

Abstract
BACKGROUND AND PURPOSE
Salvianolic acid B (Sal B), a water-soluble antioxidant derived from a Chinese medicinal herb, is known to be effective in the prevention of atherosclerosis. Here, we tested the hypothesis that the anti-atherosclerotic effect of Sal B might be mediated by suppressing maturation of human monocyte-derived dendritic cells (h-monDC).
EXPERIMENTAL APPROACH
h-monDC were derived by incubating purified human monocytes with GM-CSF and IL-4. h-monDC were pre-incubated with or without Sal B and stimulated by oxidized low-density lipoprotein (ox-LDL) in the presence or absence of PPARγ siRNA. Expression of h-monDC membrane molecules (CD40, CD86, CD1a, HLA-DR) were analysed by FACS, cytokines were measured by elisa and the TLR4-associated signalling pathway was determined by Western blotting.
KEY RESULTS
Ox-LDL promoted h-monDC maturation, stimulated CD40, CD86, CD1a, HLA-DR expression and IL-12, IL-10, TNF-α production; and up-regulated TLR4 signalling. These effects were inhibited by Sal B. Sal B also triggered PPARγ activation and promoted PPARγ nuclear translocation, attenuated ox-LDL-induced up-regulation of TLR4 and myeloid differentiation primary-response protein 88 and inhibited the downstream p38-MAPK signalling cascade. Knocking down PPARγ with the corresponding siRNA blocked these effects of Sal B.
CONCLUSIONS AND IMPLICATIONS
Our data suggested that Sal B effectively suppressed maturation of h-monDC induced by ox-LDL through PPARγ activation.
Keywords: salvianolic acid B, dendritic cell, PPARγ, TLR4, atherosclerosis
Introduction
Atherosclerosis is a chronic inflammatory disease of the arterial vessel wall and mechanisms related to immune responses play crucial roles in the initiation and progression of atherosclerosis (Ludewig et al., 2002). A growing body of evidence suggests that lipid antigens, such as monophosphoryl lipid A, could activate dendritic cells, one of the major types of antigen-presenting cells (APCs), through selective activation of the MAPK pathway (Ismaili et al., 2002; Cekic et al., 2009) and enhancement of both Th1- and Th2-specific immune responses (De Becker et al., 2000; Ismaili et al., 2002). Delivery of lipid antigens by human monocyte-derived dendritic cells (h-monDC) and secretion of inflammatory cytokines from these cells are essential for the initiation and progression of atherosclerosis (Lord and Bobryshev, 1999; Nickel et al., 2009). In mice lacking the receptor for low density lipoprotein (LDLR−/− mice), hypercholesterolaemia-induced atherosclerotic lesions could be significantly reduced by depleting intimal CD11c+ DCs (Paulson et al., 2010).
Salvianolic acid B (Sal B), a constituent of Salvia miltiorrhiza, has been previously reported to exert anti-atherosclerotic activity in vivo and in vitro, mainly through depressing the inflammatory immune response, including lower expression of inflammatory cytokines, adhesion molecules (Chen et al., 2001; Zhang and Wang, 2006) and attenuating oxidative stress-induced endothelial damage (Shi et al., 2007; Wu et al., 2009). However, the potential mechanisms and related mediators for the vascular protective effects of Sal B are not fully understood. Previous reports from our group suggested that immune maturation of h-monDC might play an important role in the process of atherosclerosis (Luo et al., 2004; Ge et al., 2005), and h-monDC suppression might be one of the anti-atherogenic mechanisms of various vasoprotective agents. Therefore, we tested the hypothesis that the anti-atherosclerotic effect of Sal B might be mediated by suppressing h-monDC maturation. Our results indicated that Sal B could suppress maturation of h-monDC, induced by oxidized LDL (ox-LDL), through activating PPARγ.
Methods
Preparation of h-monDC
Heparinized venous blood was taken from healthy volunteers from the blood bank of Zhongshan Hospital. Ethical approval was given by the Ethics Committee of Zhongshan hospital. Human peripheral blood mononuclear cells (PBMCs) were separated by density gradient centrifugation using Histopaque 1077 (Sigma Aldrich) as described previously. (Ge et al., 2005) The monocytes (over 98% pure) were then prepared from PBMCs by positive selection with anti-CD14 magnetic beads (Milteny Biotech Co., Bergisch Gladback, North Rhine-Westphalia, Germany), seeded into six-well flat-bottom plates with 106 cells per well, cultured in 2 mL RPMI-1640 (Gibco-BRL, Life Technologies, Paisley, UK) containing 100 ng·mL−1 GM-CSF (R&D Systems, Inc., Minneapolis, MN, USA), 40 ng·mL−1 IL-4 (R&D Systems, Inc.) and 10% FBS (Hyclone, Logan, UT). On culture day 6, cells were pretreated with various concentrations of Sal B (0, 10, 50, 100 µM) and then stimulated by addition of 50 µg·mL−1 ox-LDL. Cell viability was over 90% as assessed by Trypan blue staining.
H-monDC immunostaining by FACS
H-monDC were washed, resuspended in ice-cold PBS containing 5% FBS (to prevent non-specific binding) then incubated with FITC- or PE-labelled monoclonal anti-CD1a, anti-CD40, anti-CD86, anti-HLA-DR (Invitrogen, Camarillo, CA, USA), biotinylated anti-human TLR4 antibody (R&D Systems) for 30 min at 4°C. Cells were washed twice, and immunofluorescence analysis was performed using a FACScan (BD Biosciences, San Jose, CA) and analysed using Cell Quest software.
Measurement of cytokine secretion of h-monDC
TNF-α, IL-10, IL-12 levels in supernatants of cultured h-monDC were measured using elisa kits (Invitrogen) according to the manufacturer's instructions. The minimum of detectable dose is <18, <15, <18 pg·mL−1 respectively.
RNA Isolation and real-time quantitative PCR
Total RNA was extracted by Trizol (Invitrogen); 5 µg total RNA sample was reverse-transcribed using oligo-dT and SuperScript III (Invitrogen). PPARγ was amplified using the up-primer (5′-CTCATAATGCCATCAGGTTTG-3′) and down-primer (3′-CCTCGTCTCGTTTCTCCAC-5′), which were designed from the cDNA sequences for PPARγ (GenBank accession No. NM138712). Quantitative real time PCR using SYBR Green reagent (Applied Biosystems, Foster City, CA, USA) was performed on the Rotor-Gene 3000 Real-time PCR System (Corbett Research, Mortlake, New South Wales, Australia). Gene expression was analysed by corresponding software (version 6.0.19); the copies of each mRNA molecules was determined by the standard curve method.
Western blot analysis
Whole cell extracts (20 µg) were separated by electrophoresis in 12% PAGE and transferred into PVDF membranes (Millipore Corp., Billerica, MA, USA). Then, the membranes with blotted protein were blocked for 1 h in 0.1% TBS–Tween 20 (TBS-T) containing 5% bovine serum albumin, followed by probing with antibody to the myeloid differentiation primary-response protein 88(MyD88) and antibodies to phospho-p38MAPK, phospho-pJNK1/2, β-actin (Cell Signaling Technologies, Danvers, MA, USA) at 4°C overnight. The membranes were washed three times for 15 min each with TBS-T, incubated at room temperature for 2 h with diluted (1:5000) secondary HRP-marked antibodies. Immunoreactive proteins were identified using Super Signal West Pico Chemiluminescence Substrate (Thermo, Watham, MA, USA). The ChemiDoc™ XRS gel documentation system (Bio-Rad Laboratories Inc., Hercules, CA, USA) with Quantity One software was used to quantify the immunoreactive proteins. β-Actin was used as the loading control for each lane.
