

Abstract
BACKGROUND AND PURPOSE
Studies of the role of the prostaglandin EP2 receptor) have been limited by the availability of potent and selective antagonist tools. Here we describe the in vitro/in vivo pharmacological characterization of a novel EP2 receptor antagonist, PF-04418948 (1-(4-fluorobenzoyl)-3-{[(6-methoxy-2-naphthyl)oxy]methyl} azetidine-3-carboxylic acid).
EXPERIMENTAL APPROACH
Functional antagonist potency was assessed in cell-based systems expressing human EP2 receptors and native tissue preparations from human, dog and mouse. The selectivity of PF-04418948 was assessed against related receptors and a panel of GPCRs, ion channels and enzymes. The ability of PF-04418948 to pharmacologically block EP2 receptor function in vivo was tested in rats.
KEY RESULTS
PF-04418948 inhibited prostaglandin E2 (PGE2)-induced increase in cAMP in cells expressing EP2 receptors with a functional KB value of 1.8 nM. In human myometrium, PF-04418948 produced a parallel, rightward shift of the butaprost-induced inhibition of the contractions induced by electrical field stimulation with an apparent KB of 5.4 nM. In dog bronchiole and mouse trachea, PF-04418948 produced parallel rightward shifts of the PGE2-induced relaxation curve with a KB of 2.5 nM and an apparent KB of 1.3 nM respectively. Reversal of the PGE2-induced relaxation in the mouse trachea by PF-04418948 produced an IC50 value of 2.7 nM. Given orally, PF-04418948 attenuated the butaprost-induced cutaneous blood flow response in rats. PF-04418948 was selective for EP2 receptors over homologous and unrelated receptors, enzymes and channels.
CONCLUSIONS AND IMPLICATIONS
PF-04418948 is an orally active, potent and selective surmountable EP2 receptor antagonist that should aid further elaboration of EP2 receptor function.
LINKED ARTICLE
This article is commented on by Birrell and Nials, pp. 1845–1846 of this issue. To view this commentary visit http://dx.doi.org/10.1111/j.1476-5381.2011.01494.x
Keywords: PGE2 receptors, myometrium, airway smooth muscle
Introduction
Prostaglandin E2 (PGE2) is a metabolite of arachidonic acid, synthesized by the action of cyclooxygenase and PGE synthase. PGE2, which is produced in nearly all organs and tissues, has a variety of physiological effects, including mucosal protection, induction of gastric acid secretion in stomach, generation of fever, hyperalgesia, inflammation and immunity. The actions of PGE2 are mediated by four highly homologous GPCRs, EP1, EP2, EP3 and EP4 (Narumiya et al., 1999; receptor nomenclature follows Alexander et al. 2009). The EP2 and EP4 receptors preferentially couple to Gs proteins to stimulate adenylate cyclase and their activation increases intracellular cAMP levels (Narumiya et al., 1999). In contrast, the EP3 receptor couples to Gi/o proteins, which inhibit adenylate cyclase, and activation of the EP1 receptor increases intracellular Ca2+ levels through Gq (Tabata et al., 2002).
PGE2 has affinity not only for all four EP receptor subtypes but also for other prostanoid receptors, specifically the PGD2 DP1 receptor, and the PGF2α FP receptor (Abramovitz et al., 2000). Many tissues and cell types express heterogeneous prostanoid receptor populations (Sugimoto and Narumiya, 2007). Determining the particular receptor(s) involved in prostanoid-mediated events is complicated by the promiscuity of ligands, similarity in second messenger pathways (Tsuboi et al., 2002) and shortage of selective, well-characterized compounds with which to study independent receptor function. Knock-out mice have been generated for all prostanoid receptors and subtypes (Kobayashi and Narumiya, 2002) and have helped elucidate the physiological and potential pathological roles of this receptor family. However, these studies are limited to the mouse and do not address the potential developmental impact of gene deletion. Selective ligands for the prostanoid receptors are now being reported (Naganawa et al., 2006; Hall et al., 2008; Murase et al., 2008; Maubach et al., 2009; Singh et al., 2009), which will further aid understanding of this diverse receptor family. To date, however, while many groups have used the antagonist 6-isopropoxy-9-oxoxanthene-2-carboxylic acid (AH-6809) to study the EP2 receptor in combination with selective agonists and other prostanoid receptor pharmacological tools, AH-6809 is both non-selective and weakly potent (Abramovitz et al., 2000) and unsuitable for in vivo testing.
Expression of EP2 receptors has been demonstrated in a broad range of cell types and tissues, including lung, gastrointestinal tract, kidney, uterus and thymus (Bastien et al., 1994) and has been linked with PGE2-mediated vasodilation and smooth muscle relaxation in pulmonary, gastrointestinal and reproductive tracts (Coleman et al., 1990). Mouse tracheal rings pre-contracted with carbachol are relaxed by PGE2. This relaxation is absent in EP2 receptor-deficient mice (Tilley et al., 2003). Additionally, PGE2 and other PG receptor agonists have been shown to relax canine airway smooth muscle (Chand and Eyre, 1980; Abela and Daniel, 1995; Catalli et al., 2002), although the prostanoid receptor subtype involved has not been unequivocally determined. Finally, the subtype-selective agonist, butaprost (Abramovitz et al., 2000) has been used to demonstrate the presence of the EP2 receptor mediating relaxation in the human non-pregnant uterus (Senior et al., 1991; Popat and Crankshaw, 2001).
Here we report the in vitro pharmacological characterization of PF-04418948 (1-(4-fluorobenzoyl)-3-{[(6-methoxy-2-naphthyl)oxy]methyl} azetidine-3-carboxylic acid; Figure 1), a potent, selective EP2 receptor antagonist in recombinant cell-based systems, and smooth muscle preparations from human non-pregnant myometrium, dog bronchiole and mouse trachea. We have also assessed the activity of PF-04418948 in vivo in rats
Figure 1.
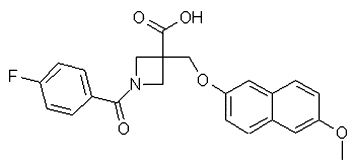
The chemical structure of PF-04418948 (1-(4-fluorobenzoyl)-3-{[(6-methoxy-2-naphthyl)oxy]methyl} azetidine-3-carboxylic acid).
Methods
Selectivity and specificity
Characterization of EP1 and EP3 receptor activity and broad spectrum specificity screening of PF-04418948 was carried out by Cerep SA (http://www.cerep.com). For these as well as other selectivity data cited in Table 1, they represent n = 1 or n = 2 as it is the practice within Pfizer not to generate large numbers of replicate data for compounds, where there is a greater than 100-fold difference with the primary potency. Furthermore, PF-04419848 emerged from an extensive screening campaign for which many more than one example was screened for selectivity, and in all cases this class of compounds were without significant activity at other prostanoid receptors. The IC50 values, EC50 values and Hill coefficients (nH) were determined by non-linear regression analysis of the concentration–response curves using Hill equation curve fitting. In each experiment, the respective reference compound was tested concurrently with PF-04418948 in order to assess the assay suitability and the data were compared with historical values determined at Cerep SA.
