

Abstract
The role of antibodies directed against the hyper variable envelope region V1 of human immunodeficiency virus type 1 (HIV-1), has not been thoroughly studied. We show that a vaccine able to elicit strain-specific non-neutralizing antibodies to this region of gp120 is associated with control of highly pathogenic chimeric SHIV89.6P replication in rhesus macaques. The vaccinated animal that had the highest titers of antibodies to the amino terminus portion of V1, prior to challenge, had secondary antibody responses that mediated cell killing by antibody-dependent cellular cytotoxicity (ADCC), as early as two weeks after infection and inhibited viral replication by antibody-dependent cell-mediated virus inhibition (ADCVI), by four weeks after infection. There was a significant inverse correlation between virus level and binding antibody titers to the envelope protein, (R = -0.83, p 0.015), and ADCVI (R = -0.84 p=0.044). Genotyping of plasma virus demonstrated in vivo selection of three SHIV89.6P variants with changes in potential N-linked glycosylation sites in V1. We found a significant inverse correlation between virus levels and titers of antibodies that mediated ADCVI against all the identified V1 virus variants. A significant inverse correlation was also found between neutralizing antibody titers to SHIV89.6 and virus levels (R = -0.72 p =0.0050). However, passive inoculation of purified immunoglobulin from animal M316, the macaque that best controlled virus, to a naïve macaque, resulted in a low serum neutralizing antibodies and low ADCVI activity that failed to protect from SHIV89.6P challenge. Collectively, while our data suggest that anti-envelope antibodies with neutralizing and non-neutralizing FcγR-dependent activities may be important in the control of SHIV replication, they also demonstrate that low levels of these antibodies alone are not sufficient to protect from infection.
INTRODUCTION
The HIV envelope gene encodes four variable regions (V1–V4) [1;2]. The V3 region is important for viral infectivity and tropism and is the principal target for neutralizing antibodies of laboratory-adapted viruses [3-8]. Similarly, the V1/V2 regions of HIV influence viral receptor and co-receptor usage and tropism [9-15]. Selection of genotypes with changes in V1/V2 occurs during the early phase of HIV infection [16-18]. HIV sequences of isolates, obtained during the chronic phase of infection, have extended V1/V2 regions and a higher number of potential N-linked glycosylation sites [12;19]. The turnover of V1 and V2, in the later stage of HIV infection, is suggestive of in vivo selection [20] and deletion or mutations that modify glycosylation sites within these regions, affect the neutralization susceptibility of HIV and SIV isolates in vitro [13;21-26]. In infected rhesus macaques, the selection of SIVmac239 strains that became resistant to neutralization has been linked to changes in N-linked and O-linked glycosylation in V1 [27]. Interestingly, deletion of the V1 region within the SIVmac239 molecular clone, results in decreased viral fitness in vivo and greater neutralization susceptibility in vitro [23]. Similarly, single amino acid changes affecting N-glycans in the V1/V2 of an HIV molecular clone, impacted viral fitness and showed resistance to antibody neutralization in vitro [28]. The plasticity of the V1/V2 region of HIV/SIV suggests its importance for viral fitness, particularly in the context of an active host immune response. However, there is no direct evidence that supports a protective role of antibodies to the V1/V2 region in HIV or SIV infection. Here, we used the SHIV89.6P rhesus macaque model and investigated the role of antibody responses to V1 in the control of viral replication. We used a vaccine based on a cDNA encoding a chimeric HIV protein, generated by an unusual splicing of the Tat open reading frame to the V1 envelope region and the last exon of Rev (Tat-Env-Rev=TEV) [29-31] in a DNA prime-protein boost regimen. The combination of these vaccines induced modest T-cell responses and antibodies to the V1 that mediated a type specific antibody-dependent cell-mediated virus inhibition in vitro. Correlative analysis suggests that ADCVI function contribute to the control of viral replication in animals that nevertheless, become infected. However, passive transfer of vaccine elicited antibodies to a neonate macaque failed to protect from infection. Thus, it is possible that other immune responses, in addition to antibodies to V1, induced by this vaccine regimen may have contributed to control of viral replication.
MATERIALS AND METHODS
DNA plasmids and protein expression
The tev genes for the HIV-1 isolates HIVBa-L, HIVSF162, and HIV89.6 were designed [32] using the published sequences for each isolate (Genbank M68893, M65024, and U39362, respectively) and were based on the published HXB2 tev sequence (Genbank M37898). The genes were synthetically constructed and cloned into pPCR-Script Amp SK (Strategene, La Jolla, CA) cloning vectors by Geneart (Regensburg, Germany). The tev genes were synthesized using human and E .coli codon bias to optimize translation in both systems.
DNA vaccine vectors were constructed for Ba-L, SF162, and SHIV89.6 Tev, pCMV Ba-L Tev, pCMV SF162 Tev, and pCMV 89.6 Tev, respectively. Briefly, for each tev construct, a SacII/EcoRI DNA fragment encoding the tev gene was ligated into a mammalian expression vector derived from pVR1332 (Vical, San Diego, CA) [33]. These plasmids are kanamycin resistant, and the expression of tev is under control of the CMV immediate-early promoter. Plasmid DNA was produced at Althea Technologies (San Diego, CA), at the concentration of 1mg/ml in sterile water. DNA was found to be >95% circular and predominantly supercoiled by gel electrophoresis, with endotoxin levels below 1.5 EU/mg DNA.
The tev vaccine vectors were tested and found to be functional by radioimmunoprecipitation assay (RIPA) of transiently transfected HEK 293 cells. Transfections were performed using Lipofectamine 2000 (Invitrogen, Carlsbad, CA), according to the manufacturer’s recommended protocol. Wells of a twenty-four-well tissue culture plate were seeded with 2×105 HEK 293 cells in 500 μl DMEM (Quality Biological, Gaithersburg, MD), supplemented with 10% heat-inactivated Fetal Bovine Serum (FBS; Hyclone, Logan, UT) and 2 mM L-glutamine (Quality Biological). Cultures were incubated for 18h at 37°C with 5% CO2. For each plasmid, 2.5 μl of Lipofectamine 2000 reagent was mixed with 47.5 μl Opti-MEM Reduced Serum Media (Invitrogen) and set at room temperature for 10 min. For each plasmid, 1 μl (1 μg) DNA was mixed with 49 μl Opti-MEM Reduced Serum Media, and set at room temperature for 5 min. DNA and Lipofectamine 2000 solutions were combined and set at room temperature for 20 min, then added to the 18h HEK 293 cultures. Cultures were further incubated at 37°C with 5% CO2 for 29h.
At 29h post-transfection, transfected and non transfected HEK 293 cells as negative control, were washed once with 1 ml D-PBS (Quality Biological) and overlaid with 500 μl L-methionine and L-cystine free DMEM (Invitrogen) supplemented with 2% heat-inactivated dialyzed FBS (Invitrogen), 2 mM L-glutamine (Quality Biological), 1 mM sodium pyruvate (Quality Biological), and 30 mg/ml L-methionine (Sigma, St. Louis, MO). Cells were labeled with 50 μCi 35 S Cysteine (Perkin Elmer, Shelton, CT) and incubated for 16h at 37°C with 5% CO2. Media was removed and cells were lysed in PBS-TD (PBS with 0.5 % Triton-X-100 and 1% deoxycholic acid) containing 1 μl/ml benzonase nuclease (EMD Biosciences, Madison, WI) and 1 μl/ml Protease Inhibitor Cocktail for Mammalian Cells (Sigma) for 10 min at room temperature. Lysates were clarified by centrifugation at 10,000 × g for 5 min. Lysates were precleared by shaking with 20 μl protein. A sepharose (PAS; Amersham Biosciences, Piscataway, NJ), and 5 μl normal rabbit serum was at room temperature for 1 hour. Ten percent glycerol was added to the precleared lysates, and lysates were immunoprecipitated with 20 μl PAS and 5 μl rabbit anti-HIV-1MN Rev serum (Advanced BioScience Laboratories, Kensington, MD), or 5 μl rabbit anti-HIV-1IIIB Tat serum (Advanced BioScience Laboratories), for 2.5h at room temperature. Precipitated proteins were separated by electrophoresis on 10% SDS-PAGE gels. The gels were subsequently soaked in destaining solution (40% methanol and 7 % acetic acid) for 45 minutes, and then soaked in Amplify (Amersham Biosciences) for 30 min. The gels were dried to filter paper and exposed to x-ray film (BioMax MR, Eastman Kodak, Rochester, NY) and developed after 20h.
Animal studies
All fifteen animals were colony-bred rhesus macaques (Macaca mulatta), obtained from Covance Research Products (Alice, TX). The animals were housed and handled in accordance with the standards of the Association for the Assessment and Accreditation of Laboratory Animal Care International, and the study was reviewed and approved by the animal care and use committees at Advanced BioScience Laboratories (Kensington, MD). The care and use of the animals were in compliance with all relevant institutional (NIH) guidelines. A total of eight animals were immunized in two rounds: M316, M218, M308, M490, and M599, M600, M607, M611, using the same DNA and protein preparation. Each immunized animal received 3 mg of the TEV89.6, TEVSF162, and TEVBa-L plasmid DNA by the intramuscular route at weeks 0, 3, and 8. Protein boost was performed using 100μg of the purified HIV89.6 TEV proteins in CpG (2 μg /ml; ODN 2006, Coley Pharmaceutical Group, Wellesley, MA). Challenge exposure of all vaccinated and control animals were performed by the intravenous route with SHIV89.6P titer XXX, kindly provided by Dr. Norman L. Letvin. The following groups of animals were challenged simultaneously with identical viral stock: animals M316, M218, M308, M490, L915, M320; animals M599, M600, M607, M611, M604; animals L941, L942, L947, and L962.
ELISA
Briefly, ELISA plates were coated overnight at 4°C with 100 μl/well 50 mM of sodium bicarbonate buffer (pH 9.6) containing 100 ng/well BSA plus: 50 ng/well of rHIV-1HXB2 Tat, rHIV-189.6 Tev, rHIV-1MN Rev proteins, a pool of 25 ng/well of each recombinant gp140 (from Ba-L, SF162 P3, or HIV89.6). Plates were blocked at room temperature for 2h with 200 μl/well 5% sucrose, 1.25% nonfat dry milk and 2.5% BSA. Serum samples were diluted in Dilsim II (BioMerieux, Durham, NC), and 100 μl/well were added to plates, in duplicate, and incubated for 1h at 37°C. Plates were washed four times with a solution of PBS at a 0.5% concentration of Tween 20. Plates were incubated at 37°C with 100μl/well goat anti-human IgG HRP conjugate diluted in PBS plus 20% NGS and 0.1% Triton-X-100 for 1h. Plates were washed four times in PBS plus 0.5% Tween 20, incubated for 30 min at room temperature with 100 μl/well TMB substrate, and stopped with 100 μl/well 2 N sulfuric acid. Absorbance was read at 450 nm. End point titers were calculated by determining the dilution at which a sample’s absorbance value equals two times the absorbance of the pre-bleed sera at 1:50 dilution.