Nuclear PPARγ detection
Cells (5 × 106) were obtained for nuclear protein extraction according to the kit protocol supplied by the manufacturer (#K0311, Fermentas Life Sciences, Burlington, ON, Canada). In brief, harvested cells were washed with ice-cold PBS twice and resuspended with 10 volumes of cell lysis buffer. After 10 min incubation on ice, the extracts were centrifuged at 500×g for 7 min at 4°C, and the supernatant were removed to separate the cytoplasmic fraction from nuclei. The nuclei pellets were washed with 500 µL nuclei washing buffer, vortexed briefly and set on ice for 2 min. After adding 50 µL nuclei lysis reagent, the nuclei pellets were rocked gently for 20 min to allow extraction of nuclear proteins. Finally, the proteins were separated on 12% PAGE and transferred into PVDF membranes (Millipore Corp.). The blots were detected by probing with anti-PPARγ (sc-7273, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA).
RNA interference
Followed by the protocols provided by Santa Cruz Biotechnology, we first seeded cells into six-well flat-bottomed plates (107 per well), cultured in 2 mL RPMI-1640 (Gibco-BRL, Life Technologies) containing 100 ng·mL−1 GM-CSF (R&D Systems, Inc.), 40 ng·mL−1 IL-4 (R&D Systems, Inc.) and 10% FBS (Hyclone). On culture day 4, the cells were washed once with 2 mL siRNA Transfection Medium (Cat. # sc-29528), then the cells were overlaid with the siRNA duplex solution, which was before mixed gently for 30 min at room temperature, from PPARγ siRNA (Cat. # sc-29455) or Control siRNA (Cat. # sc-36869) to diluted siRNA transfection reagents (Cat. # sc-29528). For each well, 0.8 mL siRNA transfection mixture was added. The cells were incubated at 37°C for 6 h. siRNA duplex solution was then removed and replaced with RPMI-1640. The cells were incubated for an additional 24 h. Twenty-four hours after transfection, cells were stimulated with ciglitazone (25 µg·mL−1). Whole cell lysates were subjected to Western blotting with anti-PPARγ antibodies (Santa Cruz Biotechnology, Inc.) to confirm the effect of siRNA on alteration of PPARγ transcriptional expression.
Transfection and luciferase assays
A glycerol kinase (GK) promoter fragment was amplified by PCR from human genomic DNA (GenBank accession no. NW001842360) using the following primers: 5′-CGCTCGAGCTTGTAAATCACAGGGCTAT-3′; 5′-CGAAGCTTCACGAGGTCAGGAGTTTG-3′. The resulting fragment was digested with XhoI and HindIII at 37°C and then was subcloned into XhoI and HindIII-restricted pGL3-Luc-Enhancer Vector (Promega, Madison, WI, USA). The reconstructed luciferase reporter system was transiently transfected into h-monDC performed by Lipofectamine 2000 reagent (Invitrogen). Briefly, cells were seeded in suspension at a density of 1 × 106 per well. On culture day 4, 1 × 107 cells were harvested and washed twice with PBS, incubated at 37°C for 6 h in 1 mL RPMI 1640 supplemented with 40 µL of Lipofectamine™ 2000, 20 µg luciferase reporter construct (pGL3-Luc-GK) or pGL3-Luc control vector, along with 5 µg CMV-β-galactosidase plasmid as internal control. Transfection mixture was removed, washed twice with PBS and re-seeded, and cells were cultured with RPMI-1640 at a density of 1 × 106 per well, and then stimulated with Sal B (50 µM) and/or ciglitazone (25 µg·mL−1). Cell extracts were collected 24 h after transfection, and luciferase activity was analysed using the Dual-Luciferase reporter assay system according to the manufacturer's instruction (Promega). Luciferase activity of each sample was normalized to its β-galactosidase activity.
Statistical analyses
Data are presented as means ± SD. Group mean values were compared with one-way anova followed by post hoc tests using Tukey's procedure for pairwise comparisons. All statistical analyses were performed with SPSS 11.5 statistical software, and a P-value < 0.05 was considered statistically significant.
Materials
Sal B was purchased from Shanghai Institute of Materia Medica (IMM), Chinese Academy of Sciences, and was dissolved in PBS (final concentration: 50 µM). Ciglitazone was purchased from Sigma Aldrich (St Louis, MO), SB203580 and SP600125 from Calbiochem (Merck KGaA, Darmstadt, Germany).
Results
Sal B effectively suppressed ox-LDL-induced h-monDC maturation
Cell surface expressions of co-stimulatory molecules CD40, CD86, CD1a and HLA-DR were examined in ox-LDL (50 µg·mL−1)-treated h-monDC, exposed to various concentrations of Sal B (0, 10, 50, 100 µM). Pilot experiments showed that Sal B at a concentration of 50 µM could inhibit, by nearly 70%, the expression of CD40, CD86, CD1a and HLA-DR (Figure S1), and that this effect could last for 24 h and was still visible after 48 h (Figure 1B). Sal B (50 µM)-induced cell apoptosis was analysed by Annexin V/7AAD double staining after 24 h incubation. Sal B (50 µM) alone was not toxic to the cells (Figure S2). Sal B at a concentration of 50 µM was therefore used in the main study. Ox-LDL-induced increased expressions of CD40, CD86, CD1a and HLA-DR were significantly reduced by Sal B (Figure 1A). In addition, ox-LDL-induced IL-12, IL-10 and TNF-α production in culture medium was also significantly reduced by Sal B (Figure 1C–E). These data suggested that Sal B suppressed h-monDC maturation and the inflammatory responses mediated by matured h-monDC.
Figure 1.
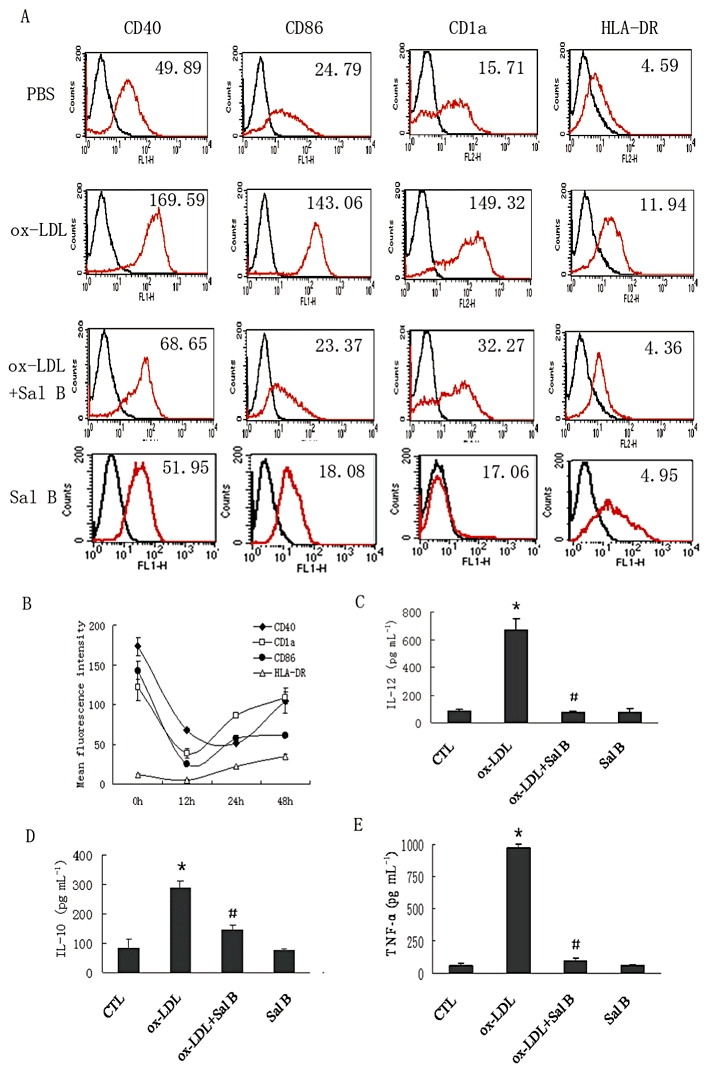
PBMCs were separated and purified with anti-CD14 magnetic beads, then cultured with GM-CSF (100 ng·mL−1) and IL-4 (40 ng·mL−1) for 5 days. On day 6, ox-LDL (50 µg·mL−1) was added to the cells 24 h before analysis in the presence or absence of Sal B (50 µM). The effect of Sal B on h-monDC phenotype changes were analysed by FACS and immune cytokines by elisa. (A) Ox-LDL-treated h-monDC were analysed for the expression of CD40, CD86, CD1a and HLA-DR in the absence or presence of Sal B by FACS. (B) Time-dependent effect of Sal B on expression of CD40, CD86, CD1a and HLA-DR. (C–E) Sal B reduced levels of IL-12, TNF-α and IL-10 in supernatants of cultured h-monDC 24 h post ox-LDL stimulation by elisa. Bar graphs show mean concentration results (mean ± SD) of three independent experiments. *P < 0.05, significantly different from Control (CTL); #P < 0.05, significantly different from ox-LDL.
