

Summary
Transposons evolve rapidly and can mobilize and trigger genetic instability. piRNAs silence these genome pathogens, but it is unclear how the piRNA pathway adapts to invasion of new transposons. In Drosophila, piRNAs are encoded by heterochromatic clusters and maternally deposited in the embryo. Paternally inherited P-element transposons thus escape silencing and trigger a hybrid sterility syndrome termed P-M hybrid dysgenesis. We show that P-M hybrid dysgenesis activates both P-elements and resident transposons, and disrupts the piRNA biogenesis machinery. As dysgenic hybrids age, however, fertility is restored, P-elements are silenced, and P-element piRNAs are produced de novo. In addition, the piRNA biogenesis machinery assembles and resident elements are silenced. Significantly, resident transposons insert into piRNA clusters, and these new insertions are transmitted to progeny, produce novel piRNAs, and are associated with reduced transposition. P-element invasion thus triggers heritable changes in genome structure that appear to enhance transposon silencing.
Introduction
Transposons are major structural components of eukaryotic genomes, and mobilization and expansion of these elements can lead to mutations that cause disease, alter gene expression, and may drive evolution (Bennetzen, 2000; Britten, 2010; Hedges and Belancio, 2011). PIWI-clade Argonaute proteins, guided by 23–30 nt piRNAs, function as sequence specific nucleases in vitro and have an evolutionarily conserved role in transposon silencing in vivo, during germline development(Aravin et al., 2007; Ghildiyal and Zamore, 2009; Khurana and Theurkauf, 2010). In Drosophila, piRNAs produced during oogenesis are maternally deposited in the embryo, where they appear to epigenetically silence transposons (Aravin et al., 2003; Brennecke et al., 2007; Malone et al., 2009; Nishida et al., 2007). Transposons that are present in the male genome but absent from the female genome thus escape silencing in the germline of hybrid progeny, leading to an adult sterility syndrome termed hybrid dysgenesis(Bucheton, 1973; Bucheton et al., 1976; Hiraizumi, 1971; Kidwell et al., 1977; Picard et al., 1972). P-elements are DNA transposons that spread through wild populations of Drosophila melanogaster after most common laboratory strains were isolated, in the early 20th century(Kidwell et al., 1977). Wild stocks carrying P-elements are referred to as P strains and lab stocks that lack these elements are referred to as M strains (Kidwell et al., 1977; Rubin et al., 1982). Crosses between P strain males and M strain females thus lead to P-M hybrid dysgenesis, which is characterized by P-element mobilization and reduced fertility in F1 progeny. Reciprocal crosses between P strain females and M strain males produce genetically identical female progeny, but these hybrids are viable and fertile due to maternal deposition of P-element piRNAs that appear to epigenetically silence target elements (Brennecke et al., 2008).
Transposons can be transmitted horizontally and spread through interbreeding (Kidwell, 1985, 1992), but it is unclear how new invading elements are silenced. The female progeny of males carrying P-element transposons and naïve females are initially sterile, but the fertility of these hybrids increases with age, suggesting that silencing can be established in a single generation (Bucheton, 1979; Bucheton and Picard, 1975; Kidwell et al., 1977). We have therefore used P-M hybrid dysgenesis in the female germline to explore the mechanisms that drive adaptation to transposon invasion.
Results
To induce hybrid dysgenesis, we crossed w1 females (an M strain) to Harwich (Har) males (a reference P strain) and analyzed the resulting female progeny (w1 × Har; Figure 1A). As a control, we crossed Har females to w1 males, which generated genetically identical F1 hybrids (Har × w1; reciprocal hybrids) that inherit P-element piRNAs from the Har mothers (Figure 1A). We then assayed egg production, eggshell patterning, and hatch rates as a function of F1 hybrid adult age (Figure 1B–D). Newly eclosed females from the reciprocal cross were fertile, produced over 50 eggs/day by day 2, and continued high-level egg production for 3 weeks (Figure 1B, upper graph). By contrast, 2–4 day old dysgenic females produced less than 0.5 eggs/day (Figure 1B, lower graph). None of these eggs hatched, and most showed fused dorsal appendages (Figure 1C, D), which can result from germline DNA damage and transposon mobilization (Chen et al., 2007; Klattenhoff et al., 2007; Pane et al., 2007). Consistent with very low egg production, most of the young dysgenic females contained only rudimentary ovaries. Over a three-week period, however, the fertility of the dysgenic females progressively improved. Between 2–4 days and 21 days, egg production increased from 0.5 eggs/female/day to 2.5 eggs/female/day (Figure 1B), embryo hatch rates increased from 3% to 52% (Figure 1C), and production of eggs with normal dorsal appendages increased from 32% to 92% (Figure 1D).
Figure 1.

Phenotypic adaptation to P-element invasion. A. Diagram of crosses used to produce dysgenic (w1 × Har) and control hybrids (Har × w1). Har is a wild type P strain harboring P-elements and w1 is a laboratory M stain that lacks these transposons. B–D. Egg production (B), hatch rate (C) and fraction of eggs showing wild type dorsal-ventral patterning (D) as a function of hybrid age. Control reciprocal hybrids (blue). Dysgenic hybrids (red). E. DNA damage in hybrid ovaries. Egg chambers were labeled for γ-H2Av, a modified histone associated with DNA breaks, and for DNA. Oocyte nuclei in 2–4 day old reciprocal hybrids do not label for γ-H2Av (Har × w1 2–4 day, arrow). Oocyte nuclei in 2–4 day old dysgenic hybrids, by contrast, show prominent γH2Av foci (w1 × Har 2–4 day, arrow). Oocyte nuclei in 21 day dysgenic hybrids do not label for γH2Av (w1 × Har 21 day, arrow). Also see Figure S1.
To determine if the sterility of young P-M dysgenic hybrid females is associated with DNA damage, which can result from P-element mobilization, we used immunofluorescence labeling and confocal microscopy to assay for γH2Av, a histone modification linked to double stranded DNA breaks (Madigan et al., 2002). Drosophila ovaries are composed of parallel bundles of ovarioles containing developmentally staged egg chambers, and oogenesis is initiated at the anterior tip of the ovariole in the germarium (Spradling, 1993). In wild type ovaries, γH2Av foci are present in region 2 of the germarium, where meiotic breaks are formed, and in nurse cell nuclei of later egg chambers, which are undergoing endo-reduplication (McKim et al., 2002). At the time egg chambers bud from the germarium (stage 2), oocyte nuclei have repaired meiotic breaks and γH2Av foci are not detected, and the somatic follicle cells show only low levels of γH2Av accumulation (Figure 1E). In 2–4 day old dysgenic egg chambers, by contrast, 26 of 28 oocyte nuclei showed prominent γH2Av foci, and the somatic follicle cells showed increased labeling for γH2Av. At 21 days, however, only 5 of 40 oocyte nuclei were positive for γH2Av foci, and follicle cell labeling was comparable to reciprocal controls (Figure 1E). Hybrid dysgenesis does not appear to mobilize P-elements in most somatic lineages, where alternative splicing suppresses production of functional transposase (Rio, 1991). However, our observations suggest that P-M dysgenesis may activate transposition in the somatic follicle cells of the ovary.