Pepscans
A library of forty-eight peptides spanning the entire length of the HIV-189.6 Tev protein was synthesized. Peptides are fifteen residues in length and overlapped the flanking peptides by eleven residues. Peptides 1 to 18 are derived from the first exon of Tat; peptides 16 to 28 are derived from the Env V1 region; peptides 26 to 48 are derived from the second exon of Rev. ELISA plates were coated at 4°C overnight with 100 μl/well 50 mM sodium bicarbonate buffer (pH 9.6) containing 100 ng/well BSA plus 1 μg/well individual peptides. Plates were blocked at room temperature for 2h with 200 μl/well 5% sucrose, 1.25% nonfat dry milk, and 2.5% BSA. Serum samples were diluted 1:100 or 1:5000 in Dilsim II, and 100 μl/well were added to plates, and incubated for 1h at 37°C. Plates were washed four times with PBS plus 0.5% Tween 20. Plates were incubated at 37°C with 100 μl/well goat anti-human IgG HRP conjugate diluted in PBS plus 20% NGS and 0.1% Triton-X-100 for 1h. Plates were washed four times in PBS + 0.5% Tween 20. Plates were incubated for 30 min at room temperature with 100 μl/well TMB substrate, and stopped with 100 μl/well 2 N sulfuric acid. Absorbance was read at 450 nm. Absorbance values from pre-bleed samples were subtracted as background from the absorbance values of reactive samples.
ELISPOT assay
ELISPOT assay was performed with the monkey-specific IFN-γ Millipore kit (Billerica, Massachusetts), according to the manufacture’s specifications. Briefly, after coating with anti-IFN-γ monoclonal antibody, plates were incubated overnight at 4°C and washed, and wells were covered with 5% human serum in PBS at room temperature for 2 hours. After washing, PBMCs 2×105 in 50μl of RPMI-1640 containing 5% heat-inactivated human serum were plated in each well and stimulated in triplicate with the HIV-I89.6 Env peptide pool at 1 μg/ml. Concanavalin A (5μg/ml) was used as a positive control. After 18h incubation at 37°C in a 5% CO2 atmosphere, plates were washed and coated with filtered rabbit polyclonal anti-IFN-γ biotinylated detector antibody for 2 hours. Plates were developed with chromogen substrate activated by alkaline phosphatase in streptavidin AP conjugate and SFC’s counted on an ImmunoSpot Analyzer (CTL LLC, Cleveland, Ohio).
Antibody-dependent cellular cytotoxicity and cell-mediated virus inhibition (ADCC and ADCVI) assays
Antibody-dependent cellular cytotoxicity (ADCC) was evaluated by a flow-cytometry-based assay as previously described [34;35]. Target cells were CEMx174 T-cells infected with SHIV89.6P and the effector cells were human PBMCs. An effector to target [E:T] ratio of 50:1 was used. Titers are defined as the reciprocal of the serum dilution at which cell killing was greater than the mean of week 0 samples of all macaques plus 3 standard deviations.
The ADCVI assay was based on methods described previously for measles virus and HIV [36]. Target cells consisted of CEM.NKR--CCR5 cells infected with the SHIV89.6P or the SHIV89.6P variants. After adsorption for 1h, target cells were washed, incubated in 5% CO2 at 37°C for 72h in medium, and washed an additional two times. Infected target cells (5×104) were next plated in 96-well round-bottom microtiter plates, to which heat-inactivated test plasma and fresh human PBMC effector cells (E:T ratio of 10:1) were added. Plasma in the absence of effector cells was also tested for anti-viral activity. After 8 days incubation at 37°C in 5% CO2, supernatant fluid was collected and assayed for p27 by ELISA (Zeptometrix, Buffalo, NY). Virus inhibition due to ADCVI was calculated as follows: % inhibition = 100(1 - ([p27p]/[p27n])), where [p27p] and [p27n] are the concentrations of p27 in supernatant fluid from wells containing SHIV-positive or -negative plasma, respectively.
HIV89.6 Tev protein purification
The HIV89.6 Tev protein was produced in E. coli and purified using immunoaffinity chromatography. The HIV89.6 tev gene was amplified from the pPCR-Script Amp SK vector containing the synthetic HIV89.6 tev gene using the following primers: HIV89.6 tev 5’ Nde (acttagcatatggaacctgtgaaccctagcctgga) containing the NdeI site and HIV89.6 tev 3’ Xho (ctaagtctcgagttactccttggtgccaggttc) containing the XhoI site. The NdeI/XhoI DNA fragment encoding the tev gene was ligated into the bacterial expression plasmid pET26 (EMD Biosciences, Madison, WI). The resulting pET26 HIV89.6 tev plasmid is kanamycin resistant, and the expression of tev is under control of the IPTG-inducible T7 lac promoter. BL21 (DE3) competent cells (EMD Biosciences) were transformed with pET26 HIV89.6 tev and the tev sequence of clones were confirmed by DNA sequence analysis.
One liter culture of the HIV89.6 tev E. coli was grown at 37°C to 0.9 OD600nm, cooled to 22°C and induced with 1 mM IPTG for 6h. Cells were harvested by centrifugation and lysed in 40 ml of a PBS-based buffer containing 0.5 M sodium chloride, 10 mM ascorbic acid, 50 mM mannitol, 2.5 % glycerol, 0.5 % Triton-X-100, protease inhibitor cocktail, 50 μg/ml lysozyme, 1 μl/ml benzonase nuclease (EMD Biosciences), and 5 mM DTT. Lysates were sonicated and clarified by centrifugation. TEV was purified from the lysate on mouse monoclonal antibody to HIV-1 Tat sepharose, using 100 mM sodium carbonate with 5 mM DTT for elution. The eluate was buffered with phosphate buffer and the pH was adjusted to 7.4 with hydrochloric acid. Protein was passed through a goat anti-mouse IgG sepharose column to remove any mouse antibody that may have leached off of the immunoaffinity column. Endotoxin was reduced to about 400 EU/mg protein using Detoxigel (Pierce Biotechnology, Rockford, IL). The final Tev product runs as a 28 kDa protein on SDS-PAGE with a purity of about 90%. In western blot, HIV89.6 Tev reacts well with rabbit anti-sera to HIV-1 gp120, HIV-1 Tat, and HIV-1 Rev, demonstrating that the Tat, Env, and Rev components that compose Tev are present and recognized immunologically.
RNA extraction and RT-PCR
Viral RNA was extracted from 140 μl of plasma using a QIAamp viral RNA kit (Qiagen, Valencia, CA) with the final elution in 60 μl of elution buffer. A Titan One-Tube RT-PCR system (Roche Molecular Biochemicals, Indianapolis, IN) was used for the viral RNA reverse transcription according to the manufacturer’s protocol. The following oligonucleotides were used for amplification of V1/V2 region: Fwd 89.6 D1 5’-GCCACACATGCCTGTGTACCCACAGA and Rev 89.6 D2 5’-CCAGCCGGGACACAATAATGTATGGGA. PCR product was treated with 1 μl of Taq polymerse (New England Biolabs, Inpswich, MA) for 15 min at 72°C and purified using QIAquick gel extraction kit (Qiagen, Valencia, CA). V1/V2 PCR products were cloned into pCR®2.1 TA TOPO vector (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol. QIAprep spin miniprep kit (Qiagen, Valencia, CA) was used for plasmids isolation. Samples were sequenced by ABI dye terminator sequencing (Perkin-Elmer Corp) and analyzed using internet available tools: http://www.expasy.ch/tools/dna.html and http://www.ebi.ac.uk/Tools/clustalw2/index.html for translation of nucleotide sequences and amino acids alignment, respectively.
Site-specific mutagenesis
The infectious molecular clone of SHIV89.6 was obtained as two plasmids (pSHIV-89.6 5’, cat. number 4130, Lot number 1 and pSHIV-89.6 3’, cat. number 4131, Lot number 3041818) through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, National Institutes of Health from Dr. Joseph Sodroski [37].
SHIV89.6 P 3’ plasmid was used as a template for generation of envelope mutants by PCR. The following site-specific mutagenic oligonucleotides were used and the sequence of plasmid clones was analyzed to confirm the mutations.
SHIV89.6 3’ PtoL Fwd 5’ GAATACTACTAATCTCACTAGTAGCAGCTGG, (aa sequence NTTNLTSSSW), SHIV89.6 3’ PtoL Rev 5’ CCAGCTGCTACTAGTGAGATTAGTAGTATTC (aa sequence NTTNLTSSSW); SHIV89.6 3’ StoN Fwd 5’ CTACTAATCTCACTAATAGCAGCTGGGGAATG (aa sequence TNLTNSSWGM), SHIV89.6 3’ StoN Rev 5’ CATTCCCCAGCTGCTATTAGTGAGATTAGTAG (aa sequence TNLTNSSWGM). 5 μg of SHIV89.6 5’ cut SphI and ClaI and 5 μg of SHIV89.6 P 3’ wild type plasmid or its envelope mutants cut SphI and AflII were purified by phenol/chloroform extraction followed by ethanol precipitation using standard procedures and ligated using T4 DNA ligase (USB Corporation, Cleveland, OH). 2 μg of linear DNA product were used for transfection of 293T-cells, seeded 24h earlier in 6-well plate, supplemented with Dulbecco’s modified Eagle medium (DMEM) containing 10% of FBS, 2 mM penicillin-streptomycin and 5 mM L-glutamine using Effectene (Qiagen, Valencia, CA) according to the manufacturer’s protocol. 48h after transfection supernatant was collected, stored at -80°C for future p27Gag ELISA measurements and infection of CEMx174 cells.
Luciferase assay
Supernatent fluid from SHIV89.6 wild type or envelope glycosylation mutant virus-infected CEMx174 cells, normalized for p27 Gag content, was used for infection of TZM-bl cells. Briefly, TZM cells were seeded in a 24-well plate and infected the following day. At the day of infection, cells were incubated in 0.5 ml of supernatant containing SHIV89.6P wild type or its envelope mutant viruses. After 4 hours, the cells were washed three times with PBS and supplemented with fresh DMEM medium containing 10% of FBS, 2 mM penicillin-streptomycin and 5 mM L-glutamine (Invitrogen, Carlsbad, CA). Cells and supernatant were collected 48h post-infection. The Dual-Luciferase Reporter Assay System was obtained from Promega (Madison, WI) and all measurements were performed according to the manufacture’s protocol. Luciferase values were normalized to protein content and protein concentration was determined using the Bio-Rad (Hercules, CA) Protein Assay.
Kinetics assay
CEMx174 cells (2×106 cells/ml) were incubated with 2 ml of supernatant containing SHIV89.6P virus or its envelope mutant viruses in 15 ml tubes at 37°C and 5% CO2. Every 15 minutes cells were gently swirled. After 2h, the cells were washed three times with PBS and aliquoted. 2×105 cells were resuspended in 1 ml fresh RPMI medium containing 10% of FBS, 2 mM penicillin-streptomycin and 5 mM L-glutamine (Invitrogen, Carlsbad, CA) and further incubated at 37°C. Supernatant was collected daily for four days, and stored at -80°C.
p27 ELISA
In experiments measuring the growth kinetics of SHIV variants, the SIV p27 Antigen Capture Assay Kit (Advanced BioScience Laboratories Inc., Kensington, MD) was used to quantify p27Gag in the supernatant. Undiluted or diluted cell culture supernatants were used for analyses to obtain values within the standard range. Experiments were repeated three times and all measurement performed according to the manufacture’s protocol. Results are shown as average and standard deviation of duplicates.
