

Abstract
BACKGROUND AND PURPOSE
Rosiglitazone is an anti-diabetic drug improving insulin sensitivity and glucose uptake in skeletal muscle and adipose tissues. However, several recent clinical trials suggest that rosiglitazone can increase the risk of cardiovascular ischaemia, although other studies failed to show such risks. Therefore, the effects of rosiglitazone on the coronary circulation and any potential vascular targets need to be elucidated. Here, we show that the vascular isoform of the ATP-sensitive K+ (KATP) channel is inhibited by rosiglitazone, impairing physiological regulation of the coronary circulation.
EXPERIMENTAL APPROACH
The KIR6.1/SUR2B channel was expressed in HEK293 cells and studied in whole-cell and inside-out patch configurations. The Langendorff heart preparation was used to evaluate rosiglitazone in the coronary circulation of wild-type (WT) and KIR6.1-null (Kcnj8−/−) mice.
KEY RESULTS
KIR6.1/SUR2B channels in HEK cells were inhibited by rosiglitazone in a membrane-delimited manner. This effect was markedly enhanced by sub-micromolar concentrations of glibenclamide and the IC50 for rosiglitazone fell to 2µM, a therapeutically achievable concentration. In the Langendorff heart preparation rosiglitazone inhibited, concentration-dependently, the coronary vasodilation induced by isoprenaline, without affecting basal coronary tone. Effects of rosiglitazone on coronary perfusion were attenuated by more than 50% in the Kcnj8−/− mice, supporting the involvement of KATP channels in this effect of rosiglitazone on the coronary circulation.
CONCLUSIONS AND IMPLICATIONS
These results indicate that the vascular KATP channel is one of the targets of rosiglitazone action, through which this drug may compromise coronary responses to circulating vasodilators and perhaps also to metabolic stress.
Keywords: thiazolidinedione, type-2 diabetes mellitus, potassium channel, vascular tones, heart, cardiovascular, mouse
Introduction
Rosiglitazone is one of the two thiazolidinediones currently available for the treatment of type-2 diabetes mellitus. Primarily by activating the peroxisome proliferator-activated receptor-gamma (PPAR-γ) (Duan et al., 2008), rosiglitazone has three major effects on the pathogenesis of type-2 diabetes mellitus and its complications: (i) it improves insulin resistance and has been successfully used to achieve glycaemic control in a manner that is at least as effective as the sulphonylureas and metformin; (ii) rosiglitazone activates PPAR-γ and regulates adipocyte proliferation and lipid storage, improving lipid profile; and (iii) through the PPAR-γ in vascular tissues, rosiglitazone interferes with the processes of foam cell formation and inflammatory responses, reduces lipid deposition in the vessel wall, and thereby attenuates the development of atherosclerosis (Barnett, 2009). Despite these beneficial outcomes, recent clinical studies have raised the issue of the potential cardiovascular risks in rosiglitazone users (Zinn et al., 2008; Kaul et al., 2010). A large meta-analysis clinical trial suggested a 43% increase in risk of myocardial infarction in patients treated with rosiglitazone (Nissen and Wolski, 2007). This study was followed by a number of additional reports using an alternative analysis of the same data, new meta-analyses and observational studies on both rosiglitazone and pioglitazone, the other clinically used thiazolidinedione (Home et al., 2007; Gerstein et al., 2008; Mannucci et al., 2008; Duckworth et al., 2009; Psaty and Furberg, 2007; Vanasse et al., 2009). The results, however, were rather variable and inconsistent. Thus, more direct evidence for any impairment of coronary circulation and the potential vascular targets is needed for an effective and appropriate application of the drug in the treatment of type-2 diabetes mellitus.
A potential target molecule of rosiglitazone on the vascular wall is the ATP-sensitive K+ (KATP) channel composed of KIR6.1/SUR2B subunits (channel nomenclature follows Alexander et al., 2009) expressed in vascular smooth muscle (VSM). Numerous vasodilator and vasoconstrictor hormones act on this K+ channel (Quayle et al., 1997; Ashcroft, 2006; Nichols, 2006; Shi et al., 2007a,b; Yang et al., 2008). The KATP channel is also regulated by several metabolites. This metabolite sensitivity allows the channel to regulate the vascular tone and regional blood flow according to the metabolic state in local tissues under both physiological and pathophysiological conditions (Ashcroft, 2006; Yang et al., 2010; 2011;). Thus, dysregulation of such a critical channel in the vasculature could affect coronary responses to circulating vasodilators, which in turn may be influenced by rosiglitazone. In order to test this hypothesis we performed these studies and our results showed that the KIR6.1/SUR2B channel was strongly inhibited by rosiglitazone and this channel inhibition compromised the vasodilatory response of the coronary circulation.
Methods
Expression of KATP channel in HEK293 cells
The HEK293 cells were cultured in DMEM/F12 medium at 37°C with 10% fetal bovine serum and penicillin/streptomycin in the presence of 5% CO2. A eukaryotic expression vector pcDNA3.1 was used to express rat KIR6.1 (GenBank Accession # D42145) in the cells together with SUR2B (GenBank # D86038, mRNA isoform NM_011511). Lipofectamine2000 (Invitrogen Inc., Carlsbad, CA) was used for transfection. Each 35 mm Petri dish containing the cells was transfected with 1 µg KIR6.1 and 3 µg SUR2B. Green fluorescent protein cDNA (0.4 µg, pEGFP-N2, Clontech, Palo Alto, CA) was included in the cDNA mixture to facilitate the identification of positively transfected cells. Cells were split and transferred to cover slips after 12–18 h of transfection. Experiments were performed on the cells on cover slips during the following 12–48 h. The HEK cells express endogenous β-adrenoceptors whose activation enhances the KATP channel activity through the PKA signalling system (Shi et al., 2007b; Yang et al., 2008). Thus, the KATP channel modulation by rosiglitazone was also studied by activating the endogenous β-adrenoceptors.
Electrophysiology
Patch clamp experiments were carried out at room temperature as described previously (Wang et al., 2003; Shi et al., 2007a,b; 2008a,b; 2010; Yang et al., 2008; 2010;). The bath solution contained (in mM): KCL 10, potassium gluconate 135, EGTA 5, glucose 5 and HEPES 10 (pH 7.4). The pipette was filled with a solution containing (in mM): KCl 10, potassium gluconate 133, EGTA 5, glucose 5, K2ATP 1, NaADP 0.5, MgCl2 1 and HEPES 10 (pH 7.4). Whole-cell currents were recorded in single-cell voltage clamp with holding potential 0 mV and step to −80 mV for 1 s. To avoid nucleotide degradation, all intracellular solutions were freshly made and used within 4 h.