To further analyze recovery of egg production in dysgenic hybrids, we quantified the population of ovarioles by egg chambers as a function of F1 hybrid age (Figure S1). Wild type ovaries are composed of 14 to 16 ovarioles. Each ovariole has a single germarium that carries germline and somatic stem cell pools. Division of these stem cells drives production of egg chambers that bud from the germarium and fill the ovariole (Spradling, 1993). 87% of the ovaries from 2–4 day old dysgenic females were rudimentary and lacked any ovarioles populated by egg chambers. Of 192 2–4 day old ovaries examined, only one contained a single ovariole with >5 egg chambers. At 21 days, 72% of ovaries were rudimentary and 28% contained ovarioles with 5 or more developmentally staged egg chambers (Figure S1). These findings are consistent with earlier studies indicating that fertility is restored in only a subpopulation of ovarioles through a process that appears to be stochastic (Bucheton, 1979). All of ovarioles that emerged contained a series of developmentally staged egg chambers, and production of these egg chambers requires ongoing germline and somatic stem cell division. Adaptation to P-element invasion thus appears to be a stochastic process that may occur in the stem cells.
Organization of the silencing machinery
A number of proteins required for piRNA biogenesis and transposon silencing are present in nuage, a germline-specific structure associated with nuclear pores (Eddy, 1974, 1975; Lim and Kai, 2007). For example, Vasa is a germline-specific DEAD box protein required for piRNA biogenesis and transposon silencing that also appears to be a core component of nuage (Liang et al., 1994; Malone et al., 2009). In ovaries from control reciprocal hybrids, Vasa localized to nuage, which forms distinct perinuclear foci (Figure 2A, 2–4 Day Har × w1). In ovaries isolated from young dysgenic hybrids, relatively few ovarioles contained well-defined egg chambers. In the egg chambers that were present, Vasa was diffusely localized to the cytoplasm (Figure 2A, 2–4 Day, w1 × Har). At 21 days, by contrast, Vasa localized to nuage in ovaries from both reciprocal and dysgenic females. In addition, Vasa localized to the posterior pole of later stage oocytes, where it assembles into pole plasm (Figure 2B).
Figure 2.

Vasa localization and expression during hybrid dysgenesis. A. At 2–4 days, only low levels of Vasa is present in the germarium of dysgenic hybrids (2–4 Day, w1 × Har), while Vasa is dispersed in the cytoplasm and concentrated in perinuclear foci (nuage) in reciprocal hybrid controls (2–4 Day, Har × w1). B. At 21 days, by contrast, Vasa is present in the cytoplasm and nuage in both dysgenic hybrids and reciprocal control. Vasa also shows the expected accumulation at the posterior pole of stage 10 oocytes (right panels). C. Western blot for Vasa in control and dysgenic ovaries. At 2–4 and 21 days, reciprocal hybrids express a species of the expected 72 kD apparent MW. 2–4 day old dysgenic hybrids, by contrast, express low levels of this species and a prominent higher mobility band. At 21 days, however, the 72 kD band is restored. In panels A and B, the distribution of Vasa is shown on the left and a merge of Vasa (green), DNA (Blue) and Lamin-C (red) is on the right. Note that Lamin-C is prominent in the terminal filament cells that occupy the anterior tip of the germarium and in the stalk cells located between egg chambers. Scale bars = 20 μm.
Figure 4.
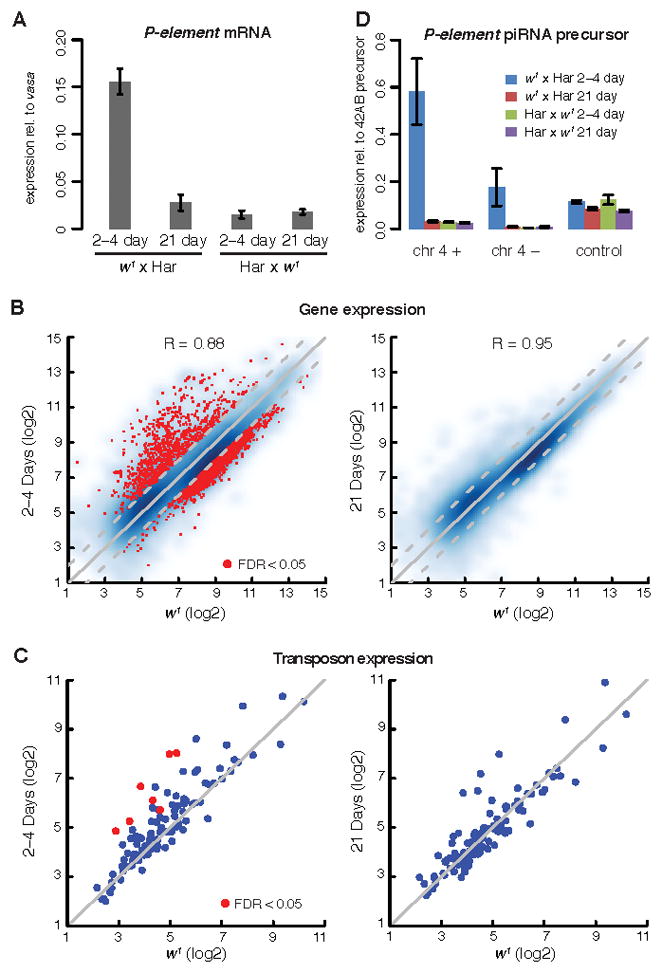
Transposon, gene and piRNA precursor expression. A. P-element transcript levels in dysgenic hybrids (w1 × Har) and reciprocal controls (Har × w1). P-element transcript levels were measured by qPCR and are expressed relative to an internal vasa mRNA control. Whole genome tiling array analysis of gene (B) and transposon family (C) expression in 2–4 and 21 day old dysgenic ovaries relative to w1 controls. Each point represents a single transposon family or protein coding gene. Points above the diagonal are over-expressed in dysgenic hybrids. Points in red indicate significant over-expression (FDR<0.05). R=Correlation coefficient. D. Transcript levels form both strands of a 4th chromosome piRNA cluster carrying a paternally inherited P-element insertion. RNA was measured by strand specific RT-qPCR using primers that span the P-element insertion site. Transcripts from both strands were significantly elevated in 2–4 day old dysgenic hybrids relative to reciprocal controls. At 21 days, by contrast, dysgenic hybrids and reciprocal controls express similar levels of cluster transcripts. Transcript levels for an unlinked piRNA cluster on chromosome 2L (control) were not altered in the dysgenic hybrids. Bar graphs display mean and standard deviation. Also see Figure S2 and Table S1.
To determine if other piRNA pathway components are disrupted during hybrid dysgenesis, we assayed localization of the PIWI clade proteins Ago3, Aub and Piwi, which bind piRNAs and catalyze sequence-specific target cleavage (Figure 3). In 2–4 day and 21-day reciprocal controls, Aub and Ago3 show the expected localization to the perinuclear nuage (Figure 3A, D, G), and Piwi accumulates in germline and somatic cell nuclei (Figure 3B, E, H). By contrast, in 2–4 day old dysgenic ovaries Aub and Ago3 were dispersed in the cytoplasm and Piwi did not accumulate in germline or somatic nuclei (Figure 3A, B, C). However, all three proteins showed wild type localization in 21-day-old dysgenic hybrids (Figure 3D–I). Hybrid dysgenesis thus transiently disrupts sub-cellular organization of the germline and somatic transposon silencing machinery.
Figure 3.

Organization of the piRNA processing machinery. Dysgenic and reciprocal control ovaries were labeled for the PIWI proteins Ago3 (A, D, G), Piwi (B, E, H), and Ago3 (C, F, I). In 2–4 day and 21 day old reciprocal Har × w1 controls, Aub and Ago3 show the expected localization to nuage, and Piwi is concentrated in nuclei. In 2–4 day old w1 × Har dysgenic ovaries, by contrast, Aub, Ago3 and Piwi are dispersed (A–C). In 21 day w1 × Har dysgenic hybrids, by contrast, localization of all three proteins is comparable to reciprocal controls. Pairs of images show the distribution of the indicated PIWI protein on the left and a merge of the PIWI protein (green), DNA (blue), and Lamin-C (red) on the right. Scale bars = 10 μm.