Neutralization assay
Neutralization was measured as a reduction in luciferase reporter gene expression after a single round of infection in TZM-bl cells as described [38;39]. TZM-bl cells were obtained from the NIH AIDS Research and Reference Reagent Program, as contributed by John Kappes and Xiaoyun Wu. Briefly, 200 TCID50 of virus was incubated with serial 3-fold dilutions of test sample in duplicate in a total volume of 150 μl for 1 hour at 37°C in 96-well flat-bottom culture plates. Freshly trypsinized cells (10,000 cells in 100 μl of growth medium containing 75 μg/ml DEAE dextran) were added to each well. One set of control wells received cells + virus (virus control) and another set received cells only (background control). After 48-hour incubation, 100 μl of cells was transferred to a 96-well black solid plate (Costar) for measurements of luminescence using the Britelite Luminescence Reporter Gene Assay System (PerkinElmer Life Sciences). Neutralization titers are the dilution at which relative luminescence units (RLU) were reduced by 50% compared to virus control wells after subtraction of background RLUs. Assay stocks of the molecularly cloned SF162.LS Env-pseudotyped virus and of the proviral NL-ADArs clone were prepared by transfection in 293T cells. An assay stock of uncloned SHIV-89.6P was prepared in human PBMC. All viruses were titrated in TZM-bl cells as described [39].
Rhesus Serum IgG Purification
IgG was purified from pooled sera collected from animal M316 at weeks 30, 34, 38 based on methods previously described [40]. Pooled serum was heat inactivated at 56° C for 1 hour and delipidated by centrifugation at 90,000 × g for 16 hours. The lower phase was removed from the lipid-containing upper phase and clarified with a 0.22 μm filter. The clarified serum was buffered with PBS and bound to Prosep® Ultra Plus (Millipore, Billerica, MA) protein A resin. Bound IgG was washed with PBS and eluted in 0.5 M Acetate buffer, pH 3. The eluate was buffered with Tris, and the pH was adjusted to 7.2 with 3 N sodium hydroxide. The eluate was concentrated using a 30 kDa molecular weight cutoff filter, and the buffer was exchanged with normal saline. The concentration of the purified IgG was calculated based on the absorbance at 280 nm and the extinction coefficient of 1.35 for a 1mg/ml solution. From 102 ml of serum, 1018 mg IgG was purified (18.5 mg/ml). Identity of purified IgG was verified by reducing and non-reducing SDS-PAGE. As described for animal sera, binding to rHIV-189.6 Tev, pooled recombinant gp140 (from Ba-L, SF162 P3, or HIV89.6P) and pooled rHIV-189.6 V1 peptides (peptides 16-28) was confirmed using ELISA. Endotoxin levels were determined to be <0.03 EU/mg using the QCL2000 assay (Lonza, Walkersville, MD).
Statistical methods
Two-group comparisons of viral load and immune response data were made using the exact Wilcoxon rank sum test. Correlations between these quantitative factors were assessed by the exact Spearman rank correlation method. Tests of mutant viral replication in vitro were performed by the analysis of variance of log-transformed activity relative to wild type. All p values are two-tailed, and p values from simultaneous tests of three viral variants were corrected by the Hochberg method.
RESULTS
Tev vaccines partially protect rhesus macaques from pathogenic SHIV89.6P challenge
Both the laboratory adapted HIVIIIB strain and the pHXB2 molecular clone derivative produce Tev protein that is encoded by a doubly spliced mRNA and that juxtaposes the first exon of Tat to the V1 region of the envelope and to the second exon of Rev [29-31]. Thus, Tev contains only the V1 region of the HIV-1 envelope gene. We investigated whether Tev would be a useful vaccine platform to present V1 to the host’s immune system. We have chosen the SHIV89.6P macaque model because we wished to test the immunogenicity and protective potential of HIV-1 epitopes within V1. In addition, it has been shown that Tat based vaccines do not protect from SHIV89.6 high virus levels so protection in this model would likely be due to V1 as opposed to Tat immune responses [41;42]. This model has been criticized because the SHIV89.6P isolate uses the CXCR4 receptor for infection of rhesus macaques, whereas most HIV-1 transmission to human occurs with HIV strains that use the CCR5 receptor [23;24;43], However, CXCR4 user HIV strains are found during the chronic HIV infection and can also be transmitted although rarely [44;45]. Importantly, is the SHIV89.6P challenge model, Tat–based vaccines alone do not confer protection from a high level of plasma virus [42;46].
We confirmed that transfection of the pHXB2 molecular clone into 293T-cells results in the expression of the 27-28 kDa Tev protein, which occurs simultaneously with the expression of Tat, Rev, Nef, and Env as expected [29-31] (Fig. 1A). The vaccines consisted of Tev cDNA plasmids derived from the HIVSF162, HIVBAL, and HIV89.6 strains whose sequence differs greatly in V1 (Fig. 1B). All three Tev cDNAs yielded the expected 28 kDa protein upon transfection in 293T-cells (Fig. 1C). Because our aim was to induce an antibody response to V1, we also expressed and purified the HIV89.6 Tev protein in bacteria (Fig. 1C) and used it in a DNA prime-protein boost approach that is known to induce good antibody responses, to vaccinate the animals with the regimen depicted in Fig. 1D.
Figure 1. Relative efficacy of Tev vaccines in macaques.
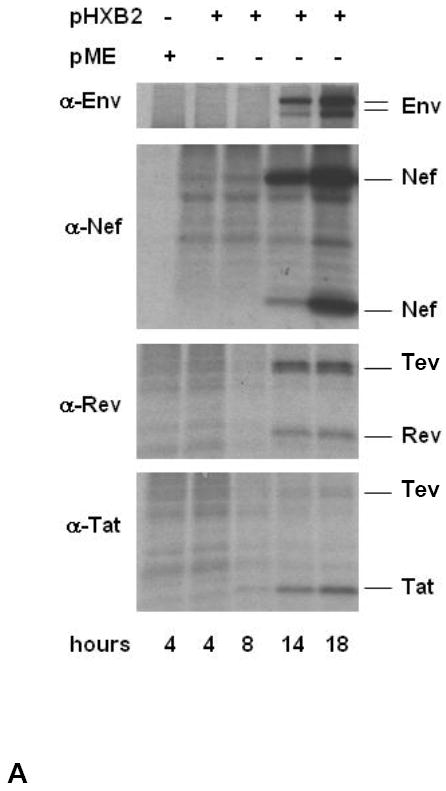

(A) The expression of Env, Nef, Rev, and Tev proteins was assessed by western blot in cell lysates at 4, 8, 14, and 18 hours post-transfection of the pHXB2 molecular clone in 293T cells.
(B) Amino acid sequence alignment of the putative HIVHXB2, HIVBa-L, HIVSF162, and HIV89.6 Tev proteins.
(C) HIVBa-L, HIVHXB2, HIVSF162, and HIV-189.6 Tev proteins expressed in HEK 293 cells. The Tev proteins produced by the cDNAs were identified using antibodies to Rev. The 89.6 Tev protein was purified from E. coli using a mouse anti-HIV-1 Tat column and separated on 10-20 % SDS-PAGE (right panel). Tev protein was recognized by Rabbit antibody to Rev (lane 1), but not with normal rabbit serum (lane 2). Molecular weight marker is shown in lane 3.
(D) Study design. The HIVBa-L, HIV89.6, and HIVSF162 Tev cDNAs were used in each DNA immunization, where “prot” stands for boosts with the purified HIV89.6 Tev protein. Challenge exposure to SHIV89.6P was performed via the intravenous route.
(E) Plasma virus levels in vaccinated and control macaques (left and center panels) following challenge exposure to SHIV89.6P. Right panel depicts mean level for both groups.
(F) CD4+ T-cell counts in vaccinated and control macaques (left and center panels) following challenge exposure to SHIV89.6P. Right panel depicts median level for both groups.
Eight macaques were immunized with the HIV89.6, Ba-L, and SF162 Tev plasmids by the conventional intradermal and intramuscular route (weeks 0, 3, 8), followed by two boosts with the HIV89.6 Tev protein in CpG adjuvant at week 12 and 18 (Fig. 1D). Of seven control animals, three were immunized with CpG (M320, L915 and M604), and four were left un-immunized. All animals were challenged intravenously with the same stock of the highly pathogenic SHIV89.6P virus at 22 weeks from the first immunization (Fig. 1D).
Virus levels following intravenous challenge with SHIV89.6P were measured by NASBA in plasma of vaccinated and control rhesus macaques. The eight vaccinated macaques had significantly lower plasma virus levels at weeks 10, 12, 14, and 16 weeks post-challenge (p<0.013 for each) than the seven unvaccinated controls (Fig. 1E). Among the eight vaccinated animals, four (M316, M490, M308, and M607) had significantly lower plasma virus levels than the other vaccinated macaques over weeks 10 to 16 (p=0.029 by the Wilcoxon rank sum test). The remaining four vaccinated rhesus macaques also had lower plasma virus levels than the control animals at all time points (p<0.05, data not shown). There was a trend toward better preservation of CD4+ T-cell counts in vaccinated animals following infection with SHIV89.6P (Fig. 1E).
Type specific antibodies to V1 but not ELISPOT inversely correlate with plasma virus levels
Vaccination elicited low levels of ELISPOT responses against 89.6 Tev overlapping peptides, which were negligible at the time of challenge (see week 0 in Fig. 2A and data not shown). In some of the vaccinated macaques however, ELISPOT responses were detected following challenge with SHIV89.6P (Fig. 2A). Animal M218 mounted a broad response to the Tev peptides derived from HIVBa-L, HIVSF162, and HIV89.6, likely because of persistent virus replication post-challenge; animal M308 responded better to peptides from HIV89.6 and less so to HIVBa-L and HIVSF162, whereas animal M316 had a poor response to all the peptides in the pool, likely because viral replication was blunted early (Fig. 1E). Interestingly, the ELISPOT responses were not elicited by infection with SHIV89.6P in the naïve control animals, even though these animals responded to Concanavalin A (Fig. 2A and data not shown). There was no significant correlation between plasma virus levels at any time and T-cell responses before or after challenge (data not shown and Table 3). Because this vaccine regimen induced poor T-cell responses, ELISPOT assays were not performed in the remaining vaccinated or control macaques.
Figure 2. Immune responses in the vaccinated/challenged macaques.

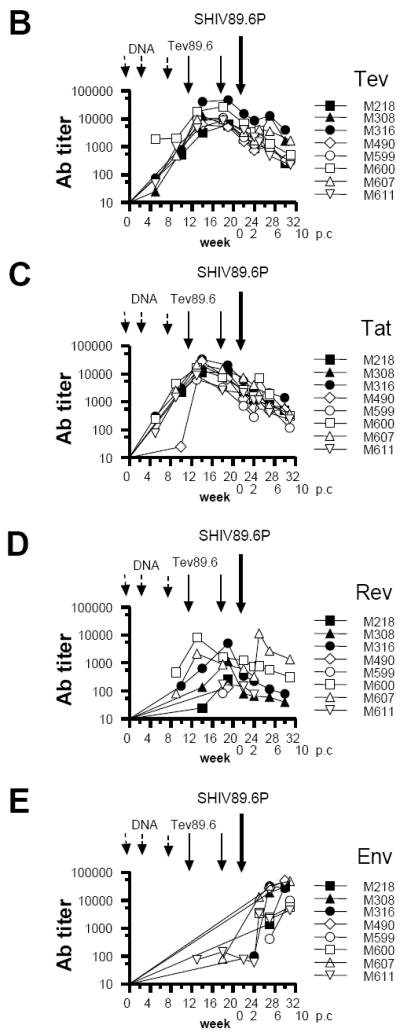

(A) ELISPOTs were performed using HIVBa-L, HIVSF162, and HIV89.6 Tev peptide pools for the stimulation of macaque blood. ELISPOT responses were also obtained using Concanavalin A, a stimulus to control for cell viability (data not shown).
