

Abstract
The title compound, C11H15BrNO+·Cl−, was obtained as a precursor within our current program for the synthesis of new β-aminoalcohols via a Mannich-type reaction. The protonated amino N atom is hydrogen bonded to the chloride anion. With exception of one methyl group, the cation is approximately planar (r.m.s. deviation for all non H-atoms = 0.069 Å).
Related literature
For (N,N-dialkylamino)propiophenones, see: Alper et al. (2002 ▶); Pupo et al. (2003 ▶); Abonia et al. (2004 ▶). For details of the synthesis, see: Brandes & Roth (1967 ▶); Vogel et al. (1978 ▶).
Experimental
Crystal data
C11H15BrNO+·Cl−
M r = 292.60
Monoclinic,
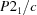
a = 10.5050 (8) Å
b = 12.5694 (5) Å
c = 10.6483 (5) Å
β = 115.594 (2)°
V = 1268.06 (12) Å3
Z = 4
Cu Kα radiation
μ = 6.16 mm−1
T = 295 K
0.44 × 0.26 × 0.26 mm
Data collection
Enraf–Nonius CAD-4 diffractometer
Absorption correction: ψ scan (CORINC; Dräger & Gattow, 1971 ▶) T min = 0.61, T max = 1.00
2564 measured reflections
2564 independent reflections
2258 reflections with I > 2σ(I)
3 standard reflections every 60 min intensity decay: 5%
Refinement
R[F 2 > 2σ(F 2)] = 0.045
wR(F 2) = 0.123
S = 1.12
2564 reflections
146 parameters
Only H-atom displacement parameters refined
Δρmax = 0.72 e Å−3
Δρmin = −0.65 e Å−3
Data collection: CAD-4 Software (Enraf–Nonius, 1989 ▶); cell refinement: CAD-4 Software; data reduction: CORINC (Dräger & Gattow, 1971 ▶); program(s) used to solve structure: SIR97 (Altomare et al., 1999 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: PLATON (Spek, 2009 ▶); software used to prepare material for publication: PLATON.
Supplementary Material
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536811041985/bt5672sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536811041985/bt5672Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536811041985/bt5672Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N10—H10⋯Cl1 | 1.00 | 1.99 | 2.983 (2) | 171 |
Acknowledgments
Financial support from COLCIENCIAS and the Universidad del Valle is gratefully acknowledged.
supplementary crystallographic information
Comment
The classical method for the synthesis of 3-(N,N-dialkylamino)propiophenone salts is the well known Mannich reaction between an alkyl aryl ketone, dialkylamine hydrochloride and polyformaldehyde in refluxing ethanol (Vogel et al., 1978). In this approach, the N,N-dialkylmethyleneammonium chloride (H2C=NR2Cl) is formed in situ, which suffers a Michael type addition from the methylene active ketone to render the expected Mannich adduct (Brandes et al., 1967).
The 3-(N,N-dialkylamino)propiophenone salts are visualized as synthetic equivalents of the less stable and more reactive α,β-unsaturated aryl vinyl ketones. For instance, they can react with nucleophiles like amines through a Michael type addition which could lead to the formation of β-aminoalcohols as is our purpose with compound (I).
In the crystal the title compound adopts an essentially planar structure with a dihedral angle of 5.0 (2)° between the almost planar aminopropane-1-one group (maximal deviation from least square plane 0.038Å at C9) and the phenyl ring. The protonated N10 atom forms a hydrogen bond to Cl1 (N10—H10···Cl1 1.99 Å).
Experimental
A mixture of dimethylamine hydrochloride (2.0 g, 25 mmol), polyformaldehyde (0.754 g, 25 mmol), p-bromoacetophenone (3.66 g, 9.2 mmol), 95% ethanol (4 mL) and conc HCl (0.02 mL) was heated at reflux in an oil bath during 3 h (Vogel et al. (1978)). After complete disappearance of the starting acetophenone, as monitored by thin-layer chromatography, the hot mixture was filtered; acetone (15 mL) was added to the filtrate and cooled into the freezer overnight. The resulting solid was filtered, washed with acetone (2 x 5 mL) and dried at ambient temperature affording the title compound (I), as white solid [yield 94%, m.p. 495 K].
Crystals of (I) suitable for single-crystal X-ray diffraction were grown by slow evaporation at ambient temperature and in air, from a 1:1 ethanol:acetone solution.
Refinement
Hydrogen atoms attached to carbons were placed at calculated positions with C—H = 0.95 Å (aromatic) or 0.98–0.99 Å (sp3 C-atom). All H atoms were refined in the riding-model approximation with isotropic displacement parameters.
Figures
Fig. 1.

View of compound I. Displacement ellipsoids are drawn at the 50% probability level.
