

Abstract
The title compound, [In2(C9H11)4(C4H10P)2], contains a centrosymmetric In2P2 core with short intermolecular In—P bonds. This core has acute P—In—P and obtuse In—P—In bond angles compared with other [R 2InPR′2]2 analogues, due to the presence of the bulky aromatic substituents on the In atom and the non-sterically demanding ethyl substituents on the P atom.
Related literature
For related dimeric phosphanylindanes, see: Alcock et al. (1989 ▶); Wells et al. (1992 ▶); Aitchison et al. (1989 ▶); Beachley et al. (1987 ▶, 1993 ▶, 1995 ▶, 2001 ▶); Culp et al. (1997 ▶); Malik et al. (1996 ▶); Thomas et al. (2002 ▶); Wells et al. (1993 ▶); von Hanisch (2001 ▶). For related trimeric phosphanylindanes, see: Burns et al. (1994 ▶); Werner & Neumüller (1996 ▶); Banks et al. (1991 ▶).
Experimental
Crystal data
[In2(C9H11)4(C4H10P)2]
M r = 884.53
Monoclinic,
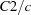
a = 22.323 (4) Å
b = 15.494 (4) Å
c = 14.331 (3) Å
β = 120.618 (4)°
V = 4265.6 (17) Å3
Z = 4
Mo Kα radiation
μ = 1.18 mm−1
T = 198 K
0.23 × 0.20 × 0.01 mm
Data collection
Bruker SMART1000/P4 diffractometer
Absorption correction: multi-scan (SADABS; Sheldrick, 2008a ▶) T min = 0.777, T max = 0.988
14538 measured reflections
4771 independent reflections
3470 reflections with I > 2σ(I)
R int = 0.038
Refinement
R[F 2 > 2σ(F 2)] = 0.031
wR(F 2) = 0.078
S = 1.09
4771 reflections
225 parameters
H-atom parameters constrained
Δρmax = 0.65 e Å−3
Δρmin = −0.34 e Å−3
Data collection: SMART (Bruker, 1999 ▶); cell refinement: SMART; data reduction: SAINT (Bruker, 2006 ▶); program(s) used to solve structure: SHELXS97 (Sheldrick, 2008b ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008b ▶); molecular graphics: DIAMOND (Brandenburg, 2011 ▶); software used to prepare material for publication: SHELXTL (Sheldrick, 2008b ▶).
Supplementary Material
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536811042668/fj2456sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536811042668/fj2456Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Acknowledgments
This work was supported by the Natural Sciences and Engineering Research Council of Canada, the New Brunswick Innovation Foundation, the Canadian Foundation for Innovation and Mount Allison University.
supplementary crystallographic information
Comment
Phosphanylindanes [R2InPR'2]n form dimeric or trimeric structures in the solid state via intermolecular In—P coordinate bonding interactions (Beachley et al., 2001; Werner & Neumuller 1996). Dimeric structures feature distorted tetrahedral geometries at indium and phosphorus, and planar four-membered In2P2 ring cores. In—P bond distances within the ring are similar and differ by less than 0.05 Å in all reported structures (Wells et al., 1993). The structure of I (Fig. 1) shows a dimer in the solid state, with a characteristic In2P2 core and distorted tetrahedral geometries at the four coordinate indium and phosphorus centres. The In—P bond distances are similar [In—P = 2.6300 (12) Å, In—Pi = 2.6364 (9) Å] and are in the range for previously reported dimeric phosphanylindanes [2.612 (1)–2.712 (1) Å] (Wells et al., 1993; Beachley et al., 1993). However, the ring P—In—Pi bond angle [81.56 (3)°] is at the lower limit of the range of reported values for [R2InPR'2]2 structures [81.80 (7)–87.53 (3)°] (Beachley et al., 1987; von Hanisch, 2001), and the In—P—Ini bond angle [98.44 (3)°] is at the upper limit of reported values [92.47 (3)–98.20 (7)°] (von Hanisch, 2001; Beachley et al., 1987). The significant distortion of the In2P2 ring is likely a result of the bulky mesityl groups on indium, which affect a compression of the P—In—Pi bond angles. Conversely, the non-bulky ethyl groups on phosphorus allows for expansion of the In—P—Ini bond angles.
