

Abstract
BACKGROUND AND PURPOSE
Inhibitors of phosphodiesterase 5 (PDE5) affect signalling pathways by elevating cGMP, which is a second messenger involved in processes of neuroplasticity. In the present study, the effects of the PDE5 inhibitor, sildenafil, on the pathological features of Alzheimer's disease and on memory-related behaviour were investigated.
EXPERIMENTAL APPROACH
Sildenafil was administered to the Tg2576 transgenic mouse model of Alzheimer's disease and to age-matched negative littermates (controls). Memory function was analysed using the Morris water maze test and fear conditioning tasks. Biochemical analyses were performed in brain lysates from animals treated with saline or with sildenafil.
KEY RESULTS
Treatment of aged Tg2576 animals with sildenafil completely reversed their cognitive impairment. Such changes were accompanied in the hippocampus by a reduction of tau hyperphosphorylation and a decrease in the activity of glycogen synthase kinase 3β (GSK3β) and of cyclin-dependent kinase 5 (CDK5) (p25/p35 ratio). Moreover, sildenafil also increased levels of brain-derived neurotrophic factor (BDNF) and the activity-regulated cytoskeletal-associated protein (Arc) in the hippocampus without any detectable modification of brain amyloid burden.
CONCLUSIONS AND IMPLICATIONS
Sildenafil improved cognitive functions in Tg2576 mice and the effect was not related to changes in the amyloid burden. These data further strengthen the potential of sildenafil as a therapeutic agent for Alzheimer's disease.
Keywords: Alzheimer's disease, p-tau, sildenafil, GSK3β, CDK5, BDNF
Introduction
Phosphodiesterases (PDEs) are enzymes that hydrolyse the cyclic nucleotides cAMP or cGMP, which act as second messengers in intracellular signalling and in processes of neuroplasticity, such as long-term potentiation (Frey et al., 1993; Son et al., 1998). PDE inhibitors affect signalling pathways by elevating cAMP and/or cGMP levels, which may ultimately lead to gene transcription through activation of cAMP response element binding (CREB) (Impey et al., 1996; Lu et al., 1999). CREB-dependent gene expression has been shown to underlie long-term memory formation in several vertebrate and invertebrate species, probably through the formation of new synaptic connections (Tully et al., 2003).
The pathological signs of Alzheimer's disease include (i) the presence of plaques (composed of deposits of amyloid filaments) and neurofibrillary tangles (composed of deposits of hyperphosphorylated tau) surrounded by altered neurite processes and glia; (ii) the loss of synapses; and (iii) a degeneration of the neurons (Selkoe, 2002). One of the earliest manifestations of Alzheimer's disease is the inability of affected individuals to form new memories. Memory impairment appears to significantly predate the death of nerve cells, implying that neuronal dysfunction is responsible for the pathophysiology of early stage Alzheimer's disease. Administration of sildenafil, a selective PDE5 inhibitor, activates the NO/cGMP pathway and significantly increases brain cGMP levels (Hartell, 1996; Prickaerts et al., 2002a,b; Zhang et al., 2002; Puerta et al., 2009). PDE5 inhibitors constitute an effective treatment for erectile dysfunction; however, the presence of PDEs in various regions of the CNS (Loughney et al., 1998; Reyes-Irisarri et al., 2005) and the fact that cGMP has been recognized as a second messenger of key neural phenomena such as synaptic plasticity (Haghikia et al., 2007; Paul et al., 2008) substantiate the potential use of PDE inhibitors for neurological disorders. Moreover, animal studies have shown that sildenafil enhances memory in several models (Prickaerts et al., 2002b; Rutten et al., 2005) and attenuates memory impairment induced by NO synthase (NOS) inhibition (Devan et al., 2006; 2007;). Another study shows that sildenafil, dose-dependently, improves performance in the object retrieval task in cynomolgus macaques (Rutten et al., 2008). Cognitive dysfunction by blockade of muscarinic cholinergic receptor (Devan et al., 2004), diabetes or electroconvulsive shock (Patil et al., 2006) is also reversed by sildenafil treatment.
Glycogen synthase kinase 3β (GSK3β) and cyclin-dependent kinase 5 (CDK5) are the most relevant kinases involved in the pathogenic mechanisms of Alzheimer's disease through the phosphorylation at multiple sites of the microtubule-binding protein, tau (Hanger et al., 1992; Mandelkow et al., 1992; Ishiguro et al., 1993; Tomidokoro et al., 2001; Elyaman et al., 2002; Liu et al., 2002; Otth et al., 2002; Tsai et al., 2004). These kinases are associated with neuronal death, the formation of paired helical filaments and neurite retraction (Plattner et al., 2006; Twomey and Mccarthy, 2006; Lopes et al., 2007). Therefore, inhibition of GSK3β and CDK5 activity has been proposed as a plausible therapeutic target for the treatment of Alzheimer's disease (Lau et al., 2002; Koh et al., 2007). Puzzo et al. (2009) have recently demonstrated that sildenafil produces an immediate and long-lasting improvement of synaptic function, CREB phosphorylation and memory in a mouse model of amyloid deposition. This effect is also associated with a long-lasting reduction of β-amyloid (Aβ) levels. In the present study, we investigated whether sildenafil could reverse the memory impairment in an aged mouse model of Alzheimer's disease with a pathology showing both Aβ deposits and hyperphosphorylated tau. Our results demonstrated that sildenafil restored cognitive deficits in this model of Alzheimer's disease, without affecting the Aβ-burden.
Methods
Mouse model and treatment
All animal care and experimental procedures were in accordance with European and Spanish regulations (86/609/CEE; RD1201/2005) and were approved by the Ethical Committee of the University of Navarra (no. 018/05). Behavioural studies were carried out during light time (from 9 am to 2 pm). In this study, Tg2576 Alzheimer's disease transgenic mice, that express the human 695-aa isoform of the amyloid precursor protein (APP) containing the Swedish double mutation (APPswe) [(APP695)Lys670→Asn, Met671→Leu] driven by a hamster prion promoter, were used. The mice were on an inbred C57BL/6/SJL genetic background. In the Tg2576 Alzheimer's disease mouse model, Aβ peptide content in the brain accumulates exponentially between 7 and 12 months of age and mice show impaired memory in the water maze test at the age of 12–15 months (Hsiao et al., 1996; Reed et al., 2010).
Based on effectiveness and toleration, the dose of sildenafil citrate (Viagra;Pfizer, New York, NY, USA) used in patients with erectile dysfunction is 25–100 mg·day−1. The dosage of sildenafil we used in the transgenic Alzheimer's disease mouse model is 15 mg·kg−1·day−1, which is equivalent to 85 mg·day−1 in humans, using the BSA-based dose calculation (Reagan-Shaw et al., 2008).
First of all, the effect of sildenafil on the modulation of memory-associated immediate early genes (IEGs), whose expression may be altered in APP transgenic mice (Dickey et al., 2004), was assessed. For this, 14- to 16-month-old female Tg2576 mice were treated once daily with sildenafil (15 mg·kg−1, i.p.) or saline for 5 days. The last injection was given 30 min before being trained in a hippocampal-dependent memory task and mice were killed 2 h after the training session.
A second group of animals was used to test the long-term effect of sildenafil in cognitive function. In this set of experiments, 14- to 16-month-old female Tg2576 mice and age-matched negative littermates (controls) were treated once daily with sildenafil (15 mg·kg−1, i.p.) or saline for 5 weeks.
Morris water maze test
The Morris water maze (MWM) test was used to evaluate spatial memory function in response to treatment with sildenafil, as previously described (Hsiao et al., 1996; Ricobaraza et al., 2009; Reed et al., 2010). After treatments, groups of animals underwent spatial reference learning and memory testing in the MWM. The water maze was a circular pool (diameter 1.2 m) filled with water maintained at 20°C and made opaque by the addition of non-toxic white paint. Mice were trained for three consecutive days (8 trials per day) swimming to a raised platform (visible-platform). No distal visible cues were present during this phase. The same platform location was used for all visible-platform sessions and was changed for the hidden-platform training (submerged 1 cm beneath the surface) conducted over 8 consecutive days (4 trials per day) with all visible distal cues present in this phase. In both visible- and hidden-platform versions, mice were placed pseudo-randomly in selected locations, facing towards the wall of the pool to eliminate the potentially confounding contribution of extramaze spatial cues. Each trial was terminated when the mouse reached the platform or after 60 s, which ever came first. To test the retention, three probe trials were performed at the beginning of 4th, 7th and the last day of the test (day 9). In the probe trials the platform was removed from the pool, and the percentage of time spent in the quadrant where the platform was previously set was recorded. All trials were monitored by a camera above the centre of the pool connected to a SMART-LD program (Panlab S.L., Barcelona, Spain) for subsequent analysis of escape latencies, swimming speed, path length and per cent time spent in each quadrant of the pool during probe trials. All experimental procedures were performed without knowledge of the treatments of the groups.
Fear conditioning test
After running the MWM, all the animals were trained in the fear conditioning test. The conditioning procedure was carried out in a StartFear system (Panlab S.L.) that allows recording and analysis of the signal generated by the animal's movement through a high sensitivity Weight Transducer system. The analogue signal is transmitted to the FREEZING and STARTLE software modules through the load cell unit for recording purposes and later analysis, in terms of activity/immobility. The conditioning box is housed inside a soundproof chamber, which minimized external stimulation during the conditioning and retention tests. The box was provided with a house light that supplied dim illumination and with a floor grid through which foot shocks could be administered.