Recordings were made with the Axopatch 200B amplifier (Axon Instruments Inc., Foster City, CA). The data were low-pass filtered (2 kHz, Bessel 4-pole filter, −3 dB), and digitized (10 kHz, 16-bit resolution) with Clampex 9 (Axon Instruments Inc.). Single-channel currents were recorded from inside-out patches with a constant single voltage of −60 mV. Higher sampling rate (20 kHz) was used to digitize the currents recorded from inside-out patch. Data were analysed using Clampfit 9 (Axon Instruments Inc.).
Langendorff-perfused hearts
All animal care and experimental procedures were in compliance with an approved protocol by the Institutional Animal Care and Use Committees (IACUC) at Georgia State University. Male wild-type (WT) C57BL/6 mice weighing 20 to 30 g (8 to 14 weeks of age) were deeply anaesthetized followed by removal of the heart. The heart was transferred to ice-cold (4°C) Krebs-Henseleit (KH) solution (composition in mM): 119.0 NaCl, 4.7 KCl, 2.5 CaCl2, 2.5 MgSO4, 25.0 NaHCO3, 1.2 KH2PO4, 0.5 disodium EDTA and 10 glucose (pH 7.4). The aortic root was quickly cannulated and flushed gently with KH solution to remove blood in the coronary arteries. The heart was then placed in an organ bath and perfused with KH solution at a constant pressure. The reservoir was maintained at a fixed height above the heart to keep the perfusion pressure at approximately 80 cm H2O. The KH solution was bubbled with a mixture of 95% O2–5% CO2 and the perfusate temperature was maintained at 35°C using a warming coil. The isolated heart was constantly bathed in a small chamber (∼2 mL) with the perfusate constantly flowing through the coronary arteries. The flow rate from the heart was measured by collecting the overflow fluid from the chamber at five minute intervals.
Data analysis
Data were evaluated using Student's t-tests and anova, and statistical significance was deemed acceptable when P < 0.05.
Materials
Rosiglitazone was purchased from Cayman Chemical Compamy (Ann Arbor, MI, USA). Pioglitazone, pinacidil, glybenclamide and isoprenaline were purchased from Sigma Chemicals (St. Louis, MO, USA).
Results
Whole-cell K+ currents were recorded from HEK cells transfected with KIR6.1/SUR2B in single-cell voltage clamp. Symmetric concentrations of K+ (145 mM) were applied to the bath and pipette solutions. The membrane potential was held at 0 mV and stepped to −80 mV for 1 s. This protocol was repeated every 3 s. Under this condition, the cells showed a very small basal current (Yang et al., 2010; 2011;). Administration of 10 µM pinacidil, a selective KATP channel activator, strongly activated the inward K+ currents that were subsequently inhibited by 10 µM glibenclamide, a KATP channel blocker. These pinacidil/glibenclamide-sensitive currents were exogenous as pinacidil/glibenclamide had very little effect on the HEK cells transfected with the expression vector alone (Figure S1A). Therefore, these KATP channel activator and inhibitor were used to determine the expression of the channel in HEK cells.
Exposure to rosiglitazone produced an inhibition of the pinacidil-activated current (Figure 1A). The channel inhibition occurred within 1 min, and showed a clear concentration dependence (Figure 1C). A complete reversal was seen after removal of rosiglitazone (Figure S1B). When the KIR6.1/SUR2B current was activated by the β-adrenoceptor agonist isoprenaline (1 µM), treatment of the cell with rosiglitazone led to a similar channel inhibition (Figure 1B). Indeed, the current activated by isoprenaline had almost the same sensitivity to rosiglitazone as the current activated by pinacidil (Figure 1C). Rosiglitazone had no evident effect on the basal current before the KIR6.1 /SUR2B channel was activated by pinacidil (Figure 1D,E). In the presence of rosiglitazone, pinacidil (10 µM or 100 µM) did not activate the KIR6.1 /SUR2B current, although the channel activity normalised after the washout of rosiglitazone (Figure S1B,C). In contrast, another thiazolidinedione, pioglitazone, caused only modest inhibition of the KIR6.1 /SUR2B current, at 3 and 30 µM (Figure 2A,B). Thus, compared with rosiglitazone, the effect of pioglitazone was much weaker (Figure 2C).
Figure 1.
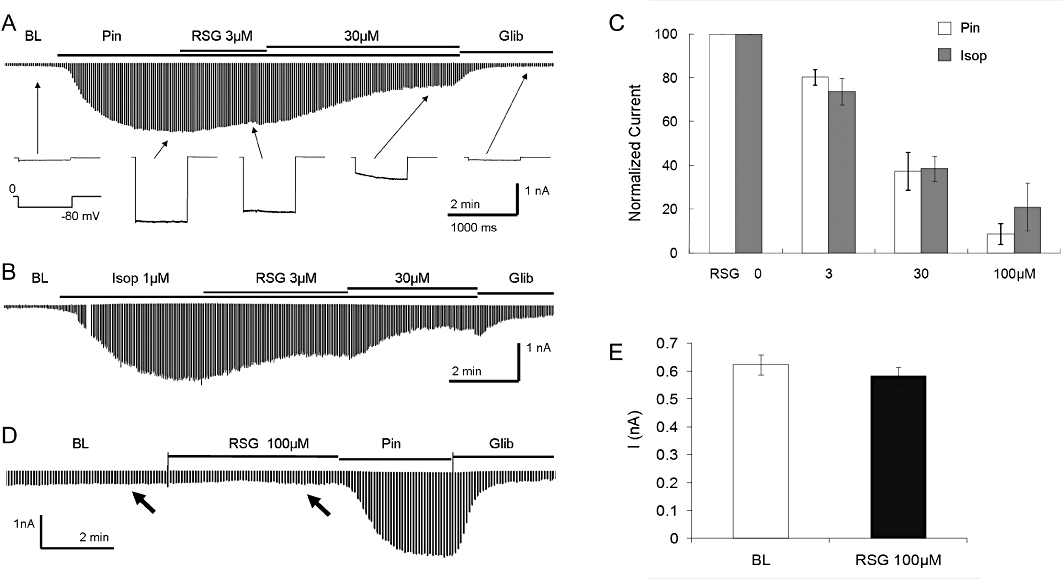
Inhibition of the KIR6.1/SUR2B channel by rosiglitazone. (A) The HEK cell was co-transfected with KIR6.1 and SUR2B. Inward currents were studied 2 days after transfection using symmetric concentrations of K+ (145 mM) applied to the bath and pipette solutions. Membrane potentials were held at 0 mV and stepped to −80 mV in every 3 s. The cell showed small currents at baseline (BL). The currents were strongly activated by 10 µM pinacidil (Pin). At the maximum channel activation the application of rosiglitazone (RSG) led to concentration-dependent inhibitions of the KIR6.1/SUR2B currents. The currents were further inhibited by 10 µM glibenclamide (Glib). (B) In the same condition, the KIR6.1/SUR2B currents were activated by isoprenaline (Isop) via the HEK cell-endogenous β-adrenoceptor as shown previously (Shi et al., 2007b). The currents had almost the same sensitivity to rosiglitazone. Note that there is a 1 min gap during isoprenaline exposure. (C) When both currents are plotted against rosiglitazone concentrations, the pinacidil-activation currents overlie the isoprenaline-activated currents. Rosiglitazone produced concentration-dependent inhibition of the KIR6.1/SUR2B currents activated by both pinacidil and isoprenaline. (D,E) Rosiglitazone did not have any evident effect on the basal KIR6.1/SUR2B currents before the currents were activated by pinacidil. The current amplitude with rosiglitazone treatment remained the same as the baseline (arrows). Similar results were found in another cell (n = 4 to 7 patches from different cells).