Experimental
Preparation of [(2,4,6-Me3C6H2)2InPEt2]2 (I). Trismesityl indium (0.520 g, 1.10 mmol) was added to a solution of diethyl phosphine (0.100 g, 1.10 mmol) in toluene (7.5 ml). The reaction mixture was stirred at 45°C for 72 h, after which time a white precipitate had formed. The precipitate was removed by filtration, dried in vacuo, and washed with toluene (2 × 5 ml) and hexane (3 ml) (yield 0.110 g, 22%). Mp: 188°C. Crystals of I were obtained by dissolving the product in dichloromethane and allowing the solution to sit at 23°C for 12 h.
Refinement
H atoms were included in calculated positions and refined using a riding model.
Figures
Fig. 1.

X-ray crystal structure of (I), with displacement ellipsoids drawn at the 50% probability level. H atoms have been omitted for clarity. Symmetry transformations used to generate equivalent atoms: (i) -x + 1/2, -y + 1/2, -z + 1. Selected bond distances (Å) and angles (°): In—P 2.6300 (12), In—Pi 2.6364 (9), In—C1 2.190 (3), In—C10 2.215 (3), P—C19 1.866 (3), P—C21 1.856 (3), P—In—Pi 81.56 (3), In—P—Ini 98.44 (3), C1—In—C10 117.6 (1), C19—P—C21 104.4 (2).
Crystal data
| [In2(C9H11)4(C4H10P)2] | F(000) = 1824 |
| Mr = 884.53 | Dx = 1.377 Mg m−3 |
| Monoclinic, C2/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -C 2yc | Cell parameters from 6933 reflections |
| a = 22.323 (4) Å | θ = 2.7–28.1° |
| b = 15.494 (4) Å | µ = 1.18 mm−1 |
| c = 14.331 (3) Å | T = 198 K |
| β = 120.618 (4)° | Plate, colourless |
| V = 4265.6 (17) Å3 | 0.23 × 0.20 × 0.01 mm |
| Z = 4 |
Data collection
| Bruker SMART1000/P4 diffractometer | 4771 independent reflections |
| Radiation source: fine-focus sealed tube, K760 | 3470 reflections with I > 2σ(I) |
| graphite | Rint = 0.038 |
| φ and ω scans | θmax = 27.5°, θmin = 1.7° |
| Absorption correction: multi-scan (SADABS; Sheldrick, 2008a) | h = −28→27 |
| Tmin = 0.777, Tmax = 0.988 | k = −19→19 |
| 14538 measured reflections | l = −18→18 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.031 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.078 | H-atom parameters constrained |
| S = 1.09 | w = 1/[σ2(Fo2) + (0.0318P)2 + 1.4614P] where P = (Fo2 + 2Fc2)/3 |
| 4771 reflections | (Δ/σ)max = 0.001 |
| 225 parameters | Δρmax = 0.65 e Å−3 |
| 0 restraints | Δρmin = −0.34 e Å−3 |
Special details
