

Abstract
Background
Alterations in intracellular Ca2+ homeostasis are an important trigger of pathological cardiac remodeling; however, mechanisms governing context-dependent changes in Ca2+ influx are poorly understood. Store-operated Ca2+ entry (SOCE) is a major mechanism regulating Ca2+ trafficking in numerous cell types, yet its prevalence in adult heart and possible role in physiology and disease are each unknown. The Ca2+-binding protein, stromal interaction molecule 1 (STIM1), is a Ca2+ sensor in the sarcoplasmic reticulum (SR), capable of triggering SOCE by interacting with plasma membrane Ca2+ channels.
Methods and Results
We report that SOCE is abundant and robust in neonatal cardiomyocytes; however, SOCE is absent from adult cardiomyocytes. Levels of STIM1 transcript and protein correlate with the amplitude of SOCE, and manipulation of STIM1 protein levels (via shRNA) or activity (via expression of constitutively active or dominant-negative mutants) reveal a critical role for STIM1 in activating SOCE in cardiac myocytes. In neonatal hearts a recently identified STIM1 splice variant (STIM1L) is predominant but diminishes with maturation, only to reemerge with agonist- or afterload-induced cardiac stress. To test for pathophysiological relevance, we evaluated both in vitro and in vivo models of cardiac hypertrophy, finding that STIM1 expression is re-activated by pathological stress to trigger significant SOCE-dependent Ca2+ influx. STIM1 amplifies agonist-induced hypertrophy via activation of the calcineurin-NFAT pathway. Importantly, inhibition of STIM1 suppresses agonist-triggered hypertrophy, pointing to a requirement for SOCE in this remodeling response.
Conclusions
Stress-triggered STIM1 re-expression, and consequent SOCE activation, are critical elements in the upstream, Ca2+-dependent control of pathological cardiac hypertrophy.
Introduction
Heart failure, the leading cause of death in industrialized nations, is increasing in incidence and prevalence around the world. For example, in the US alone, an estimated five million people have heart failure, a syndrome with 5-year mortality of ≈50% [1]. Accordingly, heart failure, the end-result of pathological cardiac remodeling elicited by a variety of stimuli, is responsible for a huge societal burden of morbidity, mortality, and cost.
Heart failure is the end-result of a complex series of remodeling events elicited by a variety of pathological signals, including biomechanical (e.g. increases in preload or afterload) or neurohormonal triggers [2]. These extracellular signals, in turn, activate a cascade of intracellular signaling events leading to cell growth, extracellular matrix deposition, contractile dysfunction, and cell death. In many instances, mechanisms whereby forces impinging on the myocyte activate intracellular signaling cascades involve alterations in cellular Ca2+ homeostasis.
In cardiomyocytes, intracellular Ca2+ vacillates widely with each contraction cycle, increasing from 0.1 μM in diastole to 1-10 μM in systole [3]. Remarkably, these vast swings in Ca2+ concentration, occurring many times a minute, are incapable of activating pathological signaling events. Rather, calcineurin and other signaling cascades are triggered by sustained increases in intracellular Ca2+ within the cell. And at a finer level of spatial detail, important changes in Ca2+ within intracellular microdomains occur at the cytoplasmic face of both plasmalemmal and sarcoplasmic reticular ion channels. Ca2+ is also sequestered within specialized organelles, such as the sarcoplasmic reticulum (SR) or mitochondria. Presently, our understanding of the relative contributions of the various Ca2+ entry pathways (e.g. L-type channels, T-type channels, ryanodine receptor, IP3 receptors) to pathological cardiac remodeling is incomplete.
In non-excitable cells, depletion of intracellular Ca2+ stores triggers a specialized means of Ca2+ influx termed store-operated Ca2+ entry (SOCE). This robust mechanism of Ca2+ influx plays an obligate role in replenishing Ca2+ stores and is best characterized in non-excitable cells. SOCE leads to sustained increases of intracellular Ca2+, and as a result, is an important proximal signaling mechanism in numerous cell types. For example, in T cells and skeletal myocytes, SOCE is crucial to cell contraction, secretion, growth, and proliferation [4-7]. Recent studies have uncovered evidence for SOCE in neonatal cardiomyocytes [8-11], and yet its regulation, its prevalence in adult heart, and its possible role in cardiac physiology and pathology are each unknown.
STIM1 (stromal interaction molecule 1), a recently described Ca2+ sensor protein, is a key molecular component required to activate SOCE channels [12]. STIM1 is characterized by a single transmembrane domain, EF-hand Ca2+-binding and sterile-alpha-motif (SAM) domains residing in the ER/SR lumen, as well as cytoplasmic ezrin-radixin-moesin (ERM), Ser/Pro-rich, and Lys-rich domains. STIM1 detects local declines in ER/SR Ca2+ and responds by clustering to regions of the ER situated near the plasma membrane, where it activates store-operated Ca2+ channels allowing Ca2+ entry. In so doing, STIM1 transduces a signal of depleted intracellular Ca2+ stores to provoke sustained influx of Ca2+ from the exterior of the cell. This, in turn, replenishes intracellular stores and is capable of activating Ca-dependent signaling events.
At present, possible roles for STIM1-dependent SOCE in adult heart physiology or pathological cardiac remodeling remain unclear. Further, regulation of SOCE by its molecular components and its effects on downstream signaling cascades in heart are similarly ill-defined. Here, we set out to elucidate the role of SOCE in clinically relevant models of load-induced heart disease and unveil molecular mechanisms of its regulation.
Results
SOCE is robust in neonatal cardiomyocytes but absent in mature cells
To test for the presence of SOCE in heart, we first studied neonatal rat ventricular myocytes (NRVMs) in culture using a “Ca2+ re-addition” protocol [13, 14]. In this protocol, cells are initially incubated in Ca2+-free medium and treated with thapsigargin (TG, 2μM), a sarcoplasmic reticulum Ca2+ ATPase (SERCA) inhibitor, to deplete SR Ca2+ stores. Ca2+ is then restored to the medium, and Ca2+ influx is recorded by Fura-2 AM fluorescence [15]. Using this approach, we were able to detect robust Ca2+ influx in NRVMs (Figures 1A, 1B). To test for specificity of SOCE, we performed additional experiments in the presence of the SOCE inhibitors SKF-96365 (10 μM) or gadolinium (25 μM). In each case, Ca2+ influx was abolished (Figures 1A-1C), confirming that the signal was indeed SOCE. Based on these studies, we concluded that SOCE is prevalent and robust in neonatal cardiomyocytes.
Figure 1. Robust SOCE in NRVMs.
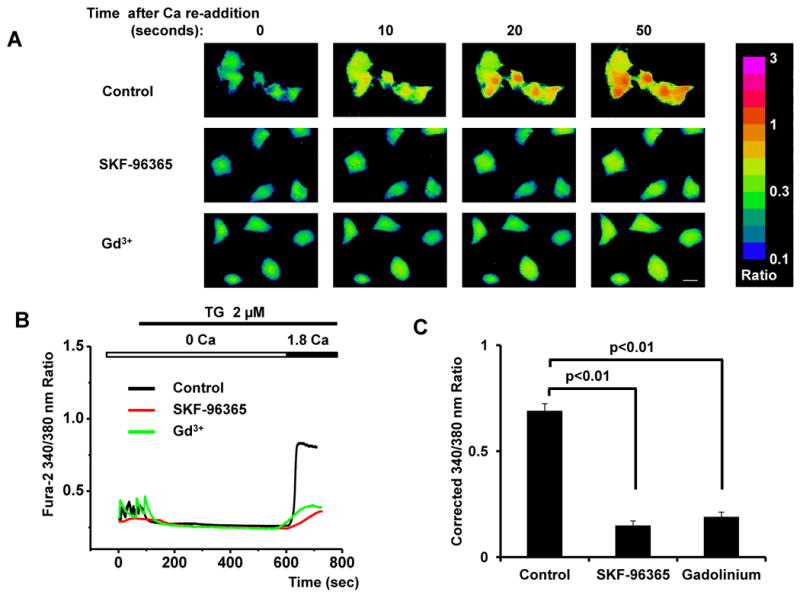
(A) Representative images of SOCE in neonatal cardiomyocytes depicted as Fura-2 ratios (340/380 nm) of Ca2+ concentration, as well as inhibition by SKF-96365 (10 μM) and Gd3+ (25 μM); time marks commence with Ca2+ re-addition; scale bar, 10 μm. (B) Representative traces of Ca2+ concentration demonstrating the time course of SOCE. (C) Quantitative analyses of SOCE shown as average peak Ca2+ levels after restoration of extracellular Ca2+ (1.8 mM) (corrected for baseline values, i.e. those before initial depletion of Ca2+ stores) from control, SKF-96365- and Gd3+-treated cardiomyocytes (n=18-22 cells) in three independent experiments.