On a training day, the mice were placed in the conditioning chamber for 2 min before the onset of a tone at 2800 Hz, 85 dB (conditioned stimulus, CS), which lasted for 30 s. The last 2 s of the CS was paired with a 0.3 mA foot shock (unconditioned stimulus, US). After the shock and 10 s of resting the same CS-US was delivered three consecutive times. Finally, 30 s after the last pair of CS-US, mice were returned to their home cages. To test the effect of sildenafil in non-transgenic mice, a lighter paradigm of training (1 CS-US pairing, Ricobaraza et al., 2010) was used to avoid an overtraining that may prevent the detection of a possible memory enhancement. Twenty-four hours after the training, the mice were placed again in the conditioning chamber; after 2 min of exposure, the tone starts for a period of 2 min and freezing time was assessed during the 4 min (no differences were found in the freezing behaviour with or without the tone). Freezing behaviour was defined as the lack of movement except for breathing for at least 2 s, and was analysed to give the percentage time freezing during exposure to the chamber. The conditioning apparatus was controlled by the experimenter with specific software (Packwin, Panlab S.L.) running on a PC computer.
Twenty-four hours after the fear conditioning test, the animals were killed and the brains removed for biochemical studies. One hemi brain was post-fixed in 4% paraformaldehyde, (PFA) followed by immersion in 2% PFA (24 h) and cytoprotected in 30% sucrose solution in phosphate buffer overnight at 4°C. Microtome sections (30µm thick) were cut coronally, collected free floating and stored in 30% ethylene glycol, 30% glycerol and 0.1 M phosphate buffer at −20°C until processed. The cortex and hippocampus from the other hemi brain were dissected, homogenized and processed as described below for subsequent Western blot.
Determination of Aβ levels
For analysis of total (soluble and insoluble) Aβ42 burden, the frontal cortex was homogenized in a buffer containing 5 M guanidine HCl and 50 mM Tris-HCl, pH 8, protease inhibitors (Complete Protease Inhibitor Cocktail, Roche, Barcelona, Spain) and phosphatase inhibitors (0.1 mM Na3VO4, 1 mM NaF). Aβ42 levels detected with 3D6 antibody, specific for amino acids 1–5 of Aβ and shows no cross-reactivity to the endogenous murine Aβ protein at concentrations up to 1 ng·mL−1 (Johnson-Wood et al., 1997), were measured using a sensitive sandwich elisa kit from Biosource (Camarillo, CA, USA) following the manufacturer's instructions.
Immunohistochemistry
Floating tissue sections comprising hippocampal formation were processed for 6E10 immunostaining following the protocol previously described by Ricobaraza et al. (2009). Briefly, brain sections were incubated in blocking solution (PBS containing 0.5% Triton X-100, 0.1% BSA and 2% normal goat serum) for 2 h at room temperature. After washing, sections were incubated in 70% formic acid for 7 min to expose the epitope. Sections were incubated with the 6E10 antibody (against amino acids 1–17 of Aβ peptide, 1:200, Chemicon) for 24 h at 4°C, washed with PBS and incubated with the secondary antibody (Alexa Fluor 488 goat anti-mouse highly cross-absorbed, Molecular Probes, Eugene, OR, USA, 1:400) for 2 h at room temperature, protected from light. Fluorescence signals were detected with confocal microscope LSM 510 Meta (Carl Zeiss, Germany); objective Plan-neofluar 40×/1.3 oil DIC. Sections were evaluated in Z-series (0.4 µm steps) using LSM 510 Meta software.
Production of protein extracts
Mice were killed by cervical dislocation and hippocampi quickly dissected from the brains. Total tissue homogenates were obtained by homogenizing the hippocampus in a cold lysis buffer with protease inhibitors (0.2 M NaCl, 0.1 M HEPES, 10% glycerol, 200 mM NaF, 2 mM Na4P2O7, 5 mM EDTA, 1 mM EGTA, 2 mM DTT, 0.5 mM PMSF, 1 mM Na3VO4, 1 mM benzamidine, 10 µg·mL−1 leupeptin, 400 U·mL−1 aprotinin), centrifuged at 14 000×g 4°C for 20 min and the supernatant was aliquoted and stored at −80°C. Total protein concentrations were determined using the Bio-Rad Bradford protein assay (Bio-Rad Laboratories).
For APP carboxy-terminal fragments determination, the prefrontal cortex was homogenized in a buffer containing SDS 2%, Tris-HCl (10 mM, pH 7.4), protease inhibitors (Complete Protease Inhibitor Cocktail, Roche) and phosphatase inhibitors (0.1 mM Na3VO4, 1 mM NaF). The homogenates were sonicated for 2 min and centrifuged at 100 000×g for 1 h. Aliquots of the supernatant were frozen at −80°C and protein concentration was determined by the Bradford method using the Bio-Rad protein assay (Bio-Rad, Hercules, CA, USA).
Immunoblotting
Protein samples were mixed with an equal volume of 2 × Laemmli sample buffer, resolved onto SDS-polyacrylamide gels and transferred to nitrocellulose membrane. The membranes were blocked with 5% milk, 0.05% Tween-20 in PBS or TBS followed by overnight incubation with the following primary antibodies: mouse monoclonal anti-p-tau AT8 (1:1000, Pierce Biotechnology, Inc. Rockford, USA), mouse monoclonal anti-tau (1:5000, clone Tau46, Sigma-Aldrich, St. Luis, MO, USA), rabbit polyclonal anti-pGSK3-Ser-9 (1:1000, Cell Signalling Technology, Beverly, MA), rabbit polyclonal anti-pAkt-Ser-473 (1:1000, Cell Signalling Technology), rabbit monoclonal anti-Akt (1:1000, Cell Signalling Technology), mouse polyclonal anti-pGSK3β-Tyr-216 (1:1000, BD Transduction Laboratories, Lexington KT) rabbit polyclonal anti-pCREB (1:500, Upstate-Millipore, Temecula, CA, USA), rabbit polyclonal anti-CREB (1:1000, Cell Signalling Technology), rabbit polyclonal anti-GSK3 (1:1000, Santa Cruz Biotechnology, Santa Cruz, CA), rabbit polyclonal anti-c-fos (1:1000, Santa Cruz Biotechnology), rabbit polyclonal anti-Arc (1:1000, Santa Cruz Biotechnology), rabbit polyclonal anti-brain-derived neurotrophic factor (BDNF) (1:1000, Osenses Pty Ltd, Flagstaff Hill, SA, Australia) rabbit polyclonal anti-p35/p25 (1:1000, Cell Signalling Technology), mouse monoclonal anti-actin (1:2000, Sigma-Aldrich, St. Louis, MO, USA) and mouse monoclonal anti-tubulin (1:10000, Sigma-Aldrich) in the corresponding buffer. Following two washes in PBS/Tween-20 or TBS/Tween-20 and one PBS or TBS alone, immunolabelled protein bands were detected by using HRP-conjugated anti-rabbit or anti-mouse antibody (Santa Cruz; dilution 1:5000) following an enhanced chemiluminescence system (ECL, GE Healthcare Bioscience, Buckinghamshire, UK), and autoradiographic exposure to Hyperfilm ECL (GE Healthcare Bioscience). Quantity One software v.4.6.3 (Bio-Rad) was used for quantification.
For Western blot analysis of APP-derived fragments, aliquots of the protein extracts were mixed with XT sample buffer plus XT reducing agent or Tricine sample buffer (Bio-Rad) and boiled for 5 min. Proteins were separated in a Criterion precast Bis-Tris 4–12% gradient precast gel (Bio-Rad) and transferred to nitrocellulose membranes. The membranes were blocked with 5% milk, 0.05% Tween-20 in TBS followed by overnight incubation with the following primary antibodies: mouse monoclonal 6E10 (amino acids 1–17 of Aβ peptide, 1:1000, Millipore, Billenica, MA), rabbit polyclonal anti-APP C-terminal (amino acids 676–695) (1:2000, Sigma-Aldrich).
Data analysis and statistical procedures
The results were processed for statistical analysis using spss package for Windows, version 15.0 (SPSS, Chicago, IL, USA). Unless otherwise indicated, results are presented as mean ± SEM. In the MWM and fear conditioning test, escape latencies during training were analysed using one-way anova followed by Scheffe's post hoc test. In the MWM, Friedman's test was performed to determine the intra-group comparisons over trials. Biochemical data were analysed using Kruskal–Wallis test followed by Mann–Whitney post hoc test. Student's t-test was used in case two groups were compared.
Results
Sildenafil facilitated the induction of memory-associated genes in Tg2576 mice
The activation of several immediate early genes (IEGs) is crucial for long-term memory formation and amyloid deposition in mice leads to impaired induction of the IEGs expressed by exposure to a novel environment (Dickey et al., 2004). To know whether sildenafil affected the induction of IEGs, 14- to 16-month-old female Tg2576 mice were treated with sildenafil (15 mg·kg−1, i.p.) or saline once a day for 5 days. Animals were given fear conditioning training 30 min after the last injection and they were killed 2 h later (Figure 1A). Gene expression in the hippocampus of transgenic mice that received saline or sildenafil were investigated and compared with a group of aged- and strain-matched non-transgenic mice that underwent the same procedure.
Figure 1.