Antibody titers to Tev (B), Tat(C), Rev(D), and the pooled HIVBa-L, SHIVSF162P3, and SHIV89.6P Envelope proteins (E). The arrows refer to DNA immunizations (weeks 0, 4, 8), protein boosts (weeks 12, 18), and challenge exposure (week 22); titers to the purified Tev, Tat, Rev, and pooled HIVBa-L, HIVSF162P3, and HIV89.6P Envelope proteins. The weeks 0, 2, 6, and 10 refer to the weeks post challenge (p.c.). Data from control macaques are not shown.
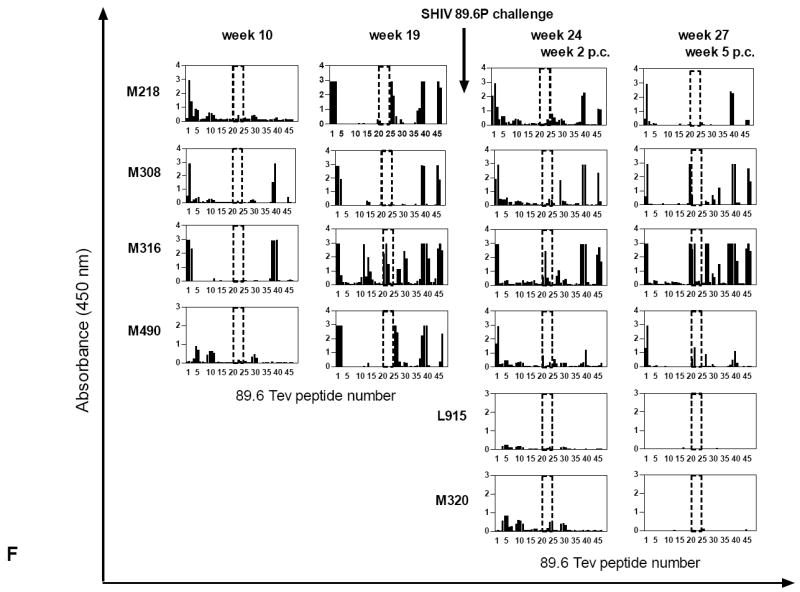
(F) Overlapping peptides of the entire HIV89.6 Tev protein whose sequence is depicted in Fig. 1B were used with the sera (1:100 dilution) of the macaques after immunization (weeks 10, 19) and challenge exposure (weeks 2, 5 p.c.) in some of the vaccinated and control macaques. The area included in the dotted line area defines the V1 peptides (16 to 23), proceeded by the peptides derived from the first exon of Tat and followed by the peptides derived from the second exon of Rev.
(G) Sera (1:100 dilution) of all immunized macaques after challenge exposure (weeks 2, 5 p.c.) and the two control macaques L915 and M320 were reacted with overlapping peptides 16–28 for the 89.6 V1.
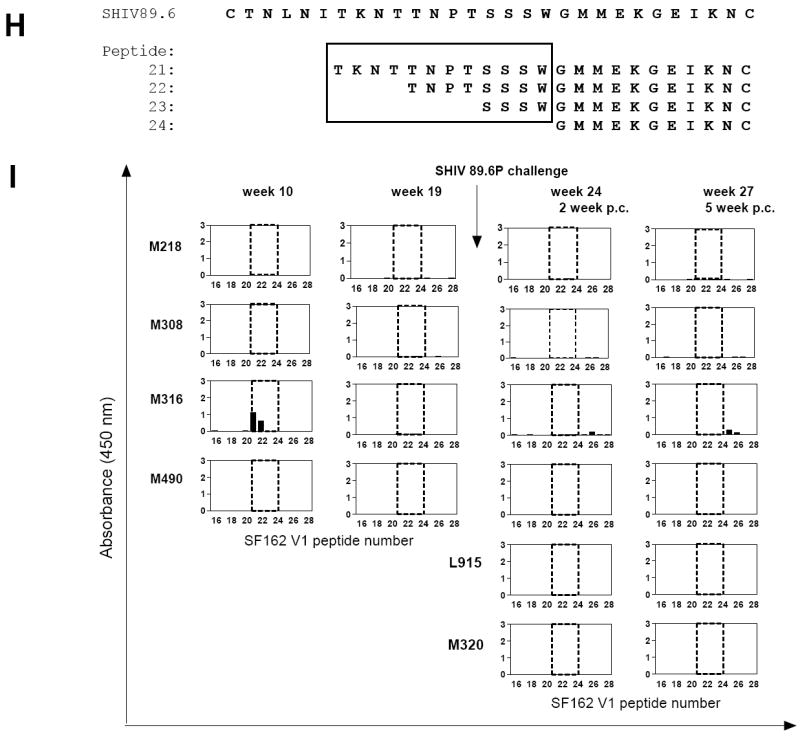
(H) Amino acid sequences of the 21-24 overlapping peptides recognized by the sera of macaques M308, M316, and M490 and M607 within the V1 region of SHIV89.6P.
(I): Sera (1:100 dilution) of the immunized macaques M218, M308, M316, and M490 before and after challenge exposure (weeks 10 and 19 and 2, 5 respectively) and the two control macaques. L915 and M320 were reacted with overlapping peptides 16–28 for the SF162 V1 region.
Table 3.
Immune correlate of protection against high plasma virus levels
| Immune Response Post challenge | Virus in plasma | Correlation |
|---|---|---|
| ELISA Ab titer to envelope (week 5) | Week 5 Post challenge | -0.83 p=0.015 |
|
| ||
| ELISA Ab titers to Tat | Any time post Post challenge | No Correlation |
|
| ||
| ELISA Ab titers to REV | Any time post Post challenge | No Correlation |
|
| ||
| Week 4 / 6 ADCVI SHIV89.6 swarm | Week 7 to 16 Post challenge | -0.84 p -0.044 |
|
| ||
| ADCV1 week 5 | Week 7 to 16 Post challenge | |
| WT SHIV89.6A | -0.079 p=0.0034 | |
| Variant SHIV89.6L | -0.72 p=0.0094 | |
| Variant SHIV89.6LN | -0.64 p=0.017 | |
|
| ||
| Week 22 | Week 7or 16 Post challenge | |
| SHIV89.6W | -0.76 p=0.015 | |
| SHIV89.6L | -0.85 p=0.0086 | |
| SHIV89.6LN | -0.78 p=0.021 | |
|
| ||
| Neutralizing Antibodies titers to SHIV89.6P Week 6 | Week 7 to 16 Post challenge | -0.72 p=0.0050 |
|
| ||
| Neutralizing Antibodies titers to HIV NL-ADArs Week 4 | Weeks 7 to 16 Post challenge | -090 p=0.0001 |
|
| ||
| Neutralizing Antibodies titers to HIV NL-ADArs | Week 7 to 16 Post challenge | -0.83 p=0.0004 |
We measured ELISA antibody titers in sera of rhesus macaques against recombinant 89.6-derived Tev, Tat, Rev, and a pool of recombinant Ba-L, SF162P3, and 89.6P Env proteins purified from the conditioned media of HEK 293 Env-expressing cell lines. All vaccinated animals developed antibody to 89.6 Tev (range of titers = 6×103 to 5×104) and to Tat (range = 6×103 to 3.5×104) (Fig. 2B and 2C). Titers to Rev were lower and peaked at 8.3×103 in animal M600 and at 5×103 in animal M316 (Fig. 2D). SHIV89.6P infection did not boost secondary responses to Tat, Tev, or Rev with the exception of animal M607, which experienced an increase in antibody titers to Rev following infection (Fig. 2D). This result suggest that there may be an insufficient expression of Tat and Rev protein in vivo to induce a clear secondary antibody response, as also observed by others [47]. Titers to the Env increased above 2 × 104 by week 25 (three weeks following challenge) in the vaccinated animals (M316, M308, M490, and M607) (Fig. 2E) and anti-Env antibody titers measured five weeks after challenge correlated inversely with plasma viremia level obtained at the same time (R = -0.83, p=0.015). Similarly, anti-Tev antibody titers inversely correlated with plasma virus level five weeks post infection (R=-0.87, p=0.0079). In contrast, there were no significant correlations between plasma virus level and titers of antibody against Tat or Rev at any time point. These data suggest that a secondary antibody response to the V1 region of the Env might have played a significant role in the controlling of the virus levels in the vaccinated rhesus macaques.
To investigate this hypothesis, we first tested serum reactivity against overlapping peptides that encompassed the entire HIV89.6 Tev amino acid sequence. Sera obtained one week after the last immunization (week 19), recognized peptide clusters at the amino terminus region of Tat and at least two regions within Rev (Fig. 2F and data not shown). By this time point, the serum from animal M316 also recognized the peptide cluster 21-24 that correspond to the V1 region. However, following challenge sera from animals M316 (weeks 24, and 27), M308 (week 27), and M490 (week 24 and 27), also recognized peptides that spanned within the V1 region, whereas, sera from animal M218 and all of the other vaccinated animals bound minimally or not at all, to this amino acid stretch (Fig 2F and data not shown). Animal M316 serum recognized V1 peptides at a 1:5000 dilution five weeks post challenge (data not shown).
To further characterize the amino acids within V1 recognized by the animal sera, we used overlapping peptides encompassing only the V1 region of HIV89.6, SF162, and Ba-L. Interestingly, only the sera from the immunized animals that were able to contain plasma virus levels (M316, M308, M490, and M607) recognized the overlapping peptides 21 to 23 derived from the SHIV89.6P V1 amino acid sequence (Fig. 2G). The sera of the remaining vaccinated or control macaques did not bind or minimally bound to these peptides (Fig. 2G and data not shown). The amino acid sequence recognized, TKNTTNPTSSSW, is depicted in Fig. 2H and corresponds to the amino terminus of the SHIV89.6 V1 region. The antibody response to V1 was strain–specific, as sera that recognized the 89.6V1 did not recognize the peptides from the equivalent region of the HIVBa-L or HIVSF162 before or after challenge exposure (Fig. 2I and data not shown). Animal M316 was an exception as its serum recognized the V1 of SF162 at week 10 before challenge exposure (Fig. 2I).
