis, the brain stem was removed at the end of the physiological experiment and fixed in 10% formaldehyde–saline solution, that contained 30% sucrose, for at least 72 h. Frozen 25 µm sections of the medulla oblongata mounted in glycerin were used for histological verification of the microinjection sites.
Statistical analysis
All values are expressed as mean ± SEM. The averaged value of MSAP or HR calculated every 20 min after administration of test agents or vehicle, the sum total of power density for the LF component in the SAP spectrum over 20 min or the activity or protein expression level in RVLM during each phase of experimental brain stem death, was used for statistical analysis. One-way or two-way anova with repeated measures was used, as appropriate, to assess group means. This was followed by the Scheffé's multiple-range test for post hoc assessment of individual means. P < 0.05 was considered to be statistically significant.
Results
Biphasic cardiovascular responses in Mev intoxication model of brain stem death
Microinjection bilaterally of Mev (10 nmol) into the RVLM of animals pretreated with vehicle elicited progressive hypotension (Figure 1) that became significant 120 min after application, accompanied by insignificant alterations in HR. Concurrent changes in power density of the LF component of SAP signals revealed two distinct phases (Yen et al., 2001; Chan et al., 2005a; Chang et al., 2009a). The pro-life phase I entailed a significantly augmented LF power (Figure 1) that endured for 100 min to reflect sustained central cardiovascular regulatory functions. The pro-death phase II, which lasted the remainder of our 180-min observation period, exhibited further and significant reduction (Figure 1) in LF power to below baseline to reflect failure of the central cardiovascular regulatory machinery that precedes brain stem death (Chan et al., 2005b).
Figure 1.
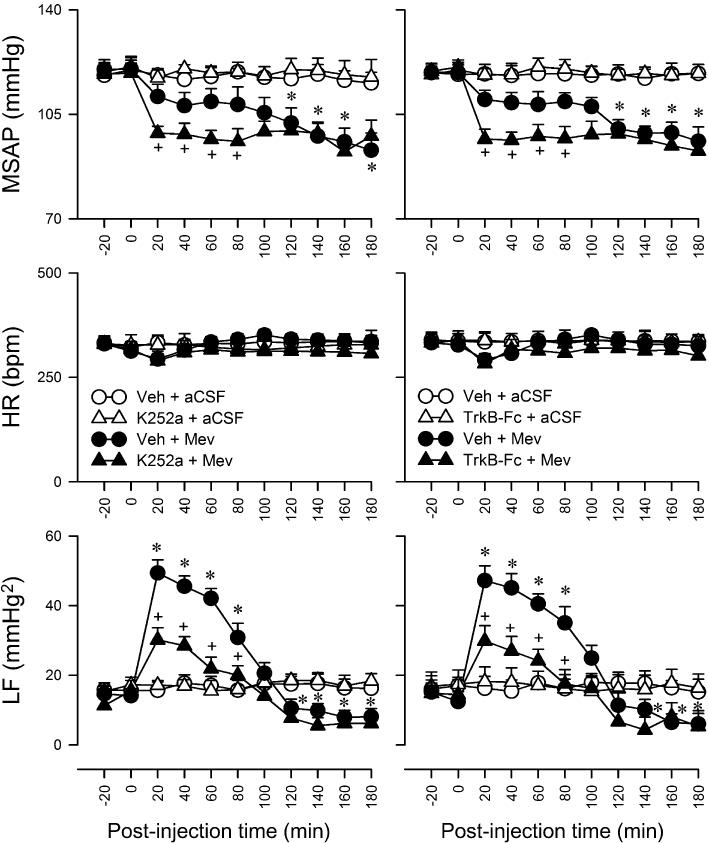
Temporal changes in MSAP, HR or power density of the LF component of SAP signals in rats that were pretreated, by microinjection bilaterally into the RVLM, with K252 (1 pmol) or recombinant mouse TrkB-Fc fusion protein (TrkB-Fc, 5 nmol), 30 min before local application (at time zero) of aCSF or Mev (10 nmol) to the bilateral RVLM. Since pretreatment with the solvents (aCSF or 0.2% DMSO) elicited comparable and minimal effects, for clarity, only one set of data labelled as vehicle (Veh) is presented in this and subsequent figures. Values are mean ± SEM (n = 5–7 animals per experimental group). *P < 0.05 versus Veh + aCSF group and +P < 0.05 versus Veh + Mev group at corresponding time-points in the Scheffé's multiple-range test.
Activation of TrkB in the RVLM ameliorates cardiovascular regulatory dysfunction during experimental brain stem death
In our first series of experiments (n = 24 rats), we determined whether a causal relationship exists between activation of TrkB in RVLM and central cardiovascular regulation during experimental brain stem death. Pretreatment by microinjection bilaterally into the RVLM of a cell-permeable and reversible inhibitor of TrkB, K252a (1 pmol), which prevents TrkB activation (Levine et al., 1995; Katsuki et al., 2009), accelerated the occurrence of significant hypotension and partially reduced the increase in power density of LF component of SAP signals during phase I Mev intoxication (Figure 1), without affecting HR. On the other hand, the hypotension and reduced LF power exhibited during phase II were not significantly affected. Pretreatment with a lower dose of K252a (0.1 pmol; data not shown) or the vehicle was ineffective, as was K252a (1 pmol) in aCSF-pretreated animals. To ascertain the specific involvement of TrkB in the RVLM in ameliorating cardiovascular regulatory dysfunction during experimental brain stem death, we carried out an additional series of experiment (n = 20 rats) using a recombinant mouse TrkB-Fc fusion protein, which sequesters endogenously released TrkB ligands and blocks TrkB receptors (Schneider and Schweiger, 1991; Huang and Reichardt, 2003). Microinjection of TrkB-Fc fusion protein (5 nmol) bilaterally into the RVLM essentially duplicated the results obtained with K252a (Figure 1). Thus, pretreatment with K252a (1 pmol) was employed in subsequent experiments to reduce the number of animals used.
Preferential activation of TrkB in the RVLM during the pro-life phase
We next established the fundamental premise that TrkB in the RVLM is activated preferentially during the pro-life phase of experimental brain stem death (n = 26 rats). Quantification by elisa revealed a significant increase in total Trk protein, TrkB protein and phosphorylated TrkB at Tyr516 in the RVLM during phase I Mev intoxication (Figure 2A). However, the level of these three determinants during phase II was comparable with the vehicle group. Pretreatment with a microinjection of K252a (1 pmol) into the bilateral RVLM significantly blunted the Mev-induced elevation in total Trk, TrkB or pTrkBY516 (Figure 2A). On the other hand, the level of TrkC in the RVLM remained unchanged during both phases of Mev intoxication and was not significantly affected by K252a pretreatment (Figure 2B).
Figure 2.