Effects of Sal B on h-monDC maturation and TLR4/MyD88 signalling
The family of Toll-like receptors (TLRs) is important for pathogen-associated molecular patterns (PAMP) recognition and antigen presenting by functional h-monDC (de Kleijn and Pasterkamp, 2003). The link between TLR4 and h-monDC maturation has been shown in previous reports (Michelsen et al., 2001; Turnbull et al., 2005). In the present study, ox-LDL increased ∼2.5-fold the expression of membrane TLR4 in h-monDC and this was attenuated by Sal B (Figure 2A). Up-regulation of MyD88, one of the TIR domain-containing adaptors recruited by TLR4 (Michelsen et al., 2004), induced by ox-LDL, was also significantly down-regulated by Sal B (Figure 2B).
Figure 2.
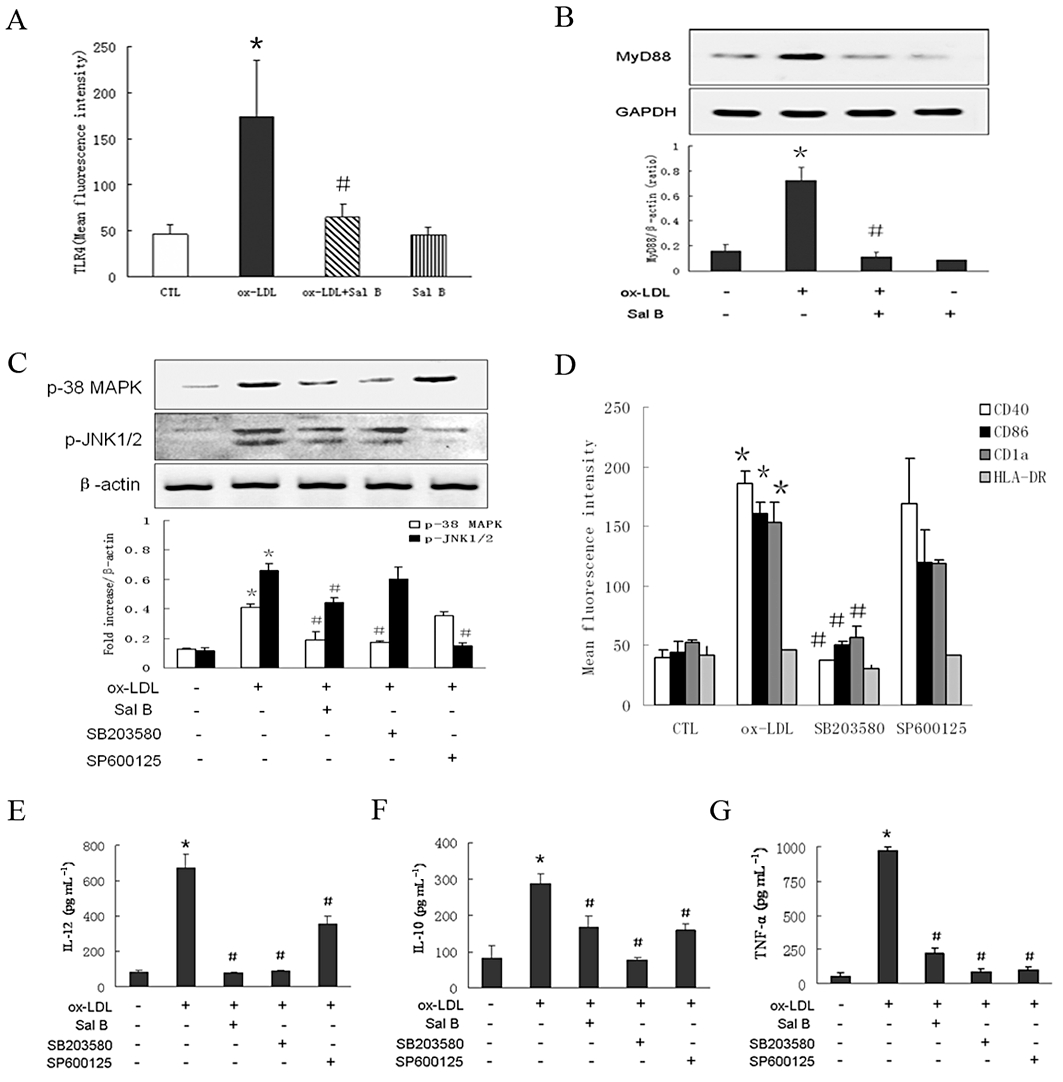
PBMCs derived h-monDC cultured with GM-CSF and IL-4 for 5 days, then stimulated by ox-LDL and/or Sal B for 24 h in the presence or absence of SB203580 (a specific p38-MAPK blocker), SP600125 (a specific JNK1/2 blocker) before analysis. (A) Effect of Sal B on TLR4 expression by FACS. (B) Effect of Sal B on MyD88 protein expression by Western blot. (C) Effect of Sal B on the phosphorylation levels of p38-MAPK and JNK1/2 by Western blot. (D) Effect of SB203580 or SP600125 on ox-LDL-stimulated expression of CD40, CD86, CD1a and HLA-DR by FACS. (E–G) Effect of Sal B on the production of IL-12, IL-10 and TNF-α compared with the application of SB203580 or SP600125 by elisa. Data were presented as means ± SD of four independent experiments. *P < 0.05, significantly different from control (CTL); #P < 0.05, significantly different from ox-LDL.
Next, we analysed the effect of Sal B on TLR4/MyD88-mediated MAPK pathway during h-monDC maturation (Ardeshna et al., 2000; Arrighi et al., 2001). As shown in Figure 2C, Sal B (50 µM) reduced ox-LDL-induced activation of p38 and JNK1/2, and ox-LDL-induced activation of p38 and JNK1/2 was also inhibited by SB203580 (a specific p38-MAPK blocker) or SP600125 (a specific JNK1/2 blocker). Subsequently, we determined the effects of SB203580 (20 µM) and SP600125 (20 µM) on ox-LDL-induced h-monDC phenotype change and IL-12, IL-10 and TNF-α production. Notably, ox-LDL-induced increased expressions of CD40, CD86, CD1a and HLA-DR were attenuated by SB203580 but not by SP600125 (Figure 2D), indicating Sal B might inhibit ox-LDL-induced h-monDC maturation through blocking the TLR4-mediated p38–MAPK signalling pathway. Interestingly, SP600125 significantly reduced IL-12, IL-10 and TNF-α production (Figure 2E–G); however, inhibiting JNK1/2 alone did not suppress ox-LDL-mediated h-monDC maturation, suggesting that the JNK1/2 pathway might be only partly involved in the process of h-monDC maturation.