| Experimental. Crystal decay was monitored by repeating the initial 50 frames at the end of the data collection and analyzing duplicate reflectionsNMR data (p.p.m., CDCl3): 1H NMR, 0.88 (m, 6H, PCH2CH3), 1.83 (q, 3J(1H-1H) = 7 Hz, 4H, PCH2CH3), 2.24 (s, 6H, 2,4,6-Me3C6H2), 2.33 (s, 12H, 2,4,6-Me3C6H2), 6.78 (s, 4H, 2,4,6-Me3C6H2); 13C{1H}11.7 (PCH2CH3), 13.8 (PCH2CH3), 21.3 (s, 2,4,6-Me3C6H2), 27.7 (s, 2,4,6-Me3C6H2), 126.8 (s, 2,4,6-Me3C6H2), 136.6 (s, 2,4,6-Me3C6H2), 144.1 (s, 2,4,6-Me3C6H2); 31P NMR, δ -44.63 (s). FT—IR: 538m, 607vw, 673w, 708w, 752w, 800w, 845m, 976w, 1030w, 1042w, 1090w, 1149vw, 1244w, 1259w, 1288w, 1402w, 1547w, 1595vw, 1712vw. |
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R-factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| In1 | 0.231901 (11) | 0.163644 (15) | 0.585903 (16) | 0.03479 (8) | |
| P1 | 0.21902 (4) | 0.33051 (6) | 0.54684 (6) | 0.03532 (19) | |
| C1 | 0.14507 (16) | 0.0724 (2) | 0.5087 (2) | 0.0355 (7) | |
| C2 | 0.07490 (17) | 0.0952 (2) | 0.4400 (2) | 0.0430 (8) | |
| C3 | 0.02405 (18) | 0.0319 (3) | 0.3942 (3) | 0.0504 (9) | |
| H3 | −0.0230 | 0.0488 | 0.3476 | 0.061* | |
| C4 | 0.03963 (19) | −0.0550 (3) | 0.4142 (3) | 0.0512 (10) | |
| C5 | 0.10908 (19) | −0.0781 (2) | 0.4811 (3) | 0.0466 (9) | |
| H5 | 0.1210 | −0.1375 | 0.4954 | 0.056* | |
| C6 | 0.16177 (17) | −0.0161 (2) | 0.5276 (2) | 0.0381 (8) | |
| C7 | 0.0530 (2) | 0.1886 (3) | 0.4137 (3) | 0.0607 (11) | |
| H7A | 0.0020 | 0.1922 | 0.3735 | 0.091* | |
| H7B | 0.0710 | 0.2124 | 0.3695 | 0.091* | |
| H7C | 0.0716 | 0.2216 | 0.4812 | 0.091* | |
| C8 | −0.0172 (2) | −0.1223 (3) | 0.3645 (4) | 0.0719 (13) | |
| H8A | −0.0539 | −0.1079 | 0.3803 | 0.108* | |
| H8B | 0.0023 | −0.1790 | 0.3951 | 0.108* | |
| H8C | −0.0369 | −0.1236 | 0.2858 | 0.108* | |
| C9 | 0.23652 (17) | −0.0451 (2) | 0.5954 (3) | 0.0464 (8) | |
| H9A | 0.2378 | −0.1068 | 0.6114 | 0.070* | |
| H9B | 0.2595 | −0.0124 | 0.6634 | 0.070* | |
| H9C | 0.2607 | −0.0350 | 0.5553 | 0.070* | |
| C10 | 0.30035 (17) | 0.1564 (2) | 0.7643 (2) | 0.0394 (8) | |
| C11 | 0.37154 (17) | 0.1357 (2) | 0.8185 (2) | 0.0427 (8) | |
| C12 | 0.41094 (19) | 0.1333 (2) | 0.9321 (3) | 0.0512 (9) | |
| H12 | 0.4590 | 0.1193 | 0.9670 | 0.061* | |
| C13 | 0.3812 (2) | 0.1508 (2) | 0.9945 (3) | 0.0541 (10) | |
| C14 | 0.3113 (2) | 0.1726 (2) | 0.9417 (3) | 0.0520 (10) | |
| H14 | 0.2903 | 0.1858 | 0.9834 | 0.062* | |
| C15 | 0.27101 (18) | 0.1755 (2) | 0.8287 (3) | 0.0437 (8) | |
| C16 | 0.40779 (19) | 0.1146 (3) | 0.7567 (3) | 0.0576 (10) | |
| H16A | 0.3855 | 0.0644 | 0.7102 | 0.086* | |
| H16B | 0.4569 | 0.1016 | 0.8080 | 0.086* | |