Next, we tested for SOCE in acutely isolated adult cardiomyocytes. Surprisingly, the presence of a detectable SOCE signal was dramatically lower in adult myocytes. In fact, in an extensive survey of SOCE in both neonatal and adult myocytes, and we detected SOCE in nearly 100% of the neonatal myocytes tested (n=78, 3 independent experiments, Figure 2A). In contrast, approximately 10% of adult cells manifested SOCE (n=89, 5 independent experiments; Figure 2A). Additionally the amplitude of Ca2+ influx in adult cells, when detectable, was markedly smaller (Figure 2B, Supplemental Figure 1). Together, these data provide strong evidence for robust SOCE in neonatal cardiomyocytes which is absent in mature cells, suggesting fundamental differences in the regulation of basal levels of SOCE in the developing and mature heart.
Figure 2. SOCE activity and STIM1 protein are nearly absent in adult heart.


(A) Prevalence of SOCE in adult and neonatal ventricular myocytes (n=78-89) from 3-5 independent experiments. (B) Representative Ca2+ traces of detectable SOCE recorded from neonatal and adult myocytes. (C) Quantitative real-time RT-PCR analysis of STIM1 mRNA levels and (D) protein levels by immunoblot in adult and neonatal heart. (E) Quantitative real-time RT-PCR analysis of STIM-L mRNA levels in neonatal and adult hearts.
STIM1 levels decrease in the adult heart
STIM1 is a Ca2+ binding/sensor protein that activates SOCE by interacting with cytoplasmic components of plasma membrane Ca2+ channels. To determine the factors underlying the differential prevalence of SOCE in neonatal and adult heart, we measured STIM1 levels. Quantitative real-time RT-PCR (qRT-PCR) of RNA harvested from neonatal and adult hearts revealed a nearly five-fold decrease in STIM1 transcript levels in the adult heart (Figure 2C). Western blot analysis of protein extracts from these same hearts revealed markedly lower steady-state levels of STIM1 protein in the adult (Figure 2D).
Consistently, two major bands were detected by the STIM1 antibody, one at around 80 kD, the expected molecular mass of the protein, and another at around 130 kD. Both bands were recognized similarly with a second, independently derived polyclonal antibody (Supplemental Figure 2), and both bands were diminished by treatment with shRNA-specific to STIM1 (Figure 3A) suggesting that the slower migrating species is a modified version of STIM1. Intriguingly, the slower migrating band was detected exclusively in NRVMs and in adult heart subjected to thoracic aortic constriction (TAC).
Figure 3. STIM1 mediates SOCE in cardiomyocytes.
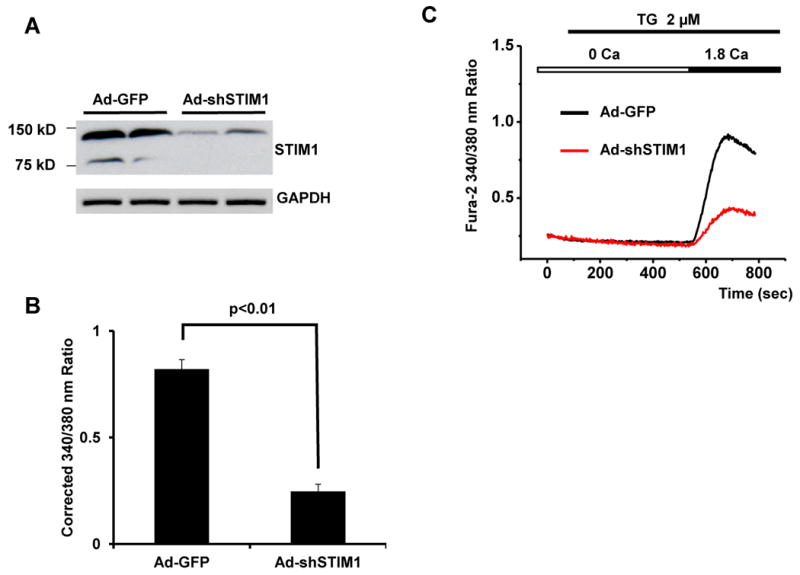

(A) Western blot analysis showing knockdown of STIM1 by Ad-shSTIM1. (B) Quantitative analysis of SOCE depicted as the corrected average peak Ca2+ levels in Ad-GFP- or Ad-shSTIM1-infected NRVMs (n=18-22) from three independent experiments. (C) Representative Ca2+ traces in Ad-GFP- or Ad-shSTIM1-infected NRVMs. (D) Representative Ca2+ traces demonstrating SOCE in cultured NRVMs from control cells or cells infected adenovirus expressing Ca-STIM1 and Dn-STIM1. (E) Quantitative analyses of SOCE shown as the corrected average peak Ca2+ levels in cells (n=19-22) shown in panel D from three independent experiments.
To determine the molecular identify of these bands, we prepared ventricular lysates from left ventricle subjected to TAC or Sham surgery and performed immunoprecipitations with anti-STIM1 antibody. The resulting immune complexes were subjected to SDS-PAGE and gel fragments encompassing 80 kD and 130 kD were excised and subjected to molecular analysis by mass spectrometry (Supplemental Figure 3). In two independent experiments, STIM1 protein was detected as the major molecular component in both the lower (80 kD, sample 1) and upper (130 kD, sample 2) bands. Specifically, Mascot scores, calculated as -10*Log(P), where P is the probability that the observed match is a random event, were 1501 (80 kD band) and 1107 (130 kD band); in other words, both bands represent bona fide STIM1 protein (Table).
Table. Mass spectrometry results for samples depicted in Supplemental Figure 3.
| Sample | 1 | 2 | 3 |
|---|---|---|---|
| Protein ID | 17368305 | 17368305 | N/A |
| STIM1 Detected | Yes | Yes | No |
| Protein Score | 1501 | 1107 | N/A |
To ensure that the STIM1 protein detected in the upper band was not an artifact from minute amounts of protein migrating spuriously within the gel and detected with a highly sensitive assay, we excised a region of the gel between the two bands (sample 3, Supplemental Figure 3). Here, no STIM1 protein was detected, consistent with the notion that the STIM1 detected in both the upper and lower bands is the major protein constituent.
A recent report by Darbellay et al [16] identified a splice variant of STIM1, STIM1L, which was highly expressed in skeletal muscle tissue. This variant harbors an extra coding sequence inserted between exons 11 and 12 and codes for a protein of size consistent with the slower migrating species of STIM1 we observe. To confirm that STIM1L was present in neonatal hearts and absent in adult cardiac tissue we used qPCR primers specific to the unique sequence region in STIM1L. Data depicted in Figure 2E demonstrate that STIM1L expression correlates with the level of the slower migrating species of STIM1.