Table 1.
Pharmacological potency of PF-04418948 in relevant recombinant systems
| Antagonist activity | Agonist activity | ||
|---|---|---|---|
| Functional reporter assay | KB (nM) | IC50 (nM) | EC50 (nM) |
| Human EP2 | 1.8 (95% CI 0.3–9.3, n = 4) | 16 (95% CI 11.7–21.8, n = 22) | >33 300 (n = 2) |
| Human EP1 | >10 000 (n = 1) | >10 000 (n = 1) | |
| Human EP3 | >10 000 (n = 1) | >10 000 (n = 1) | |
| Human EP4 | >33 300 (n = 2) | >33 300 (n = 2) | |
| Human DP1 | >32 800 (n = 8) | ND | |
| Human CRTH2 | 32 000 (n = 2) | ND |
| Binding | Ki (nM) | ||
|---|---|---|---|
| Human IP | >10 000 (n = 2) | ||
| Human FP | >10 000 (n = 2) | ||
| Human TP | >10 000 (n = 2) | ||
| Human LTB4 | 4800 (n = 1) |
ND, not determined.
Generation of human CHO-EP2, CHO-DP1 and CHO-EP4 receptor cell lines
Human EP2 receptor (GenBank accession NM_000956; Bastien et al., 1994), human DP1 receptor (GenBank accession NM_000953; Boie et al., 1995) and human EP4 receptor (GenBank accession NM_000958; Regan et al., 1994) cDNA were purchased from the UMR cDNA resource centre. A Chinese hamster ovary (CHO) cell line expressing a cAMP response element β-lactamase reporter construct was the host cell in each case. The EP2, DP1 and EP4 CHO lines were generated by screening of stable transformants and grown in Dulbecco's modified Eagle's medium (DMEM), 10% FBS, 2 mM L-glutamine, 0.1 mM non-essential amino acids (NEAA), 0.8 mg·mL−1 Geneticin, 200 µg·mL−1 Zeocin. Cells expressing EP4 receptors were grown in media as above with naproxen (1.6 µM) to prevent receptor desensitization induced by the autocrine release of PGE2.
Functional cAMP assays in recombinant CHO cells
Cells were suspended in DMEM at 1 × 106 cells·mL−1. Stocks of PGE2, BW245C and PF-04418948 (4 mM) were prepared in 100% DMSO and diluted in compound buffer (PBS; 2.5% (v/v) DMSO, 0.15% (v/v) pluronic F-127) to 2 µM final assay concentration. PGE2 was further diluted to 5 nM in PBS. PF-04418948 stock was serially diluted in 100% DMSO. Complete inhibition was determined by 10 µM of a relevant standard (data not shown).
Each independent experiment was run on a separate day with freshly processed compounds. Compounds were diluted 1:40 in compound buffer, and 5 µL transferred to a Lumitrac 200 white plate. Prepared cells (5 µL) were added and incubated at 37°C for 30 min. Agonist (5 µL) was added and incubated for 90 min at 37°C. The plates were then placed at −80°C to lyse the cells. Plates were thawed at 37°C for 15 min. Pre-warmed DiscoveRx assay reagents were added according to the manufacturer's protocol. Plates were incubated in the dark for 4–20 h and read on a LJL Analyst plate reader with visible light. The agonist assay protocol was as the antagonist protocol, except that 5 µL compound buffer was used instead of the agonist stimulation. The CRTH2 receptor antagonist assay was carried out in the DiscoveRx platform as described above, but cells were stimulated with 8 µM forskolin.
Binding assays
Binding selectivity assays for FP, BLT1 (LTB4 receptor) and TP receptors were performed at Cerep SA. Human embryonic kidneys expressing the human IP receptor (GenBank Accession number NM_000960) were generated by Pfizer. Briefly, cells were grown in DMEM, 10% FBS, 600 µg·mL−1 Geneticin, 2 mM L-glutamine and 1 mM sodium pyruvate and harvested in 10 mM HEPES, 1 mM EDTA pH 7.4 at 4°C. Membranes were prepared by Dounce homogenization in 10 mM HEPES, 1 mM EDTA (pH 7.4, 4°C). Membrane aliquots were stored at −80°C before use and diluted in assay buffer to 62.5 µg·mL−1, to give a final assay concentration of 10 µg per well. Compounds were tested at 10 µM [0.1% DMSO in assay buffer (10 mM HEPES, 10 mM MgCl2·6H2O, 1 mM EDTA, pH 7.4)] as the highest assay concentration.
Assay buffer containing 0.5% (w/v) polyethyleneimine (50 µL per well) was added to Packard GF/B filter plates, 20 µL of test compound or 20 µL assay buffer/1% DMSO (totals), or 20 µL of carbacyclin (10 µM in 0.1% (v/v) DMSO, non-specific binding) added manually. [3H] Iloprost (20 µL of 100 nM stock) and finally 160 µL of 62.5 µg·mL−1 membrane were added to start the binding reaction. The plates were centrifuged briefly to bring all the contents to the bottom of the wells (30 s, 100×g), covered and incubated at room temperature for 2 h with shaking. The binding assay was stopped rapidly by filtering through GF/B Unifilter plates using the Brandell harvester. The filters were washed with 3 × 1 mL 4°C assay buffer and then dried at 45°C for approximately 40 min in a drying oven. The bottom of the filter plates was sealed and Microscint ‘0’ (50 µL per well) added and incubated for at least 30 min before reading on an NXT Topcount.
KB methodology
PGE2 was serially diluted in buffer (PBS/0.05% (v/v) Pluronic F-127). PF-04418948 was prepared in diluent (PBS/0.05% (v/v) Pluronic F-127/2.5% DMSO) to 300, 100, 30 and 10 nM. For each concentration of PF-04418948 tested, 5 µL was added to the assay plate in duplicate. Diluent (5 µL) was added to all agonist curves and zero percentage effect control wells. Cells were prepared as described in the general cAMP assay protocol. Cell suspension (5 µL) was added to all wells of the assay plate. The plate was centrifuged at 400×g (1 min) in a benchtop centrifuge prior to incubation for 30 min in a humidified 37°C 5% (v/v) CO2 incubator. Then 5 µL of the agonist concentration–response curve were added manually. The plate was centrifuged at 400×g (1 min), before incubation for 90 min in a humidified 37°C 5% (v/v) CO2 incubator. The plate was placed at −80°C to lyse the cells and the DiscoveRx assay performed as above.
