

Abstract
In the title compound, C8H8FN3O, the semicarbazide group is close to being planar, with a maximum deviation of 0.020 (1) Å, and subtends a dihedral angle of 16.63 (9)° with its attached fluorobenzene ring. In the crystal, molecules are linked by N—H⋯O hydrogen bonds, forming layers lying parallel to the bc plane.
Related literature
For background to semicarbazides and semicarbazones, see: Dogan et al. (1999 ▶); Pandeya & Dimmock (1993 ▶); Pandeya et al. (1998 ▶); Sriram et al. (2004 ▶); Yogeeswari et al. (2004 ▶); For further synthetic details, see: Furniss et al. (1978 ▶). For related structures, see: Fun et al. (2009a
▶,b
▶). For reference bond lengths, see: Allen et al. (1987 ▶).
Experimental
Crystal data
C8H8FN3O
M r = 181.17
Monoclinic,
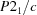
a = 16.522 (2) Å
b = 4.4381 (6) Å
c = 11.9457 (15) Å
β = 103.478 (3)°
V = 851.80 (19) Å3
Z = 4
Mo Kα radiation
μ = 0.11 mm−1
T = 296 K
0.72 × 0.18 × 0.12 mm
Data collection
Bruker APEX DUO CCD diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2009 ▶) T min = 0.923, T max = 0.987
8746 measured reflections
2418 independent reflections
1657 reflections with I > 2σ(I)
R int = 0.024
Refinement
R[F 2 > 2σ(F 2)] = 0.053
wR(F 2) = 0.209
S = 1.00
2418 reflections
130 parameters
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.31 e Å−3
Δρmin = −0.21 e Å−3
Data collection: APEX2 (Bruker, 2009 ▶); cell refinement: SAINT (Bruker, 2009 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXTL (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXTL; molecular graphics: SHELXTL; software used to prepare material for publication: SHELXTL and PLATON (Spek, 2009 ▶).
Supplementary Material
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S1600536811040797/hb6436sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536811040797/hb6436Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536811040797/hb6436Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N3—H1N3⋯O1i | 0.88 (2) | 2.07 (2) | 2.8954 (19) | 158 (2) |
| N2—H1N2⋯O1ii | 0.92 (2) | 2.00 (2) | 2.9155 (19) | 179 (2) |
Symmetry codes: (i) 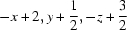 ; (ii)
; (ii) 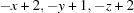 .
.
Acknowledgments
HKF and TSC thank Universiti Sains Malaysia (USM) for the Research University Grant (1001/PFIZIK/811160). TSC thanks the Malaysian Government and USM for the award of the post of Research Officer under the Structure Determination of kDa Outer Membrane Proteins From S. typhi by X-ray Protein Crystallography Grant (No. 1001/PSKBP/8630013). AMI thanks Professor Sandeep Sanchethi, Director, National Institute of Technology-Karnataka, India, for providing research facilities and the Board for Research in Nuclear Sciences, Department of Atomic Energy, Government of India, for the Young Scientist award.
supplementary crystallographic information
Comment
The semicarbazides, which are the raw material of semicarbazones, have been known to possess biological activities against many of the most common species of bacteria (Dogan et al., 1999). Semicarbazones are of much interest due to their wide spectrum of antibacterial activities (Pandeya & Dimmock, 1993). Recently some workers have reviewed the bioactivity of semicarbazones and they have exhibited anticonvulsant (Pandeya et al., 1998; Yogeeswari et al., 2004) and antitubercular (Sriram et al., 2004) properties. Accordingly and by considering the biological potential of semicarbazones, herein, we have synthesized the title compound to study its crystal structure.
The molecular structure of the title compound is shown in Fig. 1. The semicarbazone group (O1/N1–N3/C8) is essentially planar with maximum deviation of 0.020 (1) Å for atom N2. This plane makes dihedral angle of 16.63 (9)° with its terminal benzene ring (C1–C6). Bond lengths (Allen et al., 1987) and angles are within normal ranges and are comparable to related structures (Fun et al., 2009a,b).
In the crystal structure (Fig. 2), the molecules are interconnected by N3—H1N3···O1 and N2—H1N2···O1 hydrogen bonds (Table 1) forming two-dimensional networks parallel to bc plane.
Experimental
Semicarbazide hydrochloride (0.86 g, 7.70 mmol) and freshly recrystallized sodium acetate (0.77 g, 9.40 mmol) were dissolved in water (10 ml) following a literature procedure (Furniss et al., 1978). The reaction mixture was stirred at room temperature for 10 minutes. To this, 4-fluorobenzaldehyde (0.896 g, 7.23 mmol) was added and the mixture was shaken well. A little alcohol was added to dissolve the turbidity. The mixture was shaken for a further 10 minutes and allowed to stand. The title compound crystallizes out on standing for 6 h. The separated crystals were filtered, washed with cold water and recrystallized from ethanol to yield colourless needles. Yield: 0.98 g, 75.38%. M.p.: 506–508 K.
Refinement
Atoms H1N2, H1N3 and H2N3 were located in a difference map and refined freely [N—H = 0.90 (2), 0.87 (2) and 0.91 (2) Å respectively]. The remaining H atoms were positioned geometrically [C—H = 0.93 Å] and refined using a riding model with Uiso(H) = 1.2 Ueq(C).
Figures
Fig. 1.