Mutations that lead to germline DNA damage trigger Chk2-dependent phosphorylation of Vasa(Abdu et al., 2002; Klattenhoff et al., 2007). To determine if hybrid dysgenesis leads to modification of Vasa, we assayed protein mobility by SDS-PAGE and Western blotting. These studies revealed the expected 72 kD species in 2–4 day reciprocal hybrids, but only low levels of the full-length protein in the 2–4 day old dysgenic ovaries, which also expressed a prominent species with higher electrophoretic mobility (Figure 2C). At 21 days, however, similar levels of full-length Vasa were detected in both dysgenic and reciprocal hybrid samples (Figure 2C). An identical pattern was observed in three independent experiments, suggesting that P-M hybrid dysgenesis leads to a transient destabilization of Vasa.
Transposon silencing
To determine if P-elements are silenced as dysgenic F1 hybrids age, we measured transcript levels by quantitative reverse transcriptase PCR (qRT-PCR). Because germline content increases as the dysgenic hybrids age, we measured P-element transcript levels relative to vasa mRNA, which is germline specific. Consistent with the phenotypic observations described above, P-element transcript levels in 2–4 day dysgenic females were 10.4 fold higher than in reciprocal controls (Figure 4A, t-test p-value = 1.51×10−3). At 21-days, by contrast, P-element transcript levels in dysgenic hybrids had dropped and were not significantly different from reciprocal controls (Figure 4A, t-test p-value = 0.18).
To assay for global changes in gene and resident transposon expression, we used whole genome tiling arrays. In 2–4 day dysgenic ovaries, 2,158 protein coding genes showed >2-fold decreased expression (false discover rate or FDR<0.05) and 1,218 genes showed >2-fold increased expression (FDR< 0.05) (Figure 4B, left graph). The over-expressed genes were strongly enriched in the Gene Ontology (GO) terms DNA replication, nuclear division and chromosome organization, and the under-expressed genes were enriched in the GO terms extracellular matrix, plasma membrane, and mesoderm development (Table S1). These changes may reflect arrest of the dysgenic ovaries prior to germline expansion and endo-reduplication of nurse cell nuclei, and the resulting increased fraction of somatic tissue, including the muscle sheath, that surrounds the ovary. By contrast, gene expression in 21-day-old dysgenic ovaries was comparable to controls, and this correlates with expansion of the germline and increased egg production (Figure 4B, right graph; Figure S2).
These studies also revealed a global increase in resident transposon expression in 2–4 day old dysgenic females, with seven families showing significant increases relative to control ovaries (Figure 4C left, FDR<0.05). By contrast, no resident transposon families showed a statistically significant increase in expression in 21-day-old dysgenic ovaries at FDR <0.05 (Figure 4C right). An example of transient 297 element activation is illustrated in the Genome Browser screen shot shown in Figure S2. piRNAs appear to be amplified though cleavage of target elements, and the piRNAs matching 297 are highly enriched in the germline(Malone et al., 2009). Young dysgenic ovaries, by contrast, are dominated by somatic tissue. Transposon over-expression in the young dysgenic ovaries is therefore unlikely to result from changes in tissue distribution. Instead, hybrid dysgenesis appears to trigger transient over-expression of resident transposon families, which is temporally associated with defects in the organization of piRNA pathway components and reduced Vasa protein expression (Figures 2 and 3).
De novo piRNA production
Maternally deposited P-element piRNAs appear to epigenetically transmit silencing activity (Blumenstiel and Hartl, 2005; Brennecke et al., 2008; Rozhkov et al., 2010), and w1 is an M strain that lacks P-elements (see below). To determine if age-dependent silencing of P-elements in dysgenic hybrids is linked to de novo piRNA production, we therefore deep sequenced ovarian small RNAs from 2–4 day and 21-day-old dysgenic females, and from age-matched reciprocal controls (Har × w1). P-element piRNAs were abundant in 2–4 day old reciprocal hybrids, which inherited P-element piRNAs from Har mothers (Figure 5C). Ovaries from 2–4 day old dysgenic hybrids, by contrast, contained only low levels of P-element matching piRNAs (Figure 5A). At 21-days, however, dysgenic and reciprocal control ovaries expressed comparable levels of P-element piRNAs (Figure 5B and D; P-element, Figure S3A and B). In addition, the P-element piRNAs from opposite strands showed a significant bias toward a 10 nt overlap (z-score = 7.8), which is the hallmark of ping-pong amplification (P-element, Figures S3A and B). Dysgenic hybrid ovaries thus produce and amplify P-element piRNAs in the absence of a maternally supplied primary piRNA trigger.
Figure 5.

P-element and resident element piRNA expression. piRNA expression was determined by deep sequencing, normalizing for sequencing depth (see Experimental Procedures). A. 2–4 day old dysgenic ovaries (w1 × Har) express low levels of P-element piRNAs relative to reciprocal controls (C, Har × w1). At 21 days, by contrast, dysgenic (B) and control (D) ovaries express P-element piRNAs at similar levels. E. Expression of piRNAs matching shared resident elements in 2–4 and 21 day old hybrids, relative to control hybrids. Group III elements, which are enriched in somatic follicle cells, are in red. Group I elements, which are enriched in the germline, are in black. Group II elements show a sense strand bias and are in green. At 2–4 days, dysgenic ovaries over-express group III piRNA and under-express group I piRNAs. This likely reflects developmental arrest prior to germline expansion. At 21 days, by contrast, wild type piRNA expression is restored. Also see Figure S3.
Our sequencing studies also showed that 2–4 day old dysgenic hybrids and reciprocal controls expressed similar total piRNA levels. However, piRNAs linked to Group III transposon families, which are enriched in the soma, were elevated in young dysgenic hybrids (Figure 5E, 2–4 day, red points). By contrast, piRNAs linked to the germline-enriched Group I elements were reduced (Figure 5E, 2–4 day, black points). As fertility is restored and the germline expands, the balance of Group I and III piRNAs is restored (Figure 5E, 21 day). This pattern of altered piRNA expression may be linked to the over-abundance of somatic tissue in the rudimentary ovaries, which dominate young dysgenic females. However, the defects in nuage assembly may also contribute to reduced germline piRNA expression in the young dysgenic hybrids.
The piRNAs matching a subset of Group I and Group III transposon families did show a significant reduction in species from opposite strands that overlap by 10nt, indicating that ping-pong amplification is compromised (see Blood, Figure S3A). Like the changes in total piRNA abundance, these defects are largely corrected as the dysgenic hybrids age (see Blood, Figure S3B). However, this pattern was not universal and piRNAs linked to many other elements did not show a significant change in either abundance or ping-pong bias in young dysgenic ovaries. Intriguingly, this group includes piRNAs matching the 297 element, which is over-expressed in young hybrids (Figure S2). These findings, with our cytological observations, indicate that proper sub-cellular localization of the biogenesis machinery is not required to produce or amplify piRNAs that target some transposons. However, this perinuclear organization may be essential to transposon silencing.
piRNA precursor abundance
The primary piRNAs that initiate ping-pong amplification and transposon silencing appear to be produced from long precursor transcripts encoded by heterochromatic clusters (Brennecke et al., 2007; Klattenhoff et al., 2009). Har is a P strain that expresses P-element piRNAs (Brennecke et al., 2008), but there is no direct evidence that this strain carries P-element sequences in clusters. We therefore used paired-end genomic deep sequencing to define all of the transposon insertion sites in Har ovarian DNA. Insertions were defined by paired-end reads in which one end mapped uniquely to the annotated genome and the second end mapped to a consensus transposon sequence. We identified 378 uniquely mapping P-element insertions in Har (based on 50 M genome mapping read pairs; see Experimental Procedures). Two of these sites were defined by only one paired-end read, and were not detected in progeny after backcrossing to w1 (see below), indicating that these insertions are rare variants in the Har strain. The other 5 insertion sites were defined by multiple reads and were detected in progeny samples, indicating that they were present in the germline and thus represent potential sources for primary P-element piRNAs (Table S2).