Native tissue assays
All animal care and experimental procedures were in compliance with UK legislation and subject to local ethical review. Human uterine tissue was obtained with the informed consent of donors (aged 24–76) and ethical approval from local hospitals. For studies using dog bronchiole and human myometrial tissues, naproxen (10 µM) was added to the tissue bath. This concentration of naproxen was without clear benefit on the behaviour of mouse tissues and was therefore omitted.
Human myometrium
Isolated myometrial muscle was immersed in Krebs buffer (composition in mM: 118 NaCl, 4.7 KCl, 25 NaHCO3, 0.5 MgSO4·7H2O, 1.2 KH2PO4, 11 glucose, 2.5 CaCl2, 0.01 naproxen) and transported immediately to the laboratory. The myometrium was cleaned of connective tissue, and muscle strips (approximately 2 × 2 mm wide, and 15–20 mm in length) were dissected in parallel to the external capsule. Muscle strips were mounted vertically between two bipolar electrodes in a 15 mL organ bath (37°C) containing oxygenated Krebs buffer. The suture attached to the upper end of the strip was connected to the lever of an isometric force transducer (25 g Maywood Type 49034) and a resting tension of 2500 mg applied. Tissues were washed several times and then left overnight at room temperature to reduce spontaneous activity.
The following morning the baths were re-equilibrated to 37°C, the tissues re-tensioned to 1500 mg and washed at least four times over a period of 60 min. Following equilibration, an electrical field stimulation (EFS) frequency–response curve (FRC) was constructed with a 10 s stimulation every 100 s, at 15 V and 1.5 ms pulse width, at a range of frequencies (0.5, 1, 2, 5, 10, 20, 30, 40 Hz; three stimulations at each frequency). At maximum response, stimulation was halted and the tissues washed three times after 0, 15 and 30 min and left for 30 min. After this period the EFS was recommenced using a sub-maximal (90% of maximum) frequency from the FRC (15 Hz) and left to stabilize for at least 15 min. Once stable responses to EFS were observed, PF-04418948 (10, 100 nM) or vehicle were added and allowed to equilibrate for 60 min. EP2 receptor agonists [PGE2 (0.1 nM–3 µM) or butaprost (0.1 nM–10 µM)] were added cumulatively and left in contact with the muscle strips for a minimum of five EFS responses at each concentration.
Dog bronchiole
Male and female beagle dogs, aged 7–18 months, were killed by an overdose of pentobarbitone. The lungs were excised and placed in oxygenated Krebs buffer (as above with the addition of 1 µM propanolol, 10 µM phentolamine) at room temperature and transported to the laboratory. Slices of lung (3–5 mm) were cut with a scalpel, from which bronchiole rings were carefully dissected. The rings were attached to triangle hooks and connected to force displacement isometric transducers in standard 5 mL organ baths under 1 g tension. Tissues were continuously perfused with modified oxygenated Krebs buffer at 37°C for 60 min.
Then, 80 mM KCl was added to the baths and the tissues allowed to contract to plateau (∼5 min). Tissues were then washed for 30 min. Carbachol (1 µM) was added and after 15 min test, concentrations of PF-04418948 (3–100 nM) or vehicle were added to the tissues. After 1 h equilibration, tissues with a stable contraction were challenged with cumulatively increasing concentrations of PGE2. At the end of the experiment, nifedipine (10 µM) was added to each bath to determine the maximum relaxation.
Mouse trachea
Male C57BL/6 mice, aged 8–10 weeks, from Charles River, were killed using an overdose of pentobarbitone. The trachea was removed using blunt dissection of the tracheal pipe and placed in oxygenated Krebs buffer at room temperature. The connective tissue was removed and each trachea divided into two (about 2 mm in length). Tracheal rings were mounted in standard 5 mL tension myograph baths (Danish Myo Technology, Aarhus, Denmark) containing Krebs buffer aerated with 95% O2/5% CO2 at 37°C, and connected to force displacement isometric transducer heads, under a resting tension of 0.35 g. After 30 min equilibration, carbachol (10 µM) was added and following response plateau (∼35 min), tissues were washed. After 40 min the tracheal rings were again contracted with carbachol (10 µM). Upon attaining a stable plateau, half-log concentrations of PGE2 or vehicle were added cumulatively to the bath to obtain a final assay concentration range of 0.1 nM–30 µM. Following the first cumulative concentration–response curve, the tissues were washed for 1 h. Test concentrations of PF-04418948 (3 nM) or DMSO were equilibrated for 30 min prior to repeating the cumulative PGE2 relaxation curve following pre-contraction with carbachol as described above. At the end of the experiment, forskolin (10 µM) was added to each bath to determine the maximum relaxation.
Potency estimation of PF-04418948 using functional inhibition curve method
The tissues were primed and pre-contracted with carbachol as described above. Upon obtaining a stable contraction, 700 nM PGE2 was added. Once a new plateau was reached, PF-04418948 or vehicle was added to the bath in cumulatively increasing half-log concentrations (0.1 nM–1 µM) or a single concentration of 100 nM PF-04418948.
Analysis of in vitro data
The mean and standard deviation of controls were calculated for each plate; Z Prime was calculated, and then % effect calculated for each compound well as a normalized value between the two means. The % effect values were plotted against log10 concentration of the compound and an unconstrained sigmoid curve was fitted using a four-parameter logistic model. Where this was fitted, any dose–response points whose residuals (distance from curve in y-plane) were determined to be outliers from the fitted curve were excluded, and the curve re-fitted. An IC50 value was reported as the midpoint of the curve in nM. The curve fit was examined along with a number of other measures used to help in the interpretation of the IC50 including R2, sigma, degrees of freedom and monotonicity. IC50 was reported as > the highest concentration tested in the given assay.
Data generated in the cell-based KB experiments were plotted as the log10 concentration of agonist against the per cent response using an in-house Excel add-in to generate sigmoid concentration–effect curves in the absence or presence of varying concentrations of PF-04418948. The curves were unconstrained and fitted by ordinary least squares methods. Clark plots were constructed from the resultant EC50 values and KB estimates obtained using the method of Lew and Angus (1995). This method, which is more theoretically sound than the Schild analysis and gives less emphasis to the control concentration–response curves, was employed as repeat agonist curves which were not possible in this experiment format. Data acquisition from the human myometrium and dog bronchiole studies was by Notocord data capture software (Notocord, Croissy-sur-Seine, France) and from the mouse trachea study was by the Power Lab system using Chart software (ADI Instruments, East Sussex, UK). Raw values were extracted and transferred to Excel at the end of an experiment. All subsequent analyses were performed in an in-house Microsoft Excel add-in package.
Prostanoid agonist curves were normalized to the amplitude of the baseline EFS-induced contraction (human myometrium), the maximum relaxation achieved in the presence of nifedipine (dog bronchiole) or forskolin (mouse trachea). Concentration–effect curves were analysed by least squares, non-linear iterative regression using a Pfizer Excel Add-in program, from which EC50 values were extrapolated.