STIM1 regulates SOCE in cardiomyocytes
To test directly whether STIM1 regulates SOCE, we used adenovirus-based small hairpin RNA interference (shRNA) to knock down STIM1 levels in NRVMs. Western blot analysis confirmed the efficacy of RNAi-mediated protein knockdown (Figure 3A). Lower levels of STIM1 resulted in a significant decrease in SOCE activity (Figures 3B, 3C), pointing to a requisite role for this protein in this Ca2+ entry pathway.
When next took advantage of the fact that mutation of an aspartate residue to alanine (D76A) in the EF domain of STIM1 results in localization of the protein into punctae, which may then directly or indirectly trigger Ca2+ influx [17, 18]. The ERM domain, which mediates STIM1 aggregation and interaction with Orai and/or TRPC channels, is also required for SOCE activation [14, 18, 19]. Thus, we infected NRVMs with adenoviral constructs coding for constitutively active (D76A, Ca-STIM1) or dominant-negative (ΔERM, Dn-STIM1) STIM1 proteins. While infection with a GFP-expressing control had no effect on SOCE, expression of the Ca-STIM1 significantly increased SOCE (average corrected 340/380 nm ratio increased from 0.71±0.04, n=22 to 1.16±0.06, n=19, p<0.01) (Figures 3D, 3E). Similar results were observed in NRVMs infected with lentivirus over-expressing STIM1 (Supplemental Figures 4A-4B). In contrast, Dn-STIM1 markedly decreased SOCE (average corrected 340/380 nm ratio decreased from 0.71±0.04, n=22 to 0.34±0.04, n=19, p<0.01) (Figures 3D, 3E). Over-expression or knockdown of STIM1 had no effect on ORAI or TRPC channel levels (Supplemental Figures 5A-5D), nor did it alter STIM2 levels (Supplemental Figures 5E-5H). Similarly, STIM2 levels were similar in neonatal and adult hearts, as well as in Sham and TAC hearts (Supplemental Figures 5I-5J). Together, these data provide strong evidence that STIM1 is a critical regulator of SOCE in cardiomyocytes.
SOCE is re-activated in cardiomyocyte hypertrophy
In many instances, elevated intracellular Ca2+ levels drive the induction of hypertrophic response genes. Given this, we set out to test for the presence of SOCE in hypertrophied myocardium and a possible role for STIM1 therein. To accomplish this, we subjected mice to TAC using a protocol that triggers stable, compensated ventricular hypertrophy [20] (Supplemental Figure 6). In ventricular lysates from Sham- and TAC-operated hearts 3 weeks post-surgery, STIM1L protein and mRNA levels were increased significantly (Figure 4A, quantified in Supplemental Figures 7A, 7B). A similar induction of STIM1L was observed in isolated adult cardiomyocytes stimulated with the hypertrophic agonist phenylephrine (PE) (Supplemental Figure 7C). Cardiomyocytes isolated from the Sham-operated animals manifested very little SOCE; SOCE was detected in only 8% of the cells, and when detected, SOCE was weak in amplitude (average corrected 340/380 nm ratios of approximately 0.25, Figures 4B-4E), similar to that observed in unoperated animals (Figures 2A, 2B). In striking contrast, 34% of cardiomyocytes isolated from TAC hearts manifested robust SOCE with Ca2+ influxes displaying 340/380 nm fluorescence ratios of around 1.15 (amplitude 4-fold higher than Sham-operated cells) (Figures 4B-4E). Together, these data are consistent with a model in which STIM1 activates SOCE, and that re-activation of STIM1 expression accounts for the increases in SOCE seen in hypertrophy.
Figure 4. Increased SOCE activity and STIM1 levels in pressure-overload hypertrophy in vivo.
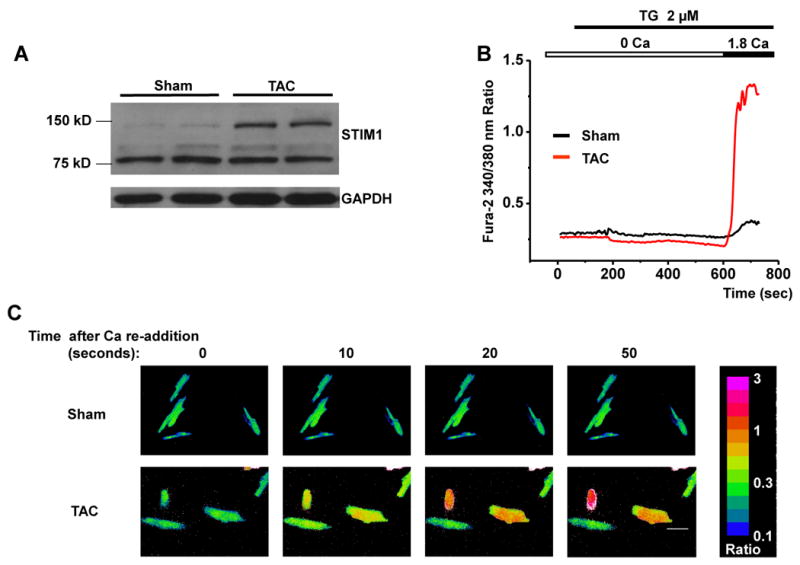

(A) Western blot analysis showing significant up-regulation of STIM1 in pressure-overload cardiac hypertrophy. (B) Representative Ca2+ traces showing SOCE detected in adult myocytes from Sham and TAC mice. (C) Representative Ca2+ images showing dynamic SOCE by Fura-2 ratios (340/380 nm) in adult cardiomyocytes from Sham and TAC mice. Time marks commence with Ca2+ re-addition; scale bar, 20 μm. (D) Quantitative analyses of SOCE shown as the corrected average peak Ca2+ levels in multiple ventricular myocytes (n=80-96) from 5-8 Sham or TAC adult hearts. (E) Prevalence of SOCE in ventricular myocytes (n=80-96) from 5-8 Sham or TAC adult hearts.
To examine this more directly, we induced hypertrophy in cultured neonatal cardiomyocytes using the hypertrophic agonists Ang II [21] or PE [22]. Treatment of NRVMs (48 hrs) with Ang II (0.1 μM) or PE (50 μM) resulted in both induction of the hypertrophic marker ANF (Figure 5A), and a substantial increase in SOCE (Figure 5B). Concomitant increases in both STIM1 mRNA (Figure 5C) and protein levels (Figure 5D) were observed. In fact, the extent of hypertrophic marker expression induced by these agonists correlated with the stimulation of STIM1 expression and the increase in SOCE (Figures 5A-5C), raising the intriguing prospect of a direct relationship between the degree of STIM1 activation and the extent of hypertrophy.
Figure 5. Increased SOCE activity and STIM1 levels in Ang II- or PE-treated cardiac myocytes in vitro.
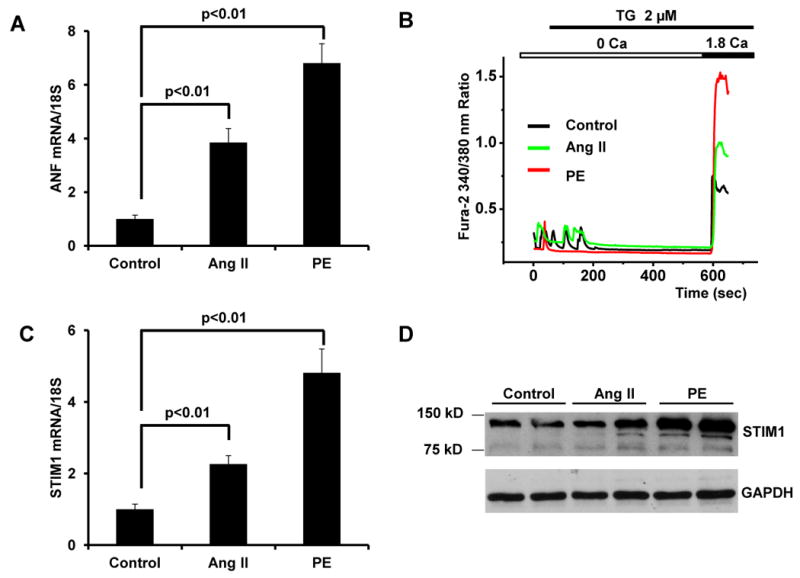
(A) Quantitative real-time RT-PCR showing increased ANF mRNA levels in cultured neonatal cardiomyocytes exposed (48h) to Ang II (0.1 μM) and PE (50 μM). (B) Representative Ca2+ traces demonstrating SOCE in control, Ang II-, or PE-treated neonatal cardiomyocytes. Quantitative real-time RT-PCR analysis demonstrating increased STIM1 mRNA levels (C) and immunoblot-detected protein levels (D) in cultured neonatal cardiomyocytes treated with Ang II or PE.