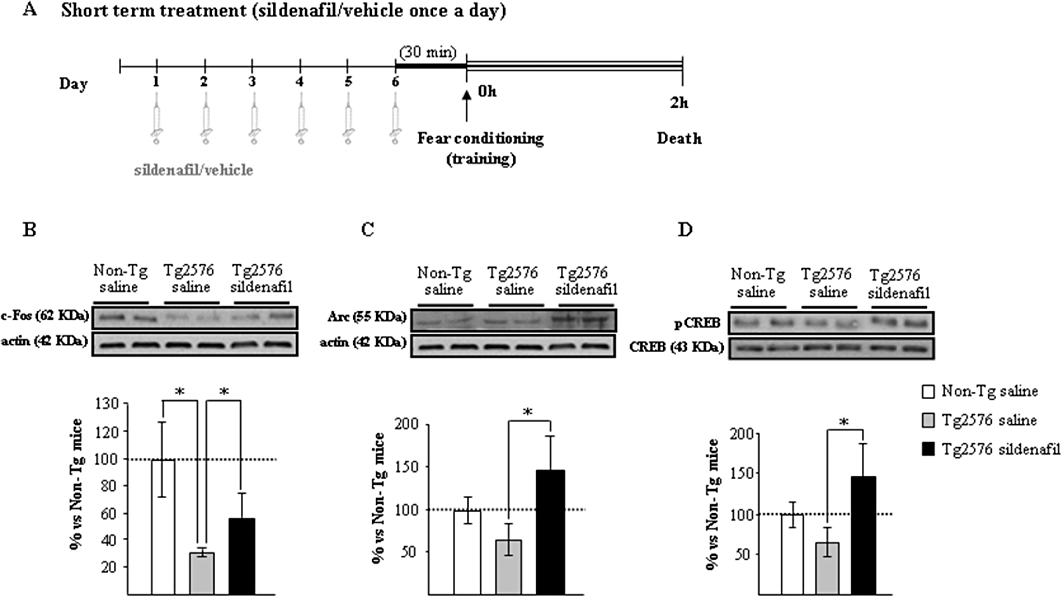
The induction of memory-related genes in the hippocampus of aged Tg2576 mice is facilitated with sildenafil treatment. (A) Scheme showing times of injection, training and death. Administration of sildenafil induces c-fos (B), Arc (C) and pCREB (D) following fear conditioning training. Representative Western blot bands from hippocampal tissues of non-transgenic mice (Non-Tg), transgenic mice treated with saline (Tg2576 saline) or with sildenafil (Tg2576 sildenafil) are shown. The histograms represent the quantification of the immunoreactive bands in the Western blot. Data are expressed as mean percentage (±SEM) of the Non-Tg results (100%). n = 5–6 in each group. *P < 0.05, significantly different from Tg2576 saline mice (Kruskal–Wallis followed by Mann–Whitney post hoc test).
The genes for the synaptic activity-dependent proteins, Arc and c-fos, are induced specifically in neurons engaged in memory-encoding processes (Guzowski et al., 1999). Tg2576 mice had a marked reduction in c-fos expression (58 ± 4% reduction; P < 0.05), which was partially reversed by sildenafil treatment (Figure 1B). Moreover, a significant increase (more than twofold) in Arc expression was observed in the sildenafil-treated group compared with Tg2576 saline-treated mice; the level of Arc was similar in wild-type and saline-treated Tg2576 mice (Figure 1C). An important mediator of transcriptional changes associated to memory is the transcription factor CREB protein (Dash et al., 1990). In the Tg2576 group receiving sildenafil there was a significant increase in the expression of hippocampal pCREB compared with that of the transgenic group receiving saline (Figure 1D). Collectively these data indicated that, in the hippocampus of transgenic mice, the induction of IEGs, related to plasticity and memory consolidation, was facilitated by sildenafil.
Sildenafil restored cognitive function in Tg2576 mice
Cognitive impairment in Tg2576 animals starts at the age of 12 months in the MWM, the memory retention measured in the probe trials being more affected (Hsiao et al., 1996; Westerman et al., 2002; Reed et al., 2010). Studies were performed comparing heterozygous transgenic Tg2576 females with age- and strain-matched transgenic negative littermates (controls) that were treated once daily with sildenafil (15 mg·kg−1, i.p.) or saline for 5 weeks. No significant differences were found among the groups during the training phase of the test (visible-platform, Figure 2B), but there were significant differences in the spatial learning during the hidden-platform between groups [F(2, 238)= 25.35; P < 0.001] (Figure 2C). Tg2576 mice treated with saline were impaired in their performance in this test (days 2–8) compared with age-matched non-transgenic mice (sildenafil- and saline-treated groups) or Tg2576 mice treated with sildenafil.
Figure 2.
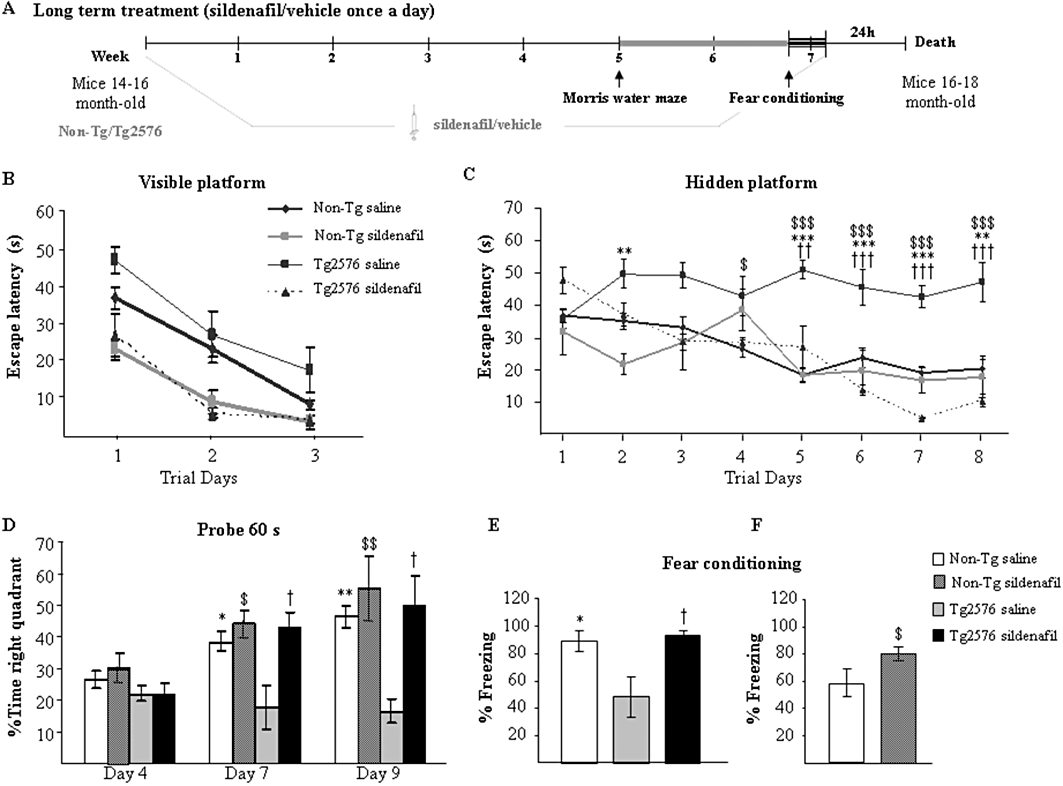
Chronic treatment with sildenafil reverses the learning deficit in aged Tg2576 mice. (A) Scheme showing times of injection, training and death. Escape latency of the visible- (B) and hidden-platform (C) in the MWM test for the non-transgenic mice and transgenic mice treated with saline (Non-Tg saline; Tg2576 saline) or sildenafil (Non-Tg sildenafil; Tg2576 sildenafil). Results are expressed as mean ± SEM (n = 10–12 in each group). Tg2576 saline mice showed significantly longer escape latencies in the hidden-platform training thatn the Non-Tg saline group (**P < 0.01, ***P < 0.001, anova with Scheffe's post hoc test), Non-Tg sildenafil ($P < 0.01, $$$P < 0.001, anova with Scheffe's post hoc test) and to Tg2576 sildenafil-treated group (††P < 0.01, †††P < 0.001, anova with Scheffe's post hoc test). (D) Percentage of time spent searching for the target quadrant of the probe test. Results are expressed as mean ± SEM n = 10–12 in each group. Tg2576 saline mice performed significantly worse than Non-Tg saline (*P < 0.05, **P < 0.01, anova with Scheffe's post hoc test), Non-Tg sildenafil ($P < 0.05, $$P < 0.01, anova with Scheffe's post hoc test) and Tg2576 sildenafil (†P < 0.05, anova with Scheffe's post hoc test) in the probe trial. (E) Tg2576 saline mice exhibited significantly less freezing than the Non-Tg control mice (*P < 0.05, anova with Scheffe's post hoc tests) and Tg2576 sildenafil mice (†P < 0.05, anova with Scheffe's post hoc tests) in the fear conditioning task. Results are expressed as mean ± SEM n = 10–12 in each group. (F) Aged wild-type (Non-Tg) mice receiving sildenafil for 5 weeks showed a significant enhancement of memory compared with Non-Tg saline-treated mice ($P < 0.05, Student's t-test). Results are expressed as mean ± SEM n = 10–12 in each group.
Intra-group comparisons were analysed by the latencies in the hidden-platform training over trials using the Friedman's repeated measure non-parametric test. The mean latencies (time spent) to reach the platform decreased over the training sessions for the non-transgenic mice treated with saline (χ2r = 30.50, P < 0.001), sildenafil (χ2r = 28.00, P < 0.001) and for the transgenic sildenafil-treated group (χ2r = 26.40, P < 0.001, Friedman's test). On the contrary, the latencies exhibited by the saline-treated transgenic group did not significantly decrease over trials (χ2r = 6.15, P > 0.05). The results indicate that non-transgenic and sildenafil-treated animals tended to learn correctly the platform location, whereas saline-treated transgenic animals did not. Specifically, intra-group comparisons of escape latencies showed a significant effect of the training for the non-transgenic and the transgenic sildenafil-treated groups. In contrast, transgenic saline-treated mice did not show any significant reduction in their escape latencies from days 2 to 8, compared with the first training day, reflecting their inability to learn the platform location.
