

Abstract
In the title compound, C16H13ClO3, the dihedral angle between the benzene rings is 80.74 (8)°. In the crystal, C—H⋯O hydrogen bonds link the molecules to form C(11) chains propagating in [010].
Related literature
For a related structure and background references to phenacyl benzoates, see: Fun et al. (2011 ▶).
Experimental
Crystal data
C16H13ClO3
M r = 288.71
Monoclinic,
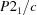
a = 5.9132 (4) Å
b = 8.5044 (6) Å
c = 27.8767 (18) Å
β = 95.880 (1)°
V = 1394.49 (16) Å3
Z = 4
Mo Kα radiation
μ = 0.28 mm−1
T = 297 K
0.51 × 0.30 × 0.06 mm
Data collection
Bruker SMART APEXII DUO CCD diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2009 ▶) T min = 0.871, T max = 0.983
21275 measured reflections
4070 independent reflections
2628 reflections with I > 2σ(I)
R int = 0.027
Refinement
R[F 2 > 2σ(F 2)] = 0.046
wR(F 2) = 0.144
S = 1.05
4070 reflections
182 parameters
H-atom parameters constrained
Δρmax = 0.20 e Å−3
Δρmin = −0.42 e Å−3
Data collection: APEX2 (Bruker, 2009 ▶); cell refinement: SAINT (Bruker, 2009 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXTL (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXTL; molecular graphics: SHELXTL; software used to prepare material for publication: SHELXTL, PLATON (Spek, 2009 ▶).
Supplementary Material
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S1600536811042851/hb6454sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536811042851/hb6454Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536811042851/hb6454Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C16—H16C⋯O1i | 0.96 | 2.46 | 3.383 (2) | 162 |
Symmetry code: (i)  .
.
Acknowledgments
HKF and WSL thank Universiti Sains Malaysia (USM) for the Research University Grant (No. 1001/PFIZIK/811160). WSL also thanks the Malaysian government and USM for the award of the post of Research Officer under the Research University Grant (No. 1001/PFIZIK/811160). AMI thanks Professor Sandeep Sanchethi, Director of the National Institute of Technology-Karnataka, India for providing the research facilities. AMI also thanks the Board for Research in Nuclear Sciences, Department of Atomic Energy, Government of India for the ‘Young Scientist’ award. MNS thanks the Department of Information Technology, Government of India for financial support.
supplementary crystallographic information
Comment
As part of our ongoing studies of phenacyl benzoates (Fun et al., 2011), we now report the synthesis and sturcture of the title compound, (I).
In the title compound (Fig. 1), the dihedral angle formed between the chloro-substituted (C1–C6) and the methyl-substituted (C10–C15) benzene rings is 80.74 (8)°. Bond lengths and angles are within the normal ranges and are comparable to a related structure (Fun et al., 2011).
In the crystal (Fig. 2), intermolecular C16—H16C···O1 hydrogen bonds (Table 1) link the molecules to form chains along the b axis.
Experimental
A mixture of 4-methylbenzoic acid (1.0 g, 0.0073 mol), potassium carbonate (1.10 g, 0.0080 mol) and 2-bromo-1-(4-chlorophenyl)ethanone (1.70 g, 0.0073 mol) in dimethylformamide (10 ml) was stirred at room temperature for 2 h. On cooling, colourless needle-shaped crystals of the title compound began to separate out. They were collected by filtration and recrystallized from ethanol to yield colourless plates of (I). Yield: 1.95 g, 92.8%. M. p: 405–406 K.
Refinement
All H atoms were positioned geometrically and refined with a riding model with Uiso(H) = 1.2 or 1.5 Ueq(C) [C–H = 0.93 or 0.97 Å]. A rotating group model was applied to the methyl group.
Figures
Fig. 1.

The molecular structure of the title compound, showing 30% probability displacement ellipsoids.
Fig. 2.

The crystal packing of the title compound, viewed along the showing the a axis. H atoms not involved in the intermolecular interactions (dashed lines) have been omitted for clarity.