Estimates of antagonist affinity in the human myometrium and mouse trachea were calculated using the equation pKB= log(CR-1) − log[B] (Schild, 1949), where CR is the concentration ratio calculated from the EC50 in the presence of the antagonist divided by the EC50 value of the agonist alone, KB is the equilibrium dissociation constant and [B] is the concentration of the antagonist. Concentration ratios were calculated between curves obtained from the same donor in human uterus studies and between repeat curves in the same tracheal ring in the mouse studies.
In the dog bronchiole study, estimates of antagonist affinity were calculated using the method of Lew and Angus (1995).
Data from cumulative inhibition curve studies in the mouse trachea were calculated as a percentage of the relaxation to PGE2 (700 nM). Concentration–effect curves were analysed by least squares, non-linear iterative regression using a Pfizer Excel Add-in program, from which IC50 values were extrapolated.
Data points and values in the text and figures represent the mean ± SEM or 95% confidence intervals of n-independent determinations from different animals/donors.
Cutaneous blood flow measurements in vivo
Technical problems associated with cutaneous blood flow studies in the mouse forced these in vivo studies to be performed in the rat. Sprague Dawley rats, supplied by Charles River UK Ltd, Margate, Kent (weight range during study 442–515 g) were dosed orally with either PF-04418948 or vehicle (0.5% w/v methylcellulose + 0.1% v/v Tween 80 in purified water), dose volume 1 mL·kg(1. PF-04418948 exhibited a low clearance (0.3 mL·min−1·kg−1) and a low volume of distribution (0.1 L·kg−1) in the rat, resulting in a terminal half-life of 8.8 h. Oral absorption was high with an oral bioavailability of 78%. Plasma protein binding was observed to be very high in rat (unbound fraction of 0.0003).
Approximately 1 h 25 min post dose, at the approximate peak of PF-04418948 exposure, rats were anaesthetized with 5% isoflurane + 2 L·min−1 oxygen and maintained on 2.5% isoflurane + 2 L·min−1 oxygen. Rats were briefly removed from the anaesthetic to have the abdomen shaved. Baseline scanning laser Doppler (moorLDI2 Imager, Moor Instruments, Axminster, Devon, UK) recordings were taken over a 2.5 × 2.5 cm area of the abdomen every 5 min for a total of 35 min. Following these baseline recordings, 10 µL of 3 µg·mL−1 s.c. of PGE2 (10% ethanol + 90% saline) or 10 µL of 10, 30, 100 or 1000 µg·mL−1 s.c. of butaprost (10% ethanol + 90% saline) was injected into the centre of the scan area. Laser Doppler recordings were taken for a further 60 min.
The Doppler response time course for each dosed rat was adjusted by subtracting the average vehicle + vehicle response at each time point from the response at each time point for the dosed rat from time points 35 min onwards. The area under the curve (AUC) for each rat was then the sum of the adjusted time course for that rat. The peak height for each rat was the maximum value of the adjusted time course for that rat. This gives a single value of AUC and peak height for each rat, which was then analysed using one-way anova.
Materials
PF-04418948 was synthesized by Pfizer Global Research & Development. A description of the discovery and synthesis will be described elsewhere. A cell reporter line expressing human chemoattractant receptor-homologous molecule expressed on Th2 cells (CRTH2) was purchased from Euroscreen. PGE2, BW245C, carbachol, phentolamine, propranolol, naproxen and dimethyl sulphoxide (DMSO), cell dissociation solution and pluronic F-127 were from Sigma (Dorset, UK). [3H] Iloprost was obtained from GE Healthcare (Little Chalfont, Buckinghamshire, UK). PBS (w/o Ca2+, Mg2+), Dulbecco's modified Eagle's medium (DMEM), Ham's F12 Medium, L-glutamine, non-essential amino acids (NEAA), Geneticin and Zeocin were from Invitrogen (Gibco, Paisley, UK). 15R, 15 methyl PGD2, butaprost (free acid) and carbacyclin were from Cayman Chemicals (Ann Arbor, MI, USA), forskolin from Tocris (Bristol, UK) and fetal bovine serum (FBS) from PAA (Pasching, Austria). The DiscoveRx HitHunterTM cAMP II assay kit was from Amersham Biosciences. All drugs were prepared in DMSO. For tissue studies, subsequent dilutions were made in Krebs buffer. Dilutions for cell-based studies are within the text.
Results
Functional activities at recombinant proteins
PGE2 caused a concentration-dependent increase in cAMP in CHO cells expressing the human EP2 receptor, with an EC50 value of 13.8 nM (95% CI 6.0–31.5 nM, n = 4). PF-04418948 did not produce any change in the levels of cAMP in the absence of PGE2 (data not shown). Increasing concentrations of PF-04418948 caused parallel rightward shifts of the PGE2 curve with no significant change in maximum response with a KB value 1.8 nM (Table 1). Analysis using the power departure model of Angus (Lew and Angus, 1995) did not produce a better fit of the data and there was no evidence to reject simple competitive antagonism.
The selectivity of PF-04418948 for the EP2 receptor was tested in functional reporter assays against a panel of homologous GPCRs. EP1, EP3, EP4, CRTH2 and DP1 receptors were selected on the basis of relatively high amino acid identity in the ligand binding domain with the EP2 receptor. PF-04418948 displayed >2000-fold functional selectivity for the human EP2 receptor over antagonist activity against the human EP1, EP3, EP4, DP1 and CRTH2 receptors, as well as binding selectivity over human IP, FP, TP and LTB4 receptors (Table 1). Additionally, the affinity of PF-04418948 over a diverse panel of GPCRs and ion channels was very weak with <30% binding at 10 µM (data not shown).
Functional activity in human myometrium
PF-04418948 was tested for its ability to antagonize the effects of butaprost and PGE2 on an EFS-induced contraction of the human myometrium. The majority of myometrial tissue strips did not show any basal phasic activity following re-equilibration to 37°C after overnight incubation at room temperature. Increasing the stimulation frequency of EFS caused an increase in the amplitude of stimulation-induced phasic contractions, with no alteration in basal tone. Sub-maximal contractions were elicited at 15 Hz; therefore this frequency was used to examine the effect of prostanoid receptor agonists. PGE2 and butaprost caused concentration-dependent inhibition of the EFS-induced contractile response with mean IC50 values of 17.3 nM (data not shown) and 27.8 nM respectively (Figure 2 and Table 2). The mean maximum inhibition of the EFS-induced stimulation produced by PGE2 and butaprost was 74.3 ± 10.4% (data not shown) and 89.7 ± 3% respectively.
Figure 2.