In vitro replication of viral variants found in the plasma of vaccinated and control macaques
Masking of B-cell epitopes by N-linked or O-linked glycosylation within V1 is thought to favor escape from immune recognition [22;25;27]. The data presented above suggests that antibodies to the amino terminus of the V1 region could play a role in protection from high-level viral replication. Therefore, we studied the genetic composition of viral variants in the plasma of vaccinated and control animals following challenge of SHIV89.6P. RNA was obtained from longitudinal plasma samples from vaccinated and control animals and from the cell free SHIV89.6P challenge stock. The RNA was reverse transcribed and the V1 DNA sequence was obtained from a minimum of 6 molecular clones per sample. Genetic analysis of the challenge virus stock demonstrated the presence of two genotype variants: the most frequent variant has at amino acid position 143 a leucine whereas, a less frequent variant has a proline (Fig. 3A-B), similar to the non pathogenic clone SHIV89.6. Of note, the V1 sequence included in the HIV89.6 Tev used to vaccinate the rhesus macaques has a proline at position 143 (Fig. 1B) [37]. Importantly, the leucine to proline change at position 143 eliminates a potential N-linked glycosylation site, as the consensus sequence of putative glycosylation sites is NXT/S and X cannot be a proline.
Figure 3. Viral variants within the V1 region following challenge exposure to SHIV89.6P.

Putative amino acid sequence of the SHIV89.6P V1 region obtained by DNA sequencing of plasma viral RNA following RT/PCR. The putative amino acid sequences of V1 in the vaccinated (A) and control (B) macaques were aligned with the V1 region of the SHIV89.6 used in the Tev vaccine. The number of viral clones sequenced is depicted on the left of each figure.
Analysis of the in vivo selection of these two variants in the plasma of infected macaques demonstrated that the leucine 143 variant predominates in the plasma of most animals by the second week of infection (Fig. 3A and B). This predominant V1 variant was substituted by the variant that has a proline at position 143 in the plasma of animal M316 during primary infection and in animals M607, L941, and L947 during the chronic phase (Fig. 3A and B). We also observed a novel V1 variant with an asparagine to serine substitution at position 145. This mutation adds a potential N-glycosylation site (KNTTNLTNSSW) and this variant was predominant by week 8 in animal L915, but constituted a small fraction of the virus present in the remaining animals (Fig. 3A and B).
Because changes in glycosylation of the envelope protein have been associated with differences in HIV plasma virus levels [12], we investigated whether these amino acid changes affected viral replication in vitro. We used the non pathogenic molecular clone of SHIV89.6 that carries the proline at position 143 and designated it as SHIV89.6P WT. The proline at position 143 was mutagenized to a leucine and this clone was designated SHIV89.6 L. The P to L change reconstitutes a potential glycosylation site within V1, and this variant predominates in all animals within two weeks from infection as demonstrated in Fig. 3. In addition, we mutagenized the asparagine at position 145 to a serine in the clone SHIV89.6LN that introduces a new potential glycosylation site within V1. These constructs were transfected into 293T-cells, and the cell supernatant fluid, normalized for p27Gag content, was used to infect TZM-bl cells, human PBMCs, and the CEMx174 T-cell line [38]. Infection of the TZM cells did not reveal significant differences between the ability of SHIV89.6 WT, SHIV89.6 L, and SHIV89.6 LN to increase HIV-LTR driven luciferase activity (Fig. 4A). Accordingly, production of p27Gag was not significantly different among these viruses in the CEMx174 T-cell line (Fig. 4B), primary human PBMCs (Fig. 4C) and in primary rhesus macaques PBMCs (data not shown).
Figure 4. In vitro replication and sensitivity to ADCVI of SHIV89.6 mutant viruses.
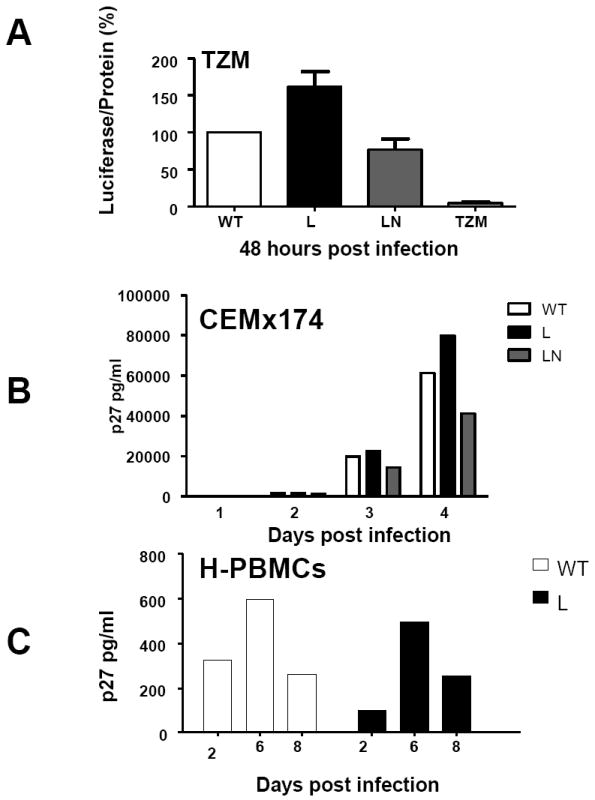
The supernatant of 293T-cells transfected with the HIV89.6WT, HIV89.6L, and HIV89.6LN mutants was normalized for p27Gag content and applied to TZM cells, CEMx174 cells, or primary human and rhesus PBMCs. Virus production was measured as activation of the HIV-! LTR Luciferase reporter gene in the TZM cells (A) or as p27Gag production in the supernatant fluid within the first 4 days of infection of the CEMx174 T-cells (B) or in human PBMCs up to day six post-infection (C).
Sensitivity to ADCVI and ADCC of SHIV 89.6 variants
Next, we investigated whether and when the sera of vaccinated macaques could mediate antibody-dependent cell-mediated inhibition of SHIV89.6P [36]. At a serum dilution of 1:100, inhibition of viral replication was observed in five of the six animals tested at time of challenge (Fig. 5A) and this activity was boosted following viral challenge exposure (Fig. 5B and 5C). Correlative analysis of ADCVI activity at week 4, with the antibody titers to the Envelope, Tat, and Rev proteins indicated a correlation that approached statistical significance only for the Envelope protein (R=0.84; p=0.067) consistent with the hypothesis that antibodies to V1 mediate ADCVI. Importantly, analysis of the geometric mean ADCVI titer at week 4 and 6 and plasma virus levels during the chronic phase of infection (weeks 7 to 16) revealed a significant inverse correlation, (R=-0.84; p=0.044), suggesting that this immune response contributed to the control of viral replication observed in these macaques.
Figure 5. Sensitivity to ADCVI of SHIV 89.6 viral variants.

SHIV89.6P-infected human CD4+ lymphocyte target cells were incubated with human PBMC effector cells (E:T = 10:1) and with plasma at the dilutions indicated. Eight days later, supernatant fluid p27Gag was determined by ELISA, and virus inhibition was determined as described in the Methods. Data are representative of several experiments, each performed in duplicate. Antibody-dependent cell-mediated virus inhibition (ADCVI) activity in plasma (1:100 dilution) of the animals was performed at first using the SHIV89.6P before immunization week -2 (data not shown), or at time of challenge (A), or thereafter at week 4 (B) and 6 (C). ADCVI titers to the SHIV89.6 viral variants a week 5 (D) and 22 (E) post challenge.
As demonstrated in Fig. 3A and 3B, the SHIV89.6P challenge stock contains at least two major genotypes that are present at different frequency in the plasma of infected macaques. Because these variants may vary in their susceptibility to ADCVI, we tested the ADCVI activity in a 1:100 dilution of the sera of the vaccinated and control macaques against the SHIV89.6 WT, SHIV89.6L, and SHIV89.6 LN viruses. As demonstrated in Fig. 5D, the sera of vaccinated animals had significantly higher ADCVI activity than control macaques at week 5 against only the SHIV89.6 WT (p=0.024). No significant differences were observed among vaccinated and control macaques in the ADCVI activity against all variants at week 22, (Fig. 5E). Importantly, a significant inverse correlation was found between plasma virus levels (week 7-16 interval) and the extent of ADCVI at week five post challenge (14 macaques) against the SHIV89.6L (R=-0.72p=0.0094), the SHIV89.6 WT (R=-0.79 p=0.0034), and the SHIV89.6 LN (R=-0.64,p=0.017) (Table 3). This inverse correlation was significant for all three variants also at week 22 SHIV89.6L (R=-0.85p=0.0086), SHIV89.6 WT (R=-0.76 p=0.015), and SHIV89.6 LN (R=-0.78,p=0.021). ADCC was also measured at week 2 and 10 post-challenge using CEMx174 cells infected with SHIV89.6P. As demonstrated in the Table 1, the only serum that had ADCC at week 2 was that obtained from animal M316. Thus, animal M316 had the highest titers to V1 and controlled plasma virus level to undetectable levels by week 6 post-infection. Only a few sera had ADCC activity at week 10 post-challenge (Table 1).
TABLE 1.
Titers of sera mediating ADCC activity in vaccinated and control macaques’ post-SHIV89.6P challenge
| ADCC titer post-challenge | ||
|---|---|---|
| Vaccinated Macaques | Week 2 | Week 10 |
| M218 | <10 | 10000 |
| M308 | <10 | <10 |
| M316 | 1000 | <10 |
| M490 | <10 | 100 |
| M599 | <10 | <10 |
| M600 | <10 | <10 |
| M607 | <10 | 10 |
| M611 | <10 | 10 |
| Control Macaques | ||
| M320 | <10 | <10 |
| L915 | <10 | 1000 |
| L962 | <10 | <10 |
Neutralizing antibody to SHIV89.6P is delayed compared to ADCVI
Rhesus macaques that mounted a secondary ADCVI antibody response by week 4 or 6 post-challenge, controlled virus replication better than those which did not, suggesting a protective role for these antibodies. To further study the functionality of these antibodies, we performed neutralization assays using TZMb1 cells [38] that expresses the CD4 and CCR5 HIV receptors. At the time of challenge, none of the animals’ sera had detectable neutralizing antibodies titers to SHIV89.6P, or the HIVSF162.LS and HIVNL-ADArs strains (Table 2). By week 4, only three of the vaccinated and none of the control macaques did develop low titers of neutralizing antibody to SHIV89.6P and none to SHIVSF162.LS (Fig. 6A-C and Table 2). By week 6 however, most vaccinated animals developed higher titers of neutralizing antibody to both SHIV89.6P, HIVSF162.SL (Table 2 and Fig. 6A-C), and titers to SHIV89.6P at week six inversely correlated with plasma virus levels (R=-0.72;p=0.005) (Table 3). Interestingly, neutralizing antibody to HIVNL-ADArs developed in most animals (Table 2) and correlated inversely with plasma virus levels (week 7-16) at both week 4 and 6 (R=-0.90;p=0.0001 and R=-83;p=0.0004 respectively, Table 3). HIVNL-ADArs is a CD4-independent virus that is highly sensitive to neutralization and in particular, appears to possess highly exposed epitopes in the V3 loop and co-receptor binding domains of gp120 [48]. Because of involvement of V1 in co-receptor usage, this virus would be expected to be a sensitive indicator of the vaccine-elicited neutralizing antibody response and of secondary V1-specific antibodies induced post-infection that are not necessarily capable of directly neutralizing the challenge virus. Thus, ADCVI was elicited as early as 4 weeks from infection, whereas a high titer of neutralizing antibody to the challenge virus SHIV89.6P, developed two weeks later (week 6), and by then both neutralizing antibody and ADCVI correlated with suppression of viral replication. Collectively, these data suggest that ADCVI mediated by non-neutralizing antibodies may be important in the early control of viral replication.