To determine the level of potential P-element piRNA precursors, we used strand specific qRT-PCR to assay RNAs that cross the unique junctions produced by P-element cluster insertions on the 4th chromosome (chr 4 + and chr 4 −, Figure 4D) and on the left arm of chromosome 2 (data not shown). As an internal control, we assayed plus strand transcripts from a distinct piRNA cluster on 2L, which does not contain a P-element insertion (Figure 4D, control). Transcripts from both strands of the control cluster were expressed at comparable levels in dysgenic and control hybrids, at both 2–4 and 21 days. By contrast, plus and minus strand precursor transcripts from the chromosome 4 cluster carrying a P-element insertion were elevated in 2–4 day old dysgenic ovaries relative to reciprocal controls (Figure 4D). The levels of these RNAs were comparable to controls in 21-day-old dysgenic ovaries, which express high levels of P-element piRNAs (Figure 5B). These findings suggested that P-element piRNAs were produced de novo through processing of precursor transcripts encoded by paternally inherited clusters.
P-element mobilization
To determine if P-element transposition into piRNAs clusters contributed to de novo P-element piRNAs production in dysgenic hybrids, we directly mapped new transposon insertions by paired-end deep sequencing of ovarian DNA. For these studies, new insertions were defined by comparing the genomic sequence of 2–4 and 21 day old dysgenic ovaries to the parental w1) and Har strains. To determine if insertions generated in the F1 hybrids were present in the germline, we backcrossed 21-day-old dysgenic females to w1 males and deep sequenced ovarian DNA from the resulting progeny ((w1 × Har) × w1). These studies identified 814 sites carrying new P-element insertions in at least one of the three progeny populations (Table S3). Fourteen of these sites mapped to piRNA clusters, but 12 of these sites were defined by only single paired-end reads and thus represent rare polymorphisms. The other two cluster insertion sites were identified by multiple reads in 21-day-old dysgenic females, but were not recovered in backcrossed F2 progeny (Table S3). Therefore, P-elements rarely transposed into clusters in the dysgenic hybrids, and none of the cluster insertions were transmitted through the germline. P-element transposition into known clusters thus did not appear to significantly contribute to de novo piRNA production in dysgenic females, or to the fertility of their backcrossed progeny. These findings, with the observations described above, strongly suggest that de novo P-element piRNAs are produced through processing of transcripts from paternal P-element containing clusters.
Resident transposon activation
Our genome sequencing studies also identified new insertions of most of the resident transposon families that are shared by the parental w1 and Har. Surprisingly, the retro-transposon roo was more active than the P-elements that triggered dysgenesis (Figure 6A). The majority of new resident element insertions were identified by single paired end reads, and these “singleton” sites are likely to reflect rare transposition events. However, a subset of insertion sites were defined by multiple paired-end reads. We defined insertion penetrance as the number of reads defining the new insertion over the sum of these reads and the reads that spanned the new insertion site. Using this definition, insertions that are homozygous in all of the individuals in the sampled population would give a penetrance of 1. In order to compare penetrance across populations, we randomly sampled all datasets to 16.8 M reads (equivalent of 18.3 fold genome coverage). Our genomic DNA libraries were generated from pairs of ovaries dissected from at least 25 females, and each ovary averages 16 ovarioles and contains 2–3 germline stem cells. Our experimental samples thus represented ~2000 germline stem cell genome equivalents. With a sampled genome coverage of 18.3-fold, insertions that occurred in single stem cell lineages would not be detected or would be defined by single reads, with a corresponding penetrance of 0.05 (=1/18.3). By contrast, insertion sites identified by multiple reads are likely to reflect integration events that took place in multiple ovaries and/or stem cell lineages, and sites detected in more than one experimental pools must have been generated in independent lineages.
Figure 6.

Transposon mobilization during hybrid dysgenesis. A. New transposon insertions were detected by paired-end genome deep sequencing and quantified following normalization to a depth of 18.3 fold. Bars indicate normalized paired end reads associated with new transposon insertions for specific transposon families in 2–4 day old (blue) and 21 day old dysgenic ovaries (red), relative to the parental Har and w1 genomes. The green bars indicate new insertions in the fertile progeny of 21 day dysgenic females mated to the parental w1 strain. These insertions are relative to the 21 day old dysgenic females. Most transposon families are active in the dysgenic hybrids, and roo is more active than the P-element trigger for hybrid dysgenesis. All transposon families show reduced activity in the progeny ovaries. B. Site specific insertion and germline transmission of an Ivk element in the 42AB cluster. Shown are the locations of paired end reads defining a new insertion in dysgenic ovaries. Blue bars indicate plus strand reads with their right end mapping to Ivk. Red bars indicate minus strand reads with left ends mapping to Ivk. Grey bars indicate reads that cross the junction between unique genomic sequences and Ivk, which define the insertion site at the nucleotide level. Ivk is not detected in the paternal Har and w1 strains, but insertion-defining reads are present at 2–4 days, increase at 21 days, and are more abundant in the progeny of 21 day dysgenic hybrids ((w1 × Har)x w1. In all three data sets, insertion spanning reads indicate transposition into precisely the same location, which defines novel junction sequences. C. The sequence of plus (blue) and minus (red) strand piRNAs reads mapping to the left hand Ivk junction, with the number of reads for each sequence indicated (reads, left columns). The number of junction specific piRNA reads increases between 2–4 and 21 days, consistent with the increase in insertion matching genome sequencing reads (B). Ivk transposition thus leads to de novo production of junction specific piRNAs. D. Total piRNAs mapping to Ivk also increase between 2–4 and 21 days (Ivk piRNAs). The number of piRNAs that overlap by 10 nt, and the bias toward this overlap, also increase with hybrid age (normalized read pair). Also see Tables S2 and S3.
The sampled data for dysgenic F1 ovaries and their F2 progeny contained 4,147 new insertion sites. 3,361 of these sites were singletons, but the remaining 786 insertion sites were defined by at least two paired-end reads, and were often detected in multiple experimental samples. For a subset of these sites, insertion penetrance increased as F1 hybrids age, suggesting that transposition was ongoing in the adult ovaries, or that cells carrying these insertions had a proliferative advantage and were therefore over-represented in the sample pool. Insertions that were heterozygous in the dysgenic germline should produce an average penetrance of 0.25 in the ovaries from the outcrossed F2 females. Remarkably, 103 insertions detected in dysgenic ovaries were recovered with 0.25 or greater penetrance in the F2 progeny (Figure 7; Table S4). Insertions in each of these sites were detected in both 2–4 and 21 day old dysgenic ovaries, demonstrating that they were produced in multiple independent lineages. Six of these sites, each carrying an insertion of a different transposon family, mapped to piRNA clusters located in pericentromeric domains of chromosome 2 (Figure 7; Table S5; Bari1, Ivk, copia, Blood, Tabor and 1731). Three of these sites, carrying insertions of Bari1, copia and Ivk, mapped to the major 42AB cluster on chromosome 2R. 2.9% of the inherited insertions thus mapped to a single cluster, which represents only 0.13% of the Drosophila genome but may encode 30% of all ovarian piRNAs(Brennecke et al., 2007).
Figure 7.