STIM1-mediated SOCE is required for hypertrophy
Data reported so far reveal robust increases in STIM1-dependent SOCE in hypertrophied myocardium. They do not, however, allow us to determine whether these events are a cause or a consequence of hypertrophy. To decipher the relationship between STIM1 and the hypertrophic response in cardiomyocytes, we manipulated both the level and activity of STIM1, and measured the effect on both SOCE and the response to hypertrophic agonists. Initially we employed an adenovirus-based shRNA construct to knock down endogenous STIM1. Western blot analysis confirmed the efficacy of RNAi-dependent STIM1 knockdown (Figure 3A). Depletion of STIM1 in neonatal myocytes reduced basal levels of SOCE by 70% (average corrected 340/380 nm ratio reduced from 0.82±0.04, n=19 to 0.25±0.03, n=18, p<0.01), and lowered Ang II (0.1 μM)-induced SOCE enhancement to a similar degree (average corrected 340/380 nm ratio reduced from 1.39±0.06, n=20 to 0.21±0.03, n=22, p<0.01) (Figures 6A-6B).
Figure 6. STIM1 is required for Ang II-induced SOCE and cardiac hypertrophy in cultured neonatal cardiomyocytes.



(A) Quantitative analyses of SOCE shown as corrected average peak Ca2+ levels in Ad-GFP- or Ad-shSTIM1-infected neonatal cardiomyocytes (n=18-22) treated (48h) with Ang II (0.1 μM) or vehicle in three independent experiments. (B) Representative Ca2+ images showing SOCE by Fura-2 ratios (340/380 nm) in Ad-GFP- or Ad-shSTIM1-infected neonatal cardiomyocytes treated with Ang II or vehicle; time marks commence with Ca2+ re-addition; scale bar, 10 μm. (C). 3H-Leucine incorporation in Ad-GFP- or Ad-shSTIM1-infected NRVMs exposed to Ang II or vehicle. (D) 3H-Leucine incorporation in Ad-GFP- or Dn-STIM1-infected NRVMs exposed to Ang II or vehicle. (E) Ca-STIM1-induced increases in NRVM 3H-Leucine incorporation is blunted by 2-APB (30 μM). (F) Quantitative analysis of SOCE depicted as the corrected average peak Ca2+ levels in cultured adult cardiomyocytes infected with Ad-GFP or Ca-STIM1 (n=17-21) from three independent experiments. (G) 3H-Leucine incorporation in Ad-GFP- or Ca-STIM1-infected ARCMs exposed to PE (50 μM) or vehicle. (H). 3H-Leucine incorporation in Ad-GFP- or Ad-shSTIM1-infected ARCMs exposed to PE (50 μM) or vehicle. N=3 independent experiments.
To test for a requirement of STIM1-dependent SOCE in myocyte hypertrophy, we quantified cell size and protein synthesis in the setting of Ang II stimulation. Here, we found that depletion of STIM1 attenuated Ang II-elicited increases in cell size (Supplemental Figures 8A, 8B) and 3H-leucine incorporation (Figure 6C). To corroborate these observations, we measured the hypertrophic response in NRVMs infected with adenovirus expressing Dn-STIM1. In agreement with the knockdown data, blocking the activity of STIM1 also suppressed the cells' ability to respond to hypertrophic agonists (Figure 6D). Conversely, expression of Ca-STIM1 in NRVMs triggered hypertrophic growth in the absence of agonist treatment, a response which was blocked by treatment with the SOCE inhibitor 2-aminoethoxy diphenylborane (2-APB, 30 μM) (Figure 6E). We corroborated these findings by infecting adult rat cardiomyocytes with ca-STIM1 or shSTIM1 and observed similar results. Specifically, ca-STIM1 elicited an increase in SOCE activity (Figure 6F) and an increase in 3H-leucine incorporation at baseline which was further enhanced by PE treatment (Figure 6G). In contrast, knockdown of STIM1 blocked PE-induced hypertrophy, as measured by 3H-leucine incorporation (Figure 6H). Collectively, these data demonstrate a critical role for STIM1 induction of SOCE in hypertrophic growth of cardiac myocytes.
STIM1-mediated SOCE triggers calcineurin-NFAT activity
The calcineurin-NFAT pathway is known to respond to tonic increases in intracellular Ca2+ and to be insensitive to the beat-by-beat changes in Ca2+ involved in excitation-contraction coupling [23]. And given that calcineurin-NFAT is a major pathway in the mediation of cardiac hypertrophy, we explored whether STIM1-mediated SOCE regulates this signaling cascade. To accomplish this, we employed an NFAT reporter assay that utilizes an RCAN promoter linked to a luciferase coding sequence. Initially, NRVMs were infected with an adenovirus encoding the reporter construct. Then, cells were treated with vehicle or TG for 6 hours to induce SOCE. TG treatment resulted in an induction of RCAN promoter activity (Figure 7A). This induction was inhibited by co-treatment with the SOCE blocker 2-APB, demonstrating specificity (Figure 7A).
Figure 7. STIM1 activity triggers calcineurin-NFAT signaling.

(A) NFAT-luciferase activity was measured in control or TG (2 μM)-treated neonatal cardiomyocytes. Increases in transcriptional activity were abrogated by 2-APB (30 μM). (B) RNAi knockdown of STIM1 (Ad-shSTIM1) in NRVMs blunted NFAT activity at both basal and TG-induced conditions. (C) Constitutively active STIM1 (Ca-STIM1) increased NFAT activity, which could be blocked by Cyclosporin A (CSA, 10 μM).
These findings suggest that induction of SOCE can activate signaling through the NFAT/calcineurin pathway. To test for a role for STIM1 in this process, we co-infected NRVMs with a STIM1-targeting shRNA adenoviral construct. Knockdown of STIM1 expression blocked TG-induced activation of NFAT signaling (Figure 7B). Conversely, co-infection with the Ca-STIM1 construct resulted in an increase in NFAT signaling (Figure 7C), consistent with the stimulation of hypertrophy in these cells (Figure 6). In aggregate, these results demonstrate that activation of SOCE is sufficient to trigger calcineurin/NFAT signaling and suggest a mechanism for the induction of pathological hypertrophy by STIM1.
Discussion
Calcium within the cardiac myocyte governs a wide array of events, including contractile, signaling, and electrophysiological processes. Remarkably, the means whereby cytoplasmic Ca2+ increases is a critical element in determining the spectrum of downstream activation events. In other words, Ca2+ entry or release by one pathway activates select downstream targets without triggering others. In the case of calcineurin, a major mechanism of pathological cardiac remodeling, it is established that the wide swings in intracellular Ca2+ occurring with each contractile cycle (>10-fold change) do not activate the protein phosphatase. At the same time, little is known regarding Ca2+ handling processes capable of activating calcineurin and thereby participating in adverse growth of the heart [23].