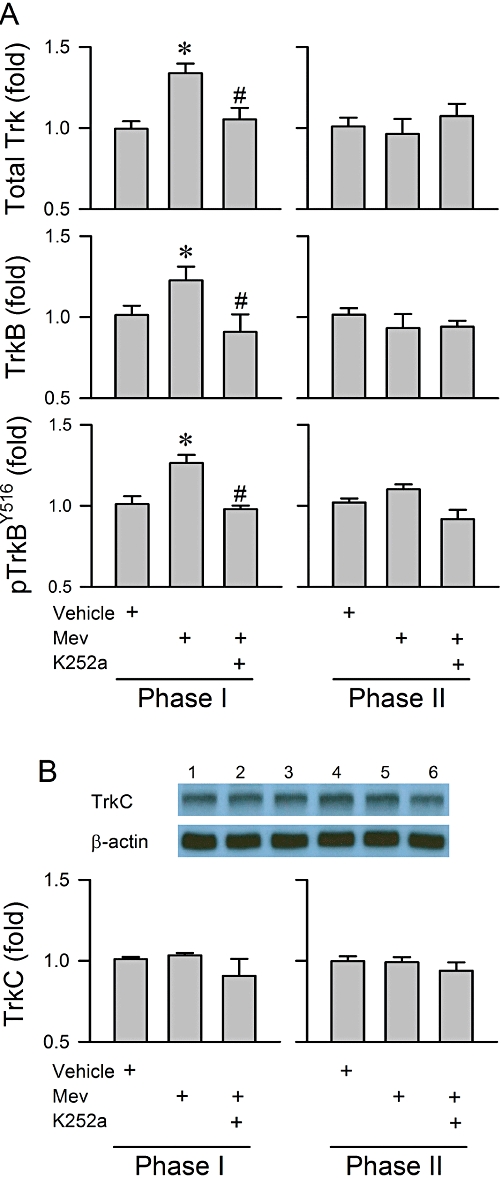
(A) Changes in total Trk, TrkB or phosphorylated TrkB at the Tyr516 site (pTrkBY516) determined by elisa and (B) illustrative gels and a summary of changes in protein expression of TrkC determined by Western blot analysis from the ventrolateral medulla obtained during phases I or II after rats had been administered, by microinjection bilaterally into the RVLM, Mev (10 nmol), given alone or after pretreatment with K252a (1 pmol). In this and subsequent figures, fold changes are determined with reference to the sham control. Values are mean ± SEM of triplicate analyses on samples from four to six animals in each group. *P < 0.05 versus vehicle group and #P < 0.05 versus Mev group in the Scheffé's multiple-range analysis. Note numbers on top of the gels correspond to columns in the data summary.
Preferential activation of Shc by TrkB in the RVLM during the pro-life phase
Our third series (n = 26 rats) of experiments assessed the engagement of Shc, an important adaptor protein that links TrkB activation to its downstream signalling cascades (Nakamura et al., 1996; You et al., 2010). Microinjection bilaterally of Mev (10 nmol) into the RVLM augmented significantly the phosphorylation of Shc at Tyr317 site during phase I, but not at the Tyr239/240 site (Figure 3). More importantly, pretreatment with K252a (1 pmol) antagonized the preferential phosphorylation of pShcY317 (Figure 3). However, the level of phosphorylation of Shc at all three sites in the RVLM during phase II was comparable with the vehicle group, and was not significantly affected by K252a pretreatment (Figure 3).
Figure 3.
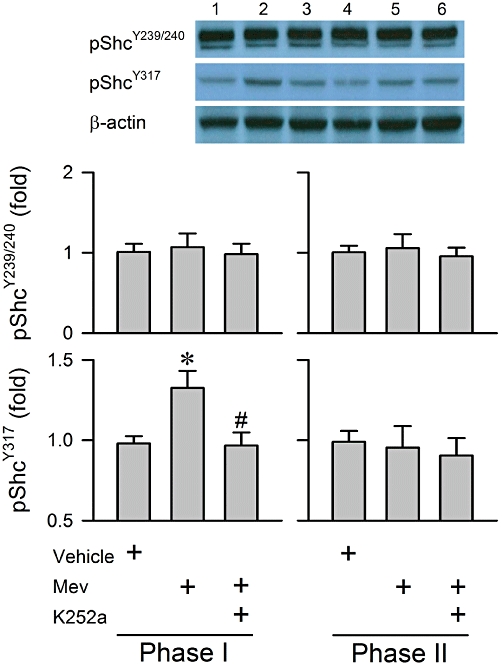
Illustrative gels and a summary of fold changes in protein expression of phosphorylated Shc at the Tyr239/240 (pShcY239/240) and Tyr317 sites (pShcY317) determined by Western blot analysis from the ventrolateral medulla obtained during phases I or II after rats had received a microinjection, bilaterally into the RVLM, of Mev (10 nmol), given alone or in addition to pretreatment with K252a (1 pmol). Values are mean ± SEM of triplicate analyses on samples from four to six animals in each group. *P < 0.05 versus vehicle group and #P < 0.05 versus Mev group in the Scheffé's multiple-range analysis. Note numbers on top of the gels correspond to columns in the data summary.
Preferential activation of Ras/ERK by TrkB in the RVLM during the pro-life phase
Figure 4 (n = 25 rats) shows that the preferential increase in Ras activity or ERK phosphorylation in the RVLM during phase I Mev intoxication was significantly blunted by pretreatment with K252a (1 pmol). Both Ras activity and pERK level returned to baseline during phase II and were not affected by K252a pretreatment (Figure 4). Similarly, K252a did not affect the essentially unaltered ERK levels in the RVLM during both phases of Mev intoxication (Figure 4).
Figure 4.

Fold changes in the activity of Ras and the protein expressions of ERK and phosphorylated ERK at the Thr202/Tyr204 site (pERK) determined by elisa from the ventrolateral medulla obtained during phases I or II after rats had received a microinjection, bilaterally into the RVLM, of Mev (10 nmol), given alone or in addition to pretreatment with K252a (1 pmol). Values are mean ± SEM of triplicate analyses on samples from four to six animals in each group. *P < 0.05 versus vehicle group and #P < 0.05 versus Mev group in the Scheffé's multiple-range analysis.
Preferential activation of NOS I/PKG by TrkB in the RVLM during the pro-life phase
We demonstrated previously (Chan et al., 2005a; Chang et al., 2009a) that up-regulation of the NOS I/PKG signalling pathway, but not the NOS II/peroxynitrite cascade, plays a crucial role in sustaining central cardiovascular regulation during the pro-life phase of experimental brain stem death. Our fifth series (n = 27 rats) of experiments investigated whether the NOS I/PKG pathway in the RVLM is activated by TrkB. Pretreatment with K252a significantly antagonized the preferentially augmented NOS I and PKG protein expression in the RVLM during phase I Mev intoxication (Figure 5). On the other hand, the progressive up-regulation of NOS II and nitrotyrosine, an experimental index of peroxynitrite, in the RVLM during both phases was minimally affected (Figure 5).
Figure 5.