TABLE 2.
Neutralizing antibody titers at time of challenge and post infection in the sera of immunized and control macaques.
| ID50 in TZM-bl cells1 | ||||
|---|---|---|---|---|
| Macaque | Week | SF162.LS | SHIV89.6P | NL-ADArs |
| M316 | -2 | <20 | <20 | <20 |
| 0 | <20 | <20 | <20 | |
| 4 | <20 | 48 | 883 | |
| 6 | 57 | 50 | 3177 | |
| M308 | -2 | <20 | <20 | <20 |
| 0 | <20 | <20 | <20 | |
| 4 | <20 | 29 | 1850 | |
| 6 | 40 | 163 | 3,611 | |
| M490 | -2 | <20 | <20 | <20 |
| 0 | <20 | <20 | <20 | |
| 4 | <20 | <20 | 1,832 | |
| 6 | 87 | 107 | 15,732 | |
| M218 | -2 | <20 | <20 | <20 |
| 0 | <20 | <20 | <20 | |
| 4 | <20 | <20 | 37 | |
| 6 | <20 | <20 | 538 | |
| L915 * | -2 | <20 | <20 | <20 |
| 0 | <20 | <20 | <20 | |
| 4 | <20 | <20 | <20 | |
| 6 | <20 | 47 | 1,949 | |
| M320* | -2 | <20 | <20 | <20 |
| 0 | <20 | <20 | <20 | |
| 4 | <20 | <20 | <20 | |
| 6 | <20 | <20 | 21 | |
| 0 | <20 | <20 | <20 | |
| M599 | 4 | <20 | <20 | <20 |
| 6 | <20 | <20 | 59 | |
| M600 | 4 | <20 | <20 | 41 |
| 6 | <20 | <20 | 47 | |
| M607 | 4 | <20 | 26 | 95 |
| 6 | <20 | 446 | 1,752 | |
| M611 | 4 | <20 | <20 | 669 |
| 6 | <20 | <20 | 285 | |
| L941* | 4 | ND | ND | ND |
| 6 | <20 | <20 | 21 | |
| L942* | 4 | <20 | <20 | 30 |
| 6 | <20 | <20 | 78 | |
| L947* | 4 | <20 | <20 | <20 |
| 6 | <20 | <20 | <20 | |
| L962* | 4 | <20 | <20 | <20 |
| 6 | <20 | <20 | <20 | |
Values are the serum dilution at which relative luminescence units (RLUs) were reduced 50% compared to virus control wells (no test sample).
ND=Not done.
Non vaccinated
Figure 6. Neutralizing antibody titers and passive transfer of purified Ig to a naïve macaque.

Neutralization antibody titers were measured as the highest serum/plasma dilution that induced a 50% reduction in luciferase reporter gene expression after a single round of infection in TZM-bl cells as described [38;39] using HIV NL-ADArs(A) SF162.LS, (B) or SHIV89.6P. (C) The data presented were obtained with plasma collected at week 4 and 6 post challenge. (D) Titers of ADCVI in macaque M316 at 24 weeks post exposure to SHIV89.6P. (E) Virus plasma levels in macaques challenged with SHIV89.6P following treatment with purified Ig from macaques M316 (macaque P155) or normal human Ig (macaque P156).
Passive transfer of immunoglobulins to naïve macaques fails to protect from SHIV89.6P infection and high virus levels
Since we demonstrated a correlation between antibodies able to mediate ADCVI, as well as neutralization and plasma virus levels, we wished to test whether purified immunoglobulins (Ig) could confer some level of protection from SHIV89.6P. To do so, we obtained serum from the vaccinated macaque M316 because this animal had ADCVI activity that was maintained up to 24 weeks after challenge exposure (Fig. 6D) even in the absence of overt plasma viremia (Fig. 1E).
Ig was purified from the serum collected from macaque M316 at time of euthanasia (week 24 post infection). At this time point, the serum of animal M316 still had ADCVI activity (Fig. 5D) as well as, neutralizing antibody titer to 89.6P of 1:263 in TZM-bl cells. We inoculated 400mg/kg of purified Ig from macaque M316 into naïve macaque P155 by the subcutaneous route and exposed this animal by the intravenous route to SHIV89.6P six hours following the Ig inoculation. A control naïve animal, macaque P156, was inoculated with the equivalent amount of a commercially available human immunoglobulin preparation. At the time of challenge the titers of neutralizing antibody and ADCVI to SHIV89.6P in the serum of macaque P155 was 68 and 1,600 respectively. At one week after infection, the neutralization titer dropped to 31, whereas ADCVI activity was sustained. Nevertheless, despite the presence of ADCVI and low neutralizing antibody titers, macaque P155 became infected following exposure to SHIV89.6P and its plasma virus level did not differ from that of the control macaque P156 (Fig. 6E).
DISCUSSION
A protective role of CD8+ T-cells [49;50], including SIV-specific CD8+ T-cells [51], in the control of SIV replication has been inferred by several studies in non-human primates. However, in rhesus macaques, vaccines able to induce T-cell responses have afforded only a limited degree of protection from high virus level and at most have delayed, rather than prevented AIDS [52-56]. The recent partial success of a combination of vaccine able to induce both T and B-cell response suggest the importance of antibody to the envelope protein [57]. Thus, there is a pressing need for continued evaluation of the role of antibody in controlling HIV/SIV replication. Early studies in chimpanzees have demonstrated that antibodies to the V3 loop, provide type-specific immunity to laboratory isolates of HIV [58] and that chimpanzees develop both neutralizing and ADCC antibodies to V3 [59]. The protective role of neutralizing antibodies has also been demonstrated in macaques, whereby passive administration of a large quantity of antibodies that neutralize a number of HIV strains has prevented infection of non human primates exposed mucosally or intravenously to high doses of SHIV [60;61]. Of note, passive administration of lgG1b12, a broadly neutralizing monoclonal antibody, has protected rhesus macaques from infection. However, the in vivo mechanism of protection was in part, Fcγ-R receptor mediated. Thus, it appears that antibody functions apart from neutralization contributed to protection [62]. Indeed, vaccine studies in non human primates [34;63-66] and analyses of sera from HIV-infected individuals [67;68] have shown an inverse correlation between the level of antibodies that mediate either ADCC or ADCVI and virus level. Antibodies directed against HIV/SIV Env, Tat, and Nef proteins [66;69;70] have been shown to mediate ADCC or ADCVI. We hypothesized that the high variability and plasticity of the HIV-1 V1 envelope region may have evolved to avoid the pressure of innate and/or adaptive host responses. Indeed, our study suggests that pre-existing antibody to V1 may affect viral replication in the immunized macaques, as also suggested by studies on viral fitness in vivo [27]. The notion that V1 may affect viral fitness is also suggested by studies in humans demonstrate that, early selection of genotypic variants in V1 occurs in humans during mucosal transmission of HIV [17;18]. Additionally, neutralizing antibodies directed to V1 have been described [71]. To test this hypothesis, we have chosen as a vaccine platform the Tev gene which encodes the V1 region of the HIV-1 envelope protein and the first and last exons of the Tat and Rev protein respectively. The Tev mRNA is generated by a rare natural mutation in the HIV envelope gene that leads to the inclusion of a cryptic exon in most HIV mRNAs and results in decreased viral production [29-31;72]. We chose a DNA prime protein boost vaccination regimen because the primary purpose was to elicit antibodies to V1. Furthermore, we used the SHIV89.6P model as it has been previously demonstrated that Tat-based vaccines do not protect rhesus macaques against SHIV89.6P challenge[41;42], and because antibodies to Rev are not known to mediate ADCC or neutralization since Rev is nuclear/nucleolar protein. Our results demonstrate that this vaccine platform elicited low level of cell mediated immune responses that did not correlate with protection. Rather, we found that this vaccine regimen induced type-specific antibody responses to the amino terminus of V1. Interestingly, the vaccinated macaque M316 that developed the highest titers to V1 and ADCC by week two post challenge also had a decrease in peak plasma virus levels and controlled virus replication to undetectable levels by week six. In the remaining immunized macaques, a substantial level of ADCVI was measured at four weeks following challenge. Consistent with this observation, ADCVI at week 4 and 6 was not associated with a decrease in plasma virus level during acute infection but was inversely correlated with plasma virus level during the chronic phase of infection. These data suggest that both the extent and the timing of the secondary antibody response to the Env may be key in curtailing primary virus replication. While the present work does not formally demonstrate that V1 is a direct target of ADCC or ADCVI, it demonstrates that the pre-existence of this strain-specific antibody response to V1 significantly correlates with the development of ADCVI by 4 weeks post- infection. ADCVI was likely directed to the envelope, as little or no secondary response to Tat or Rev was observed in the macaques that developed ADCVI. Interestingly, we found that the development of ADCVI was followed by the development of neutralizing antibodies [67], similar to the detection of ADCVI before neutralizing antibody titers in macaques immunized with adenovirus-based SIV vaccines [73]. The overall implication of our findings is that non-neutralizing antibody to HIV envelope, and possibly to V1 may contribute to viral control protection, as summarized in Table 3, suggesting that the envelope protein should be a component of an effective HIV vaccine. Our result that demonstrated the lack of protection from SHIV89.6P, mediated by the passive administration of antibody from a protected macaque to a naïve macaque, suggests the possibility that T-cell responses contribute together with ADCC, ADCVI, and neutralizing antibodies to protection from high level of virus replication [74].
Highlights.
Antibodies to the SIV V1 region
Macaque model of HIV infection
ADCC in protection
ADCVI in protection
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. We thank Teresa Habina for editorial assistance; Norman L. Letvin for providing the virus challenge stock; Maria Grazia Ferrari, Vaniambadi S., Kalyanaraman, and Ranajit Pal for help with antibody analysis, George Pavlakis and Barbara Felber for helpful discussions; Phillip D. Markham, Jim Treece, Deborah Weiss, and Sharon Orndorff for the coordination of the animal studies.