Genome distribution of inherited transposon insertions. Direct genome sequencing identified 132 transposon insertion sites in 21 day old dysgenic females that were also present in their progeny at 25% or greater penetrance. The genomic distribution of these inherited insertions is indicated, with bar height indicating penetrance in the progeny genome. The 6 insertions that map to piRNA clusters are in red. All of these sites are in pericentromeric heterochromatin or heterochromatin on chromosome 2. Three of these insertions are in the major pericentromeric cluster at 42AB. Also see Tables S4 and S5.
Over-representation of cluster insertions in dysgenic hybrids could result from an inherent bias toward transposition into these heterochromatic domains, or from random or modestly biased transposition followed by clonal expansion of cells carrying the insertions. The latter could result if cluster insertions provide a selective advantage, perhaps by enhancing transposon silencing. Inherently biased transposition would lead to new cluster insertions independent of selective pressure, while selective pressure is required for cluster insertion enrichment through clonal expansion. We therefore calculated insertion bias in dysgenic ovaries, where transposons are active and cluster insertion could enhance silencing, and in ovaries from the offspring of these hybrids, which are fertile and appear to develop in the absence of selective pressure. To quantify and statistically analyze insertion frequency and cluster bias, we randomly resampled all data sets 100 times at 18.3 fold genome coverage and calculated mean values and standard deviations of sequencing reads indicative of insertions (Table S5). In 21-day-old dysgenic females, 7.81% of all new transposon insertions mapped to clusters, which represent 3.5% of the genome. This is a modest but statistically significant 2-fold bias toward clusters (binomial p-value 8.5×10−41). For the six transposon families represented by inherited cluster insertions, by contrast, 23.74% of new insertions mapped to clusters, which represents a 3-fold increase in cluster bias relative to the total transposon pool (binomial p-value = 1.09×10−17). In the fertile F2 progeny of the dysgenic hybrids, these same six families showed only a 6.90% rate of cluster insertion, which was not significantly different from the 7.40% cluster bias of the total transposon pool (binomial p-value = 0.59; Table S5). Enhanced cluster insertion is therefore specific to a subset of transposon families within dysgenic hybrids.
To determine if inheritance of cluster insertions alters transposition frequency, we determined the normalized number of new transposition events in dysgenic hybrids and their progeny. Across all transposon families, the dysgenic ovaries averaged 4290 +/− 18 new transposon insertions, with new insertions defined by comparison to the parental w1 and Har strains (resampled data, Table 2). The ovaries of F2 progeny, by contrast, averaged only 867 new insertions relative to their F1 dysgenic parents. Total transposon activity in the F2 ovaries was therefore approximately 5 fold lower than in their dysgenic parents. For the six transposon families represented by inherited cluster insertions, the dysgenic ovaries averaged 295 new insertions while their F2 progeny averaged only 31 new insertions. This 10-fold reduction in transposon activity is significantly greater than the 5-fold reduction shown by the entire transposon pool (χ2 test p-value = 3.63×10−4). Inheritance of cluster insertions is therefore associated with reduced transposition of homologous elements.
To determine if cluster insertion leads to de novo piRNA production, we searched our deep sequencing datasets for piRNAs mapping to the unique junctions generated by transposition into clusters. Individual piRNAs are rare, but we detected species mapping specifically to the left junction of the inherited Ivk element insertion located in the 42AB cluster. Furthermore, the abundance and complexity of the junction mapping species increased between 2–4 days and 21-day-olds (Figure 6B, C), which correlated with a 3 fold increase in total piRNAs mapping across Ivk (Figure 6D, Ivk total). The increase in total and junction-specific piRNAs in the dysgenic hybrids was paralleled by an increase in Ivk insertion penetrance at the 42AB site (Figure 6B). Transposition into the 42AB piRNA cluster thus leads to de novo piRNA production, which may provide a selective advantage by enhancing target silencing.
Discussion
Transposons evolve rapidly and can mobilize to trigger genetic instability and disease-associated mutations (Callinan and Batzer, 2006; Capy et al., 1994; Daniels et al., 1990; Deininger et al., 2003; Hedges and Belancio, 2011). The piRNA pathway has a conserved role in transposon silencing, but the mechanisms by which this system adapts to new mobile elements are not understood. Using the P-M hybrid dysgenesis system, we show that introduction of P-element transposons into a naïve strain leads to mobilization of both the invading element and resident transposons, and this global activation of transposons is associated with severely reduced adult fertility. As F1 hybrid adult females age, however, fertility is restored and both the inducing P-element and resident transposons are silenced. Silencing of the invading P element is linked to de novo production of piRNAs that appear to be encoded by paternally inherited clusters. However, all of the dysgenic females inherited paternal clusters, but only a subset of the ovarioles within 26% of ovaries regained the ability to produce eggs. Silencing of the P-elements alone thus does not appear to be sufficient to restore fertility. We propose that silencing of the resident elements is a critical second step in the adaptation process, which depends on resident transposon mobilization and insertion into piRNA clusters.
Mobilization of resident elements
Our tiling array and genomic sequencing data directly show that resident elements are activated in ovaries isolated from P-M dysgenic females. By contrast, elegant genetic studies indicate that resident transposons are not activated in the testes of P-M dysgenic males (Eggleston et al., 1988). The earlier studies used genetic assays for transposon insertion over multiple generations, and thus required efficient recovery of offspring. Because the dysgenic males retain significant fertility, they were well suited to these studies. These genetic studies detected approximately one new P-element insertion/genome/generation (from Table 2 in (Eggleston et al., 1988). By contrast, our genome sequence analysis of dysgenic ovaries, which produced no viable eggs for almost a week, revealed approximately 15 new insertions in a single generation. We speculate that the 15-fold higher rate of P-element transposition in females, and the resulting DNA damage, is responsible for resident element activation. Supporting this hypothesis, pioneering studies by McClintock showed that chromosome breaks activate transposons in Maize (see McClintock, 1984), and more recent studies indicate that telomere erosion and DNA damage activate transposon in systems ranging from yeast to mammals (Beauregard et al., 2008; Bradshaw and McEntee, 1989; McClintock, 1984; Staleva Staleva and Venkov, 2001).
Studies on meiotic repair and piRNA pathway mutants, and the studies reported here, suggest that DNA damage signaling through Chk2 kinase could disrupt silencing of resident elements(Abdu et al., 2002; Chen et al., 2007; Ghabrial and Schupbach, 1999; Klattenhoff et al., 2007). Most of the piRNA machinery associates with nuage; a germline specific structure implicated in piRNA biogenesis and transposon silencing (reviewed by Klattenhoff and Theurkauf, 2007). Mutations that disrupt meiotic DNA break repair or piRNA dependent transposon silencing lead to germline DNA damage and disrupt nuage (Ghabrial and Schupbach, 1999; Klattenhoff et al., 2007; Klattenhoff et al., 2009). Chk2 kinase is activated in response to DNA breaks, and these mutations lead to Chk2-dependent phosphorylation of Vasa, which is a conserved DEAD box protein required for piRNA biogenesis and Nuage assembly (Abdu et al., 2002; Chen et al., 2007; Klattenhoff et al., 2007; Klattenhoff et al., 2009; Lim and Kai, 2007; Malone et al., 2009). Here we show that Vas protein is degraded in young dysgenic hybrids, and that this correlates with defects in the sub-cellular localization of other piRNA pathway components. The mechanism of Vasa down-regulation is not known, but Chk2 activation during the cell cycle checkpoint response leads to hyper-phosphorylation of Cdc25 and ubiquitin-dependent destruction(Falck et al., 2001). DNA damage caused by P-element mobilization may therefore trigger Chk2-dependent phosphorylation and ubiqutin-dependent destruction of Vasa, which in turn disrupts nuage organization and resident transposon silencing.