Figure 2.
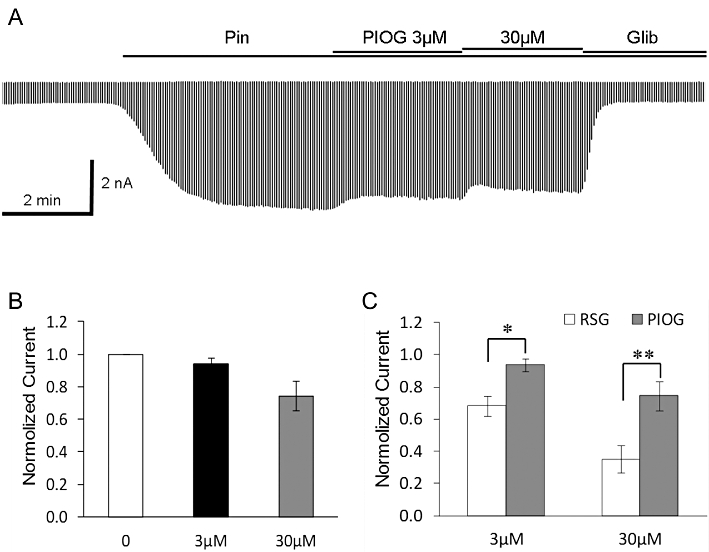
(A) Pioglitazone (PIOG) inhibited the KIR6.1/SUR2B currents only modestly. Pin, pinacidil; Glib, glibenclamide. (B) KIR6.1/SUR2B currents were insensitive to 3 µM pioglitazone and showed rather small response to 30 µM pioglitazone. (C) In comparison to rosiglitazone (RSG), the effect of pioglitazone was significantly smaller. *P < 0.05, **P < 0.01, compared with each other (n = 9 to 11).
In order to understand the biophysical mechanisms of the channel inhibition, we studied the KIR6.1/SUR2B currents in inside-out patches. The effect of rosiglitazone was mediated by suppression of the channel open state probability without affecting the unitary conductance (Figure 3A,B). The channel inhibition was reversible and showed concentration dependence. The current–concentration relationship is described using the Hill equation with IC50 of 10 µM and h of 1.2 (Figure 3C).
Figure 3.
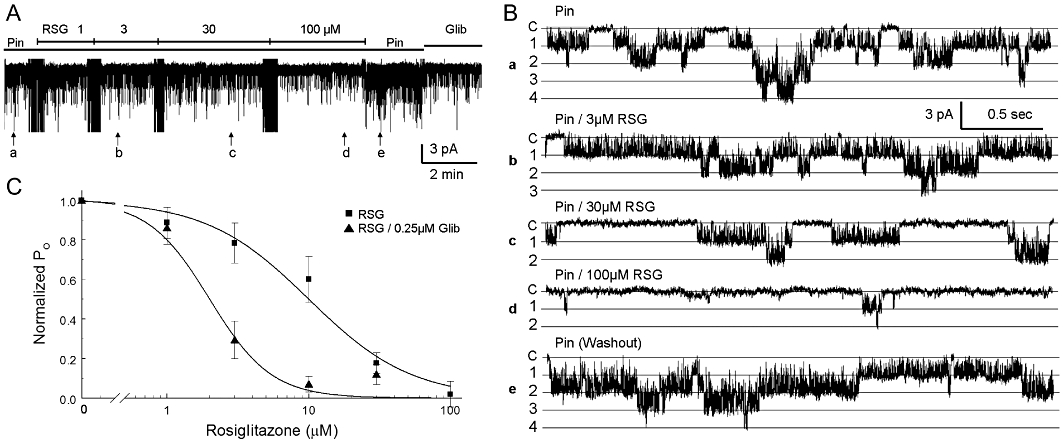
Membrane-delimited inhibition of the KIR6.1/SUR2B channel. (A) Single-channel KIR6.1/SUR2B currents were recorded in an inside-out patch from an HEK cell with symmetric K+ (145 mM) and a −60 mV membrane potential. (B) Single-channel activity shown in expanded scales (traces from top to bottom are obtained from A at times marked a – e respectively). The currents were activated by pinacidil (Pin) and inhibited dose-dependently by rosiglitazone (RSG). Washout of rosiglitazone with pinacidil-containing perfusate led to a complete recovery. Labels on the left: C, closure; 1, 2, …, n, the first, second … nth openings. (C) The channel activity is a function of rosiglitazone concentration. Their relationship is described using the Hill equation with IC50 10 µM and h 1.2 (n = 4 patches from different cells) for rosiglitazone alone and 2 µM (h 2.0) in the presence of 0.25 µM glibenclamide (Glib; n = 4).
As a number of type-2 diabetes mellitus patients are prescribed rosiglitazone together with sulphonylureas which inhibit the KATP channel, we studied the combined effect of rosiglitazone and glibenclamide. In the presence of 0.25 µM glibenclamide, the effect of rosiglitazone was markedly potentiated with the IC50 now falling to 2 µM (h 2.0) (Figure 3C). Both these rosiglitazone and glibenclamide concentrations are achieved therapeutically. Serum concentration of rosiglitazone may be raised above these levels as a result of impaired drug metabolism and genetic variations of individual patients and hence, an inhibition of the KATP channel may well take place in these patients, compromising their cardiovascular function.