Crystal data
| C11H15BrNO+·Cl− | F(000) = 592 |
| Mr = 292.60 | Dx = 1.533 Mg m−3 |
| Monoclinic, P21/c | Melting point: 495 K |
| Hall symbol: -P 2ybc | Cu Kα radiation, λ = 1.54178 Å |
| a = 10.5050 (8) Å | Cell parameters from 25 reflections |
| b = 12.5694 (5) Å | θ = 64–74° |
| c = 10.6483 (5) Å | µ = 6.16 mm−1 |
| β = 115.594 (2)° | T = 295 K |
| V = 1268.06 (12) Å3 | Block, brown |
| Z = 4 | 0.44 × 0.26 × 0.26 mm |
Data collection
| Enraf–Nonius CAD-4 diffractometer | 2258 reflections with I > 2σ(I) |
| Radiation source: rotating anode | Rint = 0.000 |
| graphite | θmax = 73.8°, θmin = 4.7° |
| θ/2ω scans | h = −11→13 |
| Absorption correction: ψ scan (CORINC; Dräger & Gattow, 1971) | k = −15→0 |
| Tmin = 0.61, Tmax = 1.00 | l = −13→0 |
| 2564 measured reflections | 3 standard reflections every 60 min |
| 2564 independent reflections | intensity decay: 5% |
Refinement
| Refinement on F2 | Secondary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Hydrogen site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.045 | Only H-atom displacement parameters refined |
| wR(F2) = 0.123 | w = 1/[σ2(Fo2) + (0.0762P)2 + 0.3958P] where P = (Fo2 + 2Fc2)/3 |
| S = 1.12 | (Δ/σ)max = 0.001 |
| 2564 reflections | Δρmax = 0.72 e Å−3 |
| 146 parameters | Δρmin = −0.65 e Å−3 |
| 0 restraints | Extinction correction: SHELXL97 (Sheldrick, 2008), Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| Primary atom site location: structure-invariant direct methods | Extinction coefficient: 0.0047 (5) |
Special details
| Experimental. IR (KBr disk): 3080, 3024, 2954, 2912, 2628, 2543 (br), 2503, 2440 (br), 1684 (C=O), 1580, 1473, 1391, 1329, 1216, 1065, 1002, 963, 787 cm-1. 1H-NMR (DMSO-d6): 2.79 (s, 6H), 3.39 (t, J = 7.5 Hz, 2H), 3.64 (t, J = 7.2 Hz,2H), 7.78 ("d", J =8.4 Hz, 2H), 7.95 ("d", J = 8.4 Hz, 2H), 10.93 (bs, 1H, NH) p.p.m.; 13C-NMR (DMSO-d6): 33.2, 42.1, 51.5, 127.8, 130.0, 131.9, 134.9, 196.0 (C=O) p.p.m.. |
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| Br1 | 0.31084 (4) | 0.13998 (3) | 0.50877 (3) | 0.04655 (18) | |
| Cl1 | 0.89690 (9) | −0.34411 (6) | 1.32816 (9) | 0.0430 (2) | |
| O1 | 0.7418 (3) | 0.0972 (2) | 1.2059 (2) | 0.0539 (6) | |
| C1 | 0.4405 (3) | 0.1090 (2) | 0.6944 (3) | 0.0374 (6) | |
| C2 | 0.5264 (3) | 0.0213 (3) | 0.7200 (3) | 0.0436 (7) | |
| H2 | 0.5200 | −0.0221 | 0.6467 | 0.058 (11)* | |
| C3 | 0.6223 (3) | −0.0025 (3) | 0.8549 (3) | 0.0411 (7) | |
| H3 | 0.6794 | −0.0623 | 0.8722 | 0.040 (9)* | |
| C4 | 0.6335 (3) | 0.0629 (2) | 0.9646 (3) | 0.0335 (6) | |
| C5 | 0.5434 (4) | 0.1497 (2) | 0.9364 (4) | 0.0419 (7) | |
| H5 | 0.5478 | 0.1922 | 1.0097 | 0.060 (11)* | |
| C6 | 0.4478 (4) | 0.1745 (3) | 0.8029 (3) | 0.0430 (7) | |
| H6 | 0.3893 | 0.2336 | 0.7854 | 0.054 (11)* | |
| C7 | 0.7377 (3) | 0.0416 (2) | 1.1111 (3) | 0.0362 (6) | |
| C8 | 0.8395 (3) | −0.0495 (2) | 1.1373 (3) | 0.0369 (6) | |
| H8A | 0.7874 | −0.1160 | 1.1125 | 0.058 (8)* | |