As a putative measurement of memory retention mice swam in the pool with the platform removed. On day 4, no significant differences were found among the groups. [F(2, 30)= 2.06, P = 0.10]. One-way anova showed significant differences between groups on days 7 and 9 [F(2, 30)= 8.96, P < 0.01 for day 7; and F(2, 30)= 5.94 P < 0.01, for day 9]. On days 7 and 9 the proportion of time spent in the target quadrant was significantly lower for transgenic mice treated with saline, compared with the non-transgenic mice or the transgenic mice that received sildenafil treatment. Transgenic sildenafil-treated animals spent a proportion of time in the target quadrant that did not differ from that of the age-matched non-transgenic group (Figure 2D). The swim speed did not differ significantly between groups and the distance data exhibited the same pattern as the escape latency data (not shown). These results indicate that sildenafil administered for 5 weeks restores the cognitive function of Tg2576 mice in the MWM test. Sildenafil did not enhance memory in non-transgenic mice in the MWM.
Mice were next given the fear conditioning test (contextual learning), another hippocampus-dependent learning task, where Tg2576 mice are impaired. As shown in Figure 2E, Tg2576 mice that received the PDE5 inhibitor showed a freezing response similar to that of the age-matched non-transgenic mice and much more than saline-treated Tg2576 animals. These data confirm that sildenafil given chronically ameliorated the memory deficits of this Alzheimer's disease mouse model.
The group of age-matched non-transgenic mice treated for 5 weeks with sildenafil or saline were subjected to the milder single CS-US pairing protocol, which allows detection of subtler learning deficits. Freezing responses were significantly enhanced by sildenafil (P < 0.05, Student's t-test; Figure 2F). These results show in non-transgenic animals that sildenafil was able to improve cognition in an associative learning paradigm.
Sildenafil effect did not correlate with a decrease in Aβ burden but with a decrease in tau pathology
The effect of sildenafil on the Aβ pathology in Tg2576 mice was explored. The levels of Aβ42 were determined in the cerebral cortex by sandwich elisa. As shown in Figure 3A, no differences were observed in Aβ42 levels in Tg2576 mice treated with saline compared with sildenafil-treated mice (no Aβ was detected in non-transgenic littermates, data not shown). In support of this finding, no marked differences between saline and sildenafil were observed when brain sections (including hippocampus and frontal cortex) from transgenic mice were labelled with the 6E10 antibody to detect Aβ load (Figure 3C). The levels of the C-terminal fragments (C83 and C99) of the APP, were analysed using Tris/tricine PAGE 16.5% gels for a better resolution. No differences in intensity of any of the fragments were found between the two groups of transgenic mice, suggesting that sildenafil does not affect APP processing (Figure 3B).
Figure 3.
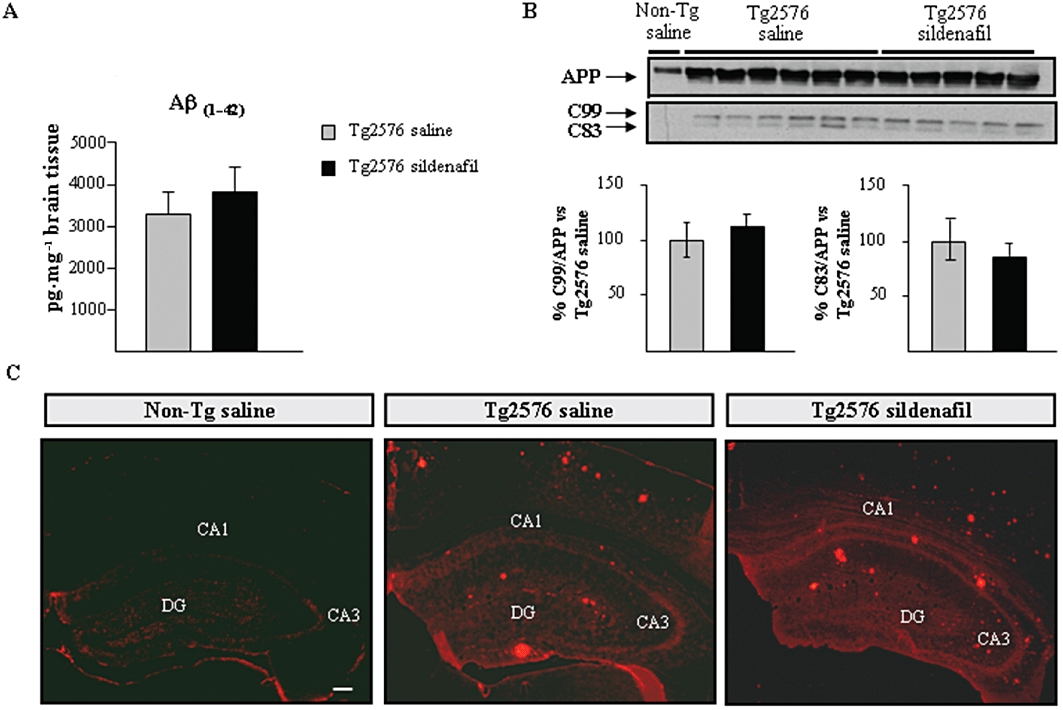
No change is detected in the amyloid levels after chronic treatment with sildenafil in aged Tg2576 mice. (A) Levels of Aβ42 in the transgenic mice by elisa. Results are expressed as mean ± SEM n = 6–8 in each group. (B) Effects of chronic administration of sildenafil on full-length APP, and on APP C-terminal fragments, C99 and C83, in aged transgenic mice. The histogram shows the quantification of the immunochemically reactive bands in the Western blot of APP, C99 and C83 (representative bands are shown). Results are expressed as mean ± SEM n = 5–6 in each group. (C) Extracellular deposits stained with 6E10 antiserum were detected in both, saline- and sildenafil-treated Tg2576 mice. Amyloid deposits were absent in age-matched (Non-Tg) control mice. Representative hippocampal sections of Non-Tg, saline- (Tg2576 saline) and sildenafil- (Tg2576 sildenafil) treated Tg2576 mice are shown. Scale bar = 100 µm.
The levels of phosphorylated tau were analysed in the mice hippocampus using a phospho-specific antibody, AT8, which recognizes aberrantly phosphorylated epitopes on Ser202/Thr205. As shown in Figure 4A, phosphorylated tau levels normalized to total tau (detected by the T46 antibody) were significantly increased (twofold) in the hippocampus of 16-month-old Tg2576 mice receiving saline compared with non-transgenic mice. Interestingly, transgenic mice treated with sildenafil showed levels of tau phosphorylated at the AT8 site similar to those in non-transgenic mice (Figure 4A). As GSK3β, among other kinases, phosphorylates tau at Ser202 (AT8 immunoreactivity) (Hanger et al., 1992; Mandelkow et al., 1992) the levels of the inactive GSK3β form, phosphorylated at Ser9 (pGSK3β-Ser9), and the active GSK3β form, phosphorylated at Tyr216 (pGSK3β-Tyr216), normalized to total GSK3β were measured. The reduced level of pGSK3β-Ser9 in saline-treated transgenic animals was fully reversed by sildenafil treatment (P < 0.05, Figure 4B). In contrast, pGSK3β-Tyr-216 levels were increased in Tg2576 saline mice and were normalized and became more similar to those found in non-transgenic animals after the treatment with sildenafil (Figure 4C). No changes were found in total GSK3β normalized to actin (data not shown). The expression of phosphorylated Akt in transgenic mice that received sildenafil was significantly different from that of the saline-treated Tg2576 mice, which showed a significant reduction in pAkt levels (normalized to total Akt) compared with non-transgenic mice (P < 0.05; Figure 4D).
Figure 4.
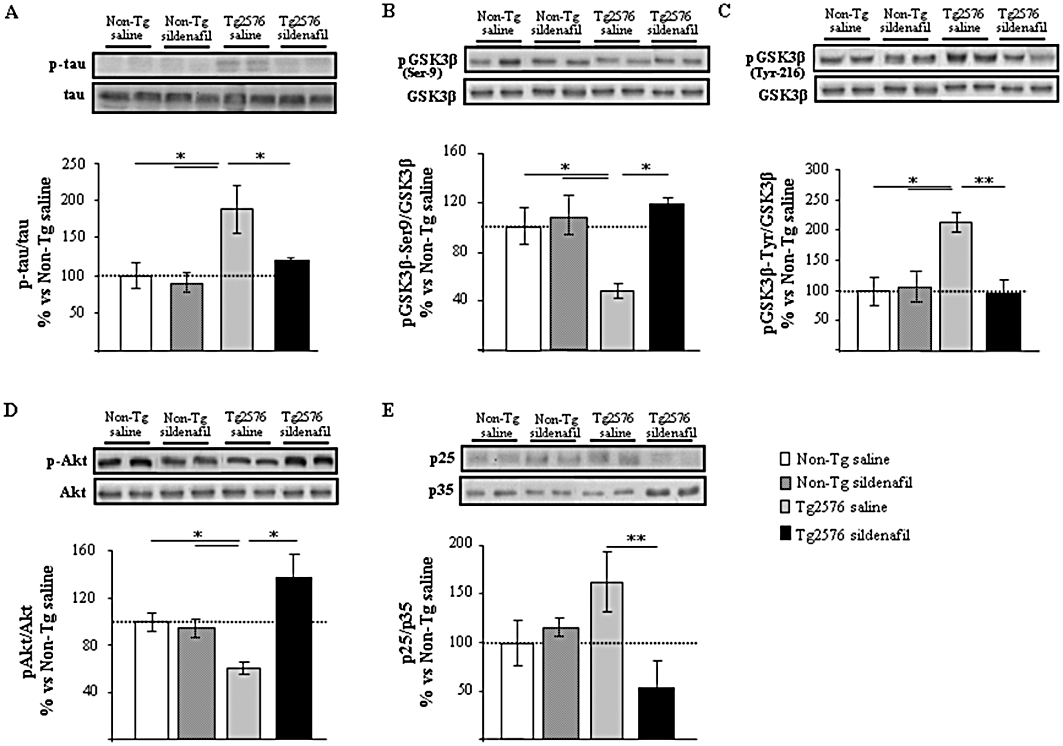
Sildenafil regulates tau phosphorylation through GSK3β and CDK5 in Tg2576 transgenic mice. Chronic treatment with sildenafil modulates phosphorylated tau (p-tau; A), pGSK3β-Ser9 (B), pGSK3β-Tyr216 (C), pAkt (D) and p25/35 (E) expression levels in aged transgenic Tg2576 mice hippocampus but not in the non-transgenic (Non-Tg) group. Representative Western blot bands from hippocampal tissues of Non-Tg mice receiving saline (Non-Tg saline) or sildenafil (Non-Tg sildenafil) and transgenic mice receiving saline (Tg2576 saline) or sildenafil (Tg2576 sildenafil) are shown. The histograms represent the quantification of the immunochemically reactive bands in the Western blot. Data are expressed as mean percentage (±SEM) of the results from the Non-Tg mice receiving saline (Non-Tg saline, 100%). n = 5–6 in each group. *P < 0.05; **P < 0.01, significantly different from Tg2576 saline mice (Kruskal–Wallis followed by Mann–Whitney post hoc test).