Since other pattern-recognition receptors (PRRs), such as CD36, might also be potential mediators of ox-LDL-mediated signalling transduction in monocyte–derived cells (Rahaman et al., 2006; Stewart et al., 2010), we further examined the possibility that other PRRs could also induce h-monDC maturation after inhibiting TLR4 receptor. First, TLR4 expression was determined by FACS after blocking TLR4 signalling using purified TLR4-neutralizing antibody. As shown in Figure S3, nearly 70% expression of TLR4 was effectively inhibited by TLR4-neutralizing antibody. Then, ox-LDL-induced h-monDC phenotypes were examined after using TLR4-neutralizing antibody. Data showed that ox-LDL-induced up-regulation of CD40, CD86, CD1a and HLA-DR were all attenuated by TLR4-neutralizing antibody (Figure S4), suggesting that TLR4 activation was critically involved in h-monDC maturation induced by ox-LDL.
Sal B enhanced PPARγ activation and nuclear translocation
Previous experiments from our group showed that the PPARγ agonist ciglitazone inhibited ox-LDL-induced h-monDC maturation and immune function (Luo et al., 2004). To elucidate if Sal B suppressed ox-LDL-induced h-monDC maturation via PPARγ signalling, we investigated the expression of PPARγ gene in the absence or presence of indicated concentrations of Sal B. As shown in Figure 3A, PPARγ mRNA level was dose-dependently up-regulated by Sal B at 2 h but was reduced to the baseline level at 24 h. However, Sal B (50–100 µM) effectively up-regulated the PPARγ protein level at 24 h in the nucleus (Figure 3B), suggesting PPARγ transcriptional activity was enhanced by Sal B.
Figure 3.
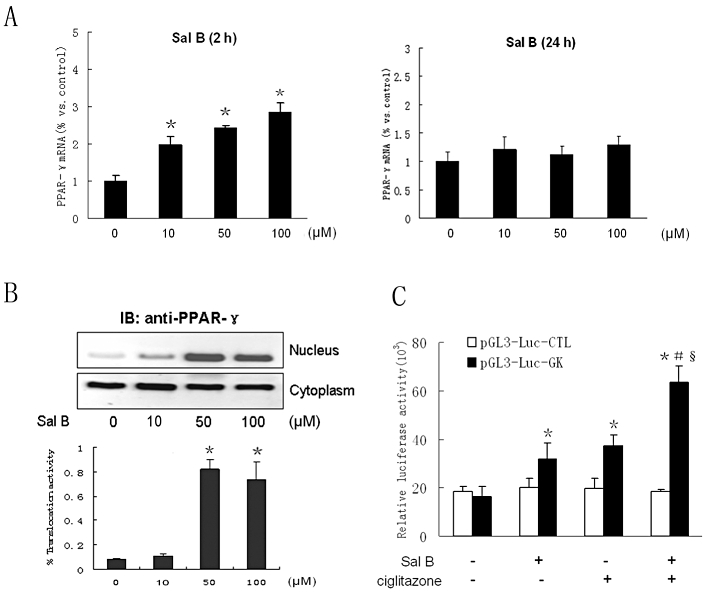
Effects of Sal B on PPARγ expression and transcriptional activity in h-monDC without ox-LDL stimulation. (A) Cultured h-monDC were treated with various concentration of Sal B for 2 h or 24 h. (B) The mRNA andprotein levels of PPARγ were examined by real-time RT-PCR and Western blot respectively. (C) pGL3-Luc-enhancer report vector with a GK promoter was transfected into h-monDC, and then the cells were treated with vehicle (PBS), Sal B (50 µM) and/or ciglitazone (25 µg·mL−1). Cell lysates were harvested, and both luciferase and β-galactosidase activities were analysed using the Dual-Luciferase reporter assay system. Data are shown as related luciferase activity in cells transfected with pGL3-Luc-GK vector or pGL3-Luc control vector after normalized to the β-galactosidase activity. Data were presented as means ± SD of four independent experiments. *P < 0.05, significantly different from control; #P < 0.05, significantly different from Sal B; §P < 0.05, significantly different from ciglitazone.
We further investigated whether Sal B could modulate DNA binding activity of PPARγ. Because PPARγ activation was linked with up-regulation of glycerol kinase expression, we determined the effects of Sal B and ciglitazone on glycerol kinase promoter activity with the luciferase reporter assay. Our result showed that both Sal B and ciglitazone significantly enhanced glycerol kinase promoter activity (Figure 3C). Transactivation capacity in pGL3-Luc-Control Vector was also reduced by Sal B, suggesting PPARγ could be activated by Sal B at the transcriptional level. Furthermore, some of the PPARγ target genes, including those for activating enhancer binding protein 2 (AP2), adipose differentiation-related protein (ADRP), PPARγ–angiopoietin related protein (PGAR) and liver X receptor α (LXRα), were identified to confirm that Sal B could actually enhance the PPARγ-mediated pathway (Figure S5). The results showed that three genes (for ADRP, PGAR and LXRα) were activated by Sal B, suggesting Sal B-induced PPARγ activation was different from that induced by ox-LDL.
Association between PPARγ signalling and H-monDC maturation in the presence of Sal B
To determine whether PPARγ activation was required for the observed effects of Sal B on ox-LDL-induced h-monDC maturation, we tested the effects of Sal B in case of PPARγ activity inhibition by knocking down PPARγ with specific siRNA (Figure S6). When PPARγ was inhibited by specific siRNA, ox-LDL-induced up-regulation of the membrane expression of CD40, CD86, CD1a and HLA-DR (Figure 4D) in h-monDC and increased secretion of IL-12, IL-10 and TNF-α in culture medium (Figure 4A–C) were no longer affected by Sal B, suggesting that PPARγ activation was essential for the inhibitory effect of Sal B on h-monDC maturation induced by ox-LDL. To exclude the effect of nuclear acids on ox-LDL-induced TLR4 signalling in h-monDC, the effects of PPARγ siRNA on TLR4 expression in h-monDC were tested. In contrast with the enhanced expression of TLR4 induced by ox-LDL, TLR4 expression was not affected by PPARγ siRNA alone (Figure 4E). As shown in Figure 4D, TLR4-mediated expression of CD40, CD86, CD1a and HLA-DR was also not affected by PPARγ siRNA or scrambled siRNA. The association between PPARγ activation and TLR4-mediated MAPK pathway after knocking down PPARγ was also investigated. As expected, enhanced expression of phosphorylated p38 in ox-LDL-treated h-monDC was down-regulated by ciglitazone or Sal B (Figure 4F, lanes 3–4) and the effects of both ciglitazone- and Sal B were reversed by PPARγ siRNA (Figure 4F, lanes 5–6), indicating Sal B-induced PPARγ activation was likely to be mediated by blocking TLR4-mediated MAPK cascade.
Figure 4.