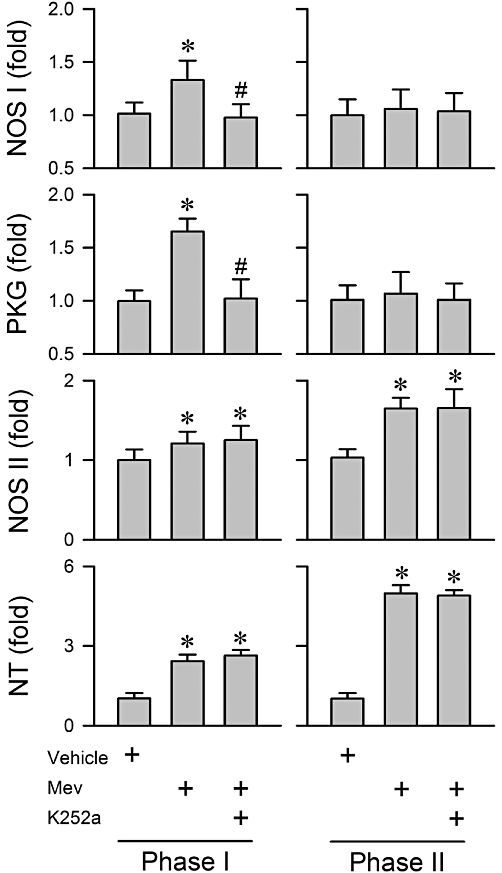
Summary of fold changes in protein expression of NOS I, NOS II or nitrotyrosine (NT; marker for peroxynitrite) or activity of PKG determined by elisa from the ventrolateral medulla obtained during phases I or II after rats had received a microinjection, bilaterally into the RVLM of Mev (10 nmol), given alone or in addition to pretreatment with K252a (1 pmol). Note that NT is presented as % relative to absorbance detected from vehicle control group. Values are mean ± SEM of triplicate analyses on samples from four to six animals in each group. *P < 0.05 versus aCSF group and #P < 0.05 versus Mev group in the Scheffé's multiple-range analysis. ND, below detection limit.
Shc mediates Ras/Raf-1/ERK activation by acting on Grb2/SOS
Phosphorylation of Shc at the Tyr317 site on TrkB activation is crucial to the recruitment and binding of the Grb2/SOS complex to TrkB, and for subsequent activation of the Ras/Raf-1/ERK signalling pathway (Ishihara et al., 1998; Patrussi et al., 2005). In our sixth series (n = 31 rats) of experiments we determined the causal relationship between Shc/Grb2/SOS signalling and Ras/Raf-1/ERK activation in the RVLM during Mev intoxication. The preferential augmentation of Ras activity (Figure 6) or ERK phosphorylation (Figure 7) in the RVLM during phase I was significantly antagonized by pretreatment, by microinjection into the bilateral RVLM, with an anti-pShcY317 antiserum (1:20) that binds specifically to this phosphorylation site to prevent its activation. A similar antagonism was elicited by SH3b-p (25 fmol), which blocks the Grb2/SOS interaction and prevents Ras activation because of its strong affinity for the N-terminal SH3 domain of Grb2 (Su et al., 1996; Upadhyay et al., 2004); or FTA (40 pmol) or manumycin A (40 pmol), which inhibits Ras activation (Amos et al., 2006; Park et al., 2006). Pretreatment with GW5074 (40 pmol), which blocks kinase activity of Raf-1 (Hughes and Brown, 2006; Park et al., 2006), inhibited Mev-induced up-regulation of phosphorylated ERK (Figure 7), but not Ras activity (Figure 6). On the other hand, immunoneutralization of pShcY239/240 in RVLM was ineffective against Mev-induced Ras (Figure 6) or ERK (Figure 7) activation during phase I. Likewise, none of the pretreatments exerted significant effects on the essentially unaltered Ras activity (Figure 6) or ERK phosphorylation (Figure 7) in the RVLM during phase II Mev intoxication. Similar observations were obtained from the minimally affected ERK protein expression (Figure 7).
Figure 6.
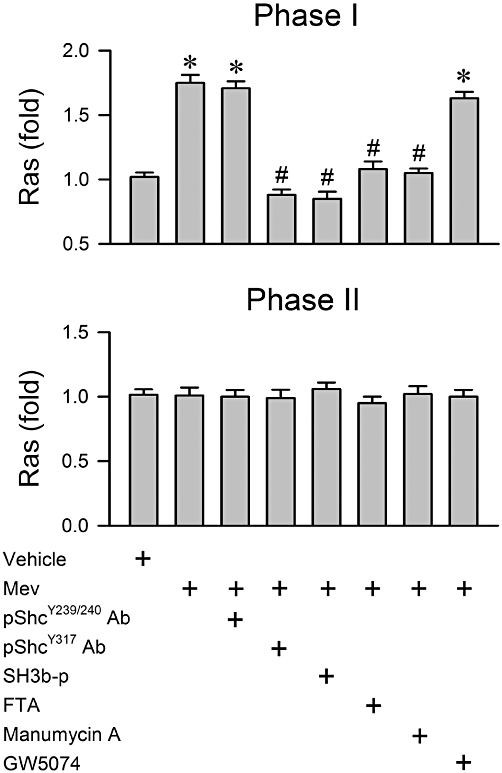
Fold changes in activity of Ras determined by elisa from the ventrolateral medulla obtained during phases I or II after rats had received a microinjection, bilaterally into RVLM, of Mev (10 nmol), given alone or in addition to pretreatment with immunoneutralization of phosphorylated Shc (pShcY239/240 Ab; 1:20 or pShcY317 Ab; 1:20) or pharmacological blockade of Grb2/SOS (SH3b-p; 25 fmol), Ras (FTA; 40 pmol or manumycin A; 40 pmol) or Raf-1 (GW5074; 40 pmol). Since comparable results were obtained from pretreatments with aCSF, 0.2% DMSO or normal goat serum, only one set of data is presented (Vehicle) in this and Figures 7–9 for clarity. Values are mean ± SEM of triplicate analyses on samples from four to six animals in each group. *P < 0.05 versus vehicle group and #P < 0.05 versus Mev group in the Scheffé's multiple-range analysis.
Figure 7.
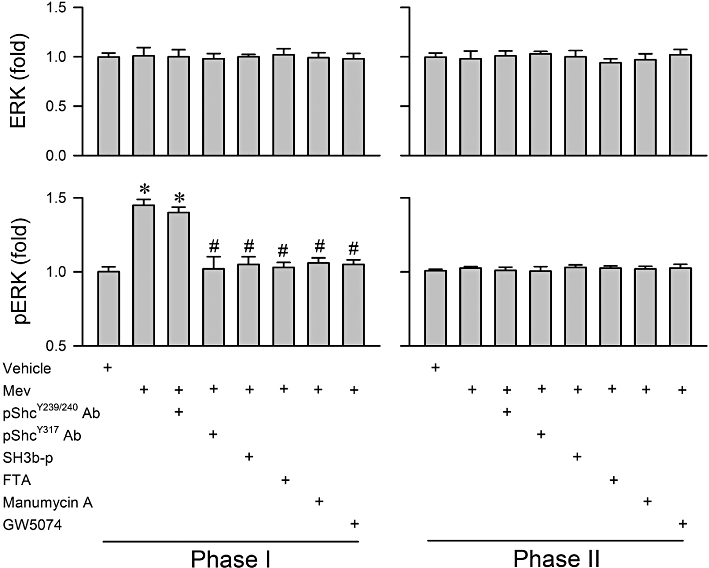
Fold changes in protein expression of ERK and phosphorylated ERK at Thr202/Tyr204 site (pERK) determined by elisa from the ventrolateral medulla obtained during phases I or II after rats had received a microinjection, bilaterally into the RVLM, of Mev (10 nmol), given alone or in addition to pretreatment with immunoneutralization of phosphorylated Shc (pShcY239/240 Ab; 1:20 or pShcY317 Ab; 1:20) or pharmacological blockade of Grb2/SOS (SH3b-p; 25 fmol), Ras (FTA; 40 pmol or Manumycin A; 40 pmol) or Raf-1 (GW5074; 40 pmol). Values are mean ± SEM of triplicate analyses on samples from four to six animals in each group. *P < 0.05 versus vehicle group and #P < 0.05 versus Mev group in the Scheffé's multiple-range analysis.