The kinase CDK5 is another kinase involved in tau phosphorylation in Alzheimer's disease and contributes to phosphorylation of tau on Ser-202, Thr-205, Ser-235 and Ser-404 (Alvarez et al., 2001; Otth et al., 2002). Several authors have shown that cleavage of p35 to p25 activates CDK5 and it is also known that the proteolytic product p25 concentrates in patients with Alzheimer's disease (Patrick et al., 1999; Ahlijanian et al., 2000). In this context, the levels of CDK5 activator, p25, referred to those of p35 were analysed, showing a significant decrease (P < 0.01) in p25/p35 expression in the protein extracts derived from Tg2576 mice receiving sildenafil compared with transgenic saline-treated mice (Figure 4E). These data suggest that Akt/GSK3 and/or p25/CDK5 pathways contribute to the modulation by sildenafil of tau phosphorylation in Tg2576 mice. Western blot analysis using protein extracts obtained from non-transgenic mice that received sildenafil treatment did not reflect significant differences in the regulation of the Akt/GSK3 and p25/CDK5 pathways compared with the non-transgenic saline group (Figure 4).
Sildenafil effect correlated with increases in BDNF and Arc expression
It has been suggested that the cGMP/PKG pathway also contributes to the phosphorylation of CREB and can be responsible for late phases of the memory consolidation processes. Prickaerts et al. (2002b) suggested that the cGMP/PKG/CREB pathway induces synthesis of proteins essential for memory consolidation.
As inhibition of PDE5 causes an increase in cGMP levels, to test if the cGMP/PKG/CREB pathway is involved in the effects of sildenafil on memory, the expression of hippocampal pCREB, that of its downstream target molecule, BDNF and that of Arc were assessed in protein hippocampal extracts. pCREB was not significantly induced in Tg2576 sildenafil-treated mice (Figure 5A). In contrast, the low BDNF (mature, 13.5 KDa band) expression level found in the Tg2576 saline-treated animals compared with non-transgenic group was partially (and significantly, P < 0.05) restored after sildenafil treatment (Figure 5B). Interestingly, in the group of non-transgenic mice receiving sildenafil, the levels of hippocampal mature BDNF were significantly increased compared with those found in the group of non-transgenic saline-treated mice (P < 0.001, Figure 5B). The level of Arc protein was significantly increased (P < 0.05) in the Tg2576 sildenafil-treated group compared with the transgenic group receiving saline (Figure 5C). Treated animals displayed a marked expression of this protein, which was higher (1.8-fold) than in non-transgenic animals.
Figure 5.
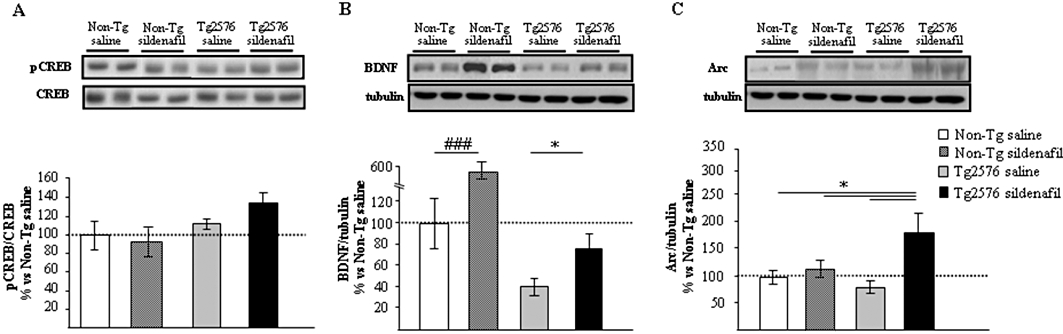
Regulation of gene expression with sildenafil in mouse hippocampus. Western blot analysis of pCREB (A), mature BDNF (B) and Arc (C) in the hippocampus of aged non-transgenic (Non-Tg) and transgenic (Tg2576) mice treated with saline or sildenafil. Representative Western blot bands from hippocampal tissues of non-transgenic mice (Non-Tg), and transgenic mice (Tg2576) receiving saline (Non-Tg saline; Tg2576 saline) or sildenafil (Non-Tg sildenafil; Tg2576 sildenafil) are shown. The histograms represent the quantification of the immunoreactive bands in the Western blot. Data are expressed as mean percentage (±SEM) of the results from versus Non-Tg mice receiving saline (Non-Tg saline, 100%). n = 5–6 in each group. *P < 0.05, significantly different from Tg2576 sildenafil mice; ###P < 0.001, significantly different from Non-Tg saline mice (Kruskal–Wallis followed by Mann–Whitney post hoc test).
Compared with the saline-treated group, no significant differences in pCREB (Figure 5A) and Arc (Figure 5C) expression levels were detected in non-transgenic mice treated with sildenafil. These data suggest that the regulation in the expression of BDNF and Arc in the hippocampus of transgenic animals may be one of the molecular and cellular mechanisms underlying the enhancement of cognition by sildenafil.
Discussion and conclusions
The main finding in this study is the reversal of memory deficits in the Tg2576 mouse model of AD using sildenafil, an effect that appeared to be unrelated to a reduction in Aβ burden but related to the modulation of the pAkt/GSK3β/p-tau and p25/CDK5 pathways (Figure 6).
Figure 6.
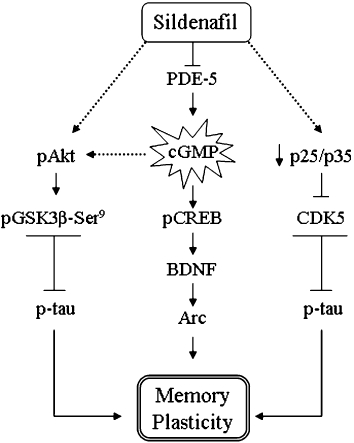
Diagram of signalling pathways indicating the possible mechanism by which sildenafil reverses the memory deficits in aged transgenic Tg2576 mice. Sildenafil increases cGMP formation by inhibiting PDE5, which can activate the pCREB pathway and its downstream gene target BDNF, which consequently mediates Arc induction. The activation of the pAkt pathway by sildenafil, and the inactivation of p25/CDK5 could have contributed to the overall effects in the memory reinstatement by decreasing phosphorylated tau in the Tg2576 mice hippocampus.
Amyloid accumulation suppresses induction of genes critical for memory consolidation in APP transgenic mice (Dickey et al., 2004). The lack of induction of IEGs has the predicted outcome of preventing memory consolidation (Josselyn et al., 2001; Han et al., 2007). Training in sildenafil-treated animals (exposure to a fear conditioning training 2 h prior to being killed) enhanced the expression of the memory-related genes, c-fos, Arc and pCREB (Figure 1) in the hippocampus of transgenic mice. The increase in Arc and pCREB expression following training is critically involved in the early neural events responsible for the acquisition and for the late phase of memory consolidation (Bernabeu et al., 1997; Guzowski, 2002). Our results suggest that sildenafil may enhance memory by extending the learning-induced Arc and pCREB interval. Although other authors have described that Aβ induces a dysregulation in Arc (Dickey et al., 2003; 2004;) and affects pCREB activation (Vitolo et al., 2002; Puzzo et al., 2009), we did not find any significant difference between transgenic mice and non-transgenic mice, in the expression/activation of these genes after training. In contrast, c-fos induced by training was impaired in transgenic mice and partially restored by sildenafil. A reduction in c-fos expression in parallel with defective memory retention in the passive avoidance test was also detected in the brain of the transgenic mouse line Tg-APP (Sw, V717F)/B6 at 13–15 months (Lee et al., 2004). Stanciu et al. (2001) have suggested that c-fos production in the hippocampus accompanies the memory consolidation of context-dependent fear conditioning. Altogether, the effect of sildenafil in the induction of c-fos, Arc and pCREB may contribute to synaptic events necessary for formation of new memories and may underlie the ability to learn, as observed in sildenafil-treated transgenic animals (Figure 2B).