In the Langendorff heart preparation, the perfusate through the coronary circulation was collected and measured at 5 min intervals under constant pressure (80 cm H2O). The viability of the preparation was confirmed by the following criteria (i) was the heart beating spontaneously or responsive to isoprenaline in the perfusate, and (ii) was the perfusion volume increased in the presence of isoprenaline. Our results showed that rosiglitazone (30 µM or 100 µM) had no significant effect on the basal perfusion volume of the heart from the WT mice (Figure S2). The perfusion volume rose rapidly when the perfusate contained isoprenaline (a 1.30 mL increase or 86.8%). During the period of isoprenaline-induced coronary vasodilation, exposure to rosiglitazone resulted in a concentration-dependent decrease in the perfusion volume (Figure 4A), that is, 30 or 100 µM rosiglitazone reduced the perfusion volume by 0.41 mL or 0.85 mL respectively and this effect of rosiglitazone was reversible (Figure 4A,B). Similar results were obtained in studies using pinacidil-induced coronary vasodilation (Figure 4C, D). Moreover, another known KIR6.1 inhibitor glibenclamide showed similar inhibitory effects on perfusion volume in the presence of isoprenaline or pinacidil (Figure S3A,B).
Figure 4.
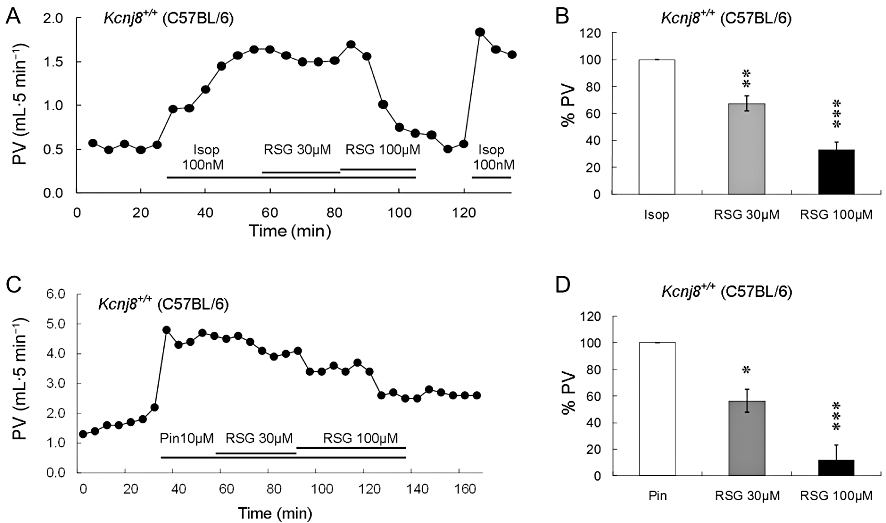
Effects of rosiglitazone on the coronary circulation of WT (Kcnj+/+) mice. (A) The perfusion volume (PV) increased markedly when there was isoprenaline (Isop; 100 nM) in the perfusate. In the presence of isoprenaline, rosiglitazone (RSG) reduced the PV in a dose-dependent manner. (B) Dose-dependent inhibition of isoprenaline-induced coronary vasorelaxation in two strains of mice in which the KIR6.1 channel is present. The PV was normalized to the maximum PV during isoprenaline exposure as 100%. The isoprenaline-induced coronary vasorelaxation was suppressed by 32.6% and 67.2% in response to 30 µM and 100 µM rosiglitazone respectively. (C,D) Dose-dependent inhibition of pinacidil (Pin)-induced coronary vasodilation. The pinacidil-induced coronary vasodilation was suppressed by 43.7% and 83.1% in response to 30 µM and 100 µM rosiglitazone respectively. Data are presented as mean ± SEM (n = 3 to 5 hearts). *P < 0.05; **P < 0.01; ***P < 0.001 compared with control.
In the Langendorff hearts of Kcnj8−/− mice (lacking the KIR6.1 channel) prepared identically to the WT mice, an exposure to 100 nM isoprenaline led to a rather small and inconsistent vasodilation, with no discernable effects of rosiglitazone. Therefore 300 nM isoprenaline was used instead of 100 nM in the hearts of the Kcnj8−/− mice and this concentration of isoprenaline produced an increase in the coronary perfusion volume (Figure 5A). The isoprenaline-induced coronary vasodilation, however, was not affected by 30 µM rosiglitazone in the KIR6.1-null hearts (Figure 5B). However, a concentration of 100 µM rosiglitazone reduced the isoprenaline-induced coronary vasodilation by 18.4 ± 9.5% (n = 6, Figure 5C in which the isoprenaline-augmented flow was reduced from 1.23 mL to 1.07 mL). This weak effect may be mediated by mechanisms other than interactions with the KATP channel. In the WT heart, the same concentration of rosiglitazone reduced coronary vasodilation by 67.2 ± 5.9% (n = 5, a 0.85 mL decrease from 1.30 mL; P < 0.001).
Figure 5.
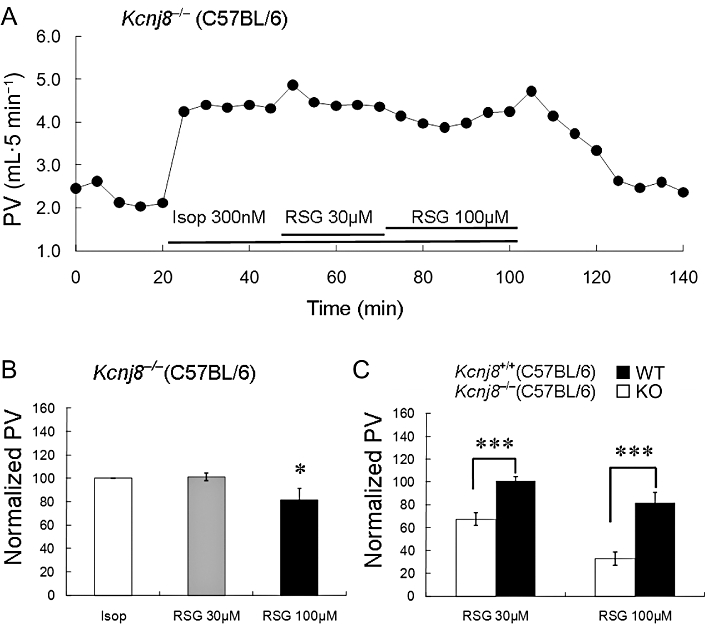
The effect of rosiglitazone on coronary PV in Kcnj8−/− mice. (A) In a Langendorf heart from a Kcnj8−/− mouse, isoprenaline (Isop; 300 nM) produced coronary vasodilation. (B) The isoprenaline-induced coronary vasorelaxation was suppressed by 18.4% in response to 100 µM rosiglitazone (RSG), while rosiglitazone at 30 µM had no evident effect on the PV. (C) In comparison with the WT hearts, the rosiglitazone effect on coronary circulation was significantly lower in 30 µM and 100 µM (*P < 0.05; ***P < 0.001; n = 6).