Activation of Shc and Raf-1 underlies the preferential augmentation of NOS I/PKG in RVLM during the pro-life phase
We next evaluated whether activation by the Shc/Grb2/SOS complex and Ras/Raf-1 cascade in the RVLM leads to the up-regulated NOS I/PKG signalling seen during the pro-life phase of experimental brain stem death (n = 31 rats). Pretreatment with SH3b-p (25 pmol) or GW5074 (40 pmol) significantly blunted the preferentially augmented NOS I or PKG protein expression in RVLM during phase I (Figure 8). However, those pretreatments exerted minimal influence on the progressive increase in NOS II or nitrotyrosine levels during both phases of Mev intoxication, or on the insignificant changes in the expression of NOS I and PKG during phase II.
Figure 8.
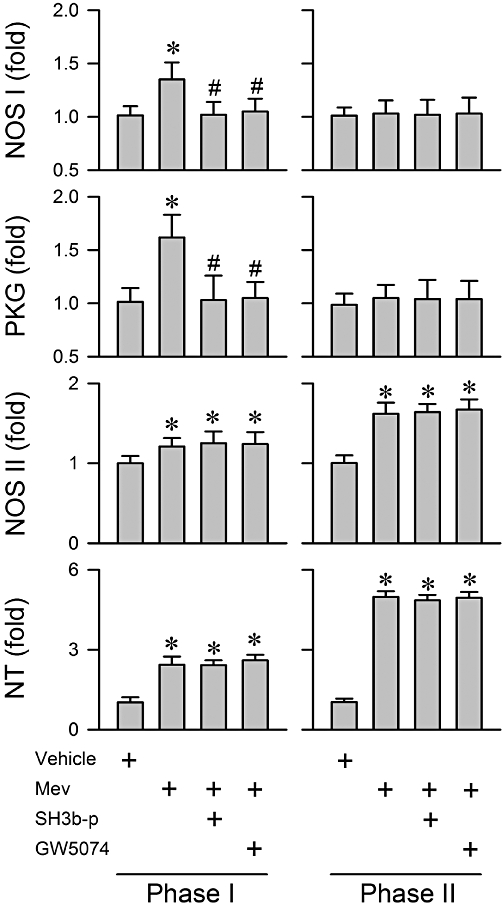
Summary of fold changes in protein expression of NOS I, NOS II and NT, and the activity of PKG determined by elisa from the ventrolateral medulla obtained during phases I or II after rats had received a microinjection, bilaterally into the RVLM, of Mev (10 nmol), given alone or in addition to pretreatment with a pharmacological blocker of Grb2/SOS (SH3b-p; 25 fmol) or Raf-1 (GW5074; 40 pmol). Note that NT is presented as % relative to absorbance detected from vehicle control group. Values are mean ± SEM of triplicate analyses on samples from four to six animals in each group. *P < 0.05 versus vehicle group and #P < 0.05 versus Mev group in the Scheffé's multiple-range analysis.
Activation of Shc/Grb2/SOS, Ras/Raf-1 in the RVLM also ameliorates cardiovascular regulatory dysfunction during Mev intoxication
In our final series (n = 42 rats) of experiments we further established a causal relationship between Shc/Grb2/SOS or Ras/Raf-1 signalling in the RVLM and central cardiovascular regulation during experimental brain stem death. In animals pretreated with anti-pShcY317 (1:20) antiserum, SH3b-p (25 pmol), FTA (40 pmol), manumycin A (40 pmol) or GW5074 (40 pmol), the Mev-induced hypotension during phase I was significantly potentiated, and the increase in power density of the LF component of SAP signals was significantly blunted (Figure 9). In contrast, pretreatment with vehicle or anti-pShcY239/240 antiserum (1:20) affected minimally the Mev-induced cardiovascular responses during both phases (Figure 9). Similarly, pretreatment with a lower dose of anti-pShcY317 antiserum (1:50), SH3b-p (2.5 pmol), manumycin A (4 pmol), FTA (4 pmol) or GW5074 (4 pmol) was ineffective (data not shown).
Figure 9.
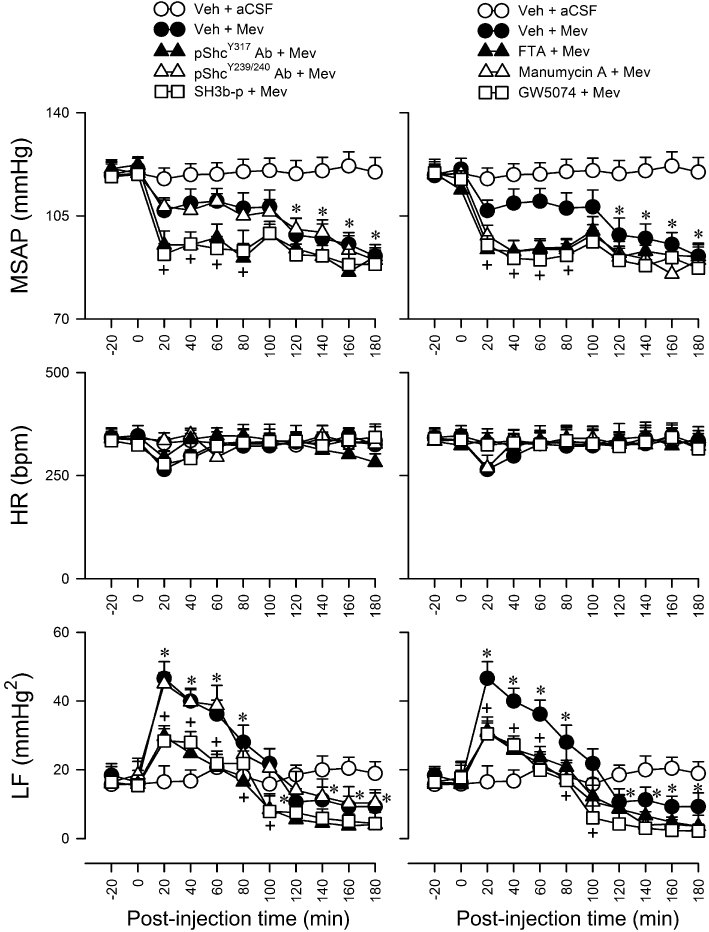
Temporal changes in MSAP, HR or power density of the LF component of SAP signals in rats that pretreated with an immunoneutralizer of phosphorylated Shc (pShcY239/240 Ab; 1:20 or pShcY317 Ab; 1:20), a pharmacological blocker of Grb2/SOS (SH3b-p; 25 fmol), Ras (FTA; 40 pmol or manumycin A; 40 pmol) or Raf-1 (GW5074; 40 pmol), or vehicle, 30 min before local application (at time zero) of aCSF or Mev (10 nmol) to the bilateral RVLM. Values are mean ± SEM, n = 5–7 animals per experimental group. *P < 0.05 versus vehicle + aCSF group, and +P < 0.05 versus vehicle + Mev group at corresponding time-points in the Scheffé's multiple-range test.