Crystal data
| C16H13ClO3 | F(000) = 600 |
| Mr = 288.71 | Dx = 1.375 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2ybc | Cell parameters from 4428 reflections |
| a = 5.9132 (4) Å | θ = 2.8–28.1° |
| b = 8.5044 (6) Å | µ = 0.28 mm−1 |
| c = 27.8767 (18) Å | T = 297 K |
| β = 95.880 (1)° | Plate, colourless |
| V = 1394.49 (16) Å3 | 0.51 × 0.30 × 0.06 mm |
| Z = 4 |
Data collection
| Bruker SMART APEXII DUO CCD diffractometer | 4070 independent reflections |
| Radiation source: fine-focus sealed tube | 2628 reflections with I > 2σ(I) |
| graphite | Rint = 0.027 |
| φ and ω scans | θmax = 30.1°, θmin = 1.5° |
| Absorption correction: multi-scan (SADABS; Bruker, 2009) | h = −8→8 |
| Tmin = 0.871, Tmax = 0.983 | k = −11→12 |
| 21275 measured reflections | l = −39→39 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.046 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.144 | H-atom parameters constrained |
| S = 1.05 | w = 1/[σ2(Fo2) + (0.0587P)2 + 0.3124P] where P = (Fo2 + 2Fc2)/3 |
| 4070 reflections | (Δ/σ)max = 0.002 |
| 182 parameters | Δρmax = 0.20 e Å−3 |
| 0 restraints | Δρmin = −0.42 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| Cl1 | 1.34410 (11) | 0.75388 (9) | 1.047675 (19) | 0.0935 (2) | |
| O1 | 0.5306 (2) | 0.69016 (17) | 0.86404 (5) | 0.0739 (4) | |
| O2 | 0.6246 (2) | 0.50027 (17) | 0.79276 (4) | 0.0612 (3) | |
| O3 | 0.3791 (2) | 0.35541 (15) | 0.83027 (4) | 0.0643 (3) | |
| C1 | 0.8059 (3) | 0.7539 (2) | 0.94970 (7) | 0.0569 (4) | |
| H1A | 0.6646 | 0.8027 | 0.9453 | 0.068* | |
| C2 | 0.9496 (4) | 0.7851 (2) | 0.99053 (7) | 0.0646 (5) | |
| H2A | 0.9056 | 0.8539 | 1.0138 | 0.078* | |
| C3 | 1.1586 (3) | 0.7138 (2) | 0.99658 (6) | 0.0589 (4) | |
| C4 | 1.2267 (3) | 0.6099 (2) | 0.96302 (6) | 0.0598 (4) | |
| H4A | 1.3684 | 0.5617 | 0.9678 | 0.072* | |
| C5 | 1.0808 (3) | 0.5784 (2) | 0.92215 (6) | 0.0530 (4) | |
| H5A | 1.1248 | 0.5083 | 0.8992 | 0.064* | |
| C6 | 0.8693 (3) | 0.65035 (18) | 0.91501 (5) | 0.0462 (3) | |
| C7 | 0.7093 (3) | 0.62133 (19) | 0.87099 (6) | 0.0487 (4) | |
| C8 | 0.7785 (3) | 0.5032 (2) | 0.83558 (6) | 0.0579 (4) | |
| H8A | 0.9298 | 0.5283 | 0.8273 | 0.070* | |
| H8B | 0.7842 | 0.3997 | 0.8503 | 0.070* | |
| C9 | 0.4256 (3) | 0.42441 (19) | 0.79494 (6) | 0.0484 (4) | |
| C10 | 0.2774 (3) | 0.43770 (17) | 0.74889 (5) | 0.0438 (3) | |
| C11 | 0.3350 (3) | 0.53418 (19) | 0.71169 (6) | 0.0496 (4) | |
| H11A | 0.4730 | 0.5876 | 0.7146 | 0.060* | |
| C12 | 0.1863 (3) | 0.55014 (19) | 0.67042 (6) | 0.0523 (4) | |
| H12A | 0.2264 | 0.6144 | 0.6456 | 0.063* | |
| C13 | −0.0210 (3) | 0.47287 (18) | 0.66499 (5) | 0.0482 (4) | |
| C14 | −0.0734 (3) | 0.3726 (2) | 0.70156 (6) | 0.0531 (4) | |
| H14A | −0.2093 | 0.3166 | 0.6982 | 0.064* | |
| C15 | 0.0749 (3) | 0.3551 (2) | 0.74308 (6) | 0.0505 (4) | |
| H15A | 0.0379 | 0.2873 | 0.7672 | 0.061* | |