Prior to this report, the prevalence and possible (patho)physiological role of cardiomyocyte SOCE were debated. Here, we report that SOCE is abundant and robust in neonatal cardiomyocytes, yet absent from adult cardiomyocytes. Our studies go on to demonstrate that STIM1 protein is a required element in the activation of SOCE in cardiac myocytes and that the magnitude of SOCE correlates closely with STIM1 protein levels. Importantly, we find that STIM1 expression, including a novel splice variant, is re-activated by pathological stress to trigger significant SOCE-dependent Ca2+ influx. We demonstrate that STIM1-dependent SOCE activates the calcineurin-NFAT signaling pathway. Finally, inhibition of STIM1 suppresses agonist-triggered hypertrophy, pointing to a requirement for SOCE for this remodeling response. Based on these data, we conclude that stress-triggered STIM1 re-expression, and consequent SOCE activation, are critical elements in the upstream, Ca2+-dependent control of pathological cardiac hypertrophy.
Distinct Ca2+ handling mechanisms in myocytes versus non-excitable cells
Cellular homeostasis relies critically on Ca2+ handling within the cytoplasm, endo(sarco)plasmic reticulum, and mitochondria. Movement of Ca2+ among these locations within the cell is tightly regulated by Ca2+ channels, transporters (pumps, exchangers), receptors, and other Ca2+-binding proteins [24]. Superimposed on these complexities are intracellular microdomains of Ca2+ accumulation or depletion and vast swings in Ca2+ occurring within excitable cells.
Ca2+ entry into cells occurs through a variety of ion channels, including those regulated by ligands or transmembrane voltage. In the case of non-excitable cells, a special class of membrane Ca2+ channels exists which is activated in response to depletion of intracellular Ca2+ stores. Indeed, in immune cells, the major cascade for Ca2+ entry occurs through store-operated Ca2+ entry (SOCE) and Ca2+ release-activated Ca2+ (CRAC) channels [4]. The resulting Ca2+ signals regulate a variety of physiological responses, including cell differentiation and activation, gene transcription, and secretion of inflammatory mediators [5]. Ca2+ signaling in non-excitable cells is initiated upon the activation of phospholipase C (PLC). Agonist binding to cell surface receptors activates PLC, leading to the elicitation of inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). IP3 triggers a transient Ca2+ release from Ca2+ stores, which is followed by a sustained increase in [Ca2+]i due to SOCE.
A role for SOCE in excitable cells – where Ca2+ handling mechanisms are quite distinct – has been unclear. We report that SOCE is nearly absent in unstressed adult cardiomyocytes and hence is unlikely to contribute to the Ca2+ fluxes mediating excitation-contraction coupling, a process which likely involves distinct Ca2+ microdomains [25]. However, we find that SOCE is highly active in cardiomyocytes under conditions that trigger cardiac hypertrophy both in vitro and in vivo. Resulting increases in the prevalence and amplitude of SOCE suggest a role for SOCE in cardiac growth and remodeling. Further, our findings uncover a critical and limiting role of STIM1 in this process. Also, given the importance of SR Ca2+ “leak” in heart failure [26], we suggest that STIM1-dependent SOCE may play a role in that disease process.
A recent report concluded that STIM1-dependent SOCE in neonatal cardiomyocytes does not activate the calcineurin-NFAT pathway, as siRNA knockdown of STIM1 was insufficient to block calcineurin-dependent hypertrophic signaling [10]. In contrast, our results revealed that STIM1 is an essential regulator of the calcineurin-NFAT signaling pathway. STIM1 activation by growth triggers (or through the artificial expression of caSTIM1) leads to activation of SOCE, provoking increased calcium levels that activate calcineurin and NFAT. Conversely, depletion of STIM1 activity, either by targeted knockdown with shRNA or by expression of dnSTIM1, results in a decrease in calcineurin signaling, both under basal conditions and in response to hypertrophic agonists.
Molecular anatomy of store-operated Ca2+ entry
Key insights into the longstanding puzzle of the molecular basis of SOCE emerged recently based on RNAi screening and molecular and cellular analyses in Drosophila [17, 27]. These studies identified STIM1 as the mechanistic ‘missing link’ between the ER and the plasma membrane, capable of launching an elaborate series of events involving association and dissociation of several protein complexes. The end-result of this intricate mechanism is the sensing of depleted Ca2+ stores, delivery of this information to the cell surface, and Ca2+ entry from the extracellular space.
Specifically, depletion of Ca2+ from intracellular stores is sensed by the ER-resident protein STIM1, leading to its oligomerization and rapid translocation to a region of the ER near the plasma membrane. There, STIM1 aggregates with plasma membrane-localized proteins as a macromolecular complex. Direct physical interactions among these molecular elements triggers clustering and conformational changes in channels of the Orai or TRP (transient receptor potential) families. Interaction of STIM1 with these channels, in turn, triggers entry of Ca2+ across the plasma membrane and consequent repletion of cytosolic Ca2+ levels required for activation of many intracellular signal transducers [12, 28, 29].
The molecular elements that participate in both the ER-resident and plasma membrane-based protein complexes involved in SOCE are not fully elucidated. Indeed, there are likely elements within this sophisticated signaling mechanism which remain undiscovered. Here, we report evidence implicating STIM1 as a required component in heart. With respect to the plasmalemmal ion channels involved, early work implicated Orai channels in store-operated Ca2+ entry [30]. More recently, however, it has been found that TRP channels are critically involved in SOCE in some cell types [28]. Indeed, ample biochemical and functional evidence points to direct interaction between TRP channels and STIM1 [14, 28], and it is clear that STIM1 can trigger SOCE through either TRP family or Orai SOC components [12]. Indeed, STIM proteins interact with a variety of signaling proteins and pathways in a cell- and tissue-type specific manner [31].
Several groups have independently reported that over-expression of TRP channels potentiates stress-induced cardiac hypertrophy [28, 32-34]. In fact, consistent with data reported here, depletion of intracellular Ca2+ stores triggers TRP channel activity, which in turn participates in the development of hypertrophy through the activation of the calcineurin/NFAT pathway [32, 35]. Conversely, inhibition of TRP channels in transgenic mice or knockdown of TRP or Orai1 channels in cultured neonatal myocytes manifests a phenotype similar to that reported here by inhibition of STIM1 [10, 34, 35]. In any event, critical questions remain regarding whether both TRP and Orai channels are opened by STIM1 in cardiac myocytes and the molecular mechanism by which STIM1 signals to them.
Regulation of STIM1 expression
We find that STIM1 protein levels track closely with the magnitude of SOCE, suggesting that this ER-resident protein is a critical, limiting step in the signaling pathway. For example, both STIM1 protein levels and SOCE decline as the neonatal heart matures, only to be up-regulated under conditions that trigger cardiac hypertrophy in vitro and in vivo. Interestingly, we observed in NRVMs and in adult hearts under stress that STIM1 migrates at both 80 kD and 130 kD. This 130kD isoform of STIM1 is consistent in size with the recently identified STIM1L splice variant, and qPCR data confirms an increase in STIM1L mRNA that correlates with the presence of this slower migrating species (Figure 2E, Supplemental Figure 7B). It is tempting to speculate that this novel STIM1L isoform is reflective of a “fetal gene program” observed in stressed myocytes. In addition, given that STIM1 is a Ca2+ sensor in the ER/SR, we tested whether manipulation of STIM1 changes ER/SR Ca2+ content. Interestingly, we found that over-expression or knockdown of STIM1 had no effect on SR Ca2+ content (Supplemental Figure 9), consistent with a recent report [11]. These data suggest that STIM1 can function in a store-independent manner, as reported in other systems [36]. Regardless, our studies demonstrate that knockdown of STIM1 or expression of a dominant-negative STIM1 mutant are each capable of blunting SOCE under both basal and stress conditions and blocks the hypertrophic response to stress. Conversely, expression of caSTIM1 results in increased SOCE and triggers cardiac hypertrophy.