| H16C | 0.4046 | 0.1642 | 0.7119 | 0.086* | |
| C17 | 0.4242 (2) | 0.1459 (3) | 1.1178 (3) | 0.0770 (14) | |
| H17A | 0.4729 | 0.1341 | 1.1407 | 0.115* | |
| H17B | 0.4062 | 0.0994 | 1.1433 | 0.115* | |
| H17C | 0.4210 | 0.2009 | 1.1488 | 0.115* | |
| C18 | 0.19456 (18) | 0.1971 (3) | 0.7786 (3) | 0.0541 (10) | |
| H18A | 0.1883 | 0.2598 | 0.7698 | 0.081* | |
| H18B | 0.1779 | 0.1764 | 0.8260 | 0.081* | |
| H18C | 0.1680 | 0.1692 | 0.7076 | 0.081* | |
| C19 | 0.29184 (17) | 0.3701 (2) | 0.6794 (2) | 0.0414 (8) | |
| H19A | 0.2829 | 0.3512 | 0.7371 | 0.050* | |
| H19B | 0.3352 | 0.3416 | 0.6930 | 0.050* | |
| C20 | 0.30422 (19) | 0.4667 (2) | 0.6901 (3) | 0.0496 (9) | |
| H20A | 0.3211 | 0.4852 | 0.6421 | 0.074* | |
| H20B | 0.3390 | 0.4808 | 0.7653 | 0.074* | |
| H20C | 0.2605 | 0.4965 | 0.6698 | 0.074* | |
| C21 | 0.14350 (17) | 0.3982 (2) | 0.5211 (3) | 0.0441 (8) | |
| H21A | 0.1544 | 0.4590 | 0.5144 | 0.053* | |
| H21B | 0.1031 | 0.3809 | 0.4505 | 0.053* | |
| C22 | 0.12205 (18) | 0.3937 (3) | 0.6068 (3) | 0.0527 (9) | |
| H22A | 0.1039 | 0.3360 | 0.6063 | 0.079* | |
| H22B | 0.0859 | 0.4369 | 0.5905 | 0.079* | |
| H22C | 0.1626 | 0.4053 | 0.6785 | 0.079* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| In1 | 0.02880 (13) | 0.04999 (15) | 0.01987 (11) | −0.01078 (11) | 0.00826 (9) | 0.00045 (10) |
| P1 | 0.0310 (4) | 0.0489 (5) | 0.0237 (4) | −0.0081 (4) | 0.0122 (3) | −0.0014 (4) |
| C1 | 0.0328 (17) | 0.053 (2) | 0.0194 (14) | −0.0126 (15) | 0.0121 (13) | −0.0020 (14) |
| C2 | 0.0355 (19) | 0.059 (2) | 0.0300 (17) | −0.0130 (17) | 0.0137 (15) | −0.0048 (16) |
| C3 | 0.0330 (19) | 0.070 (3) | 0.043 (2) | −0.0120 (18) | 0.0159 (16) | −0.0112 (18) |
| C4 | 0.042 (2) | 0.071 (3) | 0.046 (2) | −0.026 (2) | 0.0267 (18) | −0.0234 (19) |
| C5 | 0.057 (2) | 0.050 (2) | 0.045 (2) | −0.0126 (18) | 0.0349 (19) | −0.0124 (16) |
| C6 | 0.0377 (18) | 0.054 (2) | 0.0261 (15) | −0.0101 (16) | 0.0188 (14) | −0.0049 (14) |
| C7 | 0.038 (2) | 0.068 (3) | 0.052 (2) | −0.0092 (19) | 0.0052 (18) | 0.0079 (19) |
| C8 | 0.061 (3) | 0.085 (3) | 0.077 (3) | −0.038 (2) | 0.040 (2) | −0.035 (3) |
| C9 | 0.045 (2) | 0.053 (2) | 0.0365 (19) | −0.0034 (18) | 0.0176 (17) | 0.0005 (16) |
| C10 | 0.0374 (18) | 0.050 (2) | 0.0220 (15) | −0.0155 (16) | 0.0089 (13) | 0.0015 (14) |
| C11 | 0.0372 (19) | 0.057 (2) | 0.0255 (16) | −0.0129 (16) | 0.0098 (14) | 0.0013 (14) |
| C12 | 0.038 (2) | 0.068 (2) | 0.0303 (18) | −0.0126 (18) | 0.0045 (15) | 0.0032 (16) |