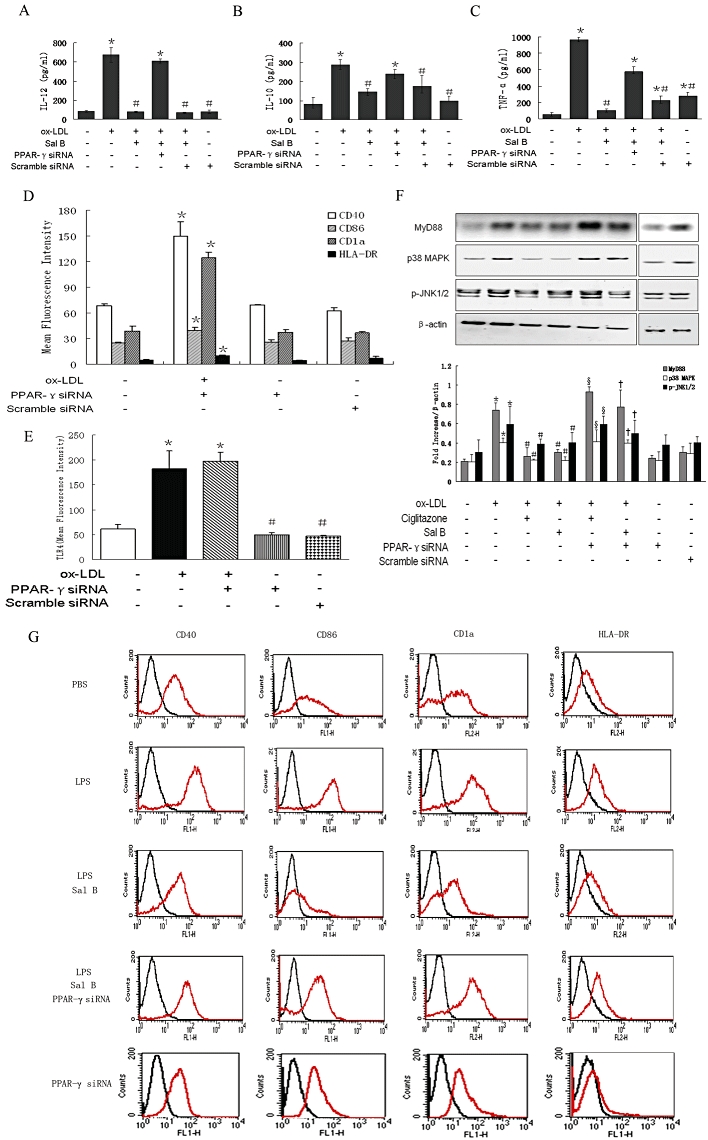
Purified h-monDCs were incubated for 5 days in the presence of GM-CSF and IL-4 with PPARγ siRNA or scrambled siRNA transfection starting from the fourth day of culture. On day 6, cells were stimulated by ox-LDL for 24 h in the absence or presence of Sal B before harvesting. Effects of PPARγ siRNA on ox-LDL-induced h-monDC were determined as follows: (A–C) production of IL-12, IL-10 and TNF-α by elisa. (D) H-monDC phenotype by FACS. (E) Membrane receptor TLR4 expression under ox-LDL stimulated or unstimulated conditions by FACS. (F) Western blot analysis of MyD88, phosphorylated p38-MAPK and JNK1/2 levels pre-incubated with Sal B/ciglitazone. (G) Effect of PPARγ siRNA on phenotype of LPS (100 ng·mL−1)-stimulated h-monDC by FACS. The results represent the means ± SD from four independent experiments. *P < 0.05, Sal B.
To elucidate whether Sal B could also inhibit h-monDC maturation stimulated by another stimulus, we examined the effect of Sal B on h-monDC maturation induced by LPS. Increased expressions of CD40, CD86, CD1a and HLA-DR in LPS (100 ng·mL−1)-treated h-monDC were also attenuated by pretreatment with Sal B, and these effects could be attenuated by PPARγ siRNA (Figure 4G). These data suggested that LPS-induced h-monDC maturation could also be attenuated by Sal B via PPARγ activation.
Discussion and conclusions
Sal B is one of the constituents of extracts of Salvia miltiorrhiza, a traditional Chinese medicinal herb. Previous studies suggested its antioxidant activities were associated with the protective effects of Sal B on the vasculature (Wu et al., 2009). Besides the oxidative injury, inflammatory and immune injury are the other major mechanisms underlying atherosclerotic vessel disease (Hansson, 1999; Ludewig et al., 2002). In the present study, we demonstrated for the first time that the possible anti-atherosclerotic effects of Sal B might be mediated by suppressing maturation of h-monDC and thus reducing the inflammatory and immune responses mediated by matured h-monDC. Our results showed that Sal B reduce ox-LDL-induced h-monDC maturation through activating PPARγ and suppressing the TLR4-mediated MAPK signalling pathway.
The TLR4-mediated signalling pathway is important for h-monDC maturation (Turnbull et al., 2005)and in the current study, Sal B attenuated the up-regulation of TLR4 signalling in h-monDC, induced by ox-LDL or LPS. Blockade of TLR4 signalling suppressed transcription of genes responsible for inflammatory chemokine production and h-monDC maturation (Turnbull et al., 2005; Wang et al., 2008), by activating the NF-κB and MAPK pathways (Appel et al., 2005; Lin et al., 2005). As expected, the enhanced expression of CD40, CD86, CD1a and HLA-DR, which reflects immune maturation of h-monDC (Carucci et al., 2000; Xia and Kao, 2002; Dopheide et al., 2007) and the increased output of pro-atherosclerotic chemokines, such as IL-12, IL-10 and TNF-α (Appel et al., 2005; Hickey et al., 2008), both induced by ox-LDL, were significantly attenuated by Sal B partly through inhibiting TLR4/MyD88 signalling.
Recent studies indicated that CD36, a membrane glycoprotein present on monocytes and macrophages, might work as a co-receptor in facilitating the formation of the TLR complex (Triantafilou et al., 2006; Stewart et al., 2010). The CD36–TLR4–TLR6 signalling complex, induced by ox-LDL, could trigger and activate MyD88-dependent signalling pathways. Both in vivo and in vitro studies showed that MyD88 deficiency reduced atherosclerosis and attenuated h-monDC maturation by decreasing IL-12 and TNF-α synthesis (Michelsen et al., 2004; Macedo et al., 2008). The role of Sal B in regulating TLR4/MyD88 signalling was explored in our study. Blocking TLR4 receptors with purified TLR4-neutralizing antibody suppressed h-monDC maturation induced by ox-LDL. In fact, blocking the TLR4 signal might also inhibit other PRR-mediated signals, which might be important for synergies between the receptor. For instance, the CD36–TLR4–TLR6 complex is suggested as the ‘first hit’ in the regulation of the innate immune response, which could initiate the progress of differentiation or apoptosis of monocytes and macrophages (Stewart et al., 2010). Recent studies also suggested that CD36 could modulate the functions of h-monDC through triggering TLR4/MyD88-dependent chemokine synthesis and downstream signalling (Taront et al., 2009; Stewart et al., 2010). Further investigations are therefore needed to clarify the role of CD36 in the h-monDC maturation process in responses to atherogenic lipids and the role and interactions of TLR4 receptor and MyD88 protein during these processes.
TLR4-mediated maturation of h-monDC is regulated by different signal transduction pathways, including PI3 kinase, p38MAPkinase and NF-kB (Ardeshna et al., 2000). LPS-induced up-regulation of TLR4 could activate all three MAPK cascades. However, the intracellular signals triggered by ox-LDL differ between h-monDC and other monocyte-derived cells because of the different uptake of ox-LDL by the different receptors (LOX–1, CD36 and TLR4). JNK1/2 phosphorylation is critically involved in macrophage activation through CD36-dependent formation of signalling complex containing Lyn and MEKK2 (Rahaman et al., 2006; Kennedy et al., 2009). In our study, ox-LDL-induced JNK1/2 phosphorylation was up-regulated in h-monDC, but ox-LDL-induced h-monDC maturation was not suppressed by blockade of JNK1/2, while blocking TLR4 signalling through the p38–MAPK pathway effectively suppressed ox-LDL-induced h-monDC maturation. Taken together, we hold the view that p38-MAPK but not the JNK1/2 cascade was involved in the inhibition by Sal B of h-monDC maturation induced by ox-LDL.
A novel finding of the current study is that PPARγ activation by Sal B might be the major working mechanism for suppressing ox-LDL-induced h-monDC maturation. There is growing evidence that the anti-atherogenic effects of HMG-CoA reductase inhibitors (statins) are partly mediated by PPARγ activation (Yano et al., 2007). PPARγ agonists, such as pioglitazone and rosiglitazone, exert their endothelium-dependent vascular protection through modulating endogenous production of stress-induced receptors such as LOX-1 or of endothelin-1 (Iglarz et al., 2003; Mehta et al., 2003). PPAR activation has been involved in the immune suppression of h-monDC, macrophages, Langerhans cells and other types of APCs (Appel et al., 2005; Dubrac et al., 2007; Yano et al., 2007). Thus, PPAR agonists play critical roles in modulating the process of h-monDC maturation, attenuating release of inflammatory cytokines and cytokine-induced migration of h-monDC (Angeli et al., 2003; Hammad et al., 2004).