The results of the 5 week treatment showed that sildenafil restored cognitive impairment in aged Tg2576 mice in the MWM and in the fear conditioning tasks. A similar effect was previously described in APP/PS1 mice treated with sildenafil. Interestingly, in non-transgenic mice, a sildenafil-triggered enhancement in contextual memory was observed in the fear conditioning test. It is noteworthy that whereas the sildenafil effect on cognitive performance correlated with a decrease of Aβ levels in the cortex of APP/PS1 mice (Puzzo et al., 2009) (plaques were not studied) this was not the case in aged Tg2576 mice. These differences are likely due to the different animal model used, and the timing of sildenafil treatment. In fact, even though the APP/PS1 mice have an accelerated rate of amyloid deposition, Puzzo et al. (2009) treated them before the onset of Aβ accumulation (Dineley et al., 2002), whereas in the present study Tg2576 mice were treated long after the onset of amyloid deposition. Nevertheless, further studies are needed to determine whether sildenafil affects soluble Aβ oligomers and/or intraneuronal Aβ. Our results point to a lack of correlation between Aβ deposits and memory function, and importantly, prove that sildenafil reinstates cognitive function in animals that display a high amyloid plaque burden that resists sildenafil treatment. Other studies have also failed in finding a correlation between a decrease in deposited forms of Aβ (such as plaques) and memory improvement in Alzheimer's disease models (Dodart et al., 2002; Gong et al., 2004; Malm et al., 2007; Ricobaraza et al., 2009). Moreover, amyloid plaques in humans do not necessarily correlate with major cognitive deficits (Price and Morris, 1999). This lack of correlation is indirectly supported by reports of negligible therapeutic benefit in Alzheimer's disease patients of Aβ42-lowering agents such as tarenflurbil (Green et al., 2009).
Other pathological features of Alzheimer's disease that correlate robustly with cognitive decline are the paired helical filaments and neurofibrillary tangles of hyperphosphorylated tau aggregates (Giannakopoulos et al., 2003; Thind and Sabbagh, 2007). Although the Tg2576 mice is a model of Alzheimer's disease amyloidosis, the presence of hyperphosphorylated tau epitopes (Ser199, Thr231/Ser235, Ser396 and Ser413) has been consistently reported (Tomidokoro et al., 2001; Otth et al., 2002; Sasaki et al., 2002 and shown here in Figure 4). Aβ amyloidosis activates the tau kinase pathway involving GSK3β (via its phosphorylation at Tyr216) and CDK5 (through calpain proteolysis of p35 to p25) and, subsequently, tau is phosphorylated at sites (Tomidokoro et al., 2001; Otth et al., 2002) that promote its accumulation and deposition.
The functional consequences of targeting the pAkt/GSK3β pathway are of special relevance to attempt the restoration of cognitive deficits associated with Alzheimer's disease pathology (Huang and Klein, 2006; Avila and Hernandez, 2007). The increase induced by sildenafil in hippocampal pAkt and pGSK3β-Ser9 levels, together with a parallel decrease in the active pGSK3β-Tyr216 form, may be instrumental in the decrease in tau phosphorylation observed in sildenafil-treated transgenic mice. On the other hand, CDK5 phosphorylates tau at Ser199 and Ser202 (Tsai et al., 2004) and its activity is deregulated when p35 is proteolytically cleaved by the protease calpain to generate p25 activator (Lee et al., 2000; Plattner et al., 2006). We found that p25/p35 ratio was increased in Tg2576 mice and was reduced by sildenafil treatment. This effect may contribute to the reduction in tau phosphorylation and could be mediated by a reduction in calpain activity, since PDE5 inhibitors reduce calpain activation (Puerta et al., 2010). Therefore, the decrease in the kinase activity of GSK3β and CDK5 due to sildenafil may lead to a decrease in tau phosphorylation (Ser202) in the hippocampus of treated mice, possibly contributing to the reinstatement of cognitive function (Hanger et al., 2009). Interestingly, sildenafil seems to restore the pAkt/GSK3β or CDK5 pathways when they are hyperactivated but not under normal conditions. In fact, these pathways were not affected by sildenafil when non-transgenic mice were analysed.
To stabilize changes in synaptic strength, neurons activate a program of gene expression that results in alterations of their molecular composition and structure. Of particular interest is the examination of signalling pathways shown to be important for the establishment of long-lasting synaptic plasticity, such as the CREB signalling pathway and that of its downstream target, BDNF. The gene for BDNF is a CREB target whose protein product regulates synaptic function (Tao et al., 1998). We found reduced BDNF levels in the hippocampus of Tg2576 saline-treated mice and this reduction was ameliorated by sildenafil treatment. Nagahara's group showed that BDNF delivery ameliorated age-related cognitive impairment in aged primates and mice even when the treatment is initiated after disease onset (Nagahara et al., 2009). These authors suggested that the mechanisms underlying BDNF actions are independent of a direct modulation of amyloid processing and include normalization of gene expression patterns, augmentation of intracellular signalling and enhancement of synaptic marker expression (Nagahara et al., 2009). In agreement with our previous study (Puerta et al., 2010), sildenafil significantly increased hippocampal BDNF levels in non-transgenic mice, which may underlie the enhancement of memory observed in the fear conditioning paradigm (Figure 2F). Thus, we hypothesized that sildenafil could replenish the pool of hippocampal BDNF. By contrast, probably due to the time mice were killed (24 h after the last sildenafil injection), no significant changes were detected in hippocampal pCREB expression levels, as pCREB induction is transient and probably had returned to basal levels when samples were taken.
Arc is required for maintenance of long-term potentiation and for memory consolidation (Lyford et al., 1995; Guzowski and McGaugh, 1997; Steward and Worley, 2001). In relation to the finding that sildenafil enhanced Arc expression only in transgenic animals, we have to take in account that Aβ peptide modulates Arc expression (Lacor et al., 2004). Thus, the presence of Aβ may interfere with sildenafil in co-modulating Arc expression levels. Moreover, activity-dependent Arc protein is involved in neuronal homeostasis to maintain neuronal activity in an optimal dynamic range (Shepherd and Bear, 2011). The scenario in wild-type mice brain is different from that in transgenic mice brain; consequently, the regulation of Arc expression by sildenafil may be different in the two groups of animals.
In conclusion, in the Tg2576 model of AD, sildenafil reversed the marked memory deficits of aged animals by regulating the Akt/GSK3β and p25/CDK5 pathways. The improvement of memory did not result from any decrease of amyloid burden but, likely from an increase in the expression of the synaptic-function regulating proteins BDNF and Arc.
Acknowledgments
This work has been supported by UTE project FIMA, Spain. We thank Maria Espelosin, Esther Gimeno and Susana Ursua for technical support.
Glossary
Abbreviations
- Aβ
β-amyloid
- Arc
activity-regulated cytoskeletal-associated protein
- BDNF
brain-derived neurotrophic factor
- CDK5
cyclin-dependent kinase 5
- CREB
cAMP response element binding
- CS
conditioned stimulus
- GSK3β
glycogen synthase kinase 3β
- IEG
immediate early gene
- MWM
Morris water maze
- US
unconditioned stimulus
Conflict of interest
The authors declare that, except for income received from our primary employer, no financial support or compensation has been received from any individual or corporate entity over the past 3 years for research or professional service and there are no personal financial holdings that could be perceived as constituting a potential conflict of interest.