| C13 | 0.056 (2) | 0.069 (3) | 0.0236 (17) | −0.021 (2) | 0.0109 (16) | 0.0005 (16) |
| C14 | 0.053 (2) | 0.072 (3) | 0.0273 (17) | −0.018 (2) | 0.0175 (16) | −0.0053 (17) |
| C15 | 0.0394 (19) | 0.058 (2) | 0.0277 (16) | −0.0146 (17) | 0.0128 (14) | 0.0006 (15) |
| C16 | 0.040 (2) | 0.091 (3) | 0.0348 (19) | −0.004 (2) | 0.0141 (17) | 0.002 (2) |
| C17 | 0.072 (3) | 0.112 (4) | 0.0230 (18) | −0.021 (3) | 0.0070 (18) | 0.002 (2) |
| C18 | 0.044 (2) | 0.080 (3) | 0.039 (2) | −0.007 (2) | 0.0216 (18) | 0.0019 (18) |
| C19 | 0.0381 (19) | 0.058 (2) | 0.0238 (16) | −0.0072 (16) | 0.0126 (14) | −0.0021 (14) |
| C20 | 0.055 (2) | 0.060 (2) | 0.0350 (19) | −0.0194 (19) | 0.0234 (17) | −0.0112 (16) |
| C21 | 0.0364 (19) | 0.057 (2) | 0.0365 (18) | −0.0003 (17) | 0.0165 (15) | 0.0003 (16) |
| C22 | 0.041 (2) | 0.078 (3) | 0.0373 (19) | −0.004 (2) | 0.0193 (17) | −0.0078 (18) |
Geometric parameters (Å, °)
| In1—C1 | 2.190 (3) | C11—C16 | 1.509 (5) |
| In1—C10 | 2.215 (3) | C12—C13 | 1.385 (5) |
| In1—P1 | 2.6300 (12) | C12—H12 | 0.9500 |
| In1—P1i | 2.6364 (9) | C13—C14 | 1.386 (5) |
| P1—C21 | 1.856 (3) | C13—C17 | 1.524 (5) |
| P1—C19 | 1.866 (3) | C14—C15 | 1.397 (4) |
| P1—In1i | 2.6364 (9) | C14—H14 | 0.9500 |
| C1—C2 | 1.406 (4) | C15—C18 | 1.514 (5) |
| C1—C6 | 1.409 (5) | C16—H16A | 0.9800 |
| C2—C3 | 1.386 (5) | C16—H16B | 0.9800 |
| C2—C7 | 1.512 (5) | C16—H16C | 0.9800 |
| C3—C4 | 1.384 (5) | C17—H17A | 0.9800 |
| C3—H3 | 0.9500 | C17—H17B | 0.9800 |
| C4—C5 | 1.392 (5) | C17—H17C | 0.9800 |
| C4—C8 | 1.511 (5) | C18—H18A | 0.9800 |
| C5—C6 | 1.398 (4) | C18—H18B | 0.9800 |
| C5—H5 | 0.9500 | C18—H18C | 0.9800 |
| C6—C9 | 1.510 (4) | C19—C20 | 1.515 (5) |
| C7—H7A | 0.9800 | C19—H19A | 0.9900 |
| C7—H7B | 0.9800 | C19—H19B | 0.9900 |
| C7—H7C | 0.9800 | C20—H20A | 0.9800 |
| C8—H8A | 0.9800 | C20—H20B | 0.9800 |
| C8—H8B | 0.9800 | C20—H20C | 0.9800 |
| C8—H8C | 0.9800 | C21—C22 | 1.530 (4) |
| C9—H9A | 0.9800 | C21—H21A | 0.9900 |
| C9—H9B | 0.9800 | C21—H21B | 0.9900 |
| C9—H9C | 0.9800 | C22—H22A | 0.9800 |
| C10—C11 | 1.405 (5) | C22—H22B | 0.9800 |
| C10—C15 | 1.408 (5) | C22—H22C | 0.9800 |
| C11—C12 | 1.402 (4) | ||
| C1—In1—C10 | 117.60 (11) | C13—C12—C11 | 121.4 (3) |
| C1—In1—P1 | 123.68 (9) | C13—C12—H12 | 119.3 |
| C10—In1—P1 | 103.40 (9) | C11—C12—H12 | 119.3 |
| C1—In1—P1i | 104.11 (7) | C12—C13—C14 | 118.2 (3) |
| C10—In1—P1i | 122.61 (9) | C12—C13—C17 | 120.9 (4) |