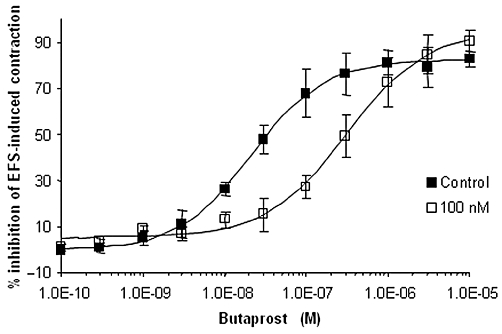
Butaprost (n = 5) caused concentration-dependent inhibition of the EFS-induced contraction of the non-pregnant human myometrium. PF-04418948 (100 nM, n = 4) produced rightward parallel shifts of the butaprost concentration–effect curve.
Table 2.
Effect of PF-04418948 on the butaprost IC50 value for inhibition of EFS-induced contraction of human myometrium
| Butaprost IC50 value (nM) | |
|---|---|
| Control | 27.8 (95% CI 9.1–85, n = 5) |
| 10 nM PF-04418948 | 74 (n = 1) |
| 54 (n = 2) | |
| 100 nM PF-04418948 | 353 (95% CI 78–1594, n = 4) |
PF-04418948 had no effect on the EFS-induced contractions. However, 60 min incubation with 10 or 100 nM PF-04418948 caused parallel rightward shifts in the butaprost concentration–effect curves, with no change in maximum relaxation (Figure 2 and summarized in Table 2) and consistent with a model of simple competitive antagonism. The mean apparent KB for PF-04418948 was 5.4 nM Table 3. PF-04418948 also appeared to competitively inhibit the PGE2-induced relaxation of an EFS-induced myometrial contraction; however, there was greater donor variability in the responses than that observed for the butaprost inhibition (data not shown).
Table 3.
Summary of pharmacological potency values of PF-04418948 in relevant native systems
| Functional native assay | KB value |
|---|---|
| Human myometrium | 5.4 nM (95% CI 1.0–27.9 nM, n = 4) |
| Dog bronchiole | 2.5 nM (95% CI 1.4–4.5 nM, n = 4) |
| Mouse trachea | 1.3 nM (95% CI 0.8–2.4 nM, n = 19) |
Functional activity in dog bronchiole
Carbachol (1 µM) was used to produce a stable contraction in dog bronchiole rings. Increasing concentrations of PGE2 relaxed the smooth muscle rings to 82 ± 5% of the smooth muscle relaxation induced by nifedipine (10 µM; Figure 3A). The EC50 value of PGE2 was 90 nM (95% CI 59.0–137 nM). In preliminary experiments, butaprost inhibited the EFS-induced contraction of the dog bronchiole with an EC50 value of 139 nM (95% CI 11.9–114 nM). This potency did not allow full characterization of PF-04418948, as full curves to butaprost could not be obtained in the presence of 100 nM PF-04418948, due to the weakness of the agonist response (data not shown). PF-04418948 caused rightward shifts of the PGE2 concentration–effect curve with no decrease in maximum response, yielding a KB value of 2.5 nM (Table 3). The effect of increasing concentrations of PF-04418948 on the PGE2-induced relaxation is represented in Figure 3. The corresponding Clark plot (Figure 3B) of the PF-04418948 data was consistent with simple competitive antagonism (Lew and Angus, 1995).
Figure 3.
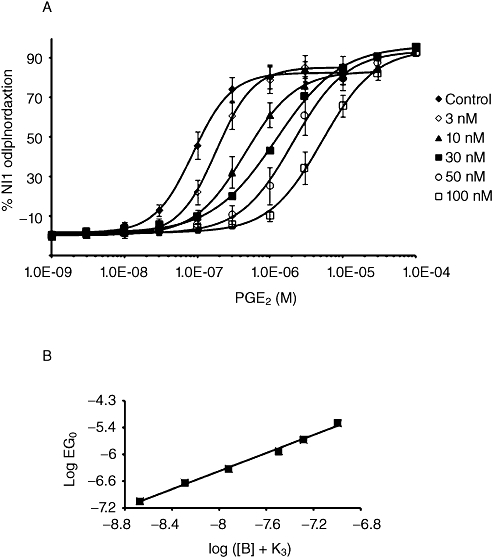
Antagonism by PF-04418948 of the PGE2-induced relaxation of carbachol pre-contracted rings of dog bronchioles (n = 4). (A) PF-04418948 (3–100 nM) produced concentration-dependent rightward parallel shifts of the PGE2 curve. (B) The corresponding Clark plot is consistent with simple competitive antagonism.
Functional activity in mouse trachea
Carbachol (10 µM) was used to produce a sustained, stable contraction in mouse tracheal rings. Increasing concentrations of PGE2 and butaprost caused concentration-dependent relaxation of the pre-contracted tracheal rings with EC50 values of 133 nM (95% CI 103–178 nM, n = 36; Figure 4) and 893 nM (95% CI 568–1408 nM, n = 9; data not shown), respectively with slopes not different from unity. PGE2 and butaprost produced a maximum relaxation, relative to the complete relaxation observed in the presence of 10 µM forskolin, of 65.4 ± 2.3% and 69.1 ± 6.9% respectively. PF-04418948 (1, 3 nM) produced parallel rightward shifts in the PGE2 concentration–effect curve, with no decrease in maximum response (Figure 4). The mean KB value, assuming simple competitive antagonism, was 1.3 nM (Table 3).
Figure 4.
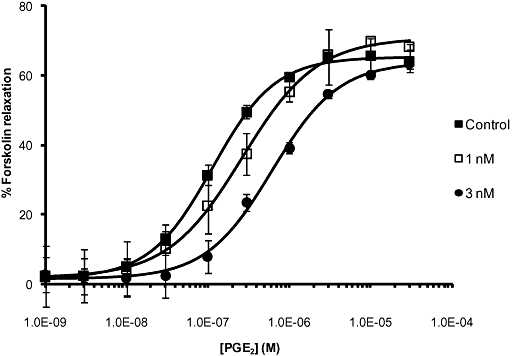
Antagonism by PF-04418948 of the PGE2-induced relaxation of carbachol pre-contracted rings of mouse trachea (n = 26). PF-04418948 (1 and 3 nM) caused concentration-dependent rightward parallel shifts of the PGE2 curve.
Increasing concentrations of PF-04418948 reversed the 700 nM PGE2-induced relaxation of mouse trachea with an IC50 value of 2.7 nM (95% CI 1.8–4.1 nM, n = 7) and a slope of −0.97 (Figure 5). The maximal reversal observed was 73.5 ± 8.8% of the carbachol pre-contractile response. Reversal experiments with 100 nM PF-04418848 demonstrated complete reversal of the PGE2-induced relaxation with a mean value of 106.8 ± 6.5% (n = 4). Vehicle control tissues, following carbachol or carbachol and 700 nM PGE2 challenge, displayed some loss of tone over the period of an inhibition curve (21.6 ± 8.8%, n = 3 and 11.0 ± 3.2% respectively), which could account for this difference. Consequently, it was not possible to calculate a Ki value from these data.