The present study shows that Sal B might suppress ox-LDL-induced h-monDC maturation through PPARγ activation. Earlier studies demonstrated that ox-LDL alone could activate PPARγ through MAPK-dependent COX-2 expression in macrophages (Taketa et al., 2008; Liu et al., 2010). Notably, ox-LDL-induced COX-2 expression in RAW264.7 macrophages was mediated by activation of ERK1/2, but not p38–MAPK. However, siRNA for COX-2 inhibited ox-LDL-induced PPARγ activation by only ∼50% (Taketa et al., 2008), suggesting ox-LDL-induced COX-2 cascade was only a part of PPARγ activation in macrophages. In the present study, we showed that PPARγ target gene ADRP could be activated by both ox-LDL and Sal B, AP2 was activated by ox-LDL, while PGAR and LXRα were only activated by Sal B, suggesting that the mechanism of regulation by ox-LDL was different from that induced by Sal B. In addition, our present data supported the proposal that ox-LDL activated TLR4-dependent h-monDC maturation through p38–MAPK signalling pathway (Xu et al., 2001; Wang et al., 2008), and TLR4-mediated activation of DCs could be attenuated by PPARγ agonists (Appel et al., 2005). Taken together, we consider that the mode of PPARγ activation induced by Sal B and ox-LDL was not the same. Moreover, p38–MAPK but not ERK1/2 were involved in h-monDC maturation and could be regulated by Sal B through PPARγ activation.
Previous studies also demonstrated that Sal B was effective in reducing lipid-related oxidative stress and in the treatment of atherosclerotic vessel disease (Chen et al., 2001; He et al., 2008; Wu et al., 2009), but the potential molecular mechanism for the vasoprotective effect of Sal B is largely unknown. From the present study, we would conclude that Sal B promotes PPARγ activation, and to some extent, Sal B enhances the process of negative regulation induced by ox-LDL. Consistent with the earlier reports of the modulating effects of PPARγ agonists on inflammatory and immune responses (Klotz et al., 2007), we have shown that the PPARγ transcriptional activity was enhanced by Sal B. In line with the effects induced by ciglitazone, Sal B also increased PPARγ nuclear translocation and target gene expression (Leroyer et al., 2006). The MAPK signalling pathway is thought to regulate through PPARγ, which controls the function of h-monDC, or by affecting local cells, such as T cells and Langerhans cells, by paracrine effects (Dubrac et al., 2007; Klotz et al., 2007). In this study, both ox-LDL- and LPS-induced TLR4-dependent MAPK pathway was inhibited by Sal B, suggesting Sal B-mediated PPARγ activation was important for h-monDC suppression. Also, Sal B-induced PPARγ activation inhibited the p38–MAPK pathway and reduced expression of CD40, CD86, CD1a, HLA-DR and production of IL-12, IL-10 and TNF-α. These effects were attenuated by PPARγ siRNA, which implied a causal relationship between the effect of Sal B on h-monDC maturation and PPARγ activation (Figure 5).
Figure 5.
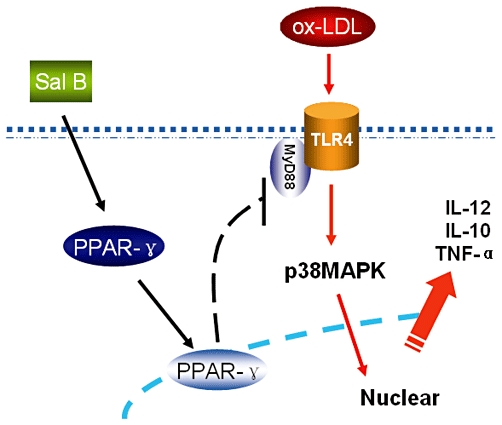
Putative mechanism for Sal B-mediated suppression of h-monDC maturation induced by ox-LDL. h-monDC maturation was stimulated by ox-LDL through TLR4/MyD88-mediated p38–MAPK signalling pathway. This signal cascade could be suppressed by Sal B through up-regulating nuclear translocation and increased transcriptional activity of PPARγ and subsequently down-regulating of TLR4/MyD88-mediated p38–MAPK pathway.
In conclusion, our present study suggests that Sal B suppressed ox-LDL-induced h-monDC maturation through activating PPARγ and subsequently down-regulating TLR4-mediated p38–MAPK pathways, which might reduce the inflammatory and immune responses of matured h-monDC. Further studies are needed to confirm that Sal B can attenuate atherosclerosis in vivo via the mechanisms we have suggested here.
Acknowledgments
This work was supported by National Basic Research Program of China [2011CB503905], 863 Program of Science and Technology Ministry [2006AA0ZA406], Outstanding Youth Grant from National Natural Science Foundation of China [30725036], and Key Projects in the National Science & Technology Pillar Program in the Eleventh Five-year Plan Period [2006BAI01A04]. We thank Dr Kai Hu and Dr Paula Arias from University of Würzburg for their help in the preparation of the present manuscript.
Glossary
Abbreviations
- ADRP
adipose differentiation-related protein
- AP2
activating enhancer binding protein 2
- APCs
antigen-presenting cells
- GK
glycerol kinase
- h-monDC
human monocyte-derived dendritic cells
- LXRα
liver X receptor α
- MyD88
myeloid differentiation primary-response protein 88
- PAMP
pathogen-associated molecular patterns
- PBMCs
peripheral blood mononuclear cells
- PGAR
PPARγ–angiopoietin related protein
- Sal B
salvianolic acid B
- TLRs
Toll-like receptors
Conflict of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Concentration curves for the effect of Sal B on DCs maturation. Ox-LDL-induced DCs were treated with 0, 10, 50 and 1007 M of Sal B, and the phenotypic changes of CD40, CD86, CD1a and HLA-DR were determined respectively.
Figure S2 PBMCs were separated and purified with anti-CD14 magnetic beads, then cultured with GM-CSF (100 ng·mL−1) and IL-4 (40 ng·mL−1) for 5 days. On day 6, cells were stimulated by Sal B (507 M), and the toxic effects of Sal B were then quantified by FACS analysis after staining with Annexin V and 7-AAD. The results represented the means ± SD of three independent experiments.
Figure S3 Purified h-monDC was incubated for 5 days in the presence of GM-CSF and IL-4. On day 6, ox-LDL (507 g·mL−1) was added, and the effect of ox-LDL-induced TLR4 expression was determined by FACS after blocking the blocking the membrane TLR4 on matured h-monDC by purified TLR4-neutralizing antibody. Data were presented as means ± SD of three independent experiments. *P < 0.05 versus control; #P < 0.05 versus ox-LDL.
Figure S4 Purified h-monDC was incubated for 5 days in the presence of GM-CSF and IL-4. On day 6, ox-LDL (507 g·mL−1) was added, and the effect of ox-LDL-induced h-monDC phenotype was determined by FACS after blocking the membrane TLR4 on matured h-monDC by purified TLR4-neutralizing antibody. Data were presented as means ± SD of three independent experiments. *P < 0.05 versus PBS; #P < 0.05 versus ox-LDL.
Figure S5 PBMCs derived h-monDC cultured with GM-CSF and IL-4 for 5 days, then stimulated by PBS, Sal B, Ciglitazone and ox-LDL, respectively, for 24 h. Then mRNA was extracted and cDNA was reverse-transcribed. The expression of PPARg target genes were analysed by RT-PCR using specific primers as follows: LXR-α, 5′-acggtgatgcttctggagac-3′ and 5′-tagcaatgagcaaggcaaact-3′ (NM_001130101.1); AP-2, 5′-gtctccgccatccctattaac-3′ and 5′-ggaatgttgtcggttgagaaa-3′ (NM_001032280.2); ADRP, 5′-agtggaaaaggagcattggata-3′ and 5′-ctgtggtacaccttggatgttg-3′ (NM_001122.3); PGAR, 5′-acaagcacctagaccatgaggt-3′ and 5′-ctgaattactgtccagcctccat-3′ (NM_001039667.1); GAPDH, 5′-agaaggctggggctcatttg-3′ and 5′-aggggccatccacagtcttc-3′ (NM_002046.3). (*P < 0.05 vs. control.)