Footnotes
Conflict-of-Interest Statement Conflict-of-interest disclosure: The authors confirm that they have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Kijak GH, McCutchan FE. HIV diversity, molecular epidemiology, and the role of recombination. Curr Infect Dis Rep. 2005 Nov;7(6):480–8. doi: 10.1007/s11908-005-0051-8. [DOI] [PubMed] [Google Scholar]
- 2.Korber B, Gaschen B, Yusim K, Thakallapally R, Kesmir C, Detours V. Evolutionary and immunological implications of contemporary HIV-1 variation. Br Med Bull. 2001;58:19–42. doi: 10.1093/bmb/58.1.19. [DOI] [PubMed] [Google Scholar]
- 3.Nolan KM, Jordan AP, Hoxie JA. Effects of partial deletions within the human immunodeficiency virus type 1 V3 loop on coreceptor tropism and sensitivity to entry inhibitors. J Virol. 2008 Jan;82(2):664–73. doi: 10.1128/JVI.01793-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Laakso MM, Lee FH, Haggarty B, et al. V3 loop truncations in HIV-1 envelope impart resistance to coreceptor inhibitors and enhanced sensitivity to neutralizing antibodies. PLoS Pathog. 2007 Aug 24;3(8):e117. doi: 10.1371/journal.ppat.0030117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kowalski M, Potz J, Basiripour L, et al. Functional regions of the envelope glycoprotein of human immunodeficiency virus type 1. Science. 1987 Sep 11;237(4820):1351–5. doi: 10.1126/science.3629244. [DOI] [PubMed] [Google Scholar]
- 6.Krachmarov CP, Kayman SC, Honnen WJ, Trochev O, Pinter A. V3-specific polyclonal antibodies affinity purified from sera of infected humans effectively neutralize primary isolates of human immunodeficiency virus type 1. AIDS Res Hum Retroviruses. 2001 Dec 10;17(18):1737–48. doi: 10.1089/08892220152741432. [DOI] [PubMed] [Google Scholar]
- 7.Gorny MK, Revesz K, Williams C, et al. The v3 loop is accessible on the surface of most human immunodeficiency virus type 1 primary isolates and serves as a neutralization epitope. J Virol. 2004 Mar;78(5):2394–404. doi: 10.1128/JVI.78.5.2394-2404.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gorny MK, Xu JY, Karwowska S, Buchbinder A, Zolla-Pazner S. Repertoire of neutralizing human monoclonal antibodies specific for the V3 domain of HIV-1 gp120. J Immunol. 1993 Jan 15;150(2):635–43. [PubMed] [Google Scholar]
- 9.Ly A, Stamatatos L. V2 loop glycosylation of the human immunodeficiency virus type 1 SF162 envelope facilitates interaction of this protein with CD4 and CCR5 receptors and protects the virus from neutralization by anti-V3 loop and anti-CD4 binding site antibodies. J Virol. 2000 Aug;74(15):6769–76. doi: 10.1128/jvi.74.15.6769-6776.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ogert RA, Lee MK, Ross W, Buckler-White A, Martin MA, Cho MW. N-linked glycosylation sites adjacent to and within the V1/V2 and the V3 loops of dualtropic human immunodeficiency virus type 1 isolate DH12 gp120 affect coreceptor usage and cellular tropism. J Virol. 2001 Jul;75(13):5998–6006. doi: 10.1128/JVI.75.13.5998-6006.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pollakis G, Kang S, Kliphuis A, Chalaby MI, Goudsmit J, Paxton WA. N-linked glycosylation of the HIV type-1 gp120 envelope glycoprotein as a major determinant of CCR5 and CXCR4 coreceptor utilization. J Biol Chem. 2001 Apr 20;276(16):13433–41. doi: 10.1074/jbc.M009779200. [DOI] [PubMed] [Google Scholar]
- 12.Shioda T, Oka S, Xin X, et al. In vivo sequence variability of human immunodeficiency virus type 1 envelope gp120: association of V2 extension with slow disease progression. J Virol. 1997 Jul;71(7):4871–81. doi: 10.1128/jvi.71.7.4871-4881.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stamatatos L, Cheng-Mayer C. An envelope modification that renders a primary, neutralization-resistant clade B human immunodeficiency virus type 1 isolate highly susceptible to neutralization by sera from other clades. J Virol. 1998 Oct;72(10):7840–5. doi: 10.1128/jvi.72.10.7840-7845.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Toohey K, Wehrly K, Nishio J, Perryman S, Chesebro B. Human immunodeficiency virus envelope V1 and V2 regions influence replication efficiency in macrophages by affecting virus spread. Virology. 1995 Oct 20;213(1):70–9. doi: 10.1006/viro.1995.1547. [DOI] [PubMed] [Google Scholar]
- 15.Walter BL, Wehrly K, Swanstrom R, Platt E, Kabat D, Chesebro B. Role of low CD4 levels in the influence of human immunodeficiency virus type 1 envelope V1 and V2 regions on entry and spread in macrophages. J Virol. 2005 Apr;79(8):4828–37. doi: 10.1128/JVI.79.8.4828-4837.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ritola K, Pilcher CD, Fiscus SA, et al. Multiple V1/V2 env variants are frequently present during primary infection with human immunodeficiency virus type 1. J Virol. 2004 Oct;78(20):11208–18. doi: 10.1128/JVI.78.20.11208-11218.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Derdeyn CA, Decker JM, Bibollet-Ruche F, et al. Envelope-constrained neutralization-sensitive HIV-1 after heterosexual transmission. Science. 2004 Mar 26;303(5666):2019–22. doi: 10.1126/science.1093137. [DOI] [PubMed] [Google Scholar]
- 18.Chohan B, Lang D, Sagar M, et al. Selection for human immunodeficiency virus type 1 envelope glycosylation variants with shorter V1-V2 loop sequences occurs during transmission of certain genetic subtypes and may impact viral RNA levels. J Virol. 2005 May;79(10):6528–31. doi: 10.1128/JVI.79.10.6528-6531.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sagar M, Wu X, Lee S, Overbaugh J. Human immunodeficiency virus type 1 V1-V2 envelope loop sequences expand and add glycosylation sites over the course of infection, and these modifications affect antibody neutralization sensitivity. J Virol. 2006 Oct;80(19):9586–98. doi: 10.1128/JVI.00141-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kitrinos KM, Hoffman NG, Nelson JA, Swanstrom R. Turnover of env variable region 1 and 2 genotypes in subjects with late-stage human immunodeficiency virus type 1 infection. J Virol. 2003 Jun;77(12):6811–22. doi: 10.1128/JVI.77.12.6811-6822.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cao J, Sullivan N, Desjardin E, et al. Replication and neutralization of human immunodeficiency virus type 1 lacking the V1 and V2 variable loops of the gp120 envelope glycoprotein. J Virol. 1997 Dec;71(12):9808–12. doi: 10.1128/jvi.71.12.9808-9812.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng-Mayer C, Brown A, Harouse J, Luciw PA, Mayer AJ. Selection for neutralization resistance of the simian/human immunodeficiency virus SHIVSF33A variant in vivo by virtue of sequence changes in the extracellular envelope glycoprotein that modify N-linked glycosylation. J Virol. 1999 Jul;73(7):5294–300. doi: 10.1128/jvi.73.7.5294-5300.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson WE, Morgan J, Reitter J, et al. A replication-competent, neutralization-sensitive variant of simian immunodeficiency virus lacking 100 amino acids of envelope. J Virol. 2002 Mar;76(5):2075–86. doi: 10.1128/jvi.76.5.2075-2086.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reitter JN, Means RE, Desrosiers RC. A role for carbohydrates in immune evasion in AIDS. Nat Med. 1998 Jun;4(6):679–84. doi: 10.1038/nm0698-679. [DOI] [PubMed] [Google Scholar]
- 25.Rudensey LM, Kimata JT, Long EM, Chackerian B, Overbaugh J. Changes in the extracellular envelope glycoprotein of variants that evolve during the course of simian immunodeficiency virus SIVMne infection affect neutralizing antibody recognition, syncytium formation, and macrophage tropism but not replication, cytopathicity, or CCR-5 coreceptor recognition. J Virol. 1998 Jan;72(1):209–17. doi: 10.1128/jvi.72.1.209-217.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blay WM, Gnanakaran S, Foley B, Doria-Rose NA, Korber BT, Haigwood NL. Consistent patterns of change during the divergence of human immunodeficiency virus type 1 envelope from that of the inoculated virus in simian/human immunodeficiency virus-infected macaques. J Virol. 2006 Jan;80(2):999–1014. doi: 10.1128/JVI.80.2.999-1014.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chackerian B, Rudensey LM, Overbaugh J. Specific N-linked and O-linked glycosylation modifications in the envelope V1 domain of simian immunodeficiency virus variants that evolve in the host alter recognition by neutralizing antibodies. J Virol. 1997 Oct;71(10):7719–27. doi: 10.1128/jvi.71.10.7719-7727.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wolk T, Schreiber M. N-Glycans in the gp120 V1/V2 domain of the HIV-1 strain NL4-3 are indispensable for viral infectivity and resistance against antibody neutralization. Med Microbiol Immunol. 2006 Sep;195(3):165–72. doi: 10.1007/s00430-006-0016-z. [DOI] [PubMed] [Google Scholar]
- 29.Benko DM, Schwartz S, Pavlakis GN, Felber BK. A novel human immunodeficiency virus type 1 protein, tev, shares sequences with tat, env, and rev proteins. J Virol. 1990 Jun;64(6):2505–18. doi: 10.1128/jvi.64.6.2505-2518.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salfeld J, Gottlinger HG, Sia RA, Park RE, Sodroski JG, Haseltine WA. A tripartite HIV-1 tat-env-rev fusion protein. EMBO J. 1990 Mar;9(3):965–70. doi: 10.1002/j.1460-2075.1990.tb08195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Furtado MR, Balachandran R, Gupta P, Wolinsky SM. Analysis of alternatively spliced human immunodeficiency virus type-1 mRNA species, one of which encodes a novel tat-env fusion protein. Virology. 1991 Nov;185(1):258–70. doi: 10.1016/0042-6822(91)90773-5. [DOI] [PubMed] [Google Scholar]
- 32.Hartikka J, Sawdey M, Cornefert-Jensen F, et al. An improved plasmid DNA expression vector for direct injection into skeletal muscle. Hum Gene Ther. 1996 Jun 20;7(10):1205–17. doi: 10.1089/hum.1996.7.10-1205. [DOI] [PubMed] [Google Scholar]
- 33.Rosati M, von Gegerfelt A, Roth P, et al. DNA vaccines expressing different forms of simian immunodeficiency virus antigens decrease viremia upon SIVmac251 challenge. J Virol. 2005 Jul;79(13):8480–92. doi: 10.1128/JVI.79.13.8480-8492.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gomez-Roman VR, Patterson LJ, Venzon D, et al. Vaccine-Elicited Antibodies Mediate Antibody-Dependent Cellular Cytotoxicity Correlated with Significantly Reduced Acute Viremia in Rhesus Macaques Challenged with SIVmac251. J Immunol. 2005 Feb 15;174(4):2185–9. doi: 10.4049/jimmunol.174.4.2185. [DOI] [PubMed] [Google Scholar]
- 35.Gomez-Roman VR, Florese RH, Patterson LJ, et al. A simplified method for the rapid fluorometric assessment of antibody-dependent cell-mediated cytotoxicity. J Immunol Methods. 2006 Jan 20;308(1-2):53–67. doi: 10.1016/j.jim.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 36.Forthal DN, Landucci G. In vitro reduction of virus infectivity by antibody-dependent cell-mediated immunity. J Immunol Methods. 1998 Nov 1;220(1-2):129–38. doi: 10.1016/s0022-1759(98)00152-5. [DOI] [PubMed] [Google Scholar]