Role of cardiomyocyte store-operated Ca2+ entry
Based on studies in animal models and in immunodeficient patients harboring mutations in STIM1, it is clear that STIM proteins are critical for the development and functioning of many cell types [29]. Consistent with this, SOCE has been shown to be a major pathway of Ca2+ influx in a variety of cell types including excitable skeletal muscle cells [6, 7]. SOCE has been described previously in neonatal cardiomyocytes [8-10], suggesting that STIM1-dependent SOCE may participate in cardiac development. Also, SOCE was detected in adult cardiomyocytes [37], and a recent report provided evidence of increased SOCE in myocytes isolated from hypertrophic hearts [35]. However, the existence of STIM1 and SOCE in adult heart and their potential contribution to adverse remodeling and growth has remained poorly characterized.
Recent studies have reported that STIM1 inhibits voltage-dependent L-type Ca2+ channels in neurons and T lymphocytes [38, 39]. These new findings uncover a reciprocal role for STIM1 in the activation of SOCE and suppression of L-type currents. In other words, STIM1 can function as a master regulator of Ca2+ entry into the cell. With respect to the heart, it is tempting to speculate that the abundance of STIM1 in neonatal cardiomyocytes contributes not only to the robustness of SOCE but to developmental changes in voltage-dependent Ca2+ currents [40]; conversely, low-level expression of STIM1 in adult myocytes leads to a near absence of SOCE and may contribute to the predominance of voltage-dependent currents.
Conclusions
In summary, our results reveal that a novel, stress-activated STIM1-SOCE-calcineurin-NFAT axis is an essential regulator of hypertrophic remodeling in heart. Together, these data lend credence to the notion that SOCE may contribute to cardiac pathology under conditions of stress.
Materials and Methods
Animal models and echocardiography
Male C57/BL6 mice (6-8 weeks old) were subjected to thoracic aortic constriction (TAC) [41] for 3 weeks as previously described [20]. Control animals underwent sham operations. All animal care and procedures were approved by the Animal Care and Use Committee of the University of Texas Southwestern Medical Center at Dallas. Echocardiograms were performed using a Vevo 770 High Resolution Imaging System and an RMV-707B Scanhead probe (VisualSonics Inc) [42]. Measurements of left parasternal long and short axes and M-mode images were obtained at a heart rate of 500-550 bpm.
Adult mouse cardiomyocytes
Adult mouse ventricular myocytes were isolated after enzymatic dissociation as described [43] with slight modification. Briefly, after retrograde perfusion with Krebs-Ringer solution (2 mL/min, 5 min), the heart was perfused with fresh solution containing 0.8 mg/mL collagenase (Worthington type II) for another 12-15 min. The LV was removed and cut into small pieces in KB solution [(mM) taurine 10, glutamic acid 70, KCl 25, KH2PO4 10, Glucose 22, EGTA 0.5, pH 7.2]. After filtration, cells were maintained in KB buffer and studied within 4-6 hours. All isolation steps were carried out at 36°C with continuous gassing with 95% O2 + 5% CO2. Only Ca2+-tolerant, quiescent and rod-shaped cells with clear cross striations were used.
Primary culture of neonatal cardiomyocytes and adenovirus infection
Neonatal cardiomyocytes were isolated and cultured as described previously [44]. Forty-eight hours after seeding, cells were transferred to serum-free medium and used for adenovirus infection (multiplicity of infection 10).
Adenoviral and lentiviral constructs
Short hairpin STIM1 RNA (shRNA-STIM1) was engineered [45] to target rat and mouse STIM1 transcripts (ACAGUGAAACACAGCACCUUCCAUGGUGA). A non-silencing shRNA with no homology with mammalian genes (GGUUCUCCACCUUUAUAGGUGGCUU) was used as negative control [46]. Oligonucleotides were annealed and ligated via Mlu I/Xho I into shuttle vector pRNAT-H1.1 (GenScript). The recombinant vector was then transformed into competent DH5α cells. The linearized DNA fragment was inserted into pAdEasy viral DNA following the manufacturer's instructions (Stratagene). The adenovirus was packaged and propagated in HEK 293A cells as described [47]. Recombinant adenoviruses encoding a constitutively active mutant of STIM1 (D76A) and a dominant negative mutant of STIM1 (ERM deletion) were gifts from Dr. Shmuel Muallem (UT Southwestern Medical Center, Dallas, TX).
A mouse STIM1 expression clone (Stratagene) was amplified, the ORF was linearized by KpnI digestion, and then cloned into pLVX-IRES-hygromycin lentivirus vector with the IRES-hygromycin removed (Clonetec). To generate lentivirus expressing STIM1, we used a second generation packaging system with pCD/NL-BH and pMD2.VSVG. Briefly, HEK 293T cells were cultured and transfected with pCD/NL-BH, pMD2.VSVG and pLVX-mSTIM1, culture medium was collected, and virus was harvested and stored at -80°C. All constructs expressed GFP allowing verification of infection and expression in the individual cells studied.
Real-time RT-PCR
Total RNA was harvested from NRVMs or mouse LV using TRIzol (Invitrogen) according to the manufacturer's protocol. cDNA was prepared from RNA using a high capacity cDNA reverse transcription kit (Applied Biosystems). Real-time PCR was performed using SYBR green on an ABI 7000 Prism Sequence Detection System (Applied Biosystems). To confirm amplification specificity, the PCR products were subjected to melting curve analysis. Negative controls containing water instead of cDNA were run concomitantly. Data for each transcript were normalized to reactions performed using GAPDH or 18S rRNA primers, and fold change was determined using the comparative threshold method [48].
Western blot analysis
LVs were either homogenized immediately or quick frozen in liquid nitrogen and stored at -80°C for later use. To harvest protein, tissues were homogenized at 4°C in extraction buffer containing Tris-buffered saline, 0.1% Triton X-100, 4% glycerol, 1mM DTT, 1mM EDTA, protease inhibitors (Roche), and phosphatase inhibitors (Sigma). Homogenates were passed over glass wool to remove DNA. Whole cell lysates from cultured neonatal myocytes were prepared by directly harvesting cells in M-PER® mammalian protein extraction reagent (Thermo Scientific). The lysates were run on SDS-PAGE and transferred to PVDF membrane (Millipore). Polyclonal STIM1 antibody (#54680) was purchased from AnaSpec Inc. GAPDH (sc-25778) antibody was purchased from Santa Cruz. Polyclonal RCAN1 antibody was described previously [49].
In vitro cardiomyocyte hypertrophy
After culturing for 24 hour in serum-free medium, cells were treated with Ang II (0.1 μM) or PE (50 μM) for an additional 48 hours. Cell hypertrophy was evaluated by cell cross-sectional area (CSA), 3H-Leucine incorporation, and atrial natriuretic factor (ANF) expression. CSA was quantified from manually outlined cells in digitized microscopic images (recorded by an Olympus fluorescence microscope) of randomly chosen cell fields using ImageJ software. A minimum of 50 cells over 4 wells/treatment were measured in each independent experiment, and the experiments were repeated 3 times. For 3H-Leucine incorporation, upon the onset of treatment with Ang II or PE, cells were incubated with 3H-Leucine (1.0 μCi/ml, Amersham Biosciences) for 48 hours. Cells were washed with cold PBS and incubated in 10% trichloroacetic acid (TCA). The resultant pellet was solubilized in 0.2 N NaOH and counted in a scintillation counter. ANF expression was measured by real-time RT-PCR as described above.