In this model, P-element mobilization leads to DNA damage that activates resident transposons, further destabilizing the genome. As dysgenic hybrids age, however, the nuage reassembles, full length Vasa protein is expressed, and transposons are silenced. How is the proposed DNA damage-transposon activation cycle terminated? DNA damage activates checkpoints that arrest cell cycle progression to provide time for repair (Lazzaro et al., 2009). When damage persists, checkpoints adapt and the cell cycle progresses, despite the presence of unrepaired lesions (Clemenson and Marsolier-Kergoat, 2009). We speculate that a similar process allows adaptation to persistent transposition-induced damage, leading to Vasa accumulation, nuage assembly, piRNA production and transposon silencing.
A two-step model for adaptation to transposon invasion
Based on the observations reported here we propose a multi-step model for adaptation to P-element transposon invasion. In this model, the invading P-element escapes silencing due to the absence of matching maternal piRNAs, and mobilization of these elements leads to DNA damage(Kaufman and Rio, 1992) and Chk-2 activation, which directly or indirectly disrupts nuage organization and resident element silencing. The resulting DNA damage blocks stem cell proliferation and oogenesis, but also provides time for resident element transposition. Persistent damage signaling is eventually overcome, perhaps through a mechanism related to checkpoint adaptation, restoring Vasa expression and nuage organization, which permits de novo production of P-element piRNAs from paternally inherited clusters. However, all of the stem cells in dysgenic hybrids have the genetic capacity to produce P-element piRNAs, but only a subset of ovarioles resume egg production. P-element silencing thus does not appear to be sufficient to restore fertility. We propose fertility is restored after a subset of resident elements transpose into piRNA clusters, where they template piRNA production. This may occur through random or modestly biased transposition, followed by selective division of the rare cells that show enhanced piRNA production and transposon silencing. These cells populate the germline and transmit the new cluster insertions to the next generation.
P-elements are DNA transposons that move by a cut and paste mechanism, and we speculate that damage linked to this process is the trigger for resident element activation. In the Drosophila melanogaster I-R system of hybrid dysgenesis, however, the invading I element is a retrotransposon that moves by a copy and paste mechanism (Bucheton, 1990; Van De Bor et al., 2005). It is unclear if resident elements are activated by I element invasion. However, the Penelope retrotransposon induces hybrid dysgenesis in D. virilis, which leads to mobilization of at least four additional resident transposon families(Petrov et al., 1995). Retrotransposon mobilization has been reported to induce DNA breaks(Belgnaoui et al., 2006), raising the possibility that DNA damage signaling activates resident transposons after invasion of DNA elements or retrotransposons.
McClintock discovered transposition as a response to telomere deletions that destabilize the genome by initiating a break-bridge-fusion cycle, and speculated that the genome would prove to be a “…highly sensitive organ of the cell that monitors genomic activities and corrects common errors, senses unusual and unexpected events, and responds to them, often by restructuring the genome”(McClintock, 1984). Within this framework, transposon invasion represents is an “unexpected event” and resident element transposition into clusters, which appears to enhance piRNA silencing capacity, is the genome restructuring response to this event. We speculate that this interplay between invading and resident elements, which produces heritable changes in heterochromatin organization, has a significant role in genome evolution.
Experimental procedures
Fly husbandry
All the stocks and crosses were maintained at 25° C on cornmeal medium using standard conditions. The Harwich stock was obtained from Stephane Ronsseray. w1 was obtained from Bloomington Stock Center. For the dysgenic cross, w1 females were mated to Harwich males and in the reciprocal cross, Harwich females were mated to w1 males.
Immunohistochemistry and Western blotting
Ovaries were immunolabled as described earlier using the Buffer A staining protocol(Liu et al., 2002). DNA DSBs were indirectly detected by labeling with a rabbit polyclonal antibody against γ-H2Av (at 1:500, Rockland). Vas protein was visualized using a rabbit polycolonal anti-Vasa antibody (a gift from Dr. Paul Lasko) at 1:5000 (Liang et al., 1994). Ago3, Aub and Piwi were localized using rabbit polyclonal antibodies at 1:250, 1:1000 and 1:1000, respectively (Brennecke et al., 2007). TOTO-3 dye (Molecular Probes) was used at 1:500 to label DNA. Lamin C was detected with mouse monoclonal antibody LC28.28 (Developmental Studies Hybridoma Bank) at 1:15. For Western blots, the rabbit anti-Vasa antibody was used at 1:5000.
Small RNA and tiling array analyses
Total RNA was extracted from 2–4 day old and 21 day old ovaries from dysgenic and reciprocal control hybrids using MirVana kit (Ambion). 18–29 nt small RNAs were gel purified following 2S rRNA depletion and treated using previously published protocol(Li et al., 2009). Small RNA libraries were prepared for sequencing with a Solexa Genome Analyzer (Illumina, San Diego, CA). Total RNA was extracted from ovaries using the RNeasy Kit (Qiagen) and the manufacturer’s instructions. For each genotype, RNA samples from three biological replicates were assayed (Klattenhoff et al., 2009). Tiling array data are available in the NCBI GEO data base (GSE31813) and small RNA and genomic deep sequencing data are available through the NCBI SRA data base (SRP007937).
Strand-specific Reverse Transcriptase PCR
Strand-specific RT-PCR for cluster transcripts was performed as described previously(Klattenhoff et al., 2009). Signal linked to paternal P-element cluster insertions was normalized to plus strand piRNA precursor RNA from the 42AB cluster (cl1A-rt-plus)(Klattenhoff et al., 2009). P-values (cited in the text) for RT-PCR quantification were calculated from at least three independent biological replicates using a two-tailed unpaired t-test.
Illumina genomic DNA library preparation
Whole ovaries were dissected and genomic DNA was prepared using DNeasy Blood and Tissue DNA extraction Kit (Qiagen). After fragmentation to an average length of 300 nt using a Bioruptor (Diagenode Inc.), the DNA was processed for the paired-end Illumina sequencing according to the manufacturer’s protocol. Sequencing statistics are presented in Table S6.
piRNA sequence analyses
For each sequence read, the first occurrence of the hexamer perfectly matching the 5′ end of the 3′ linker was identified. The extracted inserts for sequences that contained the 3′ linker were then mapped to the Drosophila melanogaster genome (Release R5.5) using Bowtie31, and the corresponding genomic coordinates were determined for downstream functional analysis. piRNAs were defined as 23–30 nt genome-mapping reads that did not map to pre-miRNA hairpins (miRBase(Griffiths-Jones et al., 2008) version 13.0) or ncRNAs. Gene sequences were retrieved from FlyBase(Tweedie et al., 2009) (R5.5). In order to compare the reads among different data sets, we normalize to the total number of perfectly matching genome-mapping, non-ncRNA reads and expressed levels in parts per million (ppm). piRNA clusters are from Brennecke et al.5, with 141 clusters (excluding the chrX_TAS) in total, occupying 4,924,944 bp in the genome.
Computational analysis of tiling arrays
Tiling array analysis was performed as described previously(Klattenhoff et al., 2009; Li et al., 2009).
Computational identification of transposon insertion sites
We aligned paired-end reads that were of sufficiently high sequencing quality against the unmasked Drosophila reference genome using the BWA algorithm(Li and Durbin, 2009), allowing insertions, deletions and up to two mismatches per 76-nt read. The average size of genomic DNA in the sequencing libraries was 500 bp, thus we expected most of the read pairs to map to two locations in the reference genome that were approximately 500 bp apart. We defined discordant pairs as those that mapped to two locations that were more than 1 kb apart, or those with only one genome mapping read. In order to detect transposon insertions that were in an experimental genome but not in a reference genome, we identified discordant read pairs for which one read mapped to a location in the reference genome while the other read maps to a transposon sequence. The transposon sequences were defined by the full-length consensus sequence and transposon fragment sequences that exist anywhere in the reference genome. Transposon insertion reads that suggested insertion positions within 1 kb were clustered and collectively called an insertion site.