- 37.Karlsson GB, Halloran M, Li J, et al. Characterization of molecularly cloned simian-human immunodeficiency viruses causing rapid CD4+ lymphocyte depletion in rhesus monkeys. J Virol. 1997 Jun;71(6):4218–25. doi: 10.1128/jvi.71.6.4218-4225.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Montefiori DC. Evaluating neutralizing antibodies against HIV, SIV, and SHIV in luciferase reporter gene assays. Curr Protoc Immunol. 2005 Jan;Chapter 12 doi: 10.1002/0471142735.im1211s64. Unit. [DOI] [PubMed] [Google Scholar]
- 39.Li M, Gao F, Mascola JR, et al. Human immunodeficiency virus type 1 env clones from acute and early subtype B infections for standardized assessments of vaccine-elicited neutralizing antibodies. J Virol. 2005 Aug;79(16):10108–25. doi: 10.1128/JVI.79.16.10108-10125.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haigwood NL, Montefiori DC, Sutton WF, et al. Passive immunotherapy in simian immunodeficiency virus-infected macaques accelerates the development of neutralizing antibodies. J Virol. 2004 Jun;Nov;78:5983–95. doi: 10.1128/JVI.78.11.5983-5995.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liang X, Casimiro DR, Schleif WA, et al. Vectored Gag and Env but not Tat show efficacy against simian-human immunodeficiency virus 89.6P challenge in Mamu-A*01-negative rhesus monkeys. J Virol. 2005 Oct;79(19):12321–31. doi: 10.1128/JVI.79.19.12321-12331.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Silvera P, Richardson MW, Greenhouse J, et al. Outcome of simian-human immunodeficiency virus strain 89.6p challenge following vaccination of rhesus macaques with human immunodeficiency virus Tat protein. J Virol. 2002 Apr;76(8):3800–9. doi: 10.1128/JVI.76.8.3800-3809.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moore JP, Wallace LA, Follett EA, McKeating JA. An enzyme-linked immunosorbent assay for antibodies to the envelope glycoproteins of divergent strains of HIV-1. AIDS. 1989 Mar;3(3):155–63. doi: 10.1097/00002030-198903000-00006. [DOI] [PubMed] [Google Scholar]
- 44.Wang YH, Han XX, An MH, et al. Coreceptor usage of human immunodeficiency virus-1 in primary infection through sexual contact transmission. Zhonghua Yi Xue Za Zhi. 2011 Jun 7;91(21):1457–62. [PubMed] [Google Scholar]
- 45.Cicala C, Arthos J, Fauci AS. HIV-1 envelope, integrins and co-receptor use in mucosal transmission of HIV. J Transl Med. 2011;9(Suppl 1):S2. doi: 10.1186/1479-5876-9-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liang X, Casimiro DR, Schleif WA, et al. Vectored Gag and Env but not Tat show efficacy against simian-human immunodeficiency virus 89.6P challenge in Mamu-A*01-negative rhesus monkeys. J Virol. 2005 Oct;79(19):12321–31. doi: 10.1128/JVI.79.19.12321-12331.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Demberg T, Florese RH, Heath MJ, et al. A replication-competent adenovirus-human immunodeficiency virus (Ad-HIV) tat and Ad-HIV env priming/Tat and envelope protein boosting regimen elicits enhanced protective efficacy against simian/human immunodeficiency virus SHIV89.6P challenge in rhesus macaques. J Virol. 2007 Apr;81(7):3414–27. doi: 10.1128/JVI.02453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kolchinsky P, Kiprilov E, Sodroski J. Increased neutralization sensitivity of CD4-independent human immunodeficiency virus variants. J Virol. 2001 Mar;75(5):2041–50. doi: 10.1128/JVI.75.5.2041-2050.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Matano T, Shibata R, Siemon C, Connors M, Lane H, Martin MA. Administration of an anti-CD8 monoclonal antibody interferes with the clearance of chimeric simian/human immunodeficiency virus during primary infections of rhesus macaques. J Virol. 1998;72(1):164–9. doi: 10.1128/jvi.72.1.164-169.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schmitz JE, Johnson RP, McClure HM, et al. Effect of CD8+ lymphocyte depletion on virus containment after simian immunodeficiency virus SIVmac251 challenge of live attenuated SIVmac239delta3-vaccinated rhesus macaques. J Virol. 2005 Jul;79(13):8131–41. doi: 10.1128/JVI.79.13.8131-8141.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vaccari M, Mattapallil J, Song K, et al. Reduced protection from simian immunodeficiency virus SIVmac251 infection afforded by memory CD8+ T cells induced by vaccination during CD4+ T-cell deficiency. J Virol. 2008 Oct;82(19):9629–38. doi: 10.1128/JVI.00893-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Amara RR, Villinger F, Altman JD, et al. Control of a mucosal challenge and prevention of AIDS by a multiprotein DNA/MVA vaccine. Science. 2001 Apr 6;292(5514):69–74. doi: 10.1126/science.1058915. [DOI] [PubMed] [Google Scholar]
- 53.Malkevitch N, Rohne D, Pinczewski J, et al. Evaluation of combination DNA/replication-competent Ad-SIV recombinant immunization regimens in rhesus macaques. AIDS Res Hum Retroviruses. 2004 Feb;20(2):235–44. doi: 10.1089/088922204773004969. [DOI] [PubMed] [Google Scholar]
- 54.Patterson LJ, Malkevitch N, Venzon D, et al. Protection against Mucosal Simian Immunodeficiency Virus SIV(mac251) Challenge by Using Replicating Adenovirus-SIV Multigene Vaccine Priming and Subunit Boosting. J Virol. 2004 Mar 1;78(5):2212–21. doi: 10.1128/JVI.78.5.2212-2221.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shiver JW, Fu TM, Chen L, et al. Replication-incompetent adenoviral vaccine vector elicits effective anti-immunodeficiency-virus immunity. Nature. 2002 Jan 17;415(6869):331–5. doi: 10.1038/415331a. [DOI] [PubMed] [Google Scholar]
- 56.Vaccari M, Boasso A, Ma ZM, et al. CD4+ T-cell loss and delayed expression of modulators of immune responses at mucosal sites of vaccinated macaques following SIV(mac251) infection. Mucosal Immunol. 2008 Nov;1(6):497–507. doi: 10.1038/mi.2008.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rerks-Ngarm S, Pitisuttithum P, Nitayaphan S, et al. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N Engl J Med. 2009 Dec 3;361(23):2209–20. doi: 10.1056/NEJMoa0908492. [DOI] [PubMed] [Google Scholar]
- 58.Girard M, Kieny MP, Pinter A, et al. Immunization of chimpanzees confers protection against challenge with human immunodeficiency virus. Proc Natl Acad Sci U S A. 1991 Jan 15;88(2):542–6. doi: 10.1073/pnas.88.2.542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Belo M, Yagello M, Girard M, et al. Antibody-dependent cellular cytotoxicity against HIV-1 in sera of immunized chimpanzees. AIDS. 1991 Feb;5(2):169–76. doi: 10.1097/00002030-199102000-00006. [DOI] [PubMed] [Google Scholar]
- 60.Baba TW, Liska V, Hofmann-Lehmann R, et al. Human neutralizing monoclonal antibodies of the IgG1 subtype protect against mucosal simian-human immunodeficiency virus infection. Nat Med. 2000;6(2):200–6. doi: 10.1038/72309. [DOI] [PubMed] [Google Scholar]
- 61.Mascola JR, Lewis MG, Stiegler G, et al. Protection of Macaques against pathogenic simian/human immunodeficiency virus 89.6PD by passive transfer of neutralizing antibodies. J Virol. 1999 May;73(5):4009–18. doi: 10.1128/jvi.73.5.4009-4018.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hessell AJ, Hangartner L, Hunter M, et al. Fc receptor but not complement binding is important in antibody protection against HIV. Nature. 2007 Sep 6;449(7158):101–4. doi: 10.1038/nature06106. [DOI] [PubMed] [Google Scholar]
- 63.Hidajat R, Xiao P, Zhou Q, et al. Correlation of vaccine-elicited systemic and mucosal nonneutralizing antibody activities with reduced acute viremia following intrarectal simian immunodeficiency virus SIVmac251 challenge of rhesus macaques. J Virol. 2009 Jan;83(2):791–801. doi: 10.1128/JVI.01672-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Banks ND, Kinsey N, Clements J, Hildreth JE. Sustained antibody-dependent cell-mediated cytotoxicity (ADCC) in SIV-infected macaques correlates with delayed progression to AIDS. AIDS Res Hum Retroviruses. 2002 Nov 1;18(16):1197–205. doi: 10.1089/08892220260387940. [DOI] [PubMed] [Google Scholar]
- 65.Ohkawa S, Xu K, Wilson LA, Montelaro R, Martin LN, Murphey-Corb M. Analysis of envelope glycoprotein-specific antibodies from SIV-infected and gp110-immunized monkeys in ACC and ADCC assays. AIDS Res Hum Retroviruses. 1995 Mar;11(3):395–403. doi: 10.1089/aid.1995.11.395. [DOI] [PubMed] [Google Scholar]
- 66.Chung A, Rollman E, Johansson S, Kent SJ, Stratov I. The utility of ADCC responses in HIV infection. Curr HIV Res. 2008 Nov;6(6):515–9. doi: 10.2174/157016208786501472. [DOI] [PubMed] [Google Scholar]
- 67.Forthal DN, Landucci G, Daar ES. Antibody from patients with acute human immunodeficiency virus (HIV) infection inhibits primary strains of HIV type 1 in the presence of natural-killer effector cells. J Virol. 2001 Aug;75(15):6953–61. doi: 10.1128/JVI.75.15.6953-6961.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Forthal DN, Landucci G, Cole KS, Marthas M, Becerra JC, van Rompay K. Rhesus macaque polyclonal and monoclonal antibodies inhibit simian immunodeficiency virus in the presence of human or autologous rhesus effector cells. J Virol. 2006 Sep;80(18):9217–25. doi: 10.1128/JVI.02746-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yamada T, Watanabe N, Nakamura T, Iwamoto A. Antibody-dependent cellular cytotoxicity via humoral immune epitope of Nef protein expressed on cell surface. J Immunol. 2004 Feb 15;172(4):2401–6. doi: 10.4049/jimmunol.172.4.2401. [DOI] [PubMed] [Google Scholar]
- 70.Florese RH, Demberg T, Xiao P, et al. Contribution of nonneutralizing vaccine-elicited antibody activities to improved protective efficacy in rhesus macaques immunized with tat/env compared with multigenic vaccines. J Immunol. 2009 Mar 15;182(6):3718–27. doi: 10.4049/jimmunol.0803115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.He Y, Honnen WJ, Krachmarov CP, et al. Efficient isolation of novel human monoclonal antibodies with neutralizing activity against HIV-1 from transgenic mice expressing human Ig loci. J Immunol. 2002 Jul 1;169(1):595–605. doi: 10.4049/jimmunol.169.1.595. [DOI] [PubMed] [Google Scholar]
- 72.Wentz MP, Moore BE, Cloyd MW, Berget SM, Donehower LA. A naturally arising mutation of a potential silencer of exon splicing in human immunodeficiency virus type 1 induces dominant aberrant splicing and arrests virus production. J Virol. 1997 Nov;71(11):8542–51. doi: 10.1128/jvi.71.11.8542-8551.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu J, O’Brien KL, Lynch DM, et al. Immune control of an SIV challenge by a T-cell-based vaccine in rhesus monkeys. Nature. 2009 Jan 1;457(7225):87–91. doi: 10.1038/nature07469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Florese RH, Van Rompay KK, Aldrich K, et al. Evaluation of passively transferred, nonneutralizing antibody-dependent cellular cytotoxicity-mediating IgG in protection of neonatal rhesus macaques against oral SIVmac251 challenge. J Immunol. 2006 Sep 15;177(6):4028–36. doi: 10.4049/jimmunol.177.6.4028. [DOI] [PubMed] [Google Scholar]