| P1—In1—P1i | 81.56 (3) | C14—C13—C17 | 120.9 (4) |
| C21—P1—C19 | 104.40 (16) | C13—C14—C15 | 121.4 (3) |
| C21—P1—In1 | 125.97 (11) | C13—C14—H14 | 119.3 |
| C19—P1—In1 | 99.36 (11) | C15—C14—H14 | 119.3 |
| C21—P1—In1i | 120.44 (11) | C14—C15—C10 | 120.9 (3) |
| C19—P1—In1i | 104.85 (10) | C14—C15—C18 | 117.5 (3) |
| In1—P1—In1i | 98.44 (3) | C10—C15—C18 | 121.6 (3) |
| C2—C1—C6 | 118.1 (3) | C11—C16—H16A | 109.5 |
| C2—C1—In1 | 125.1 (2) | C11—C16—H16B | 109.5 |
| C6—C1—In1 | 116.8 (2) | H16A—C16—H16B | 109.5 |
| C3—C2—C1 | 120.3 (3) | C11—C16—H16C | 109.5 |
| C3—C2—C7 | 118.4 (3) | H16A—C16—H16C | 109.5 |
| C1—C2—C7 | 121.3 (3) | H16B—C16—H16C | 109.5 |
| C4—C3—C2 | 122.1 (3) | C13—C17—H17A | 109.5 |
| C4—C3—H3 | 119.0 | C13—C17—H17B | 109.5 |
| C2—C3—H3 | 119.0 | H17A—C17—H17B | 109.5 |
| C3—C4—C5 | 117.9 (3) | C13—C17—H17C | 109.5 |
| C3—C4—C8 | 120.8 (4) | H17A—C17—H17C | 109.5 |
| C5—C4—C8 | 121.3 (4) | H17B—C17—H17C | 109.5 |
| C4—C5—C6 | 121.5 (3) | C15—C18—H18A | 109.5 |
| C4—C5—H5 | 119.3 | C15—C18—H18B | 109.5 |
| C6—C5—H5 | 119.3 | H18A—C18—H18B | 109.5 |
| C5—C6—C1 | 120.1 (3) | C15—C18—H18C | 109.5 |
| C5—C6—C9 | 119.1 (3) | H18A—C18—H18C | 109.5 |
| C1—C6—C9 | 120.8 (3) | H18B—C18—H18C | 109.5 |
| C2—C7—H7A | 109.5 | C20—C19—P1 | 116.6 (2) |
| C2—C7—H7B | 109.5 | C20—C19—H19A | 108.1 |
| H7A—C7—H7B | 109.5 | P1—C19—H19A | 108.1 |
| C2—C7—H7C | 109.5 | C20—C19—H19B | 108.1 |
| H7A—C7—H7C | 109.5 | P1—C19—H19B | 108.1 |
| H7B—C7—H7C | 109.5 | H19A—C19—H19B | 107.3 |
| C4—C8—H8A | 109.5 | C19—C20—H20A | 109.5 |
| C4—C8—H8B | 109.5 | C19—C20—H20B | 109.5 |
| H8A—C8—H8B | 109.5 | H20A—C20—H20B | 109.5 |
| C4—C8—H8C | 109.5 | C19—C20—H20C | 109.5 |
| H8A—C8—H8C | 109.5 | H20A—C20—H20C | 109.5 |
| H8B—C8—H8C | 109.5 | H20B—C20—H20C | 109.5 |
| C6—C9—H9A | 109.5 | C22—C21—P1 | 116.1 (2) |
| C6—C9—H9B | 109.5 | C22—C21—H21A | 108.3 |
| H9A—C9—H9B | 109.5 | P1—C21—H21A | 108.3 |
| C6—C9—H9C | 109.5 | C22—C21—H21B | 108.3 |
| H9A—C9—H9C | 109.5 | P1—C21—H21B | 108.3 |
| H9B—C9—H9C | 109.5 | H21A—C21—H21B | 107.4 |
| C11—C10—C15 | 117.3 (3) | C21—C22—H22A | 109.5 |
| C11—C10—In1 | 124.8 (2) | C21—C22—H22B | 109.5 |
| C15—C10—In1 | 117.9 (2) | H22A—C22—H22B | 109.5 |
| C12—C11—C10 | 120.8 (3) | C21—C22—H22C | 109.5 |
| C12—C11—C16 | 118.0 (3) | H22A—C22—H22C | 109.5 |
| C10—C11—C16 | 121.2 (3) | H22B—C22—H22C | 109.5 |
Symmetry codes: (i) −x+1/2, −y+1/2, −z+1.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: FJ2456).