References
- Ahlijanian MK, Barrezueta NX, Williams RD, Jakowski A, Kowsz KP, Mccarthy S, et al. Hyperphosphorylated tau and neurofilament and cytoskeletal disruptions in mice overexpressing human p25, an activator of cdk5. Proc Natl Acad Sci USA. 2000;97:2910–2915. doi: 10.1073/pnas.040577797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez A, Munoz JP, Maccioni RB. A Cdk5-p35 stable complex is involved in the beta-amyloid-induced deregulation of Cdk5 activity in hippocampal neurons. Exp Cell Res. 2001;264:266–274. doi: 10.1006/excr.2001.5152. [DOI] [PubMed] [Google Scholar]
- Avila J, Hernandez F. GSK-3 inhibitors for Alzheimer's disease. Expert Rev Neurother. 2007;7:1527–1533. doi: 10.1586/14737175.7.11.1527. [DOI] [PubMed] [Google Scholar]
- Bernabeu R, Bevilaqua L, Ardenghi P, Bromberg E, Schmitz P, Bianchin M, et al. Involvement of hippocampal cAMP/cAMP-dependent protein kinase signaling pathways in a late memory consolidation phase of aversively motivated learning in rats. Proc Natl Acad Sci USA. 1997;94:7041–7046. doi: 10.1073/pnas.94.13.7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash PK, Hochner B, Kandel ER. Injection of the cAMP-responsive element into the nucleus of Aplysia sensory neurons blocks long-term facilitation. Nature. 1990;345:718–721. doi: 10.1038/345718a0. [DOI] [PubMed] [Google Scholar]
- Devan BD, Sierra-Mercado D, Jr, Jimenez M, Bowker JL, Duffy KB, Spangler EL, et al. Phosphodiesterase inhibition by sildenafil citrate attenuates the learning impairment induced by blockade of cholinergic muscarinic receptors in rats. Pharmacol Biochem Behav. 2004;79:691–699. doi: 10.1016/j.pbb.2004.09.019. [DOI] [PubMed] [Google Scholar]
- Devan BD, Bowker JL, Duffy KB, Bharati IS, Jimenez M, Sierra-Mercado D, et al. Phosphodiesterase inhibition by sildenafil citrate attenuates a maze learning impairment in rats induced by nitric oxide synthase inhibition. Psychopharmacology (Berl) 2006;183:439–445. doi: 10.1007/s00213-005-0232-z. [DOI] [PubMed] [Google Scholar]
- Devan BD, Pistell PJ, Daffin LW, Jr, Nelson CM, Duffy KB, Bowker JL, et al. Sildenafil citrate attenuates a complex maze impairment induced by intracerebroventricular infusion of the NOS inhibitor Nomega-nitro-L-arginine methyl ester. Eur J Pharmacol. 2007;563:134–140. doi: 10.1016/j.ejphar.2007.02.008. [DOI] [PubMed] [Google Scholar]
- Dickey CA, Loring JF, Montgomery J, Gordon MN, Eastman PS, Morgan D. Selectively reduced expression of synaptic plasticity-related genes in amyloid precursor protein + presenilin-1 transgenic mice. J Neurosci. 2003;23:5219–5526. doi: 10.1523/JNEUROSCI.23-12-05219.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickey CA, Gordon MN, Mason JE, Wilson NJ, Diamond DM, Guzowski JF, et al. Amyloid suppresses induction of genes critical for memory consolidation in APP + PS1 transgenic mice. J Neurochem. 2004;88:434–442. doi: 10.1111/j.1471-4159.2004.02185.x. [DOI] [PubMed] [Google Scholar]
- Dineley KT, Xia X, Bui D, Sweatt JD, Zheng H. Accelerated plaque accumulation, associative learning deficits, and up-regulation of alpha 7 nicotinic receptor protein in transgenic mice co-expressing mutant human presenilin 1 and amyloid precursor proteins. J Biol Chem. 2002;277:22768–22780. doi: 10.1074/jbc.M200164200. [DOI] [PubMed] [Google Scholar]
- Dodart JC, Bales KR, Gannon KS, Greene SJ, Demattos RB, Mathis C, et al. Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer's disease model. Nat Neurosci. 2002;5:452–457. doi: 10.1038/nn842. [DOI] [PubMed] [Google Scholar]
- Elyaman W, Terro F, Wong NS, Hugon J. In vivo activation and nuclear translocation of phosphorylated glycogen synthase kinase-3beta in neuronal apoptosis: links to tau phosphorylation. Eur J Neurosci. 2002;15:651–660. doi: 10.1046/j.1460-9568.2002.01899.x. [DOI] [PubMed] [Google Scholar]
- Frey U, Huang YY, Kandel ER. Effects of cAMP simulate a late stage of LTP in hippocampal CA1 neurons. Science. 1993;260:1661–1664. doi: 10.1126/science.8389057. [DOI] [PubMed] [Google Scholar]
- Giannakopoulos P, Herrmann FR, Bussiere T, Bouras C, Kovari E, Perl DP, et al. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer's disease. Neurology. 2003;60:1495–1500. doi: 10.1212/01.wnl.0000063311.58879.01. [DOI] [PubMed] [Google Scholar]
- Gong B, Vitolo OV, Trinchese F, Liu S, Shelanski M, Arancio O. Persistent improvement in synaptic and cognitive functions in an Alzheimer mouse model after rolipram treatment. J Clin Invest. 2004;114:1624–1634. doi: 10.1172/JCI22831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RC, Schneider LS, Amato DA, Beelen AP, Wilcock G, Swabb EA, et al. Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: a randomized controlled trial. JAMA. 2009;302:2557–2564. doi: 10.1001/jama.2009.1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzowski JF. Insights into immediate-early gene function in hippocampal memory consolidation using antisense oligonucleotide and fluorescent imaging approaches. Hippocampus. 2002;12:86–104. doi: 10.1002/hipo.10010. [DOI] [PubMed] [Google Scholar]
- Guzowski JF, Mcgaugh JL. Antisense oligodeoxynucleotide-mediated disruption of hippocampal cAMP response element binding protein levels impairs consolidation of memory for water maze training. Proc Natl Acad Sci USA. 1997;94:2693–2698. doi: 10.1073/pnas.94.6.2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzowski JF, Mcnaughton BL, Barnes CA, Worley PF. Environment-specific expression of the immediate-early gene Arc in hippocampal neuronal ensembles. Nat Neurosci. 1999;2:1120–1124. doi: 10.1038/16046. [DOI] [PubMed] [Google Scholar]
- Haghikia A, Mergia E, Friebe A, Eysel UT, Koesling D, Mittmann T. Long-term potentiation in the visual cortex requires both nitric oxide receptor guanylyl cyclases. J Neurosci. 2007;27:818–823. doi: 10.1523/JNEUROSCI.4706-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JH, Kushner SA, Yiu AP, Cole CJ, Matynia A, Brown RA, et al. Neuronal competition and selection during memory formation. Science. 2007;316:457–460. doi: 10.1126/science.1139438. [DOI] [PubMed] [Google Scholar]
- Hanger DP, Hughes K, Woodgett JR, Brion JP, Anderton BH. Glycogen synthase kinase-3 induces Alzheimer's disease-like phosphorylation of tau: generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci Lett. 1992;147:58–62. doi: 10.1016/0304-3940(92)90774-2. [DOI] [PubMed] [Google Scholar]
- Hanger DP, Anderton BH, Noble W. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol Med. 2009;15:112–119. doi: 10.1016/j.molmed.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Hartell NA. Inhibition of cGMP breakdown promotes the induction of cerebellar long-term depression. J Neurosci. 1996;16:2881–2890. doi: 10.1523/JNEUROSCI.16-09-02881.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, et al. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Huang HC, Klein PS. Multiple roles for glycogen synthase kinase-3 as a drug target in Alzheimer's disease. Curr Drug Targets. 2006;7:1389–1397. doi: 10.2174/1389450110607011389. [DOI] [PubMed] [Google Scholar]
- Impey S, Mark M, Villacres EC, Poser S, Chavkin C, Storm DR. Induction of CRE-mediated gene expression by stimuli that generate long-lasting LTP in area CA1 of the hippocampus. Neuron. 1996;16:973–982. doi: 10.1016/s0896-6273(00)80120-8. [DOI] [PubMed] [Google Scholar]
- Ishiguro K, Shiratsuchi A, Sato S, Omori A, Arioka M, Kobayashi S, et al. Glycogen synthase kinase 3 beta is identical to tau protein kinase I generating several epitopes of paired helical filaments. FEBS Lett. 1993;325:167–172. doi: 10.1016/0014-5793(93)81066-9. [DOI] [PubMed] [Google Scholar]
- Johnson-Wood K, Lee M, Motter R, Hu K, Gordon G, Barbour R, et al. Amyloid precursor protein processing and A beta42 deposition in a transgenic mouse model of Alzheimer disease. Proc Natl Acad Sci USA. 1997;94:1550–1555. doi: 10.1073/pnas.94.4.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josselyn SA, Shi C, Carlezon WA, Jr, Neve RL, Nestler EJ, Davis M. Long-term memory is facilitated by cAMP response element-binding protein overexpression in the amygdala. J Neurosci. 2001;21:2404–2412. doi: 10.1523/JNEUROSCI.21-07-02404.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh SH, Kim Y, Kim HY, Hwang S, Lee CH, Kim SH. Inhibition of glycogen synthase kinase-3 suppresses the onset of symptoms and disease progression of G93A-SOD1 mouse model of ALS. Exp Neurol. 2007;205:336–346. doi: 10.1016/j.expneurol.2007.03.004. [DOI] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, et al. Synaptic targeting by Alzheimer's-related amyloid beta oligomers. J Neurosci. 2004;24:10191–10200. doi: 10.1523/JNEUROSCI.3432-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau LF, Seymour PA, Sanner MA, Schachter JB. Cdk5 as a drug target for the treatment of Alzheimer's disease. J Mol Neurosci. 2002;19:267–273. doi: 10.1385/JMN:19:3:267. [DOI] [PubMed] [Google Scholar]
- Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature. 2000;405:360–364. doi: 10.1038/35012636. [DOI] [PubMed] [Google Scholar]
- Lee KW, Lee SH, Kim H, Song JS, Yang SD, Paik SG, et al. Progressive cognitive impairment and anxiety induction in the absence of plaque deposition in C57BL/6 inbred mice expressing transgenic amyloid precursor protein. J Neurosci Res. 2004;76:572–580. doi: 10.1002/jnr.20127. [DOI] [PubMed] [Google Scholar]
- Liu F, Iqbal K, Grundke-Iqbal I, Gong CX. Involvement of aberrant glycosylation in phosphorylation of tau by cdk5 and GSK-3beta. FEBS Lett. 2002;530:209–214. doi: 10.1016/s0014-5793(02)03487-7. [DOI] [PubMed] [Google Scholar]