Pinacidil had no vasodilatory effects in the Kcnj8−/− hearts (Figure S3A). In comparison with the 84.6% inhibition seen in the WT hearts, glibenclamide did not reduce the isoprenaline-induced coronary dilation in these mutant hearts (Figure S3A,B). These results suggest strongly that the KIR6.1/SUR2B channel is a major vascular target of rosiglitazone in the coronary circulation's vasodilator response to isoprenaline.
Discussion and conclusions
These studies are the first demonstration that rosiglitazone, at therapeutically-achieved concentrations, prevented coronary vasodilatory responses to the β-adrenoceptor agonist isoprenaline. The effect is likely to involve the KIR6.1/SUR2B channel that is expressed primarily in the VSM. Channel activity was strongly inhibited by rosiglitazone when the channel was activated by pinacidil or isoprenaline using the whole-cell configuration. Cytosolic soluble factors did not seem necessary for the channel inhibition as the rosiglitazone effect was seen in excised inside-out patches. Indeed the IC50 of rosiglitazone was lower in inside-out patches than in the whole-cell configuration.
Previous clinical studies have suggested that type-2 diabetes mellitus patients treated with rosiglitazone are at increased risk of cardiovascular ischaemic events, (Nissen and Wolski, 2007; Zinn et al., 2008; Kaul et al., 2010), although such observations are not consistent (Home et al., 2007; Psaty and Furberg, 2007; Gerstein et al., 2008; Mannucci et al., 2008; Duckworth et al., 2009; Vanasse et al., 2009). The discrepancies may be related to the many factors involved in clinical studies, such as placebo- versus active-controlled trials, patient demographics and treatment variations.
However, data from animal experiments suggest that rosiglitazone has more beneficial than harmful cardiovascular effects. The beneficial effects are likely to be due to the improvement of metabolic profile and VSM remodelling, whilst the deleterious effects might involve oxidative stress and endothelial damage. (Wang et al., 2006; How et al., 2007; Lu et al., 2008b; Kanda et al., 2009; Savoia et al., 2010; Torres Tda et al., 2010; Yu et al., 2010). Some cardiovascular effects of rosiglitazone have been studied using various animal models of ischaemia and cardiac protection (Knock et al., 1999; Khandoudi et al., 2002; Abe et al., 2008; Kilter et al., 2009; Potenza et al., 2009; Wang et al., 2009; 2010;). But direct effects of rosiglitazone on the vessel tone and cardiac regional blood flow has not been demonstrated.
As ion channel activity determines membrane potential, excitability and contractility in VSM cells, the potential involvement of these channels in the effects of rosiglitazone has been studied and reported previously (Knock et al., 1999; Mishra and Aaronson, 1999; Eto et al., 2001; Lu et al., 2008a; Chang et al., 2009). Rosiglitazone inhibits Ca2+ currents and voltage-activated K+ currents that play a role in cAMP-mediated vasodilation (Eto et al., 2001; Li et al., 2003). Moreover, rosiglitazone activates Ca2+-activated K+ currents in acutely dissociated mesenteric VSM cells (Eto et al., 2001; Lu et al., 2008a). However, rosiglitazone does not induce vasorelaxation of human subcutaneous small arterial rings preconstricted with noradrenaline (Walker et al., 1998). Also, rosiglitazone inhibits glibenclamide-sensitive K+ currents in freshly isolated aortic myocytes (Chang et al., 2009), but does not affect the vasorelaxation of endothelium-denuded human internal mammary artery rings preconstricted with noradrenaline and KCl (Irat et al., 2006). These inconsistent findings may be due to differences in the experimental preparations employed and/or the presence of multiple ion channel species in the preparations. Focusing on a specific subunit, rosiglitazone has been shown to stimulate insulin secretion in pancreatic beta cells via phosphorylation of the KIR6.2 channel by AMP-dependent protein kinase (Chang et al., 2009). In other studies, rosiglitazone has been shown to block cardiac KATP channels and promote the onset of ventricular fibrillation during severe ischaemia (Mishra and Aaronson, 1999).
Our current studies indicate that the KIR6.1/SUR2B channel is indeed a target of rosiglitazone as the Kir6.1/SUR2B channel is inhibited by rosiglitazone. In WT and KIR6.1-null mice, inhibition of the KIR6.1/SUR2B channel leads to an impairment of coronary vasodilatory responses. This KIR6.1/SUR2B channel is a common target of both vasodilator and vasoconstrictor hormones and transmitters that activate and inhibit the channel via distinct protein phosphorylation (Ashcroft, 2006; Shi et al., 2007a,b; Yang et al., 2008; Orie et al., 2009). Experimental genetic disruption of either KIR6.1 or SUR2B subunits leads to a defective coronary circulation and sudden death (Chutkow et al., 2002; Miki et al., 2002). We have shown that the inhibition of this KATP channel by rosiglitazone disrupted coronary vasodilatory responses to a β-adrenoceptor agonist. As the basal level of channel activity is low under physiological conditions (Quayle et al., 1997; Nichols, 2006), the inhibition of the KATP channel in VSM cells may not produce depolarization sufficient to cause muscle contraction, which could explain the lack of effect of rosiglitazone on basal vascular tone, as shown in previous studies (Walker et al., 1998; Irat et al., 2006). Our results have also shown that rosiglitazone did not affect the basal KIR6.1/SUR2B currents. However, during stimulation of the KIR6.1/SUR2B channel by activation with vasoactive agents, rosiglitazone did impair coronary vasodilation. Under patho-physiological conditions where critical and continuous regulation of the coronary circulation is controlled by the sympathetic/adrenal gland systems, it is very likely that KATP channel inhibition induced by rosiglitazone therapy may compromise coronary circulation under circumstances when vasodilation is essential, such as during elevated metabolic activity, exercise and stress.