Figure 5.
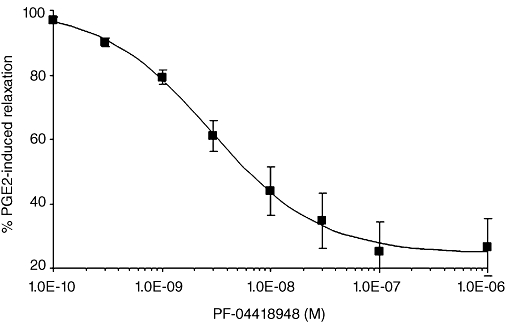
Increasing concentrations of PF-04418948 reversed the relaxation in rings of mouse trachea induced by 700 nM PGE2 (n = 7).
In vivo functional effect on cutaneous blood flow in the rat
The effect of PF-04418948 on PGE2 and butaprost-induced cutaneous blood flow response was investigated in the rat. Local administration of PGE2 induced a significant dose-dependent increase in rat cutaneous blood flow compared with a corresponding vehicle response (Figure 6A and data not shown). PF-04418948 (10 mg·kg−1) was unable to attenuate the cutaneous blood flow response to PGE2 (Figure 6A). In order to eliminate the potential compensation from other receptors sensitive to PGE2, the effect of butaprost on the cutaneous blood flow response was also examined. Butaprost induced a significant dose-dependent increase in rat cutaneous blood flow compared with a corresponding vehicle response, with a maximal effect of 30 µg·mL−1 injected subcutaneously (Figure 6B and data not shown). Escalating oral doses of PF-04418948, administered approximately 1.5 h prior to the butaprost challenge, in order to coincide with the Tmax of PF-04418948 exposure, reduced the peak and AUC butaprost-induced cutaneous blood flow response in a dose-dependent fashion (Figure 6B). PF-04418948 (10 mg·kg−1) reduced the mean cutaneous blood flow peak response and AUC0–60 by 41% and 61% compared with vehicle control respectively. The mean interpolated free plasma exposure of PF-04418948 at this dose was 170 and 94 nM at 1.5 h (at the time of butaprost challenge) and 3 h post-PF-04418948 dose respectively.
Figure 6.
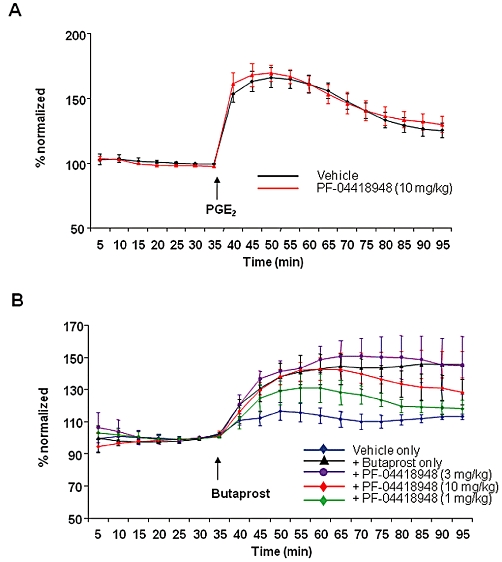
The effects of orally administered PF-04418948 or vehicle on PGE2 (A) and butaprost (B) induced cutaneous blood flow responses in the rat. (A) PGE2 (3 µg·mL−1, s.c.) was used to induce a cutaneous vasodilatory response and the effect of PF-04418948 (10 mg·kg−1, p.o.) on this response was compared with that of vehicle. Data are normalized to the mean of the final three baseline measurements (mean ± SEM, n = 6). (B) Butaprost (30 µg·mL−1, s.c.) or vehicle was used to induce a cutaneous blood flow response and the effect of PF-04418948 (1 mg·kg−1, 3 mg·kg−1, and 10 mg·kg−1, p.o.), compared with vehicle. Data are normalized to the mean of the final three baseline measurements (mean ± SEM, n = 4). The change in cutaneous blood flow response becomes statistically significant (P < 0.05) over vehicle control at 30 min post-butaprost dose.
Discussion
The antagonist AH-6809 has been widely used to study EP2 receptor function, but has both weak affinity and selectivity for the EP2 receptor [EP1 (1.2 µM), EP2 (1.5 µM), EP3 (1.6 µM), DP1 (1.5 µM) and TP (4 µM) according to Abramovitz et al. 2000]. In addition it has poor utility in vivo after oral administration to dissect EP2 receptor involvement in physiological and pathological function. PF-04418948 was identified as a potent and selective EP2 receptor antagonist through a lead identification and medicinal chemistry optimization programme. Human recombinant receptors and relevant smooth muscle tissue preparations from human and other animal species (mouse, rat and dog) were used to characterize the pharmacological profile of PF-04418948. PF-04418948 was shown to be devoid of any agonist activity at the recombinant human EP2 receptor, to competitively antagonize PGE2-mediated cAMP accumulation in an EP2 receptor-specific manner as well as demonstrating greater than 2000-fold potency over homologous receptors. PF-04418948 represents the first potent and selective EP2 receptor antagonist described.
PF-04418948 was shown to potently antagonize the actions of the endogenous ligand, PGE2 as well as the selective EP2 receptor agonist, butaprost, in human myometrium, in a surmountable fashion commensurate with simple, competitive antagonism. Butaprost-induced relaxation of the non-pregnant human myometrium has been used previously to characterize the prostanoid-induced relaxation as being mediated by EP2 receptors (Senior et al., 1991). However, the greater variation seen in the responses to PGE2 over butaprost in this study may be due to the reported heterogeneous responses to PGE2 in the myometrium (Popat and Crankshaw, 2001), reflecting the presence of other prostanoid receptors including EP1 (Senior et al., 1991) and DP (Hillock and Crankshaw, 1999) receptors. The KB value obtained (5.4 nM) was not significantly different to the value obtained in the cAMP accumulation studies with recombinant EP2 receptors (1.8 nM).
Inhaled PGE2 has been shown to inhibit both exercise-induced bronchoconstriction and allergen-induced early and late asthmatic responses and has been considered a potential asthma therapy (Sweatman and Collier, 1968; Pavord et al., 1993). However, these studies have also highlighted the pro-tussive (Costello et al., 1985) and in some instances bronchoconstrictor effects of PGE2 (Mathé and Hedqvist, 1975). Studies with wild-type and EP2 receptor-deficient mice have shown that PGE2 only induces relaxation of denervated tracheal rings and that PGE2-mediated bronchoconstriction occurs indirectly through activation of neural pathways (Tilley et al., 2003). This present study confirmed that the prostanoid receptor mediating relaxation and located post-junctionally in mouse bronchial smooth muscle is the EP2 receptor, as butaprost relaxes the carbachol-contracted smooth muscle and the selective EP2 antagonist PF-0418948 inhibited the PGE2-induced relaxation. Butaprost has a lower affinity than PGE2 for the EP2 receptor (Abramovitz et al., 2000). This was reflected in a lower potency in relaxing the mouse trachea and dog bronchiole, making butaprost an unsuitable agonist for characterizing the pharmacology of PF-04418948.