| C16 | −0.1878 (3) | 0.5006 (2) | 0.62043 (6) | 0.0604 (4) | |
| H16A | −0.1102 | 0.4892 | 0.5920 | 0.091* | |
| H16B | −0.2492 | 0.6049 | 0.6215 | 0.091* | |
| H16C | −0.3089 | 0.4252 | 0.6197 | 0.091* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Cl1 | 0.0873 (4) | 0.1316 (6) | 0.0578 (3) | −0.0190 (4) | −0.0114 (3) | −0.0225 (3) |
| O1 | 0.0601 (8) | 0.0737 (8) | 0.0831 (9) | 0.0200 (7) | −0.0156 (7) | −0.0148 (7) |
| O2 | 0.0487 (6) | 0.0890 (9) | 0.0452 (6) | −0.0116 (6) | 0.0008 (5) | −0.0060 (6) |
| O3 | 0.0746 (8) | 0.0656 (8) | 0.0513 (7) | −0.0104 (7) | −0.0006 (6) | 0.0087 (6) |
| C1 | 0.0517 (9) | 0.0576 (10) | 0.0617 (10) | 0.0007 (8) | 0.0071 (8) | −0.0088 (8) |
| C2 | 0.0714 (12) | 0.0683 (11) | 0.0555 (10) | −0.0103 (10) | 0.0130 (9) | −0.0163 (8) |
| C3 | 0.0615 (10) | 0.0705 (11) | 0.0438 (8) | −0.0165 (9) | 0.0011 (7) | −0.0034 (8) |
| C4 | 0.0510 (9) | 0.0736 (11) | 0.0526 (9) | 0.0014 (8) | −0.0044 (7) | −0.0015 (8) |
| C5 | 0.0518 (9) | 0.0582 (9) | 0.0480 (8) | 0.0033 (7) | 0.0011 (7) | −0.0065 (7) |
| C6 | 0.0466 (8) | 0.0462 (8) | 0.0457 (8) | −0.0043 (6) | 0.0039 (6) | −0.0001 (6) |
| C7 | 0.0452 (8) | 0.0482 (8) | 0.0518 (8) | −0.0021 (7) | −0.0002 (7) | 0.0017 (7) |
| C8 | 0.0470 (9) | 0.0747 (11) | 0.0503 (9) | 0.0005 (8) | −0.0042 (7) | −0.0102 (8) |
| C9 | 0.0497 (8) | 0.0485 (8) | 0.0469 (8) | 0.0006 (7) | 0.0046 (7) | −0.0075 (7) |
| C10 | 0.0455 (8) | 0.0443 (7) | 0.0418 (7) | −0.0007 (6) | 0.0059 (6) | −0.0054 (6) |
| C11 | 0.0483 (8) | 0.0488 (8) | 0.0522 (8) | −0.0082 (7) | 0.0071 (7) | 0.0001 (7) |
| C12 | 0.0617 (10) | 0.0492 (8) | 0.0467 (8) | −0.0037 (7) | 0.0081 (7) | 0.0046 (7) |
| C13 | 0.0532 (9) | 0.0478 (8) | 0.0431 (8) | 0.0039 (7) | 0.0030 (6) | −0.0083 (6) |
| C14 | 0.0483 (8) | 0.0598 (10) | 0.0508 (8) | −0.0109 (7) | 0.0040 (7) | −0.0052 (7) |
| C15 | 0.0518 (9) | 0.0555 (9) | 0.0449 (8) | −0.0097 (7) | 0.0082 (7) | 0.0004 (7) |
| C16 | 0.0638 (11) | 0.0631 (10) | 0.0524 (9) | 0.0048 (9) | −0.0030 (8) | −0.0047 (8) |
Geometric parameters (Å, °)
| Cl1—C3 | 1.7400 (17) | C8—H8A | 0.9700 |
| O1—C7 | 1.2062 (19) | C8—H8B | 0.9700 |
| O2—C9 | 1.349 (2) | C9—C10 | 1.483 (2) |
| O2—C8 | 1.4250 (18) | C10—C15 | 1.383 (2) |
| O3—C9 | 1.203 (2) | C10—C11 | 1.392 (2) |
| C1—C2 | 1.375 (2) | C11—C12 | 1.381 (2) |
| C1—C6 | 1.387 (2) | C11—H11A | 0.9300 |
| C1—H1A | 0.9300 | C12—C13 | 1.385 (2) |
| C2—C3 | 1.371 (3) | C12—H12A | 0.9300 |
| C2—H2A | 0.9300 | C13—C14 | 1.388 (2) |
| C3—C4 | 1.377 (3) | C13—C16 | 1.523 (2) |
| C4—C5 | 1.383 (2) | C14—C15 | 1.387 (2) |
| C4—H4A | 0.9300 | C14—H14A | 0.9300 |
| C5—C6 | 1.388 (2) | C15—H15A | 0.9300 |
| C5—H5A | 0.9300 | C16—H16A | 0.9600 |
| C6—C7 | 1.491 (2) | C16—H16B | 0.9600 |
| C7—C8 | 1.495 (2) | C16—H16C | 0.9600 |
| C9—O2—C8 | 117.12 (13) | O3—C9—O2 | 122.98 (15) |
| C2—C1—C6 | 120.75 (17) | O3—C9—C10 | 125.56 (15) |
| C2—C1—H1A | 119.6 | O2—C9—C10 | 111.46 (14) |