Using the Kcnj8−/− mice allowed us to estimate the contributions of both the KATP channel-dependent and the KATP channel-independent effects of rosiglitazone on the coronary circulation. Rosiglitazone (100 µM) inhibited coronary perfusion by ∼19% in Kcnj8−/− mice and ∼68% in the WT mice. This difference indicates that >50% of the isoprenaline-induced coronary vasodilation was inhibited by rosiglitazone via the KATP channel, which is consistent with the idea that the vascular KATP channel is the major target of rosiglitazone.
Our results indicated that the Kir6.1/SUR2B channel was inhibited by rosiglitazone alone with an IC50 of 10 µM but the IC50 was lowered to 2 µM, in the presence of 0.25 µM glibenclamide. Both of these concentrations are within the range of therapeutic concentrations in the treatment of type-2 diabetes mellitus (Coppack et al., 1990; Cox et al., 2000). Many diabetics are prescribed combinations of rosiglitazone and glibenclamide, hence it is likely that the vascular KATP channel is inhibited, at least partially, in these patients.
Interestingly, we found that pioglitazone at 30 µM produced only a modest inhibition of the vascular KATP channel. We were unable to use pioglitazone in higher concentrations as the compound from Sigma and LKT Lab were insoluble even with DMSO as a solvent. As 30 µM pioglitazone is a higher concentration than the blood level in patients given this drug, our results suggest that the vascular KATP channel would not be significantly suppressed by therapeutic concentrations of pioglitazone, consistent with data from several clinical trials (Wong et al., 2004).
In conclusion, the VSM isoform of the KATP channel was inhibited by rosiglitazone. The channel inhibition is membrane-delimited and seems to occur by direct interaction with the channel protein. In the isolated perfused heart, rosiglitazone did not affect the basal coronary circulation but inhibited the β-adrenoceptor agonist-mediated and KIR6.1/SUR2B channel activator-induced coronary vasodilation. The effect is likely to be mediated via the KIR6.1 channel as it was markedly attenuated in hearts from Kcnj8−/− mice. Therefore, KIR6.1/SUR2B channel inhibition may be an important contributory factor in myocardial ischaemia in diabetics who have an underlying cardiovascular condition.
Acknowledgments
This work was supported by the NIH (HD060959), the American Heart Association (09GRNT2010037). L.Y. was partially supported by a scholarship of Harbin Medical University. Y.Y. is a Brains & Behavior fellow of Georgia State University. Thanks for Timothy C. Trower's technique support.
Glossary
Abbreviations
- KATP
ATP-sensitive K+
- VSM
vascular smooth muscles
- WT
wild-type
Conflict of interest
There is no conflict of interest for any author.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 (A) Pinacidil did not activate currents in non-transfected HEK cells. (B) Reversible inhibition of Kir6.1/SUR2B currents. Whole-cell recording was performed in an HEK cell under the conditions shown in Figure 1. The currents activated by pinacidil were inhibited by 30 μM rosiglitazone by ∼60%. The exposure to 10 μM glibenclamide produced further current inhibition. The currents returned to almost the pre-exposure level after washout with pinacidil-containing solution, indicating that inhibition does not result from channel rundown. (C) pinacidil (10 μM and 100 μM did not activate the Kir6.1/SUR2B currents in presence of rosiglitazone, and the channel activation occurred immediately after washout of rosiglitazone.
Figure S2 (A,B) Rosiglitazone in either 30 μM or 100 μM had very little effect on basal perfusion volume.
Figure S3 (A,B) Glibenclamide produced about 85% inhibition of the isoprenaline-induced coronary dilation in WT hearts. (C,D) In the Kcnj8−/− mouse heart, pinacidil did not produce coronary dilation although isoprenaline was capable of increasing the coronary perfusion to a less degree. Glibenclamide failed to reduce the perfusion volume. ***P < 0.001; n = 3.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Abe M, Takiguchi Y, Ichimaru S, Kaji S, Tsuchiya K, Wada K. Different effect of acute treatment with rosiglitazone on rat myocardial ischemia/reperfusion injury by administration method. Eur J Pharmacol. 2008;589:215–219. doi: 10.1016/j.ejphar.2008.05.005. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft FM. From molecule to malady. Nature. 2006;440:440–447. doi: 10.1038/nature04707. [DOI] [PubMed] [Google Scholar]
- Barnett AH. Redefining the role of thiazolidinediones in the management of type 2 diabetes. Vasc Health Risk Manag. 2009;5:141–151. doi: 10.2147/vhrm.s4664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang TJ, Chen WP, Yang C, Lu PH, Liang YC, Su MJ, et al. Serine-385 phosphorylation of inwardly rectifying K+ channel subunit (Kir6.2) by AMP-dependent protein kinase plays a key role in rosiglitazone-induced closure of the K(ATP) channel and insulin secretion in rats. Diabetologia. 2009;52:1112–1121. doi: 10.1007/s00125-009-1337-4. [DOI] [PubMed] [Google Scholar]
- Chutkow WA, Pu J, Wheeler MT, Wada T, Makielski JC, Burant CF, et al. Episodic coronary artery vasospasm and hypertension develop in the absence of Sur2 K(ATP) channels. J Clin Invest. 2002;110:203–208. doi: 10.1172/JCI15672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppack SW, Lant AF, McIntosh CS, Rodgers AV. Pharmacokinetic and pharmacodynamic studies of glibenclamide in non-insulin dependent diabetes mellitus. Br J Clin Pharmacol. 1990;29:673–684. doi: 10.1111/j.1365-2125.1990.tb03688.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox PJ, Ryan DA, Hollis FJ, Harris AM, Miller AK, Vousden M, et al. Absorption, disposition, and metabolism of rosiglitazone, a potent thiazolidinedione insulin sensitizer, in humans. Drug Metab Dispos. 2000;28:772–780. [PubMed] [Google Scholar]