The heterogeneous expression of prostanoid receptors is evident in the dog airway smooth muscle. It has long been known that prostanoid receptor activation with PGE2 inhibited EFS-induced acetylcholine release in the dog (Zhao et al., 1994; Abela and Daniel, 1995) as well as human (Reinheimer et al., 1998) and guinea pig (Spicuzza et al., 1998), where this has been suggested to be via the EP3 subtype. However, a direct relaxation of cholinergic tone in the dog bronchus by alpha-11-deoxy-11 alpha-(2-hydroxyethylthio)-PGE2 methyl ester (Birnbaum et al., 1981) and 8-isoprostane E1 and E2 (Catalli et al., 2002) suggests the presence of a post-junctional prostanoid receptor mediating the relaxation. Butaprost was not potent enough to allow characterization of the pharmacology of PF-04418948, as was the case with the mouse trachea and consequently studies in the isolated dog bronchiole were evaluated with PGE2. The resultant KB value of PF-04418948 (2.5 nM) was in agreement with that seen in the other studies, confirming the PGE2-induced relaxation of the carbachol pre-contracted dog bronchiole is mediated via the EP2 receptor.
It has been established that PGE2 induces smooth muscle relaxation and contributes to a cutaneous vasodilatory blood flow response on skin which can be measured directly by Doppler flow (Neisius et al., 2002; McCord et al., 2006; Kabashima et al., 2007). The cutaneous PGE2 receptor has not been fully characterized but studies in EP2 receptor knock-out mice have indicated that the cutaneous response to PGE2 might be in part mediated by EP2 receptors (Kabashima et al., 2007). PF-04418948 was tested for its ability to modulate a cutaneous blood flow response induced by PGE2 and butaprost in vivo in the rat. Both agents produced a robust and reproducible cutaneous blood flow response, but PF-04418948 was only able to modulate the response to butaprost, suggesting the cutaneous blood flow response to PGE2 is only in part mediated by EP2 receptors in the rat.
In conclusion, PF-04418948 is the first potent and selective EP2 receptor antagonist described, which demonstrated consistent potency at EP2 receptors across a number of assays, tissues and species. This profile, together with the demonstration that the in vivo butaprost-induced cutaneous blood flow response can be attenuated with PF-04418948, suggests the compound has suitable kinetics and tissue penetration to be a useful tool in elucidating the role of the EP2 receptor in physiological and pathological processes.
Acknowledgments
The authors wish to acknowledge Drs J Gale and P Bungay for critical reading of the manuscript.
Glossary
Abbreviations
- AUC
area under the curve
- CHO
Chinese hamster ovary
- CRTH2
chemoattractant receptor-homologous expressed on Th2 cells receptor
- DMEM
Dulbecco's modified Eagle's medium
- DMSO
dimethyl sulphoxide
- DP1
prostaglandin D2 receptor-1
- EFS
electrical field stimulation
- FBS
fetal bovine serum
- FRC
frequency–response curve
- LTB4
leukotriene B4
- NEAA
non-essential amino acids
Conflicts of interest
KJ af Forselles, J Root, T Clarke, D Davey, K Aughton, K Dack and N Pullen are employees of Pfizer. PF-04418948 is a Pfizer clinical development candidate.
References
- Abela AP, Daniel EE. Neural and myogenic effects of cyclooxygenase products on canine bronchial smooth muscle. Am J Physiol. 1995;268:L47–L55. doi: 10.1152/ajplung.1995.268.1.L47. Part 1. [DOI] [PubMed] [Google Scholar]
- Abramovitz M, Adam M, Boie Y, Carrière M, Denis D, Godbout C, et al. The utilization of recombinant prostanoid receptors to determine the affinities and selectivities of prostaglandins and related analogs. Biochim Biophys Acta. 2000;1483:285–293. doi: 10.1016/s1388-1981(99)00164-x. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edition. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastien L, Sawyer N, Grygorczyk R, Metters KM, Adam M. Cloning, functional expression and characterization of the human prostaglandin E2 receptor EP2 subtype. J Biol Chem. 1994;269:11873–11877. [PubMed] [Google Scholar]
- Birnbaum JE, Birkhead NC, Oronsky AL, Dessy F, Rihoux JP, VanHumbeeck L. Bronchodilator activity of a PGE2 analog in animals and in man. Prostaglandins. 1981;21:457–469. doi: 10.1016/0090-6980(81)90091-5. [DOI] [PubMed] [Google Scholar]
- Boie Y, Sawyer N, Slipetz DM, Metters KM, Abramovitz M. Molecular cloning and characterization of the human prostanoid DP receptor. J Biol Chem. 1995;270:18910–18916. doi: 10.1074/jbc.270.32.18910. [DOI] [PubMed] [Google Scholar]
- Catalli A, Zhang D, Janssen LJ. Receptors and signaling pathway underlying relaxations to isoprostanes in canine and porcine airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2002;283:L1151–L1159. doi: 10.1152/ajplung.00038.2002. [DOI] [PubMed] [Google Scholar]
- Chand N, Eyre P. Further studies of dog tracheobronchial smooth muscle. Res Commun Chem Pathol Pharmacol. 1980;28:245–254. [PubMed] [Google Scholar]
- Coleman RA, Kennedy I, Humphrey PPA, Bunce K, Lumley P. Prostanoids and their receptors. In: Emmett JC, editor. Comprehensive Medicinal Chemistry, Membranes and Receptors. Oxford: Pergamon Press; 1990. p. 643. [Google Scholar]
- Costello JF, Dunlop LS, Gardiner PJ. Characteristics of prostaglandin induced cough in man. Br J Clin Pharmacol. 1985;20:355–359. doi: 10.1111/j.1365-2125.1985.tb05077.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall A, Billinton A, Brown SH, Clayton NM, Chowdhury A, Giblin GM, et al. Non-acidic pyrazole EP1 receptor antagonists with in vivo analgesic efficacy. Bioorg Med Chem Lett. 2008;18:3392–3399. doi: 10.1016/j.bmcl.2008.04.018. [DOI] [PubMed] [Google Scholar]
- Hillock CJ, Crankshaw DJ. Inhibitory prostanoid EP receptors in human non-pregnant myometrium. Eur J Pharmacol. 1999;378:99–108. doi: 10.1016/s0014-2999(99)00442-2. [DOI] [PubMed] [Google Scholar]