Ca2+ imaging and SOCE measurements
Ca2+ imaging was performed using a PTI (Photon Technology International) Ca2+ Imaging System (Birmingham, NJ) with an automated fluorescence microscope and CCD camera [15]. Neonatal cardiomyocytes were plated on 0.01% polylysine plus 0.1% gelatin-coated Assistant-Präzision coverslips (Deckgläser, Germany). Isolated adult mouse myocytes were loaded on laminin-coated coverslips. Cytosolic Ca2+ concentration ([Ca2+]i) was measured using the fluorescent Ca2+ indicator Fura-2 AM as described previously [15]. Cells were loaded with 5 μM Fura-2 AM in extracellular buffer (in mM,140 NaCl, 5 KCl, 1.8 CaCl2, 1 MgCl2, 10 glucose, and 10 Hepes) containing 0.1% BSA and 1% pyruvate for 30 min (RT) while shielded from light. Next, cells were washed with extracellular buffer and kept in this buffer with 0.1% BSA and 1% pyruvate until use. The glass coverslip was inserted into the bottom of a perfusion chamber. The cells were perfused with extracellular buffer (RT), and agonists/blockers were delivered with the perfusate. [Ca2+]i in individual cells by exciting Fura-2 alternately at 340 and 380 nm, and recording emitted fluorescence at 510 nm. A standard protocol referred as “Ca2+ re-addition” was used to trigger SOCE [13, 14], in which cells are exposed to Ca2+-free medium containing thapsigargin (TG, 2μM) to deplete SR Ca2+ stores. Then, Ca2+ (1.8 mM) is restored to the medium, and Ca2+ influx is recorded. Data were analyzed using ImageMaster software and shown as 340/380 nm ratios at indicated time points or as time-ratio traces translated from the images.
NFAT-Luciferase activity assay
Adenovirus harboring a construct in which a luciferase reporter gene is controlled by the promoter region of exon 4 of the RCAN1 gene with 15 NFAT sites (Ad-RCAN1-Luc), was used to evaluate calcineurin/NFAT transcriptional activity [32, 50]. Cells were infected by Ad-RCAN1-Luc for 48 hours, harvested, and luciferase and control β-galactosidase activities were measured according to the manufacturer's instructions (Promega). All assays were performed at least twice with triplicates for each group.
Statistics
Data are presented as mean±SEM. The unpaired Student's t test was used for comparison between two groups, and ANOVA with a post hoc Fisher's test was used for comparison among multiple groups. Values of p<0.05 were considered significant.
Supplementary Material
Highlights.
SOCE is abundant and robust in neonatal cardiomyocytes, yet absent from adult cardiomyocytes.
STIM1 is a required element in the activation of SOCE.
STIM1 expression – including a novel splicing isoform – is re-activated by pathological stress.
STIM1-dependent SOCE activates the calcineurin-NFAT signaling pathway.
STIM1-dependent SOCE is a critical element in the upstream control of pathological hypertrophy.
Acknowledgments
We thank Yongli Kong, Herman May, Janet Johnstone, Guojing Huang, Hua Zhang, Jun Wu, Suya Sun, and Wei-Zhong Zeng for assistance and support. We thank the Protein Chemistry Technology Center at UT Southwestern for expert assistance with mass spectrometry. We gratefully acknowledge Shmuel Muallem and Ilya Bezprozvanny for reagents and access to equipment.
Source of Funding: This work was supported by grants from the NIH (HL-075173, JAH; HL-080144, JAH; HL-090842, JAH), AHA (0640084N, JAH), ADA (7-08-MN-21-ADA, JAH), and the AHA-Jon Holden DeHaan Foundation (0970518N, JAH).
Footnotes
Conflicts of Interest Disclosures: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Roger VL, Go AS, Lloyd-Jones DM, Adams RJ, Berry JD, Brown TM, et al. Heart Disease and Stroke Statistics--2011 Update: A Report From the American Heart Association. Circulation. 2011;123:e18–e209. doi: 10.1161/CIR.0b013e3182009701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hill JA, Olson EN. Cardiac plasticity. The New England journal of medicine. 2008;358:1370–80. doi: 10.1056/NEJMra072139. [DOI] [PubMed] [Google Scholar]
- 3.Bers DM. Calcium fluxes involved in control of cardiac myocyte contraction. Circ Res. 2000;87:275–81. doi: 10.1161/01.res.87.4.275. [DOI] [PubMed] [Google Scholar]
- 4.Feske S, Giltnane J, Dolmetsch R, Staudt LM, Rao A. Gene regulation mediated by calcium signals in T lymphocytes. Nat Immunol. 2001;2:316–24. doi: 10.1038/86318. [DOI] [PubMed] [Google Scholar]
- 5.Parekh AB, Putney JW., Jr Store-operated calcium channels. Physiol Rev. 2005;85:757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- 6.Lyfenko AD, Dirksen RT. Differential dependence of store-operated and excitation-coupled Ca2+ entry in skeletal muscle on STIM1 and Orai1. J Physiol. 2008;586:4815–24. doi: 10.1113/jphysiol.2008.160481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stiber J, Hawkins A, Zhang ZS, Wang S, Burch J, Graham V, et al. STIM1 signalling controls store-operated calcium entry required for development and contractile function in skeletal muscle. Nat Cell Biol. 2008;10:688–97. doi: 10.1038/ncb1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hunton DL, Lucchesi PA, Pang Y, Cheng X, Dell'Italia LJ, Marchase RB. Capacitative calcium entry contributes to nuclear factor of activated T-cells nuclear translocation and hypertrophy in cardiomyocytes. J Biol Chem. 2002;277:14266–73. doi: 10.1074/jbc.M107167200. [DOI] [PubMed] [Google Scholar]
- 9.Uehara A, Yasukochi M, Imanaga I, Nishi M, Takeshima H. Store-operated Ca2+ entry uncoupled with ryanodine receptor and junctional membrane complex in heart muscle cells. Cell Calcium. 2002;31:89–96. doi: 10.1054/ceca.2001.0257. [DOI] [PubMed] [Google Scholar]
- 10.Voelkers M, Salz M, Herzog N, Frank D, Dolatabadi N, Frey N, et al. Orai1 and Stim1 regulate normal and hypertrophic growth in cardiomyocytes. J Mol Cell Cardiol. 2010;48:1329–34. doi: 10.1016/j.yjmcc.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hulot JS, Fauconnier J, Ramanujam D, Chaanine A, Aubart F, Sassi Y, et al. Critical role for stromal interaction molecule 1 in cardiac hypertrophy. Circulation. 2011;124:796–805. doi: 10.1161/CIRCULATIONAHA.111.031229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cahalan MD. STIMulating store-operated Ca(2+) entry. Nat Cell Biol. 2009;11:669–77. doi: 10.1038/ncb0609-669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bird GS, DeHaven WI, Smyth JT, Putney JW., Jr Methods for studying store-operated calcium entry. Methods. 2008;46:204–12. doi: 10.1016/j.ymeth.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liao Y, Erxleben C, Abramowitz J, Flockerzi V, Zhu MX, Armstrong DL, et al. Functional interactions among Orai1, TRPCs, and STIM1 suggest a STIM-regulated heteromeric Orai/TRPC model for SOCE/Icrac channels. Proc Natl Acad Sci U S A. 2008;105:2895–900. doi: 10.1073/pnas.0712288105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luo X, Shin DM, Wang X, Konieczny SF, Muallem S. Aberrant localization of intracellular organelles, Ca2+ signaling, and exocytosis in Mist1 null mice. J Biol Chem. 2005;280:12668–75. doi: 10.1074/jbc.M411973200. [DOI] [PubMed] [Google Scholar]
- 16.Darbellay B, Arnaudeau S, Bader CR, Konig S, Bernheim L. STIM1L is a new actin-binding splice variant involved in fast repetitive Ca2+ release. The Journal of cell biology. 2011;194:335–46. doi: 10.1083/jcb.201012157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169:435–45. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang GN, Zeng W, Kim JY, Yuan JP, Han L, Muallem S, et al. STIM1 carboxyl-terminus activates native SOC, I(crac) and TRPC1 channels. Nat Cell Biol. 2006;8:1003–10. doi: 10.1038/ncb1454. [DOI] [PubMed] [Google Scholar]