- Duan SZ, Usher MG, Mortensen RM. Peroxisome proliferator-activated receptor-gamma-mediated effects in the vasculature. Circ Res. 2008;102:283–294. doi: 10.1161/CIRCRESAHA.107.164384. [DOI] [PubMed] [Google Scholar]
- Duckworth W, Abraira C, Moritz T, Reda D, Emanuele N, Reaven PD, et al. Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med. 2009;360:129–139. doi: 10.1056/NEJMoa0808431. [DOI] [PubMed] [Google Scholar]
- Eto K, Ohya Y, Nakamura Y, Abe I, Fujishima M. Comparative actions of insulin sensitizers on ion channels in vascular smooth muscle. Eur J Pharmacol. 2001;423:1–7. doi: 10.1016/s0014-2999(01)01047-0. [DOI] [PubMed] [Google Scholar]
- Gerstein HC, Miller ME, Byington RP, Goff DC, Jr, Bigger JT, Buse JB, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008;358:2545–2559. doi: 10.1056/NEJMoa0802743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Home PD, Pocock SJ, Beck-Nielsen H, Gomis R, Hanefeld M, Jones NP, et al. Rosiglitazone evaluated for cardiovascular outcomes–an interim analysis. N Engl J Med. 2007;357:28–38. doi: 10.1056/NEJMoa073394. [DOI] [PubMed] [Google Scholar]
- How OJ, Larsen TS, Hafstad AD, Khalid A, Myhre ES, Murray AJ, et al. Rosiglitazone treatment improves cardiac efficiency in hearts from diabetic mice. Arch Physiol Biochem. 2007;113:211–220. doi: 10.1080/13813450701783281. [DOI] [PubMed] [Google Scholar]
- Irat AM, Aslamaci S, Karasu C, Ari N. Alteration of vascular reactivity in diabetic human mammary artery and the effects of thiazolidinediones. J Pharm Pharmacol. 2006;58:1647–1653. doi: 10.1211/jpp.58.12.0012. [DOI] [PubMed] [Google Scholar]
- Kanda T, Brown JD, Orasanu G, Vogel S, Gonzalez FJ, Sartoretto J, et al. PPARgamma in the endothelium regulates metabolic responses to high-fat diet in mice. J Clin Invest. 2009;119:110–124. doi: 10.1172/JCI36233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul S, Bolger AF, Herrington D, Giugliano RP, Eckel RH. Thiazolidinedione drugs and cardiovascular risks: a science advisory from the American Heart Association and American College Of Cardiology Foundation. J Am Coll Cardiol. 2010;55:1885–1894. doi: 10.1016/j.jacc.2010.02.014. [DOI] [PubMed] [Google Scholar]
- Khandoudi N, Delerive P, Berrebi-Bertrand I, Buckingham RE, Staels B, Bril A. Rosiglitazone, a peroxisome proliferator-activated receptor-gamma, inhibits the Jun NH(2)-terminal kinase/activating protein 1 pathway and protects the heart from ischemia/reperfusion injury. Diabetes. 2002;51:1507–1514. doi: 10.2337/diabetes.51.5.1507. [DOI] [PubMed] [Google Scholar]
- Kilter H, Werner M, Roggia C, Reil JC, Schafers HJ, Kintscher U, et al. The PPAR-gamma agonist rosiglitazone facilitates Akt rephosphorylation and inhibits apoptosis in cardiomyocytes during hypoxia/reoxygenation. Diabetes Obes Metab. 2009;11:1060–1067. doi: 10.1111/j.1463-1326.2009.01097.x. [DOI] [PubMed] [Google Scholar]
- Knock GA, Mishra SK, Aaronson PI. Differential effects of insulin-sensitizers troglitazone and rosiglitazone on ion currents in rat vascular myocytes. Eur J Pharmacol. 1999;368:103–109. doi: 10.1016/s0014-2999(99)00020-5. [DOI] [PubMed] [Google Scholar]
- Li H, Chai Q, Gutterman DD, Liu Y. Elevated glucose impairs cAMP-mediated dilation by reducing Kv channel activity in rat small coronary smooth muscle cells. Am J Physiol Heart Circ Physiol. 2003;285:H1213–H1219. doi: 10.1152/ajpheart.00226.2003. [DOI] [PubMed] [Google Scholar]
- Lu L, Reiter MJ, Xu Y, Chicco A, Greyson CR, Schwartz GG. Thiazolidinedione drugs block cardiac KATP channels and may increase propensity for ischaemic ventricular fibrillation in pigs. Diabetologia. 2008a;51:675–685. doi: 10.1007/s00125-008-0924-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YL, Jimbu YM, Chen Y, Zhao JB, Ye TT, Yang H. The effects of rosiglitazione on renal artery endothelium in diabetic rats. Exp Clin Endocrinol Diabetes. 2008b;116:537–540. doi: 10.1055/s-2008-1058087. [DOI] [PubMed] [Google Scholar]
- Mannucci E, Monami M, Lamanna C, Gensini GF, Marchionni N. Pioglitazone and cardiovascular risk. A comprehensive meta-analysis of randomized clinical trials. Diabetes Obes Metab. 2008;10:1221–1238. doi: 10.1111/j.1463-1326.2008.00892.x. [DOI] [PubMed] [Google Scholar]
- Miki T, Suzuki M, Shibasaki T, Uemura H, Sato T, Yamaguchi K, et al. Mouse model of Prinzmetal angina by disruption of the inward rectifier Kir6.1. Nat Med. 2002;8:466–472. doi: 10.1038/nm0502-466. [DOI] [PubMed] [Google Scholar]
- Mishra SK, Aaronson PI. Differential block by troglitazone and rosiglitazone of glibenclamide-sensitive K(+) current in rat aorta myocytes. Eur J Pharmacol. 1999;386:121–125. doi: 10.1016/s0014-2999(99)00713-x. [DOI] [PubMed] [Google Scholar]
- Nichols CG. KATP channels as molecular sensors of cellular metabolism. Nature. 2006;440:470–476. doi: 10.1038/nature04711. [DOI] [PubMed] [Google Scholar]
- Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457–2471. doi: 10.1056/NEJMoa072761. [DOI] [PubMed] [Google Scholar]
- Orie NN, Thomas AM, Perrino BA, Tinker A, Clapp LH. Ca2+/calcineurin regulation of cloned vascular K ATP channels: crosstalk with the protein kinase A pathway. Br J Pharmacol. 2009;157:554–564. doi: 10.1111/j.1476-5381.2009.00221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potenza MA, Gagliardi S, De Benedictis L, Zigrino A, Tiravanti E, Colantuono G, et al. Treatment of spontaneously hypertensive rats with rosiglitazone ameliorates cardiovascular pathophysiology via antioxidant mechanisms in the vasculature. Am J Physiol Endocrinol Metab. 2009;297:E685–E694. doi: 10.1152/ajpendo.00291.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psaty BM, Furberg CD. The record on rosiglitazone and the risk of myocardial infarction. N Engl J Med. 2007;357:67–69. doi: 10.1056/NEJMe078116. [DOI] [PubMed] [Google Scholar]