References
- Aitchison, K. A., Julius Backer-Dirks, J. D., Bradley, D. C., Faktor, M. M., Frigo, D. M., Hursthouse, M. B., Hussain, B. & Short, R. L. (1989). J. Organomet. Chem. 366, 11–23.
- Alcock, N. W., Degnam, I. A., Wallbridge, M. G. H., Powell, H. R., McPartlin, M. & Sheldrick, G. M. (1989). J. Organomet. Chem. 361, C33–C36.
- Banks, M. A., Beachley, O. T. Jr, Buttrey, L. A., Churchill, M. R. & Fettinger, J. C. (1991). Organometallics, 10, 1901–1906.
- Beachley, O. T. Jr, Chao, S.-H. L., Churchill, M. R. & Lake, C. H. (1993). Organometallics, 12, 3992–3997.
- Beachley, O. T. Jr, Chao, S.-H. L., Churchill, M. R. & Lake, C. H. (2001). Organometallics, 20, 4896–4902.
- Beachley, O. T. Jr, Kopasz, J. P., Zhang, H., Hunter, W. E. & Atwood, J. L. (1987). J. Organomet. Chem. 325, 69–81.
- Beachley, O. T. Jr, Maloney, J. D., Banks, M. A. & Rogers, R. D. (1995). Organometallics, 14, 3448–3454.
- Brandenburg, K. (2011). DIAMOND. Crystal Impact GbR, Bonn, Germany.
- Bruker (1999). SMART Bruker AXS Inc., Madison, Wisconsin, USA.
- Bruker (2006). SAINT Bruker AXS inc., Madison, Wisconsin, USA.
- Burns, J. A., Dillingham, M. D. B., Hill, J. B., Gripper, K. D., Pennington, W. T. & Robinson, G. H. (1994). Organometallics, 13, 1514–1517.
- Culp, R. D., Cowley, A. H., Decken, A., Jones, R. A., Bond, M. R., Mokry, L. M. & Carrano, C. J. (1997). Inorg. Chem. 36, 5165-5172.
- Hanisch, C. von (2001). Z. Anorg. Allg. Chem. 627, 68–72.
- Malik, M. A., Haggata, S. W., Motevalli, M. & O’Brien, P. (1996). J. Organomet. Chem. 524, 95–101.
- Sheldrick, G. M. (2008a). SADABS Bruker AXS Inc., Madison, Wisconsin, USA.
- Sheldrick, G. M. (2008b). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Thomas, F., Schulz, S. & Nieger, M. (2002). Z. Anorg. Allg. Chem. 628, 235–242.
- Wells, R. L., McPhail, A. T., Jones, L. J. & Self, M. F. (1993). J. Organomet. Chem. 449, 85–94.
- Wells, R. L., McPhail, A. T. & Self, M. F. (1992). Organometallics, 11, 221–225.
- Werner, B. & Neumüller, B. (1996). Organometallics, 15, 4258–4263.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536811042668/fj2456sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536811042668/fj2456Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report