| H8B | 0.8885 | −0.0414 | 1.0788 | 0.058 (8)* | |
| C9 | 0.9456 (3) | −0.0535 (2) | 1.2878 (3) | 0.0360 (6) | |
| H9A | 0.9986 | 0.0126 | 1.3113 | 0.046 (7)* | |
| H9B | 0.8957 | −0.0590 | 1.3459 | 0.046 (7)* | |
| N10 | 1.0466 (3) | −0.14452 (17) | 1.3199 (3) | 0.0358 (6) | |
| H10 | 0.9959 | −0.2136 | 1.3123 | 0.053 (11)* | |
| C11 | 1.1447 (5) | −0.1452 (3) | 1.4695 (4) | 0.0578 (11) | |
| H11A | 1.2084 | −0.2042 | 1.4889 | 0.082 (10)* | |
| H11B | 1.0919 | −0.1518 | 1.5236 | 0.082 (10)* | |
| H11C | 1.1974 | −0.0800 | 1.4932 | 0.082 (10)* | |
| C12 | 1.1245 (4) | −0.1462 (3) | 1.2322 (4) | 0.0526 (9) | |
| H12A | 1.0585 | −0.1462 | 1.1357 | 0.101 (11)* | |
| H12B | 1.1817 | −0.2092 | 1.2524 | 0.101 (11)* | |
| H12C | 1.1838 | −0.0845 | 1.2518 | 0.101 (11)* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Br1 | 0.0519 (3) | 0.0423 (2) | 0.0400 (2) | 0.00146 (13) | 0.01472 (18) | 0.00820 (13) |
| Cl1 | 0.0541 (5) | 0.0294 (4) | 0.0461 (4) | −0.0091 (3) | 0.0223 (4) | −0.0025 (3) |
| O1 | 0.0644 (15) | 0.0500 (14) | 0.0408 (12) | 0.0130 (12) | 0.0166 (12) | −0.0113 (11) |
| C1 | 0.0398 (15) | 0.0351 (15) | 0.0376 (15) | −0.0020 (12) | 0.0170 (13) | 0.0041 (12) |
| C2 | 0.0492 (18) | 0.0441 (18) | 0.0381 (15) | 0.0058 (14) | 0.0193 (14) | −0.0037 (13) |
| C3 | 0.0436 (16) | 0.0380 (16) | 0.0418 (16) | 0.0102 (13) | 0.0187 (14) | −0.0025 (13) |
| C4 | 0.0364 (14) | 0.0294 (14) | 0.0363 (14) | 0.0003 (11) | 0.0170 (12) | −0.0011 (11) |
| C5 | 0.0505 (18) | 0.0309 (16) | 0.0444 (18) | 0.0058 (12) | 0.0206 (15) | −0.0055 (12) |
| C6 | 0.0519 (19) | 0.0291 (15) | 0.0471 (18) | 0.0082 (13) | 0.0206 (15) | −0.0004 (13) |
| C7 | 0.0401 (15) | 0.0284 (13) | 0.0406 (15) | −0.0010 (11) | 0.0181 (13) | −0.0009 (12) |
| C8 | 0.0418 (15) | 0.0318 (15) | 0.0347 (14) | 0.0019 (12) | 0.0142 (12) | −0.0018 (11) |
| C9 | 0.0467 (16) | 0.0270 (14) | 0.0339 (14) | −0.0007 (12) | 0.0170 (13) | 0.0003 (11) |
| N10 | 0.0458 (14) | 0.0228 (11) | 0.0342 (13) | −0.0019 (9) | 0.0130 (11) | 0.0029 (9) |
| C11 | 0.070 (2) | 0.0371 (19) | 0.0411 (19) | 0.0028 (16) | 0.0002 (18) | 0.0031 (14) |
| C12 | 0.057 (2) | 0.044 (2) | 0.063 (2) | 0.0121 (15) | 0.0319 (19) | 0.0101 (16) |
Geometric parameters (Å, °)
| Br1—C1 | 1.894 (3) | C2—H2 | 0.9300 |
| O1—C7 | 1.213 (4) | C3—H3 | 0.9300 |
| C1—C2 | 1.375 (4) | C5—H5 | 0.9300 |
| C1—C6 | 1.394 (4) | C6—H6 | 0.9300 |
| C2—C3 | 1.385 (4) | C8—H8A | 0.9700 |
| C3—C4 | 1.391 (4) | C8—H8B | 0.9700 |
| C4—C5 | 1.390 (4) | C9—H9A | 0.9700 |
| C4—C7 | 1.493 (4) | C9—H9B | 0.9700 |
| C5—C6 | 1.376 (5) | C11—H11A | 0.9600 |
| C7—C8 | 1.509 (4) | C11—H11B | 0.9600 |
| C8—C9 | 1.507 (4) | C11—H11C | 0.9600 |
| C9—N10 | 1.496 (4) | C12—H12A | 0.9600 |
| N10—C11 | 1.477 (4) | C12—H12B | 0.9600 |
| N10—C12 | 1.484 (5) | C12—H12C | 0.9600 |
| N10—H10 | 1.0000 | ||
| C2—C1—C6 | 120.9 (3) | C1—C6—H6 | 121.00 |
| C2—C1—Br1 | 119.1 (2) | C5—C6—H6 | 121.00 |