Discussion and conclusions
Based on a Mev intoxication model (Chan et al., 2005b), in conjunction with pharmacological blockade or immunoneutralization, the present study provided the first demonstration that activation of TrkB, followed by Shc phosphorylation and formation of the Grb2/SOS complex in the RVLM ameliorates central cardiovascular regulatory dysfunction during experimental brain stem death. We further showed that the latter effect is mediated by the Ras/Raf-1/ERK signalling pathway, which preferentially engages the pro-life NOS I/PKG cascade in the RVLM (Figure 10).
Figure 10.
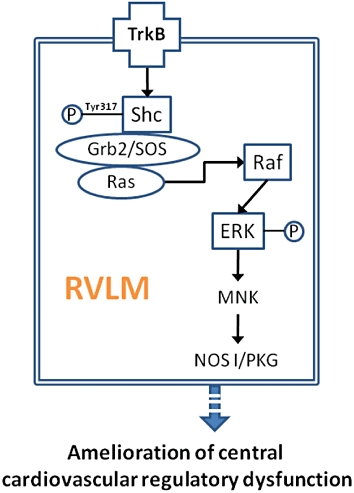
Signalling pathways in the RVLM that link activation of TrkB to amelioration of central cardiovascular regulatory dysfunction via the pro-life NOS I/PKG cascade during experimental brain stem death.
Our findings revealed that preferential activation of TrkB in the RVLM sustains central cardiovascular regulation during the pro-life phase of experimental brain stem death. Because TrkB is the cognate receptor for brain-derived neurotrophic factor (BDNF) (Liang et al., 1998; Liu et al., 2007), our results imply that in addition to its classical neurotrophic effects (Frost, 2001; Tang et al., 2008), BDNF in the RVLM plays a pro-life role during experimental brain stem death. The exacerbation of hypotension and antagonism of the increase in power density of LF component of SAP signals or the elevated levels of total Trk, TrkB or pTrkBY516 in the RVLM during the pro-life phase induced by K252a or TrkB-Fc pretreatment further support this notion.
A novel finding in the present study is the amelioration of central cardiovascular regulatory dysfunction during experimental brain stem death by Shc/Grb2/SOS signalling and its downstream target Ras in the RVLM. Shc is an important adaptor protein that links TrkB to the docking Grb2/SOS complex for the maintenance of cell survival, differentiation and migration (Egan et al., 1993; Nakamura et al., 1996; Patrussi et al., 2005). Ras activity is determined by the guanine nucleotide status and is regulated by guanine nucleotide exchange factors such as SOS (Li et al., 1995; Rao, 1996), which binds to the N-terminal SH3 domain of Grb2. Although both Tyr239/240 and Tyr317 residues are potential sites for Shc phosphorylation, it is TrkB-induced phosphorylation of the Shc Tyr317 residue that is important for its binding to Grb2, which forms a complex with SOS that leads to Ras activation (Sasaoka and Kobayashi, 2000). Our results from systematic pharmacological dissection showed that this repertoire of cellular events indeed takes place in the RVLM during the pro-life phase, leading to amelioration of central cardiovascular regulatory dysfunction during experimental brain stem death.
The Ras/Raf-1/ERK pathway is generally taken to be an important mediator for neuronal survival, differentiation and protection (Marais and Marshall, 1996; Zebisch et al., 2007; McCubrey et al., 2008). The present study further assigned a novel pro-life role to this signalling cascade in the RVLM during experimental brain stem death. Ras activity in the RVLM is significantly elevated in stroke-prone spontaneously hypertensive rats (Kishi et al., 2010), and activation of Ras and Raf-1 in the brain stem underlies angiotensin II-induced hypertension (Yang and Raizada, 1998; Chan et al., 2007c). Activation of TrkB mediates cell survival or protection via the MAPK/ERK signalling (Zhu et al., 2005), and formation of the Grb2/SOS complex is best known for its ability to link TrkB to Ras activation and its downstream ERK1/2 pathway (Rao, 1996). Of interest is that phosphorylation of Shc at Tyr317, which we demonstrated to be crucially involved in sustaining central cardiovascular regulation during experimental brain stem death, selectively activates the MAPK/ERK pathway (Nakamura et al., 1996). In contrast, pShcY239/240, which plays only a minor role in our model of brain stem death, mediates mainly Myc activation (Gotoh et al., 1996; Patrussi et al., 2005). We demonstrated previously (Chan et al., 2010) that activation of the MEK/ERK/MNK signalling pathway in the RVLM plays a preferential pro-life role by sustaining the central cardiovascular regulatory machinery during brain stem death via up-regulation of the NOS I/PKG signalling cascade. Results from the present study further suggest that the Shc/Grb2/SOS/Ras/Raf-1 cascade acts as the interposing cellular signals between TrkB, ERK and NOS I/PKG signalling in this pro-life process in the RVLM.
We are aware of the potential influence of anaesthesia on the circulatory responses evaluated in the present study. In this regard, we demonstrated previously (Yang et al., 1995) that the anaesthetic maintenance scheme used in our study induced minimal depressive action on the central cardiovascular machinery. Our results further indicated that stable MSAP, HR or LF power was manifested across all experimental groups. We also recognize that since the design of our study is based primarily on pharmacological treatments, the specificity of our test agents, particularly K252a, becomes crucial to our conclusion. In this regard, we chose to use a low concentration of K252a, 1 pmol. This is because K252a not only mediates specific inhibition of TrkB activity at low concentrations (IC50, 3 nM) (Tapley et al., 1992) but also inhibits other protein kinases, including MAP kinase (Morotti et al., 2002) at higher concentrations (Ki, 20–500 nM). We further ascertained the specific engagement of TrkB in the RVLM by showing that pretreatment with a recombinant mouse TrkB-Fc fusion protein, which depletes endogenous TrkB ligands and blocks TrkB receptors (Schneider and Schweiger, 1991; Huang and Reichardt, 2003), essentially duplicated the effects of K252a. Similarly, we applied manumycin A at 40 pmol because it is a potent and specific inhibitor of p21ras that not only prevents Ras activation (Sugita et al., 2007) at low doses but also inhibits MAP kinase activation at higher concentrations, 10–30 µM (Nagase et al., 1996). FTA also prevents Ras activation because of its potent and specific inhibition of methylation of farneylated ras proteins (Park et al., 2006). GW5074 is a potent, specific and cell-permeable c-Raf1 kinase inhibitor (Park et al., 2006). SH3b-p is a synthetic peptide that corresponds to amino acids 689–701 of human SH3BP1 and interferes with TrkB and its adaptor Grb2/SOS complex (Su et al., 1996). The anti-pShcY317 or anti-pShcY239/240 antiserum was raised against a short amino acids sequence that contains the corresponding 317 or both 239 and 240 phosphorylation sites of Shc.