Supplementary Material
Acknowledgments
We thank the UMass Medical School RNA biology community for stimulating discussions and constructive criticisms. Thanks to Stephane Ronsseray and the Bloomington Stock Center for fly strains, Julius Brennecke and Paul Lasko for antibodies, and the UMass Deep Sequencing Core for technical assistance. This work was supported by grants from the NIH to WET, ZW and PDZ (R01HD049116) and to PDZ (GM62862 and GM65236).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdu U, Brodsky M, Schupbach T. Activation of a meiotic checkpoint during Drosophila oogenesis regulates the translation of Gurken through Chk2/Mnk. Curr Biol. 2002;12:1645–1651. doi: 10.1016/s0960-9822(02)01165-x. [DOI] [PubMed] [Google Scholar]
- Aravin AA, Hannon GJ, Brennecke J. The Piwi-piRNA pathway provides an adaptive defense in the transposon arms race. Science (New York, NY) 2007;318:761–764. doi: 10.1126/science.1146484. [DOI] [PubMed] [Google Scholar]
- Aravin AA, Lagos-Quintana M, Yalcin A, Zavolan M, Marks D, Snyder B, Gaasterland T, Meyer J, Tuschl T. The small RNA profile during Drosophila melanogaster development. Dev Cell. 2003;5:337–350. doi: 10.1016/s1534-5807(03)00228-4. [DOI] [PubMed] [Google Scholar]
- Beauregard A, Curcio MJ, Belfort M. The take and give between retrotransposable elements and their hosts. Annu Rev Genet. 2008;42:587–617. doi: 10.1146/annurev.genet.42.110807.091549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belgnaoui SM, Gosden RG, Semmes OJ, Haoudi A. Human LINE-1 retrotransposon induces DNA damage and apoptosis in cancer cells. Cancer cell international. 2006;6:13. doi: 10.1186/1475-2867-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennetzen JL. Transposable element contributions to plant gene and genome evolution. Plant Mol Biol. 2000;42:251–269. [PubMed] [Google Scholar]
- Blumenstiel JP, Hartl DL. Evidence for maternally transmitted small interfering RNA in the repression of transposition in Drosophila virilis. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15965–15970. doi: 10.1073/pnas.0508192102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw VA, McEntee K. DNA damage activates transcription and transposition of yeast Ty retrotransposons. Mol Gen Genet. 1989;218:465–474. doi: 10.1007/BF00332411. [DOI] [PubMed] [Google Scholar]
- Brennecke J, Aravin AA, Stark A, Dus M, Kellis M, Sachidanandam R, Hannon GJ. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell. 2007;128:1089–1103. doi: 10.1016/j.cell.2007.01.043. [DOI] [PubMed] [Google Scholar]
- Brennecke J, Malone C, Aravin A, Sachidanandam R, Stark A, Hannon G. An epigenetic role for maternally inherited piRNAs in transposon silencing. Science (New York, NY) 2008;322:1387–1392. doi: 10.1126/science.1165171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britten RJ. Transposable element insertions have strongly affected human evolution. Proc Natl Acad Sci U S A. 2010;107:19945–19948. doi: 10.1073/pnas.1014330107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucheton A. Study of non Mendelian female sterility in Drosophila melanogaster. Hereditary transmission of the degree of efficacy of the reactor factor. Comptes rendus hebdomadaires des séances de l’Académie des sciences Série D: Sciences naturelles. 1973;276:641–644. [PubMed] [Google Scholar]
- Bucheton A. Non-Mendelian female sterility in Drosophila melanogaster: influence of aging and thermic treatments. III. Cumulative effects induced by these factors. Genetics. 1979;93:131–142. doi: 10.1093/genetics/93.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucheton A. I transposable elements and I-R hybrid dysgenesis in Drosophila. Trends Genet. 1990;6:16–21. doi: 10.1016/0168-9525(90)90044-7. [DOI] [PubMed] [Google Scholar]
- Bucheton A, Lavige JM, Picard G, L’Heritier P. Non-mendelian female sterility in Drosophila melanogaster: quantitative variations in the efficiency of inducer and reactive strains. Heredity. 1976;36:305–314. doi: 10.1038/hdy.1976.38. [DOI] [PubMed] [Google Scholar]
- Bucheton A, Picard G. A partially inheritable aging influence on a non-Mendelian female sterility in Drosophila melanogaster. Comptes rendus hebdomadaires des séances de l’Académie des sciences Série D: Sciences naturelles. 1975;281:1035–1038. [PubMed] [Google Scholar]
- Callinan PA, Batzer MA. Retrotransposable elements and human disease. Genome Dyn. 2006;1:104–115. doi: 10.1159/000092503. [DOI] [PubMed] [Google Scholar]
- Capy P, Langin T, Bigot Y, Brunet F, Daboussi MJ, Periquet G, David JR, Hartl DL. Horizontal transmission versus ancient origin: mariner in the witness box. Genetica. 1994;93:161–170. doi: 10.1007/BF01435248. [DOI] [PubMed] [Google Scholar]
- Chen Y, Pane A, Schüpbach T. Cutoff and aubergine mutations result in retrotransposon upregulation and checkpoint activation in Drosophila. Current biology: CB. 2007;17:637–642. doi: 10.1016/j.cub.2007.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemenson C, Marsolier-Kergoat MC. DNA damage checkpoint inactivation: adaptation and recovery. DNA Repair (Amst) 2009;8:1101–1109. doi: 10.1016/j.dnarep.2009.04.008. [DOI] [PubMed] [Google Scholar]
- Daniels SB, Peterson KR, Strausbaugh LD, Kidwell MG, Chovnick A. Evidence for horizontal transmission of the P transposable element between Drosophila species. Genetics. 1990;124:339–355. doi: 10.1093/genetics/124.2.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deininger PL, Moran JV, Batzer MA, Kazazian HH., Jr Mobile elements and mammalian genome evolution. Curr Opin Genet Dev. 2003;13:651–658. doi: 10.1016/j.gde.2003.10.013. [DOI] [PubMed] [Google Scholar]
- Eddy EM. Fine structural observations on the form and distribution of nuage in germ cells of the rat. The Anatomical record. 1974;178:731–757. doi: 10.1002/ar.1091780406. [DOI] [PubMed] [Google Scholar]
- Eddy EM. Germ plasm and the differentiation of the germ cell line. International review of cytology. 1975;43:229–280. doi: 10.1016/s0074-7696(08)60070-4. [DOI] [PubMed] [Google Scholar]
- Eggleston WB, Johnson-Schlitz DM, Engels WR. P-M hybrid dysgenesis does not mobilize other transposable element families in D. melanogaster. Nature. 1988;331:368–370. doi: 10.1038/331368a0. [DOI] [PubMed] [Google Scholar]
- Falck J, Mailand N, Syljuasen RG, Bartek J, Lukas J. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. 2001;410:842–847. doi: 10.1038/35071124. 101038/nature04917. [DOI] [PubMed] [Google Scholar]
- Ghabrial A, Schupbach T. Activation of a meiotic checkpoint regulates translation of Gurken during Drosophila oogenesis. Nat Cell Biol. 1999;1:354–357. doi: 10.1038/14046. java/Propub/cellbio/ncb1099_1354.fulltextjava/Propub/cellbio/ncb1099_1354.abstract. [DOI] [PubMed]