- Lopes JP, Oliveira CR, Agostinho P. Amyloid-Beta and prion peptides: implications for Alzheimer's disease and prion-related encephalopathies. Cell Mol Neurobiol. 2007;27:943–957. doi: 10.1007/s10571-007-9224-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loughney K, Hill TR, Florio VA, Uher L, Rosman GJ, Wolda SL, et al. Isolation and characterization of cDNAs encoding PDE5A, a human cGMP-binding, cGMP-specific 3′,5′-cyclic nucleotide phosphodiesterase. Gene. 1998;216:139–147. doi: 10.1016/s0378-1119(98)00303-5. [DOI] [PubMed] [Google Scholar]
- Lu YF, Kandel ER, Hawkins RD. Nitric oxide signaling contributes to late-phase LTP and CREB phosphorylation in the hippocampus. J Neurosci. 1999;19:10250–10261. doi: 10.1523/JNEUROSCI.19-23-10250.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyford GL, Yamagata K, Kaufmann WE, Barnes CA, Sanders LK, Copeland NG, et al. Arc, a growth factor and activity-regulated gene, encodes a novel cytoskeleton-associated protein that is enriched in neuronal dendrites. Neuron. 1995;14:433–445. doi: 10.1016/0896-6273(95)90299-6. [DOI] [PubMed] [Google Scholar]
- Malm TM, Iivonen H, Goldsteins G, Keksa-Goldsteine V, Ahtoniemi T, Kanninen K, et al. Pyrrolidine dithiocarbamate activates Akt and improves spatial learning in APP/PS1 mice without affecting beta-amyloid burden. J Neurosci. 2007;27:3712–3721. doi: 10.1523/JNEUROSCI.0059-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelkow EM, Drewes G, Biernat J, Gustke N, Van Lint J, Vandenheede JR, et al. Glycogen synthase kinase-3 and the Alzheimer-like state of microtubule-associated protein tau. FEBS Lett. 1992;314:315–321. doi: 10.1016/0014-5793(92)81496-9. [DOI] [PubMed] [Google Scholar]
- Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroeder BE, Shaked GM, et al. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer's disease. Nat Med. 2009;15:331–337. doi: 10.1038/nm.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otth C, Concha I, Arendt T, Stieler J, Schliebs R, Gonzalez-Billault C, et al. AbetaPP induces cdk5-dependent tau hyperphosphorylation in transgenic mice Tg2576. J Alzheimers Dis. 2002;4:417–430. doi: 10.3233/jad-2002-4508. [DOI] [PubMed] [Google Scholar]
- Patil CS, Singh VP, Kulkarni SK. Modulatory effect of sildenafil in diabetes and electroconvulsive shock-induced cognitive dysfunction in rats. Pharmacol Rep. 2006;58:373–380. [PubMed] [Google Scholar]
- Patrick GN, Zukerberg L, Nikolic M, De La Monte S, Dikkes P, Tsai LH. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402:615–622. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- Paul C, Schoberl F, Weinmeister P, Micale V, Wotjak CT, Hofmann F, et al. Signaling through cGMP-dependent protein kinase I in the amygdala is critical for auditory-cued fear memory and long-term potentiation. J Neurosci. 2008;28:14202–14212. doi: 10.1523/JNEUROSCI.2216-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plattner F, Angelo M, Giese KP. The roles of cyclin-dependent kinase 5 and glycogen synthase kinase 3 in tau hyperphosphorylation. J Biol Chem. 2006;281:25457–25465. doi: 10.1074/jbc.M603469200. [DOI] [PubMed] [Google Scholar]
- Price JL, Morris JC. Tangles and plaques in nondemented aging and ‘preclinical’ Alzheimer's disease. Ann Neurol. 1999;45:358–368. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Prickaerts J, De Vente J, Honig W, Steinbusch HW, Blokland A. cGMP, but not cAMP, in rat hippocampus is involved in early stages of object memory consolidation. Eur J Pharmacol. 2002a;436:83–87. doi: 10.1016/s0014-2999(01)01614-4. [DOI] [PubMed] [Google Scholar]
- Prickaerts J, Van Staveren WC, Sik A, Markerink-Van Ittersum M, Niewohner U, Van Der Staay FJ, et al. Effects of two selective phosphodiesterase type 5 inhibitors, sildenafil and vardenafil, on object recognition memory and hippocampal cyclic GMP levels in the rat. Neuroscience. 2002b;113:351–361. doi: 10.1016/s0306-4522(02)00199-9. [DOI] [PubMed] [Google Scholar]
- Puerta E, Hervias I, Goni-Allo B, Lasheras B, Jordan J, Aguirre N. Phosphodiesterase 5 inhibitors prevent 3,4-methylenedioxymethamphetamine-induced 5-HT deficits in the rat. J Neurochem. 2009;108:755–766. doi: 10.1111/j.1471-4159.2008.05825.x. [DOI] [PubMed] [Google Scholar]
- Puerta E, Hervias I, Barros-Minones L, Jordan J, Ricobaraza A, Cuadrado-Tejedor M, et al. Sildenafil protects against 3-nitropropionic acid neurotoxicity through the modulation of calpain, CREB, and BDNF. Neurobiol Dis. 2010;38:237–245. doi: 10.1016/j.nbd.2010.01.013. [DOI] [PubMed] [Google Scholar]
- Puzzo D, Staniszewski A, Deng SX, Privitera L, Leznik E, Liu S, et al. Phosphodiesterase 5 inhibition improves synaptic function, memory, and amyloid-beta load in an Alzheimer's disease mouse model. J Neurosci. 2009;29:8075–8086. doi: 10.1523/JNEUROSCI.0864-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2008;22:659–661. doi: 10.1096/fj.07-9574LSF. [DOI] [PubMed] [Google Scholar]
- Reed MN, Liu P, Kotilinek LA, Ashe KH. Effect size of reference memory deficits in the Morris water maze in Tg2576 mice. Behav Brain Res. 2010;212:115–120. doi: 10.1016/j.bbr.2010.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Irisarri E, Perez-Torres S, Mengod G. Neuronal expression of cAMP-specific phosphodiesterase 7B mRNA in the rat brain. Neuroscience. 2005;132:1173–1185. doi: 10.1016/j.neuroscience.2005.01.050. [DOI] [PubMed] [Google Scholar]
- Ricobaraza A, Cuadrado-Tejedor M, Perez-Mediavilla A, Frechilla D, Del Rio J, Garcia-Osta A. Phenylbutyrate ameliorates cognitive deficit and reduces tau pathology in an Alzheimer's disease mouse model. Neuropsychopharmacology. 2009;34:1721–1732. doi: 10.1038/npp.2008.229. [DOI] [PubMed] [Google Scholar]
- Ricobaraza A, Cuadrado-Tejedor M, Marco S, Perez-Otano I, Garcia-Osta A. Phenylbutyrate rescues dendritic spine loss associated with memory deficits in a mouse model of Alzheimer disease. Hippocampus. 2010 doi: 10.1002/hipo.20883. DOI: 10.1002/hipo.20883 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Rutten K, Vente JD, Sik A, Ittersum MM, Prickaerts J, Blokland A. The selective PDE5 inhibitor, sildenafil, improves object memory in Swiss mice and increases cGMP levels in hippocampal slices. Behav Brain Res. 2005;164:11–16. doi: 10.1016/j.bbr.2005.04.021. [DOI] [PubMed] [Google Scholar]
- Rutten K, Basile JL, Prickaerts J, Blokland A, Vivian JA. Selective PDE inhibitors rolipram and sildenafil improve object retrieval performance in adult cynomolgus macaques. Psychopharmacology. 2008;196:643–648. doi: 10.1007/s00213-007-0999-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki A, Shoji M, Harigaya Y, Kawarabayashi T, Ikeda M, Naito M, et al. Amyloid cored plaques in Tg2576 transgenic mice are characterized by giant plaques, slightly activated microglia, and the lack of paired helical filament-typed, dystrophic neurites. Virchows Arch. 2002;441:358–367. doi: 10.1007/s00428-002-0643-8. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Shepherd JD, Bear MF. New views of Arc, a master regulator of synaptic plasticity. Nat Neurosci. 2011;14:279–284. doi: 10.1038/nn.2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son H, Lu YF, Zhuo M, Arancio O, Kandel ER, Hawkins RD. The specific role of cGMP in hippocampal LTP. Learn Mem. 1998;5:231–245. [PMC free article] [PubMed] [Google Scholar]
- Stanciu M, Radulovic J, Spiess J. Phosphorylated cAMP response element binding protein in the mouse brain after fear conditioning: relationship to Fos production. Brain Res Mol Brain Res. 2001;94:15–24. doi: 10.1016/s0169-328x(01)00174-7. [DOI] [PubMed] [Google Scholar]
- Steward O, Worley PF. Selective targeting of newly synthesized Arc mRNA to active synapses requires NMDA receptor activation. Neuron. 2001;30:227–240. doi: 10.1016/s0896-6273(01)00275-6. [DOI] [PubMed] [Google Scholar]
- Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron. 1998;20:709–726. doi: 10.1016/s0896-6273(00)81010-7. [DOI] [PubMed] [Google Scholar]
- Thind K, Sabbagh MN. Pathological correlates of cognitive decline in Alzheimer's disease. Panminerva Med. 2007;49:191–195. [PubMed] [Google Scholar]
- Tomidokoro Y, Ishiguro K, Harigaya Y, Matsubara E, Ikeda M, Park JM, et al. Abeta amyloidosis induces the initial stage of tau accumulation in APP(Sw) mice. Neurosci Lett. 2001;299:169–172. doi: 10.1016/s0304-3940(00)01767-5. [DOI] [PubMed] [Google Scholar]
- Tsai LH, Lee MS, Cruz J. Cdk5, a therapeutic target for Alzheimer disease? Biochim Biophys Acta. 2004;1697:137–142. doi: 10.1016/j.bbapap.2003.11.019. [DOI] [PubMed] [Google Scholar]
- Tully T, Bourtchouladze R, Scott R, Tallman J. Targeting the CREB pathway for memory enhancers. Nat Rev Drug Discov. 2003;2:267–277. doi: 10.1038/nrd1061. [DOI] [PubMed] [Google Scholar]
- Twomey C, Mccarthy JV. Presenilin-1 is an unprimed glycogen synthase kinase-3beta substrate. FEBS Lett. 2006;580:4015–4020. doi: 10.1016/j.febslet.2006.06.035. [DOI] [PubMed] [Google Scholar]
- Vitolo OV, Sant'Angelo A, Costanzo V, Battaglia F, Arancio O, Shelanski M. Amyloid beta -peptide inhibition of the PKA/CREB pathway and long-term potentiation: reversibility by drugs that enhance cAMP signaling. Proc Natl Acad Sci USA. 2002;99:13217–13221. doi: 10.1073/pnas.172504199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerman MA, Cooper-Blacketer D, Mariash A, Kotilinek L, Kawarabayashi T, Younkin LH, et al. The relationship between Aβeta and memory in the Tg2576 mouse model of Alzheimer's disease. J Neurosci. 2002;1:1858–1867. doi: 10.1523/JNEUROSCI.22-05-01858.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Wang Y, Zhang L, Zhang Z, Tsang W, Lu M, et al. Sildenafil (Viagra) induces neurogenesis and promotes functional recovery after stroke in rats. Stroke. 2002;33:2675–2680. doi: 10.1161/01.str.0000034399.95249.59. [DOI] [PubMed] [Google Scholar]