We noted that total Trk or TrkB expression in the RVLM was significantly augmented 30 min after application of Mev, concomitant with an enhanced phosphorylation of TrkB. This short latency for up-regulation of Trk is not uncommon, given that a significant elevation in TrkB mRNA, that peaks at 30 min, occurs in the hippocampus 15 min after stress (Shi et al., 2010). Finally, the thrust of the present study is on the pro-life role of the downstream signalling cascades in the RVLM triggered by TrkB during experimental brain stem death, which are based on cardiovascular evaluations. Recent studies from our laboratory (Chang et al., 2009a,b; Dai et al., 2010) indicated that, in response to transient hypoxia in the RVLM caused by respiratory depression, transcriptional up-regulation of haeme oxygenase-1 by hypoxia-inducible factor-1α also plays a pro-life role by sustaining central cardiovascular regulatory function via activation of heat shock protein 70. Since both signalling pathways converge on the NOS I/PKG cascade, we are currently delineating whether cross-talks exist between these two signalling pathways and the role of respiratory functions in this process.
In conclusion, the results of the present study revealed that up-regulation of the Shc/Grb2/SOS complex on activation of TrkB in the RVLM ameliorates central cardiovascular regulatory dysfunction via the Ras/Raf-1/ERK cascade that triggers the pro-life NOS I/PKG signalling in our Mev intoxication model of brain stem death. This information should provide further insights into the deterioration of central cardiovascular regulation during the advancement towards brain stem death and offer novel leads for devising clinical management or developing therapeutic strategies against this fatal eventuality.
Acknowledgments
Supported by research grants NSC98-2321-B-182A-004 and NSC99-2321-B-182A-005 (SHHC), NSC98-2321-B-075B-001 (JYHC) and NSC96-2320-B-182A-016-MY3, NSC98-2321-B-182A-003 and NSC99-2321-B-182A-006 (AYWC) from the National Science Council, Taiwan, Republic of China. The authors acknowledged Jing-Yun Wu and Chi-Ming Liu for their skilful assistance in elisa.
Glossary
Abbreviations
- aCSF
artificial CSF
- BDNF
brain-derived trophic factor
- FTA
farnesylthioacetic acid
- Grb2
growth factor receptor-bound protein 2
- GW5074
3-(3,5-dibromo-4-hydroxybenzylidine)-5-iodo-1,3-dihydro-indol-2-one
- HR
heart rate
- LF
component, low-frequency component
- MEK
MAPK kinase
- Mev
Mevinphos
- MNK
MAPK signal-interacting kinase
- MSAP
mean systemic arterial pressure
- NGS
normal goat serum
- RVLM
rostral ventrolateral medulla
- SAP
systemic arterial pressure
- SOS
Son of Sevenless
- SH3b-p
Src homology 3 binding peptide
- TrkB
tropomyocin receptor kinase B
- TrkC
tropomyocin receptor kinase C
Conflicts of interest
The authors state no conflicts of interests.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Representative photomicrograph of the medulla oblongata stained with hematoxyline and eosin showing the punched area of RVLM. Scale bar = 250 μm. Abbreviations: IO, inferior olive; NA, nucleus ambiguus; py, pyramidal tract; RVLM, rostral ventrolateral medulla, Sp5, spinal trigeminal nucleus.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Amos S, Redpath GT, Polar G, McPheson R, Schiff D, Hussaini IM. Farnesylthiosalicylic acid induces caspase activation and apoptosis in glioblastoma cells. Cell Death Differ. 2006;13:642–651. doi: 10.1038/sj.cdd.4401783. [DOI] [PubMed] [Google Scholar]
- Anonymous. Diagnosis of brain death. Statement issued by the honorary secretary of the Conference of Medical Royal Colleges and their Faculties in the United Kingdom on 11 October 1976. Br Med J. 1976;2:1187–1188. doi: 10.1136/bmj.2.6045.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anonymous. Diagnosis of death. Memorandum issued by the honorary secretary of the Conference of Medical Royal Colleges and their Faculties in the United Kingdom on 15 January 1979. Br Med J. 1979;1:332. doi: 10.1136/bmj.1.6159.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anonymous. Report of the medical consultants on the diagnosis of death to the president's commission for the study of ethical problems in medicine and biomedical and behavioral research. J Am Med Assoc. 1981;246:2184–2186. [PubMed] [Google Scholar]
- Chan JYH, Chan SHH, Li FC, Tsai CY, Cheng HL, Chang AYW. Phasic cardiovascular responses to mevinphos are mediated through differential activation of cGMP/PKG cascade and peroxynitrite via nitric oxide generated in the rat rostral ventrolateral medulla by NOS I and II isoforms. Neuropharmacology. 2005a;48:161–172. doi: 10.1016/j.neuropharm.2004.08.012. [DOI] [PubMed] [Google Scholar]
- Chan JYH, Chang AYW, Chan SHH. New insights on brain stem death: from bedside to bench. Prog Neurobiol. 2005b;77:396–425. doi: 10.1016/j.pneurobio.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Chan JYH, Cheng HL, Chou JL, Li FC, Dai KY, Chan SHH, et al. Heat shock protein 60 or 70 activates nitric-oxide synthase (NOS) I- and inhibits NOS II-associated signaling and depresses the mitochondrial apoptotic cascade during brain stem death. J Biol Chem. 2007a;282:4585–4600. doi: 10.1074/jbc.M603394200. [DOI] [PubMed] [Google Scholar]
- Chan JYH, Wu CH, Tsai CY, Cheng HL, Dai KY, Chan SHH, et al. Transcriptional up-regulation of nitric oxide synthase II by nuclear factor-κB at rostral ventrolateral medulla in a rat mevinphos intoxication model of brain stem death. J Physiol. 2007b;581:1293–1307. doi: 10.1113/jphysiol.2007.130872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SHH, Wang LL, Tseng HL, Chan JYH. Upregulation of AT1 receptor gene on activation of protein kinase Cβ/nicotinamide adenine dinucleotide diphosphate oxidase/ERK1/2/c-fos signaling cascade mediates long-term pressor effect of angiotensin II in rostral ventrolateral medulla. J Hypertens. 2007c;25:1845–1861. doi: 10.1097/HJH.0b013e328217b286. [DOI] [PubMed] [Google Scholar]
- Chan SHH, Sun EY, Chang AYW. Extracellular signal-regulated kinase 1/2 plays a pro-life role in experimental brain stem death via MAPK signal-interacting kinase at rostral ventrolateral medulla. J Biomed Sci. 2010;17:17–25. doi: 10.1186/1423-0127-17-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang AYW, Chan JYH, Cheng HL, Tsai CY, Chan SHH. Hypoxia-inducible factor 1/heme oxygenase 1 cascade as upstream signals in the prolife role of heat shock protein 70 at rostral ventrolateral medulla during experimental brain stem death. Shock. 2009a;32:651–658. doi: 10.1097/SHK.0b013e3181a71027. [DOI] [PubMed] [Google Scholar]