- Ghildiyal M, Zamore P. Small silencing RNAs: an expanding universe. Nature reviews Genetics. 2009;10:94–108. doi: 10.1038/nrg2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36:D154–158. doi: 10.1093/nar/gkm952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedges DJ, Belancio VP. Restless genomes humans as a model organism for understanding host-retrotransposable element dynamics. Adv Genet. 2011;73:219–262. doi: 10.1016/B978-0-12-380860-8.00006-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraizumi Y. Spontaneous recombination in Drosophila melanogaster males. Proceedings of the National Academy of Sciences of the United States of America. 1971;68:268–270. doi: 10.1073/pnas.68.2.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman PD, Rio DC. P element transposition in vitro proceeds by a cut-and-paste mechanism and uses GTP as a cofactor. Cell. 1992;69:27–39. doi: 10.1016/0092-8674(92)90116-t. [DOI] [PubMed] [Google Scholar]
- Khurana JS, Theurkauf W. piRNAs, transposon silencing, and Drosophila germline development. J Cell Biol. 2010;191:905–913. doi: 10.1083/jcb.201006034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidwell MG. Hybrid dysgenesis in Drosophila melanogaster: nature and inheritance of P element regulation. Genetics. 1985;111:337–350. doi: 10.1093/genetics/111.2.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidwell MG. Horizontal transfer. Curr Opin Genet Dev. 1992;2:868–873. doi: 10.1016/s0959-437x(05)80109-1. [DOI] [PubMed] [Google Scholar]
- Kidwell MG, Kidwell JF, Sved JA. Hybrid Dysgenesis in DROSOPHILA MELANOGASTER: A Syndrome of Aberrant Traits Including Mutation, Sterility and Male Recombination. Genetics. 1977;86:813–833. doi: 10.1093/genetics/86.4.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klattenhoff C, Bratu DP, McGinnis-Schultz N, Koppetsch BS, Cook HA, Theurkauf WE. Drosophila rasiRNA pathway mutations disrupt embryonic axis specification through activation of an ATR/Chk2 DNA damage response. Developmental cell. 2007;12:45–55. doi: 10.1016/j.devcel.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Klattenhoff C, Theurkauf W. Biogenesis and germline functions of piRNAs. Development. 2007;135 doi: 10.1242/dev.006486. dev-006486. [DOI] [PubMed] [Google Scholar]
- Klattenhoff C, Xi H, Li C, Lee S, Xu J, Khurana JS, Zhang F, Schultz N, Koppetsch BS, Nowosielska A, et al. The Drosophila HP1 Homolog Rhino Is Required for Transposon Silencing and piRNA Production by Dual-Strand Clusters. Cell. 2009;138:1137–1149. doi: 10.1016/j.cell.2009.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzaro F, Giannattasio M, Puddu F, Granata M, Pellicioli A, Plevani P, Muzi-Falconi M. Checkpoint mechanisms at the intersection between DNA damage and repair. DNA Repair (Amst) 2009;8:1055–1067. doi: 10.1016/j.dnarep.2009.04.022. [DOI] [PubMed] [Google Scholar]
- Li C, Vagin VV, Lee S, Xu J, Ma S, Xi H, Seitz H, Horwich MD, Syrzycka M, Honda BM, et al. Collapse of germline piRNAs in the absence of Argonaute3 reveals somatic piRNAs in flies. Cell. 2009;137:509–521. doi: 10.1016/j.cell.2009.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang L, Diehl-Jones W, Lasko P. Localization of vasa protein to the Drosophila pole plasm is independent of its RNA-binding and helicase activities. Development. 1994;120:1201–1211. doi: 10.1242/dev.120.5.1201. [DOI] [PubMed] [Google Scholar]
- Lim A, Kai T. Unique germ-line organelle, nuage, functions to repress selfish genetic elements in Drosophila melanogaster. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:6714–6719. doi: 10.1073/pnas.0701920104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Jang JK, Kato N, McKim KS. mei-P22 encodes a chromosome-associated protein required for the initiation of meiotic recombination in Drosophila melanogaster. Genetics. 2002;162:245–258. doi: 10.1093/genetics/162.1.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madigan JP, Chotkowski HL, Glaser RL. DNA double-strand break-induced phosphorylation of Drosophila histone variant H2Av helps prevent radiation-induced apoptosis. Nucleic acids research. 2002;30:3698–3705. doi: 10.1093/nar/gkf496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone CD, Brennecke J, Dus M, Stark A, McCombie WR, Sachidanandam R, Hannon GJ. Specialized piRNA pathways act in germline and somatic tissues of the Drosophila ovary. Cell. 2009;137:522–535. doi: 10.1016/j.cell.2009.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClintock B. The significance of responses of the genome to challenge. Science. 1984;226:792–801. doi: 10.1126/science.15739260. [DOI] [PubMed] [Google Scholar]
- McKim KS, Jang JK, Manheim EA. Meiotic recombination and chromosome segregation in Drosophila females. Annu Rev Genet. 2002;36:205–232. doi: 10.1146/annurev.genet.36.041102.113929. [DOI] [PubMed] [Google Scholar]
- Nishida KM, Saito K, Mori T, Kawamura Y, Nagami-Okada T, Inagaki S, Siomi H, Siomi MC. Gene silencing mechanisms mediated by Aubergine piRNA complexes in Drosophila male gonad. RNA (New York, NY) 2007;13:1911–1922. doi: 10.1261/rna.744307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pane A, Wehr K, Schüpbach T. zucchini and squash encode two putative nucleases required for rasiRNA production in the Drosophila germline. Developmental cell. 2007;12:851–862. doi: 10.1016/j.devcel.2007.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrov DA, Schutzman JL, Hartl DL, Lozovskaya ER. Diverse transposable elements are mobilized in hybrid dysgenesis in Drosophila virilis. Proc Natl Acad Sci U S A. 1995;92:8050–8054. doi: 10.1073/pnas.92.17.8050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard G, Bucheton A, Lavige JM, Fleuriet A. A sterility phenomenon of nonmendelian determinism in Drosophila melanogaster. Comptes rendus hebdomadaires des séances de l’Académie des sciences Série D: Sciences naturelles. 1972;275:933–936. [PubMed] [Google Scholar]
- Rio DC. Regulation of Drosophila P element transposition. Trends Genet. 1991;7:282–287. doi: 10.1016/0168-9525(91)90309-E. [DOI] [PubMed] [Google Scholar]
- Rozhkov NV, Aravin AA, Zelentsova ES, Schostak NG, Sachidanandam R, McCombie WR, Hannon GJ, Evgen’ev MB. Small RNA-based silencing strategies for transposons in the process of invading Drosophila species. RNA. 2010;16:1634–1645. doi: 10.1261/rna.2217810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin GM, Kidwell MG, Bingham PM. The molecular basis of P-M hybrid dysgenesis: the nature of induced mutations. Cell. 1982;29:987–994. doi: 10.1016/0092-8674(82)90462-7. [DOI] [PubMed] [Google Scholar]
- Spradling AC. Developmental Genetics of Oogenesis. In: Bate M, Arias AM, editors. The Development of Drosophila Melanogaster. 1993. pp. 1–70. [Google Scholar]
- Staleva Staleva L, Venkov P. Activation of Ty transposition by mutagens. Mutat Res. 2001;474:93–103. doi: 10.1016/s0027-5107(00)00165-2. [DOI] [PubMed] [Google Scholar]
- Tweedie S, Ashburner M, Falls K, Leyland P, McQuilton P, Marygold S, Millburn G, Osumi-Sutherland D, Schroeder A, Seal R, et al. FlyBase: enhancing Drosophila Gene Ontology annotations. Nucleic Acids Res. 2009;37:D555–559. doi: 10.1093/nar/gkn788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van De Bor V, Hartswood E, Jones C, Finnegan D, Davis I. gurken and the I factor retrotransposon RNAs share common localization signals and machinery. Dev Cell. 2005;9:51–62. doi: 10.1016/j.devcel.2005.04.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.