| C6—C1—Br1 | 120.0 (2) | C7—C8—H8A | 109.00 |
| C1—C2—C3 | 120.0 (3) | C7—C8—H8B | 109.00 |
| C2—C3—C4 | 120.2 (3) | C9—C8—H8A | 109.00 |
| C5—C4—C3 | 118.7 (3) | C9—C8—H8B | 109.00 |
| C5—C4—C7 | 119.3 (3) | H8A—C8—H8B | 108.00 |
| C3—C4—C7 | 122.0 (3) | N10—C9—H9A | 109.00 |
| C6—C5—C4 | 121.7 (3) | N10—C9—H9B | 109.00 |
| C5—C6—C1 | 118.5 (3) | C8—C9—H9A | 109.00 |
| O1—C7—C4 | 120.9 (3) | C8—C9—H9B | 109.00 |
| O1—C7—C8 | 121.1 (3) | H9A—C9—H9B | 108.00 |
| C4—C7—C8 | 118.0 (2) | N10—C11—H11A | 109.00 |
| C9—C8—C7 | 111.2 (2) | N10—C11—H11B | 109.00 |
| N10—C9—C8 | 113.2 (2) | N10—C11—H11C | 109.00 |
| C11—N10—C12 | 111.2 (3) | H11A—C11—H11B | 109.00 |
| C11—N10—C9 | 110.2 (2) | H11A—C11—H11C | 110.00 |
| C12—N10—C9 | 113.4 (2) | H11B—C11—H11C | 109.00 |
| C1—C2—H2 | 120.00 | N10—C12—H12A | 110.00 |
| C3—C2—H2 | 120.00 | N10—C12—H12B | 109.00 |
| C2—C3—H3 | 120.00 | N10—C12—H12C | 110.00 |
| C4—C3—H3 | 120.00 | H12A—C12—H12B | 109.00 |
| C4—C5—H5 | 119.00 | H12A—C12—H12C | 109.00 |
| C6—C5—H5 | 119.00 | H12B—C12—H12C | 109.00 |
| C6—C1—C2—C3 | −0.5 (5) | C5—C4—C7—O1 | 2.1 (5) |
| Br1—C1—C2—C3 | 179.7 (3) | C3—C4—C7—O1 | −177.0 (3) |
| C1—C2—C3—C4 | −0.8 (5) | C5—C4—C7—C8 | −176.6 (3) |
| C2—C3—C4—C5 | 2.3 (5) | C3—C4—C7—C8 | 4.2 (4) |
| C2—C3—C4—C7 | −178.6 (3) | O1—C7—C8—C9 | −4.1 (4) |
| C3—C4—C5—C6 | −2.5 (5) | C4—C7—C8—C9 | 174.6 (2) |
| C7—C4—C5—C6 | 178.4 (3) | C7—C8—C9—N10 | 178.4 (2) |
| C4—C5—C6—C1 | 1.2 (5) | C8—C9—N10—C11 | −178.4 (3) |
| C2—C1—C6—C5 | 0.3 (5) | C8—C9—N10—C12 | 56.2 (4) |
| Br1—C1—C6—C5 | −179.9 (2) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N10—H10···Cl1 | 1.00 | 1.99 | 2.983 (2) | 171 |
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: BT5672).
References
- Abonia, R., Insuasty, B., Quiroga, J., Nogueras, M. & Meier, H. (2004). Mini-Rev. Org. Chem. 1, 387–402.
- Alper, K., Barry, J. & Balabanov, A. (2002). Epilepsy Behav. 3, 13–18. [DOI] [PubMed]
- Altomare, A., Burla, M. C., Camalli, M., Cascarano, G. L., Giacovazzo, C., Guagliardi, A., Moliterni, A. G. G., Polidori, G. & Spagna, R. (1999). J. Appl. Cryst. 32, 115–119.
- Brandes, R. & Roth, H. (1967). Arch. Pharm. 300, 1005–1007. [DOI] [PubMed]
- Dräger, M. & Gattow, G. (1971). Acta Chem. Scand. 25, 761–762.
- Enraf–Nonius (1989). CAD-4 Software Enraf–Nonius, Delft, The Netherlands.
- Pupo, A., Uberti, M. & Minneman, K. (2003). Eur. J. Pharmacol. 462, 1–8. [DOI] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
- Vogel, A. I., Tatchell, A. R., Furnis, B. S., Hannaford, A. J. & Smith, P. G. W. (1978). Textbook of Practical Organic Chemistry including Qualitative Organic Analysis, 4th ed., p. 773. New York: Longman Group Inc.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536811041985/bt5672sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536811041985/bt5672Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536811041985/bt5672Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report