- Chang AYW, Chan JYH, Chuang YC, Chan SHH. Brain stem death as the vital determinant for resumption of spontaneous circulation after cardiac arrest in rats. PLoS ONE. 2009b;4:e7744. doi: 10.1371/journal.pone.0007744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai KY, Chan SHH, Chang AYW. Heme oxygenase-1 plays a pro-life role in experimental brain stem death via nitric oxide synthase I/protein kinase G signaling at rostral ventrolateral medulla. J Biomed Sci. 2010;17:72–83. doi: 10.1186/1423-0127-17-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan SE, Giddings BW, Brooks MW, Buday L, Sizeland AM, Weinberg RA. Association of Sos Ras exchange protein with Grb2 is implicated in tyrosine kinase signal transduction and transformation. Nature. 1993;363:45–51. doi: 10.1038/363045a0. [DOI] [PubMed] [Google Scholar]
- Frost DO. BDNF/trkB signaling in the developmental sculpting of visual connections. Prog Brain Res. 2001;134:35–49. doi: 10.1016/s0079-6123(01)34004-9. [DOI] [PubMed] [Google Scholar]
- Gotoh N, Tojo A, Shibuya M. A novel pathway from phosphorylation of tyrosine residues 239/240 of Shc, contributing to suppress apoptosis by IL-3. EMBO J. 1996;15:6197–6204. [PMC free article] [PubMed] [Google Scholar]
- Haupt WF, Rudolf J. European brain death codes: a comparison of national guidelines. J Neurol. 1999;246:432–437. doi: 10.1007/s004150050378. [DOI] [PubMed] [Google Scholar]
- Hollis ER, II, Jamshidi P, Löw K, Blesch A, Tuszynski MH. Induction of corticospinal regeneration by lentiviral trkB-induced Erk activation. Proc Natl Acad Sci USA. 2009;106:7215–7220. doi: 10.1073/pnas.0810624106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Hughes PJ, Brown G. 1Alpha,25-dihydroxyvitamin D3-mediated stimulation of steroid sulphatase activity in myeloid leukaemic cell lines requires VDRnuc-mediated activation of the RAS/RAF/ERK-MAP kinase signalling pathway. J Cell Biochem. 2006;98:590–617. doi: 10.1002/jcb.20787. [DOI] [PubMed] [Google Scholar]
- Hung TP, Chen ST. Prognosis of deeply comatose patients on ventilators. J Neurol Neurosurg Psychiatry. 1995;58:75–80. doi: 10.1136/jnnp.58.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara H, Sasaoka T, Wada T, Ishiki M, Haruta T, Usui I, et al. Relative involvement of Shc tyrosine 239/240 and tyrosine 317 on insulin induced mitogenic signaling in rat1 fibroblasts expressing insulin receptors. Biochem Biophys Res Commun. 1998;252:139–144. doi: 10.1006/bbrc.1998.9621. [DOI] [PubMed] [Google Scholar]
- Katsuki H, Kurimoto E, Takemori S, Kurauchi Y, Hisatsune A, Isohama Y, et al. Retinoic acid receptor stimulation protects midbrain dopaminergic neurons from inflammatory degeneration via BDNF-mediated signaling. J Neurochem. 2009;110:707–718. doi: 10.1111/j.1471-4159.2009.06171.x. [DOI] [PubMed] [Google Scholar]
- Kishi T, Hirooka Y, Konno S, Ogawa K, Sunagawa K. Angiotensin II type 1 receptor-activated caspase-3 through ras/mitogen-activated protein kinase/extracellular signal-regulated kinase in the rostral ventrolateral medulla is involved in sympathoexcitation in stroke-prone spontaneously hypertensive rats. Hypertension. 2010;55:291–297. doi: 10.1161/HYPERTENSIONAHA.109.138636. [DOI] [PubMed] [Google Scholar]
- Krikov M, Thone-Reineke C, Müller S, Villringer A, Unger T. Candesartan but not ramipril pretreatment improves outcome after stroke and stimulates neurotrophin BNDF/TrkB system in rats. J Hypertens. 2008;26:544–552. doi: 10.1097/HJH.0b013e3282f2dac9. [DOI] [PubMed] [Google Scholar]
- Kuo TBJ, Chan SHH. Continuous, on-line, real-time spectral analysis of systemic arterial pressure signals. Am J Physiol. 1993;264:H2208–H2213. doi: 10.1152/ajpheart.1993.264.6.H2208. [DOI] [PubMed] [Google Scholar]
- Kuo TBJ, Yang CCH, Chan SHH. Selective activation of vasomotor component of SAP spectrum by nucleus reticularis ventrolateralis in rats. Am J Physiol. 1997a;272:H485–H492. doi: 10.1152/ajpheart.1997.272.1.H485. [DOI] [PubMed] [Google Scholar]
- Kuo TBJ, Yien HW, Hseu SS, Yang CCH, Lin YY, Lee LC, et al. Diminished vasomotor component of systemic arterial pressure signals and baroreflex in brain death. Am J Physiol. 1997b;273:H1291–H1298. doi: 10.1152/ajpheart.1997.273.3.H1291. [DOI] [PubMed] [Google Scholar]
- Levine ES, Dreyfus CF, Black IB, Plummer MR. Brain-derived neurotrophic factor rapidly enhances synaptic transmission in hippocampal neurons via postsynaptic tyrosine kinase receptors. Proc Natl Acad Sci USA. 1995;92:8074–8077. doi: 10.1073/pnas.92.17.8074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li BQ, Wang MH, Kung HF, Ronsin C, Breathnach R, Leonard EJ, et al. Macrophage-stimulating protein activates Ras by both activation and translocation of SOS nucleotide exchange factor. Biochem Biophys Res Commun. 1995;216:110–118. doi: 10.1006/bbrc.1995.2598. [DOI] [PubMed] [Google Scholar]
- Li PL, Chao YM, Chan SHH, Chan JYH. Potentiation of baroreceptor reflex response by heat shock protein 70 in nucleus tractus solitarii confers cardiovascular protection during heatstroke. Circulation. 2001;103:2114–2119. doi: 10.1161/01.cir.103.16.2114. [DOI] [PubMed] [Google Scholar]
- Liang FQ, Sohrabji F, Miranda R, Earnest B, Earnest D. Expression of brain-derived neurotrophic factor and its cognate receptor, TrkB, in the rat suprachiasmatic nucleus. Exp Neurol. 1998;151:184–193. doi: 10.1006/exnr.1998.6804. [DOI] [PubMed] [Google Scholar]
- Liu X, Grishanin RN, Tolwani RJ, Rentería RC, Xu B, et al. Brain-derived neurotrophic factor and TrkB modulate visual experience-dependent refinement of neuronal pathways in retina. J Neurosci. 2007;27:7256–7267. doi: 10.1523/JNEUROSCI.0779-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marais R, Marshall CJ. Control of the ERK MAP kinase cascade by Ras and Raf. Cancer Surv. 1996;27:101–125. [PubMed] [Google Scholar]
- McCubrey JA, Steelman LS, Abrams SL, Bertrand FE, Ludwig DE, Bäsecke J, et al. Targeting survival cascades induced by activation of Ras/Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways for effective leukemia therapy. Leukemia. 2008;22:708–722. doi: 10.1038/leu.2008.27. [DOI] [PubMed] [Google Scholar]
- Morotti A, Mila S, Accornero P, Tagliabue E, Ponzetto C. K252a inhibits the oncogenic properties of Met, the HGF receptor. Oncogene. 2002;21:4885–4893. doi: 10.1038/sj.onc.1205622. [DOI] [PubMed] [Google Scholar]