| C6—C1—H1A | 119.6 | C15—C10—C11 | 119.15 (14) |
| C3—C2—C1 | 119.28 (17) | C15—C10—C9 | 119.38 (14) |
| C3—C2—H2A | 120.4 | C11—C10—C9 | 121.45 (14) |
| C1—C2—H2A | 120.4 | C12—C11—C10 | 119.66 (15) |
| C2—C3—C4 | 121.52 (16) | C12—C11—H11A | 120.2 |
| C2—C3—Cl1 | 119.91 (15) | C10—C11—H11A | 120.2 |
| C4—C3—Cl1 | 118.57 (15) | C11—C12—C13 | 121.67 (15) |
| C3—C4—C5 | 118.90 (17) | C11—C12—H12A | 119.2 |
| C3—C4—H4A | 120.5 | C13—C12—H12A | 119.2 |
| C5—C4—H4A | 120.5 | C12—C13—C14 | 118.22 (14) |
| C4—C5—C6 | 120.60 (16) | C12—C13—C16 | 120.48 (15) |
| C4—C5—H5A | 119.7 | C14—C13—C16 | 121.28 (15) |
| C6—C5—H5A | 119.7 | C15—C14—C13 | 120.59 (15) |
| C1—C6—C5 | 118.94 (15) | C15—C14—H14A | 119.7 |
| C1—C6—C7 | 118.97 (15) | C13—C14—H14A | 119.7 |
| C5—C6—C7 | 122.08 (14) | C10—C15—C14 | 120.61 (15) |
| O1—C7—C6 | 121.57 (15) | C10—C15—H15A | 119.7 |
| O1—C7—C8 | 120.98 (15) | C14—C15—H15A | 119.7 |
| C6—C7—C8 | 117.46 (13) | C13—C16—H16A | 109.5 |
| O2—C8—C7 | 111.72 (14) | C13—C16—H16B | 109.5 |
| O2—C8—H8A | 109.3 | H16A—C16—H16B | 109.5 |
| C7—C8—H8A | 109.3 | C13—C16—H16C | 109.5 |
| O2—C8—H8B | 109.3 | H16A—C16—H16C | 109.5 |
| C7—C8—H8B | 109.3 | H16B—C16—H16C | 109.5 |
| H8A—C8—H8B | 107.9 | ||
| C6—C1—C2—C3 | 0.6 (3) | C8—O2—C9—O3 | −3.1 (2) |
| C1—C2—C3—C4 | −0.9 (3) | C8—O2—C9—C10 | 176.99 (14) |
| C1—C2—C3—Cl1 | 179.20 (14) | O3—C9—C10—C15 | −5.7 (2) |
| C2—C3—C4—C5 | 0.6 (3) | O2—C9—C10—C15 | 174.20 (14) |
| Cl1—C3—C4—C5 | −179.55 (14) | O3—C9—C10—C11 | 172.90 (16) |
| C3—C4—C5—C6 | 0.1 (3) | O2—C9—C10—C11 | −7.2 (2) |
| C2—C1—C6—C5 | 0.0 (3) | C15—C10—C11—C12 | 2.2 (2) |
| C2—C1—C6—C7 | −179.28 (16) | C9—C10—C11—C12 | −176.38 (15) |
| C4—C5—C6—C1 | −0.4 (3) | C10—C11—C12—C13 | 0.3 (3) |
| C4—C5—C6—C7 | 178.91 (16) | C11—C12—C13—C14 | −2.6 (2) |
| C1—C6—C7—O1 | 2.9 (3) | C11—C12—C13—C16 | 176.20 (15) |
| C5—C6—C7—O1 | −176.38 (18) | C12—C13—C14—C15 | 2.3 (2) |
| C1—C6—C7—C8 | −177.07 (16) | C16—C13—C14—C15 | −176.49 (15) |
| C5—C6—C7—C8 | 3.6 (2) | C11—C10—C15—C14 | −2.5 (2) |
| C9—O2—C8—C7 | −77.7 (2) | C9—C10—C15—C14 | 176.13 (15) |
| O1—C7—C8—O2 | 7.0 (3) | C13—C14—C15—C10 | 0.2 (3) |
| C6—C7—C8—O2 | −173.03 (14) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C16—H16C···O1i | 0.96 | 2.46 | 3.383 (2) | 162 |
Symmetry codes: (i) −x, y−1/2, −z+3/2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: HB6454).
References
- Bruker (2009). APEX2, SAINT and SADABS Bruker AXS Inc., Madison, Wisconsin, USA.
- Fun, H.-K., Loh, W.-S., Garudachari, B., Isloor, A. M. & Satyanarayana, M. N. (2011). Acta Cryst. E67, o2854. [DOI] [PMC free article] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S1600536811042851/hb6454sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536811042851/hb6454Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536811042851/hb6454Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report