- 19.Dziadek MA, Johnstone LS. Biochemical properties and cellular localisation of STIM proteins. Cell Calcium. 2007;42:123–32. doi: 10.1016/j.ceca.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 20.Hill JA, Karimi M, Kutschke W, Davisson RL, Zimmerman K, Wang Z, et al. Cardiac hypertrophy is not a required compensatory response to short-term pressure overload. Circulation. 2000;101:2863–9. doi: 10.1161/01.cir.101.24.2863. [DOI] [PubMed] [Google Scholar]
- 21.Sadoshima J, Malhotra R, Izumo S. The role of the cardiac renin-angiotensin system in load-induced cardiac hypertrophy. J Card Fail. 1996;2:S1–6. doi: 10.1016/s1071-9164(96)80052-2. [DOI] [PubMed] [Google Scholar]
- 22.Deng XF, Rokosh DG, Simpson PC. Autonomous and growth factor-induced hypertrophy in cultured neonatal mouse cardiac myocytes. Comparison with rat. Circ Res. 2000;87:781–8. doi: 10.1161/01.res.87.9.781. [DOI] [PubMed] [Google Scholar]
- 23.Molkentin JD. Dichotomy of Ca2+ in the heart: contraction versus intracellular signaling. J Clin Invest. 2006;116:623–6. doi: 10.1172/JCI27824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dibb KM, Graham HK, Venetucci LA, Eisner DA, Trafford AW. Analysis of cellular calcium fluxes in cardiac muscle to understand calcium homeostasis in the heart. Cell Calcium. 2007;42:503–12. doi: 10.1016/j.ceca.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 25.Parekh AB. Ca2+ microdomains near plasma membrane Ca2+ channels: impact on cell function. J Physiol. 2008;586:3043–54. doi: 10.1113/jphysiol.2008.153460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wehrens XH, Lehnart SE, Marks AR. Intracellular calcium release and cardiac disease. Annu Rev Physiol. 2005;67:69–98. doi: 10.1146/annurev.physiol.67.040403.114521. [DOI] [PubMed] [Google Scholar]
- 27.Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, et al. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437:902–5. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yuan JP, Kim MS, Zeng W, Shin DM, Huang G, Worley PF, et al. TRPC channels as STIM1-regulated SOCs. Channels (Austin) 2009;3:221–5. doi: 10.4161/chan.3.4.9198. [DOI] [PubMed] [Google Scholar]
- 29.Feske S. CRAC channelopathies. Pflugers Arch. 2010;460:417–35. doi: 10.1007/s00424-009-0777-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang SL, Yeromin AV, Zhang XH, Yu Y, Safrina O, Penna A, et al. Genome-wide RNAi screen of Ca(2+) influx identifies genes that regulate Ca(2+) release-activated Ca(2+) channel activity. Proc Natl Acad Sci U S A. 2006;103:9357–62. doi: 10.1073/pnas.0603161103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnstone LS, Graham SJ, Dziadek MA. STIM proteins: integrators of signalling pathways in development, differentiation and disease. J Cell Mol Med. 2010;14:1890–903. doi: 10.1111/j.1582-4934.2010.01097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuwahara K, Wang Y, McAnally J, Richardson JA, Bassel-Duby R, Hill JA, et al. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. The Journal of clinical investigation. 2006;116:3114–26. doi: 10.1172/JCI27702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Onohara N, Nishida M, Inoue R, Kobayashi H, Sumimoto H, Sato Y, et al. TRPC3 and TRPC6 are essential for angiotensin II-induced cardiac hypertrophy. EMBO J. 2006;25:5305–16. doi: 10.1038/sj.emboj.7601417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seth M, Zhang ZS, Mao L, Graham V, Burch J, Stiber J, et al. TRPC1 channels are critical for hypertrophic signaling in the heart. Circulation research. 2009;105:1023–30. doi: 10.1161/CIRCRESAHA.109.206581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu X, Eder P, Chang B, Molkentin JD. TRPC channels are necessary mediators of pathologic cardiac hypertrophy. Proc Natl Acad Sci U S A. 2010;107:7000–5. doi: 10.1073/pnas.1001825107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mignen O, Thompson JL, Shuttleworth TJ. STIM1 regulates Ca2+ entry via arachidonate-regulated Ca2+-selective (ARC) channels without store depletion or translocation to the plasma membrane. The Journal of physiology. 2007;579:703–15. doi: 10.1113/jphysiol.2006.122432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hunton DL, Zou L, Pang Y, Marchase RB. Adult rat cardiomyocytes exhibit capacitative calcium entry. Am J Physiol Heart Circ Physiol. 2004;286:H1124–32. doi: 10.1152/ajpheart.00162.2003. [DOI] [PubMed] [Google Scholar]
- 38.Park CY, Shcheglovitov A, Dolmetsch R. The CRAC channel activator STIM1 binds and inhibits L-type voltage-gated calcium channels. Science. 2010;330:101–5. doi: 10.1126/science.1191027. [DOI] [PubMed] [Google Scholar]
- 39.Wang Y, Deng X, Mancarella S, Hendron E, Eguchi S, Soboloff J, et al. The calcium store sensor, STIM1, reciprocally controls Orai and CaV1.2 channels. Science. 2010;330:105–9. doi: 10.1126/science.1191086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schroder EA, Wei Y, Satin J. The developing cardiac myocyte: maturation of excitability and excitation-contraction coupling. Ann N Y Acad Sci. 2006;1080:63–75. doi: 10.1196/annals.1380.006. [DOI] [PubMed] [Google Scholar]
- 41.Rockman HA, Ross RS, Harris AN, Knowlton KU, Steinhelper ME, Field LJ, et al. Segregation of atrial-specific and inducible expression of an atrial natriuretic factor transgene in an in vivo murine model of cardiac hypertrophy. Proc Natl Acad Sci U S A. 1991;88:8277–81. doi: 10.1073/pnas.88.18.8277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Berry JM, Le V, Rotter D, Battiprolu PK, Grinsfelder B, Tannous P, et al. Reversibility of adverse, calcineurin-dependent cardiac remodeling. Circulation research. 2011;109:407–17. doi: 10.1161/CIRCRESAHA.110.228452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Y, Cheng J, Joyner RW, Wagner MB, Hill JA. Remodeling of early-phase repolarization: a mechanism of abnormal impulse conduction in heart failure. Circulation. 2006;113:1849–56. doi: 10.1161/CIRCULATIONAHA.106.615682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ni YG, Berenji K, Wang N, Oh M, Sachan N, Dey A, et al. Foxo transcription factors blunt cardiac hypertrophy by inhibiting calcineurin signaling. Circulation. 2006;114:1159–68. doi: 10.1161/CIRCULATIONAHA.106.637124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jones BF, Boyles RR, Hwang SY, Bird GS, Putney JW. Calcium influx mechanisms underlying calcium oscillations in rat hepatocytes. Hepatology. 2008;48:1273–81. doi: 10.1002/hep.22461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Spassova MA, Soboloff J, He LP, Xu W, Dziadek MA, Gill DL. STIM1 has a plasma membrane role in the activation of store-operated Ca(2+) channels. Proc Natl Acad Sci U S A. 2006;103:4040–5. doi: 10.1073/pnas.0510050103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luo J, Deng ZL, Luo X, Tang N, Song WX, Chen J, et al. A protocol for rapid generation of recombinant adenoviruses using the AdEasy system. Nat Protoc. 2007;2:1236–47. doi: 10.1038/nprot.2007.135. [DOI] [PubMed] [Google Scholar]
- 48.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 49.Bush E, Fielitz J, Melvin L, Martinez-Arnold M, McKinsey TA, Plichta R, et al. A small molecular activator of cardiac hypertrophy uncovered in a chemical screen for modifiers of the calcineurin signaling pathway. Proc Natl Acad Sci U S A. 2004;101:2870–5. doi: 10.1073/pnas.0308723101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang J, Rothermel B, Vega RB, Frey N, McKinsey TA, Olson EN, et al. Independent signals control expression of the calcineurin inhibitory proteins MCIP1 and MCIP2 in striated muscles. Circ Res. 2000;87:E61–8. doi: 10.1161/01.res.87.12.e61. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.