- Kabashima K, Nagamachi M, Honda T, Nishigori C, Miyachi Y, Tokura Y, et al. Prostaglandin E2 is required for ultraviolet B-induced skin inflammation via EP2 and EP4 receptors. Lab Invest. 2007;87:49–55. doi: 10.1038/labinvest.3700491. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Narumiya S. Function of prostanoid receptors: studies on knockout mice. Prostaglandins Other Lipid Mediat. 2002;507:557–573. doi: 10.1016/s0090-6980(02)00055-2. [DOI] [PubMed] [Google Scholar]
- Lew MJ, Angus JA. Analysis of competitive agonist-antagonist interactions by nonlinear regression. Trends Pharmacol Sci. 1995;16:328–337. doi: 10.1016/s0165-6147(00)89066-5. [DOI] [PubMed] [Google Scholar]
- Mathé AA, Hedqvist P. Effect of prostaglandins F2 alpha and E2 on airway conductance in healthy subjects and asthmatic patients. Am Rev Respir Dis. 1975;111:313–320. doi: 10.1164/arrd.1975.111.3.313. [DOI] [PubMed] [Google Scholar]
- Maubach KA, Davis RJ, Clark DE, Fenton G, Lockey PM, Clark KL, et al. BGC20-1531, a novel, potent and selective prostanoid EP receptor antagonist: a putative new treatment for migraine headache. Br J Pharmacol. 2009;156:316–327. doi: 10.1111/j.1476-5381.2009.00027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCord GR, Cracowski JL, Minson CT. Prostanoids contribute to cutaneous active vasodilation in humans. Am J Physiol Regul Integr Comp Physiol. 2006;291:R596–R602. doi: 10.1152/ajpregu.00710.2005. [DOI] [PubMed] [Google Scholar]
- Murase A, Taniguchi Y, Tonai-Kachi H, Nakao K, Takada J. In vitro pharmacological characterization of CJ-042794, a novel, potent, and selective prostaglandin EP(4) receptor antagonist. Life Sci. 2008;82:226–232. doi: 10.1016/j.lfs.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Naganawa A, Matsui T, Ima M, Saito T, Murota M, Aratani Y, et al. Further optimization of sulfonamide analogs as EP1 receptor antagonists: synthesis and evaluation of bioisosteres for the carboxylic acid group. Bioorg Med Chem. 2006;16:7121–7137. doi: 10.1016/j.bmc.2006.06.067. [DOI] [PubMed] [Google Scholar]
- Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiol Rev. 1999;79:1193–1226. doi: 10.1152/physrev.1999.79.4.1193. [DOI] [PubMed] [Google Scholar]
- Neisius U, Olsson R, Rukwied R, Lischetzki G, Schmelz M. Prostaglandin E2 induces vasodilation and pruritus, but no protein extravasation in atopic dermatitis and controls. J Am Acad Dermatol. 2002;47:28–32. doi: 10.1067/mjd.2002.120462. [DOI] [PubMed] [Google Scholar]
- Pavord ID, Wong CS, Williams J, Tattersfield AE. Effect of inhaled prostaglandin E2 on allergen-induced asthma. Am Rev Respir Dis. 1993;148:87–90. doi: 10.1164/ajrccm/148.1.87. [DOI] [PubMed] [Google Scholar]
- Popat A, Crankshaw DJ. Variable responses to prostaglandin E in human non-pregnant myometrium. Eur J Pharmacol. 2001;416:145–152. doi: 10.1016/s0014-2999(01)00852-4. [DOI] [PubMed] [Google Scholar]
- Regan JW, Bailey TJ, Pepperl DJ, Pierce KL, Bogardus AM, Donello JE, et al. Cloning of a novel human prostaglandin receptor with characteristics of the pharmacologically defined EP2 subtype. Mol Pharmacol. 1994;46:213–220. [PubMed] [Google Scholar]
- Reinheimer T, Harnack E, Racke K, Wessler I. Prostanoid receptors of the EP3 subtype mediate inhibition of evoked [3H]acetylcholine release from isolated human bronchi. Br J Pharmacol. 1998;125:271–276. doi: 10.1038/sj.bjp.0702057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schild HO. pAx and competitive drug antagonism. Br J Pharmacol Chemother. 1949;4:277–280. doi: 10.1111/j.1476-5381.1949.tb00548.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senior J, Marshall K, Sangha R, Baxter GS, Clayton JK. In vitro characterization of prostanoid EP-receptors in the non-pregnant human myometrium. Br J Pharmacol. 1991;102:747–753. doi: 10.1111/j.1476-5381.1991.tb12244.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J, Zeller W, Zhou N, Hategen G, Mishra R, Polozov A, et al. Antagonists of the EP(3) receptor for prostaglandin E(2) are novel antiplatelet agents that do not prolong bleeding. ACS Chem Biol. 2009;4:115–126. doi: 10.1021/cb8002094. [DOI] [PubMed] [Google Scholar]
- Spicuzza L, Giembycz MA, Barnes PJ, Belvisi MG. Prostaglandin E2 suppression of acetylcholine release from parasympathetic nerves innervating guinea-pig trachea by interacting with prostanoid receptors of the EP3-subtype. Br J Pharmacol. 1998;123:1246–1252. doi: 10.1038/sj.bjp.0701720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto Y, Narumiya S. Prostaglandin E receptors. J Biol Chem. 2007;282:11613–11617. doi: 10.1074/jbc.R600038200. [DOI] [PubMed] [Google Scholar]
- Sweatman WJ, Collier HO. Effects of prostaglandins on human bronchial muscle. Nature. 1968;217:69. doi: 10.1038/217069a0. [DOI] [PubMed] [Google Scholar]
- Tabata H, Tanaka S, Sugimoto Y, Kanki H, Kaneko S, Ichikawa A. Possible coupling of prostaglandin E receptor EP(1) to TRP5 expressed in Xenopus laevis oocytes. Biochem Biophys Res Commun. 2002;298:398–402. doi: 10.1016/s0006-291x(02)02455-5. [DOI] [PubMed] [Google Scholar]
- Tilley SL, Hartney JM, Erikson CJ, Jania C, Nguyen M, Stock J, et al. Receptors and pathways mediating the effects of prostaglandin E2 on airway tone. Am J Physiol Lung Cell Mol Physiol. 2003;284:L599–L606. doi: 10.1152/ajplung.00324.2002. [DOI] [PubMed] [Google Scholar]
- Tsuboi K, Sugimoto Y, Ichikawa A. Prostanoid receptor subtypes. Prostaglandins Other Lipid Mediat. 2002;68–69:535–556. doi: 10.1016/s0090-6980(02)00054-0. [DOI] [PubMed] [Google Scholar]
- Zhao WW, Robinson NE, Yu MF. PGE2 inhibits acetylcholine release from cholinergic nerves in canine but not equine airways. Prostaglandins Leukot Essent Fatty Acids. 1994;51:347–355. doi: 10.1016/0952-3278(94)90007-8. [DOI] [PubMed] [Google Scholar]