- Quayle JM, Nelson MT, Standen NB. ATP-sensitive and inwardly rectifying potassium channels in smooth muscle. Physiol Rev. 1997;77:1165–1232. doi: 10.1152/physrev.1997.77.4.1165. [DOI] [PubMed] [Google Scholar]
- Savoia C, Ebrahimian T, Lemarie CA, Paradis P, Iglarz M, Amiri F, et al. Countervailing vascular effects of rosiglitazone in high cardiovascular risk mice: role of oxidative stress and PRMT-1. Clin Sci (Lond) 2010;118:583–592. doi: 10.1042/CS20090289. [DOI] [PubMed] [Google Scholar]
- Shi W, Cui N, Shi Y, Zhang X, Yang Y, Jiang C. Arginine vasopressin inhibits Kir6.1/SUR2B channel and constricts the mesenteric artery via V1a receptor and protein kinase C. Am J Physiol Regul Integr Comp Physiol. 2007a;293:R191–R199. doi: 10.1152/ajpregu.00047.2007. [DOI] [PubMed] [Google Scholar]
- Shi Y, Wu Z, Cui N, Shi W, Yang Y, Zhang X, et al. PKA phosphorylation of SUR2B subunit underscores vascular KATP channel activation by beta-adrenergic receptors. Am J Physiol Regul Integr Comp Physiol. 2007b;293:R1205–R1214. doi: 10.1152/ajpregu.00337.2007.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Chen X, Wu Z, Shi W, Yang Y, Cui N, et al. cAMP-dependent protein kinase phosphorylation produces interdomain movement in SUR2B leading to activation of the vascular KATP channel. J Biol Chem. 2008a;283:7523–7530. doi: 10.1074/jbc.M709941200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Cui N, Shi W, Jiang C. A short motif in Kir6.1 consisting of four phosphorylation repeats underlies the vascular KATP channel inhibition by protein kinase C. J Biol Chem. 2008b;283:2488–2494. doi: 10.1074/jbc.M708769200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W, Cui N, Wu Z, Yang Y, Zhang S, Gai H, et al. Lipopolysaccharides up-regulate Kir6.1/SUR2B channel expression and enhance vascular KATP channel activity via NF-kappaB-dependent signaling. J Biol Chem. 2010;285:3021–3029. doi: 10.1074/jbc.M109.058313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres Tda S, Aguila MB, Mandarim-de-Lacerda CA. Rosiglitazone reverses cardiac adverse remodeling (fibrosis and vascularization) in perinatal low protein rat offspring. Pathol Res Pract. 2010;206:642–646. doi: 10.1016/j.prp.2010.03.007. [DOI] [PubMed] [Google Scholar]
- Vanasse A, Carpentier AC, Courteau J, Asghari S. Stroke and cardiovascular morbidity and mortality associated with rosiglitazone use in elderly diabetic patients. Diab Vasc Dis Res. 2009;6:87–93. doi: 10.1177/1479164109336047. [DOI] [PubMed] [Google Scholar]
- Walker AB, Naderali EK, Chattington PD, Buckingham RE, Williams G. Differential vasoactive effects of the insulin sensitizers rosiglitazone (BRL 49653) and troglitazone on human small arteries in vitro. Diabetes. 1998;47:810–814. doi: 10.2337/diabetes.47.5.810. [DOI] [PubMed] [Google Scholar]
- Wang X, Wu J, Li L, Chen F, Wang R, Jiang C. Hypercapnic acidosis activates KATP channels in vascular smooth muscles. Circ Res. 2003;92:1225–1232. doi: 10.1161/01.RES.0000075601.95738.6D. [DOI] [PubMed] [Google Scholar]
- Wang K, Zhou Z, Zhang M, Fan L, Forudi F, Zhou X, et al. Peroxisome proliferator-activated receptor gamma down-regulates receptor for advanced glycation end products and inhibits smooth muscle cell proliferation in a diabetic and nondiabetic rat carotid artery injury model. J Pharmacol Exp Ther. 2006;317:37–43. doi: 10.1124/jpet.105.095125. [DOI] [PubMed] [Google Scholar]
- Wang CX, Ding X, Noor R, Pegg C, He C, Shuaib A. Rosiglitazone alone or in combination with tissue plasminogen activator improves ischemic brain injury in an embolic model in rats. J Cereb Blood Flow Metab. 2009;29:1683–1694. doi: 10.1038/jcbfm.2009.87. [DOI] [PubMed] [Google Scholar]
- Wang Y, Lau WB, Gao E, Tao L, Yuan Y, Li R, et al. Cardiomyocyte-derived adiponectin is biologically active in protecting against myocardial ischemia-reperfusion injury. Am J Physiol Endocrinol Metab. 2010;298:E663–E670. doi: 10.1152/ajpendo.00663.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong H, Ozalp Y, Lainesse A, Alpan RS. In vivo bioequivalence of oral antidiabetic agents: pioglitazone tablets. Arzneimittelforschung. 2004;54:618–624. doi: 10.1055/s-0031-1297059. [DOI] [PubMed] [Google Scholar]
- Yang Y, Shi Y, Guo S, Zhang S, Cui N, Shi W, et al. PKA-dependent activation of the vascular smooth muscle isoform of KATP channels by vasoactive intestinal polypeptide and its effect on relaxation of the mesenteric resistance artery. Biochim Biophys Acta. 2008;1778:88–96. doi: 10.1016/j.bbamem.2007.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Shi W, Cui N, Wu Z, Jiang C. Oxidative stress inhibits vascular K(ATP) channels by S-glutathionylation. J Biol Chem. 2010;285:38641–38648. doi: 10.1074/jbc.M110.162578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Shi W, Chen X, Cui N, Konduru AS, Shi Y, et al. Molecular basis and structural insight of vascular KATP channel gating by S-glutathionylation. J Biol Chem. 2011;286:9298–9307. doi: 10.1074/jbc.M110.195123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Zhang Z, Li Z, Feng X, He L, Liu S, et al. Peroxisome proliferator-activated receptor-gamma(PPARgamma) agonist improves coronary artery endothelial function in diabetic patients with coronary artery disease. J Int Med Res. 2010;38:86–94. doi: 10.1177/147323001003800110. [DOI] [PubMed] [Google Scholar]
- Zinn A, Felson S, Fisher E, Schwartzbard A. Reassessing the cardiovascular risks and benefits of thiazolidinediones. Clin Cardiol. 2008;31:397–403. doi: 10.1002/clc.20312. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.