- Nagase T, Kawata S, Tamura S, Matsuda Y, Inui Y, Yamasaki E, et al. Inhibition of cell growth of human hepatoma cell line (Hep G2) by a farnesyl protein transferase inhibitor: a preferential suppression of ras farnesylation. Int J Cancer. 1996;65:620–626. doi: 10.1002/(SICI)1097-0215(19960301)65:5<620::AID-IJC11>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Sanokawa R, Sasaki Y, Ayusawa D, Oishi M, Mori N. N-Shc: a neural-specific adapter molecule that mediates signaling from neurotrophin/Trk to Ras/MAPK pathway. Oncogene. 1996;13:1111–1121. [PubMed] [Google Scholar]
- Pallis C. ABC of Brain Stem Death. London: British Medical Journal Press; 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park PH, Aroor AR, Shukla SD. Role of Ras in ethanol modulation of angiotensin II activated p42/p44 MAP kinase in rat hepatocytes. Life Sci. 2006;79:2357–2363. doi: 10.1016/j.lfs.2006.07.037. [DOI] [PubMed] [Google Scholar]
- Patrussi L, Savino MT, Pellegrini M, Paccani SR, Migliaccio E, Plyte S, et al. Cooperation and selectivity of the two Grb2 binding sites of p52Shc in T-cell antigen receptor signaling to Ras family GTPases and Myc-dependent survival. Oncogene. 2005;24:2218–2228. doi: 10.1038/sj.onc.1208384. [DOI] [PubMed] [Google Scholar]
- Rao GN. Hydrogen peroxide induces complex formation of SHC-Grb2-SOS with receptor tyrosine kinase and activates Ras and extracellular signal-regulated protein kinases group of mitogen-activated protein kinases. Oncogene. 1996;13:713–719. [PubMed] [Google Scholar]
- Sahenk Z, Galloway G, Edwards C, Malik V, Kaspar BK, Eagle A, et al. TrkB and TrkC agonist antibodies improve function, electrophysiologic and pathologic features in Trembler J mice. Exp Neurol. 2010;224:495–506. doi: 10.1016/j.expneurol.2010.05.013. [DOI] [PubMed] [Google Scholar]
- Sasaoka T, Kobayashi M. The functional significance of Shc in insulin signaling as a substrate of the insulin receptor. Endocr J. 2000;47:373–381. doi: 10.1507/endocrj.47.373. [DOI] [PubMed] [Google Scholar]
- Schneider R, Schweiger M. A novel modular mosaic of cell adhesion motifs in the extracellular domains of the neurogenic trk and trkB tyrosine kinase receptors. Oncogene. 1991;6:1807–1811. [PubMed] [Google Scholar]
- Shi SS, Shao SH, Yuan BP, Pan F, Li ZL. Acute stress and chornic stress change brain-derived neurotrophic factor (BDNF) and tyrosine kinase-coupled receptor (TrkB) expression in both young and aged rat hippocampus. Yonsei Med J. 2010;51:661–671. doi: 10.3349/ymj.2010.51.5.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spyer KM. Central nervous mechanisms contributing to cardiovascular control. J Physiol. 1994;474:1–19. doi: 10.1113/jphysiol.1994.sp019997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su J, Yang LT, Sap J. Association between receptor protein-tyrosine phosphatase RPTPalpha and the Grb2 adaptor. Dual Src homology (SH) 2/SH3 domain requirement and functional consequences. J Biol Chem. 1996;271:28086–28096. doi: 10.1074/jbc.271.45.28086. [DOI] [PubMed] [Google Scholar]
- Sugita M, Sugita H, Kaneki M. Farnesyltransferase inhibitor, manumycin A, prevents atherosclerosis development and reduces oxidative stress in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2007;27:1390–1395. doi: 10.1161/ATVBAHA.107.140673. [DOI] [PubMed] [Google Scholar]
- Tang S, Machaalani R, Waters KA. Brain-derived neurotrophic factor (BDNF) and TrkB in the piglet brainstem after post-natal nicotine and intermittent hypercapnic hypoxia. Brain Res. 2008;1232:195–205. doi: 10.1016/j.brainres.2008.07.039. [DOI] [PubMed] [Google Scholar]
- Tapley P, Lamballe F, Barbacid M. K252a is a selective inhibitor of the tyrosine protein kinase activity of the trk family of oncogenes and neurotrophin receptors. Oncogene. 1992;7:371–381. [PubMed] [Google Scholar]
- Upadhyay D, Bundesmann M, Panduri V, Correa-Meyer E, Kamp DW. Fibroblast growth factor-10 attenuates H2O2-induced alveolar epithelial cell DNA damage: role of MAPK activation and DNA repair. Am J Respir Cell Mol Biol. 2004;31:107–113. doi: 10.1165/rcmb.2003-0064OC. [DOI] [PubMed] [Google Scholar]
- Xu L, Zhang Y, Cohen SB, DiPetrillo K. TrkB agonist antibody dose-dependently raises blood pressure in mice with diet-induced obesity. Am J Hypertens. 2010;23:732–736. doi: 10.1038/ajh.2010.49. [DOI] [PubMed] [Google Scholar]
- Yang CH, Shyr MH, Kuo TBJ, Tan PPC, Chan SHH. Effects of propofol on nociceptive response and power spectra of electroencephalographic and systemic arterial pressure signals in the rat: correlation with plasma concentration. J Pharmacol Exp Ther. 1995;275:1568–1574. [PubMed] [Google Scholar]
- Yang H, Raizada MK. MAP kinase-independent signaling in angiotensin II regulation of neuromodulation in SHR neurons. Hypertension. 1998;32:473–481. doi: 10.1161/01.hyp.32.3.473. [DOI] [PubMed] [Google Scholar]
- Yen DHT, Yien HW, Wang LM, Lee CH, Chan SHH. Spectral analysis of systemic arterial pressure and heart rate signals of patients with acute respiratory failure induced by severe organophosphate poisoning. Crit Care Med. 2000;28:2805–2811. doi: 10.1097/00003246-200008000-00021. [DOI] [PubMed] [Google Scholar]
- Yen DHT, Yen JC, Len WB, Wang LM, Lee CH, Chan SHH. Spectral changes in systemic arterial pressure signals during acute mevinphos intoxication in the rat. Shock. 2001;15:35–41. doi: 10.1097/00024382-200115010-00006. [DOI] [PubMed] [Google Scholar]
- Yien HW, Hseu SS, Lee LC, Kuo TBJ, Lee TY, Chan SHH. Spectral analysis of systemic arterial pressure and heart rate signals as a prognostic tool for the prediction of patient outcome in intensive care unit. Crit Care Med. 1997;25:258–266. doi: 10.1097/00003246-199702000-00011. [DOI] [PubMed] [Google Scholar]
- You Y, Li W, Gong Y, Yin B, Qiang B, Yuan J, et al. ShcD interacts with TrkB via its PTB and SH2 domains and regulates BDNF-induced MAPK activation. BMB Rep. 2010;43:485–490. doi: 10.5483/bmbrep.2010.43.7.485. [DOI] [PubMed] [Google Scholar]
- Zebisch A, Czernilofsky AP, Keri G, Smigelskaite J, Sill H, Troppmair J. Signaling through RAS-RAF-MEK-ERK: from basics to bedside. Curr Med Chem. 2007;14:601–623. doi: 10.2174/092986707780059670. [DOI] [PubMed] [Google Scholar]
- Zhu D, Wu X, Strauss KI, Lipsky RH, Qureshi Z, Terhakopian A, et al. N-methyl-D-aspartate and TrkB receptors protect neurons against glutamate excitotoxicity through an extracellular signal-regulated kinase pathway. J Neurosci Res. 2005;80:104–113. doi: 10.1002/jnr.20422. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.