Figure S6 PBMCs derived h-monDC cultured with GM-CSF and IL-4 for 5 days, then cells were transfected with mock conditions (no siRNA), PPARg siRNA or scramble siRNA for 24 h respectively.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Angeli V, Hammad H, Staels B, Capron M, Lambrecht BN, Trottein F. Peroxisome proliferator-activated receptor gamma inhibits the migration of dendritic cells: consequences for the immune response. J Immunol. 2003;170:5295–5301. doi: 10.4049/jimmunol.170.10.5295. [DOI] [PubMed] [Google Scholar]
- Appel S, Mirakaj V, Bringmann A, Weck MM, Grunebach F, Brossart P. PPARgamma agonists inhibit toll-like receptor-mediated activation of dendritic cells via the MAP kinase and NF-kappaB pathways. Blood. 2005;106:3888–3894. doi: 10.1182/blood-2004-12-4709. [DOI] [PubMed] [Google Scholar]
- Ardeshna KM, Pizzey AR, Devereux S, Khwaja A. The PI3 kinase, p38 SAP kinase, and NF-kappaB signal transduction pathways are involved in the survival and maturation of lipopolysaccharide-stimulated human monocyte-derived dendritic cells. Blood. 2000;96:1039–1046. [PubMed] [Google Scholar]
- Arrighi JF, Rebsamen M, Rousset F, Kindler V, Hauser C. A critical role for p38 mitogen-activated protein kinase in the maturation of human blood-derived dendritic cells induced by lipopolysaccharide, TNF-alpha, and contact sensitizers. J Immunol. 2001;166:3837–3845. doi: 10.4049/jimmunol.166.6.3837. [DOI] [PubMed] [Google Scholar]
- Carucci JA, Ignatius R, Wei Y, Cypess AM, Schaer DA, Pope M, et al. Calcitonin gene-related peptide decreases expression of HLA-DR and CD86 by human dendritic cells and dampens dendritic cell-driven T cell-proliferative responses via the type I calcitonin gene-related peptide receptor. J Immunol. 2000;164:3494–3499. doi: 10.4049/jimmunol.164.7.3494. [DOI] [PubMed] [Google Scholar]
- Cekic C, Casella CR, Eaves CA, Matsuzawa A, Ichijo H, Mitchell TC. Selective activation of the p38 MAPK pathway by synthetic monophosphoryl lipid A. J Biol Chem. 2009;284:31982–31991. doi: 10.1074/jbc.M109.046383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YH, Lin SJ, Ku HH, Shiao MS, Lin FY, Chen JW, et al. Salvianolic acid B attenuates VCAM-1 and ICAM-1 expression in TNF-alpha-treated human aortic endothelial cells. J Cell Biochem. 2001;82:512–521. doi: 10.1002/jcb.1176. [DOI] [PubMed] [Google Scholar]
- De Becker G, Moulin V, Pajak B, Bruck C, Francotte M, Thiriart C, et al. The adjuvant monophosphoryl lipid A increases the function of antigen-presenting cells. Int Immunol. 2000;12:807–815. doi: 10.1093/intimm/12.6.807. [DOI] [PubMed] [Google Scholar]
- Dopheide JF, Sester U, Schlitt A, Horstick G, Rupprecht HJ, Munzel T, et al. Monocyte-derived dendritic cells of patients with coronary artery disease show an increased expression of costimulatory molecules CD40, CD80 and CD86 in vitro. Coron Artery Dis. 2007;18:523–531. doi: 10.1097/MCA.0b013e3282eff1ad. [DOI] [PubMed] [Google Scholar]
- Dubrac S, Stoitzner P, Pirkebner D, Elentner A, Schoonjans K, Auwerx J, et al. Peroxisome proliferator-activated receptor-alpha activation inhibits Langerhans cell function. J Immunol. 2007;178:4362–4372. doi: 10.4049/jimmunol.178.7.4362. [DOI] [PubMed] [Google Scholar]
- Ge J, Jia Q, Liang C, Luo Y, Huang D, Sun A, et al. Advanced glycosylation end products might promote atherosclerosis through inducing the immune maturation of dendritic cells. Arterioscler Thromb Vasc Biol. 2005;25:2157–2163. doi: 10.1161/01.ATV.0000181744.58265.63. [DOI] [PubMed] [Google Scholar]
- Hammad H, de Heer HJ, Soullie T, Angeli V, Trottein F, Hoogsteden HC, et al. Activation of peroxisome proliferator-activated receptor-gamma in dendritic cells inhibits the development of eosinophilic airway inflammation in a mouse model of asthma. Am J Pathol. 2004;164:263–271. doi: 10.1016/s0002-9440(10)63116-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson GK. Inflammation and immune response in atherosclerosis. Curr Atheroscler Rep. 1999;1:150–155. doi: 10.1007/s11883-999-0011-0. [DOI] [PubMed] [Google Scholar]
- He H, Shi M, Zeng X, Yang J, Li Y, Wu L, et al. Cardioprotective effect of salvianolic acid B on large myocardial infarction mediated by reversing upregulation of leptin, endothelin pathways, and abnormal expression of SERCA2a, phospholamban in rats. J Ethnopharmacol. 2008;118:35–45. doi: 10.1016/j.jep.2008.03.006. [DOI] [PubMed] [Google Scholar]
- Hickey FB, Brereton CF, Mills KH. Adenylate cycalse toxin of Bordetella pertussis inhibits TLR-induced IRF-1 and IRF-8 activation and IL-12 production and enhances IL-10 through MAPK activation in dendritic cells. J Leukoc Biol. 2008;84:234–243. doi: 10.1189/jlb.0208113. [DOI] [PubMed] [Google Scholar]
- Iglarz M, Touyz RM, Amiri F, Lavoie MF, Diep QN, Schiffrin EL. Effect of peroxisome proliferator-activated receptor-alpha and -gamma activators on vascular remodeling in endothelin-dependent hypertension. Arterioscler Thromb Vasc Biol. 2003;23:45–51. doi: 10.1161/01.atv.0000047447.67827.cd. [DOI] [PubMed] [Google Scholar]
- Ismaili J, Rennesson J, Aksoy E, Vekemans J, Vincart B, Amraoui Z, et al. Monophosphoryl lipid A activates both human dendritic cells and T cells. J Immunol. 2002;168:926–932. doi: 10.4049/jimmunol.168.2.926. [DOI] [PubMed] [Google Scholar]
- Kennedy DJ, Kuchibhotla SD, Guy E, Park YM, Nimako G, Vanegas D, et al. Dietary cholesterol plays a role in CD36-mediated atherogenesis in LDLR-knockout mice. Arterioscler Thromb Vasc Biol. 2009;29:1481–1487. doi: 10.1161/ATVBAHA.109.191940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kleijn D, Pasterkamp G. Toll-like receptors in cardiovascular diseases. Cardiovasc Res. 2003;60:58–67. doi: 10.1016/s0008-6363(03)00348-1. [DOI] [PubMed] [Google Scholar]
- Klotz L, Dani I, Edenhofer F, Nolden L, Evert B, Paul B, et al. Peroxisome proliferator-activated receptor gamma control of dendritic cell function contributes to development of CD4+ T cell anergy. J Immunol. 2007;178:2122–2131. doi: 10.4049/jimmunol.178.4.2122. [DOI] [PubMed] [Google Scholar]
- Leroyer SN, Tordjman J, Chauvet G, Quette J, Chapron C, Forest C, et al. Rosiglitazone controls fatty acid cycling in human adipose tissue by means of glyceroneogenesis and glycerol phosphorylation. J Biol Chem. 2006;281:13141–13149. doi: 10.1074/jbc.M512943200. [DOI] [PubMed] [Google Scholar]
- Lin YL, Liang YC, Lee SS, Chiang BL. Polysaccharide purified from Ganoderma lucidum induced activation and maturation of human monocyte-derived dendritic cells by the NF-kappaB and p38 mitogen-activated protein kinase pathways. J Leukoc Biol. 2005;78:533–543. doi: 10.1189/jlb.0804481. [DOI] [PubMed] [Google Scholar]
- Liu Q, Dai Z, Liu Z, Liu X, Tang C, Wang Z, et al. Oxidized low-density lipoprotein activates adipophilin through ERK1/2 signal pathway in RAW264.7 cells. Acta Biochim Biophys Sin (Shanghai) 2010;42:635–645. doi: 10.1093/abbs/gmq070. [DOI] [PubMed] [Google Scholar]
- Lord RS, Bobryshev YV. Clustering of dendritic cells in athero-prone areas of the aorta. Atherosclerosis. 1999;146:197–198. doi: 10.1016/s0021-9150(99)00119-7. [DOI] [PubMed] [Google Scholar]
- Ludewig B, Zinkernagel RM, Hengartner H. Arterial inflammation and atherosclerosis. Trends Cardiovasc Med. 2002;12:154–159. doi: 10.1016/s1050-1738(01)00166-9. [DOI] [PubMed] [Google Scholar]
- Luo Y, Liang C, Xu C, Jia Q, Huang D, Chen L, et al. Ciglitazone inhibits oxidized-low density lipoprotein induced immune maturation of dendritic cells. J Cardiovasc Pharmacol. 2004;44:381–385. doi: 10.1097/01.fjc.0000138164.88740.f8. [DOI] [PubMed] [Google Scholar]
- Macedo GC, Magnani DM, Carvalho NB, Bruna-Romero O, Gazzinelli RT, Oliveira SC. Central role of MyD88-dependent dendritic cell maturation and proinflammatory cytokine production to control Brucella abortus infection. J Immunol. 2008;180:1080–1087. doi: 10.4049/jimmunol.180.2.1080. [DOI] [PubMed] [Google Scholar]
- Mehta JL, Hu B, Chen J, Li D. Pioglitazone inhibits LOX-1 expression in human coronary artery endothelial cells by reducing intracellular superoxide radical generation. Arterioscler Thromb Vasc Biol. 2003;23:2203–2208. doi: 10.1161/01.ATV.0000094411.98127.5F. [DOI] [PubMed] [Google Scholar]
- Michelsen KS, Aicher A, Mohaupt M, Hartung T, Dimmeler S, Kirschning CJ, et al. The role of toll-like receptors (TLRs) in bacteria-induced maturation of murine dendritic cells (DCS). Peptidoglycan and lipoteichoic acid are inducers of DC maturation and require TLR2. J Biol Chem. 2001;276:25680–25686. doi: 10.1074/jbc.M011615200. [DOI] [PubMed] [Google Scholar]
- Michelsen KS, Wong MH, Shah PK, Zhang W, Yano J, Doherty TM, et al. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc Natl Acad Sci USA. 2004;101:10679–10684. doi: 10.1073/pnas.0403249101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickel T, Schmauss D, Hanssen H, Sicic Z, Krebs B, Jankl S, et al. oxLDL uptake by dendritic cells induces upregulation of scavenger-receptors, maturation and differentiation. Atherosclerosis. 2009;205:442–450. doi: 10.1016/j.atherosclerosis.2009.01.002. [DOI] [PubMed] [Google Scholar]
- Paulson KE, Zhu SN, Chen M, Nurmohamed S, Jongstra-Bilen J, Cybulsky MI. Resident intimal dendritic cells accumulate lipid and contribute to the initiation of atherosclerosis. Circ Res. 2010;106:383–390. doi: 10.1161/CIRCRESAHA.109.210781. [DOI] [PubMed] [Google Scholar]
- Rahaman SO, Lennon DJ, Febbraio M, Podrez EA, Hazen SL, Silverstein RL. A CD36-dependent signaling cascade is necessary for macrophage foam cell formation. Cell Metab. 2006;4:211–221. doi: 10.1016/j.cmet.2006.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi CS, Huang HC, Wu HL, Kuo CH, Chang BI, Shiao MS, et al. Salvianolic acid B modulates hemostasis properties of human umbilical vein endothelial cells. Thromb Res. 2007;119:769–775. doi: 10.1016/j.thromres.2006.06.008. [DOI] [PubMed] [Google Scholar]
- Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. 2010;11:155–161. doi: 10.1038/ni.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taketa K, Matsumura T, Yano M, Ishii N, Senokuchi T, Motoshima H, et al. Oxidized low density lipoprotein activates peroxisome proliferator-activated receptor-alpha (PPARalpha) and PPARgamma through MAPK-dependent COX-2 expression in macrophages. J Biol Chem. 2008;283:9852–9862. doi: 10.1074/jbc.M703318200. [DOI] [PubMed] [Google Scholar]
- Taront S, Dieudonne A, Blanchard S, Jeannin P, Lassalle P, Delneste Y, et al. Implication of scavenger receptors in the interactions between diesel exhaust particles and immature or mature dendritic cells. Part Fibre Toxicol. 2009;6:9. doi: 10.1186/1743-8977-6-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triantafilou M, Gamper FG, Haston RM, Mouratis MA, Morath S, Hartung T, et al. Membrane sorting of toll-like receptor (TLR)-2/6 and TLR2/1 heterodimers at the cell surface determines heterotypic associations with CD36 and intracellular targeting. J Biol Chem. 2006;281:31002–31011. doi: 10.1074/jbc.M602794200. [DOI] [PubMed] [Google Scholar]
- Turnbull EL, Yrlid U, Jenkins CD, Macpherson GG. Intestinal dendritic cell subsets: differential effects of systemic TLR4 stimulation on migratory fate and activation in vivo. J Immunol. 2005;174:1374–1384. doi: 10.4049/jimmunol.174.3.1374. [DOI] [PubMed] [Google Scholar]
- Wang L, Li D, Yang K, Hu Y, Zeng Q. Toll-like receptor-4 and mitogen-activated protein kinase signal system are involved in activation of dendritic cells in patients with acute coronary syndrome. Immunology. 2008;125:122–130. doi: 10.1111/j.1365-2567.2008.02827.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HL, Li YH, Lin YH, Wang R, Li YB, Tie L, et al. Salvianolic acid B protects human endothelial cells from oxidative stress damage: a possible protective role of glucose-regulated protein 78 induction. Cardiovasc Res. 2009;81:148–158. doi: 10.1093/cvr/cvn262. [DOI] [PubMed] [Google Scholar]
- Xia CQ, Kao KJ. Heparin induces differentiation of CD1a+ dendritic cells from monocytes: phenotypic and functional characterization. J Immunol. 2002;168:1131–1138. doi: 10.4049/jimmunol.168.3.1131. [DOI] [PubMed] [Google Scholar]
- Xu XH, Shah PK, Faure E, Equils O, Thomas L, Fishbein MC, et al. Toll-like receptor-4 is expressed by macrophages in murine and human lipid-rich atherosclerotic plaques and upregulated by oxidized LDL. Circulation. 2001;104:3103–3108. doi: 10.1161/hc5001.100631. [DOI] [PubMed] [Google Scholar]
- Yano M, Matsumura T, Senokuchi T, Ishii N, Murata Y, Taketa K, et al. Statins activate peroxisome proliferator-activated receptor gamma through extracellular signal-regulated kinase 1/2 and p38 mitogen-activated protein kinase-dependent cyclooxygenase-2 expression in macrophages. Circ Res. 2007;100:1442–1451. doi: 10.1161/01.RES.0000268411.49545.9c. [DOI] [PubMed] [Google Scholar]
- Zhang HS, Wang SQ. Salvianolic acid B from Salvia miltiorrhiza inhibits tumor necrosis factor-alpha (TNF-alpha)-induced MMP-2 upregulation in human aortic smooth muscle cells via suppression of NAD(P)H oxidase-derived reactive oxygen species. J Mol Cell Cardiol. 2006;41:138–148. doi: 10.1016/j.yjmcc.2006.03.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.