The molecular structure of the title compound with atom labels with 50% probability displacement ellipsoids.
Fig. 2.

The crystal packing of the title compound. The dashed lines represent the hydrogen bonds.
Crystal data
| C8H8FN3O | F(000) = 376 |
| Mr = 181.17 | Dx = 1.413 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2ybc | Cell parameters from 2666 reflections |
| a = 16.522 (2) Å | θ = 2.5–29.4° |
| b = 4.4381 (6) Å | µ = 0.11 mm−1 |
| c = 11.9457 (15) Å | T = 296 K |
| β = 103.478 (3)° | Needle, colourless |
| V = 851.80 (19) Å3 | 0.72 × 0.18 × 0.12 mm |
| Z = 4 |
Data collection
| Bruker APEX DUO CCD diffractometer | 2418 independent reflections |
| Radiation source: fine-focus sealed tube | 1657 reflections with I > 2σ(I) |
| graphite | Rint = 0.024 |
| φ and ω scans | θmax = 29.9°, θmin = 1.3° |
| Absorption correction: multi-scan (SADABS; Bruker, 2009) | h = −23→23 |
| Tmin = 0.923, Tmax = 0.987 | k = −6→6 |
| 8746 measured reflections | l = −16→15 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.053 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.209 | H atoms treated by a mixture of independent and constrained refinement |
| S = 1.00 | w = 1/[σ2(Fo2) + (0.1446P)2 + 0.0418P] where P = (Fo2 + 2Fc2)/3 |
| 2418 reflections | (Δ/σ)max < 0.001 |
| 130 parameters | Δρmax = 0.31 e Å−3 |
| 0 restraints | Δρmin = −0.21 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| F1 | 0.51936 (9) | 1.2636 (4) | 1.15848 (18) | 0.1170 (6) | |
| O1 | 1.01013 (7) | 0.7059 (3) | 0.87156 (9) | 0.0508 (3) | |
| N1 | 0.83497 (7) | 0.8821 (3) | 0.97516 (10) | 0.0448 (3) | |
| N2 | 0.90494 (8) | 0.7354 (3) | 0.96093 (11) | 0.0470 (3) | |
| N3 | 0.91237 (8) | 1.0690 (3) | 0.81619 (11) | 0.0493 (4) | |
| C1 | 0.71134 (11) | 0.8578 (5) | 1.18840 (15) | 0.0637 (5) | |
| H1A | 0.7461 | 0.7333 | 1.2413 | 0.076* | |
| C2 | 0.63885 (11) | 0.9701 (6) | 1.21288 (16) | 0.0705 (5) | |
| H2A | 0.6244 | 0.9217 | 1.2814 | 0.085* | |
| C3 | 0.58977 (12) | 1.1518 (5) | 1.1342 (2) | 0.0740 (6) | |
| C4 | 0.60851 (13) | 1.2285 (6) | 1.0325 (2) | 0.0855 (7) | |
| H4A | 0.5735 | 1.3543 | 0.9805 | 0.103* | |
| C5 | 0.68021 (11) | 1.1161 (5) | 1.00849 (17) | 0.0669 (5) | |
| H5A | 0.6937 | 1.1662 | 0.9394 | 0.080* | |
| C6 | 0.73235 (9) | 0.9300 (4) | 1.08559 (13) | 0.0493 (4) | |
| C7 | 0.80796 (9) | 0.8014 (4) | 1.06141 (13) | 0.0504 (4) | |
| H7A | 0.8371 | 0.6560 | 1.1109 | 0.060* | |
| C8 | 0.94581 (8) | 0.8362 (3) | 0.88136 (11) | 0.0405 (3) | |
| H1N3 | 0.9423 (12) | 1.145 (5) | 0.7725 (18) | 0.064 (5)* | |
| H1N2 | 0.9306 (12) | 0.599 (5) | 1.0133 (16) | 0.061 (5)* | |
| H2N3 | 0.8717 (14) | 1.181 (5) | 0.8357 (19) | 0.074 (6)* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| F1 | 0.0854 (10) | 0.1393 (14) | 0.1524 (15) | 0.0374 (9) | 0.0807 (10) | 0.0080 (10) |
| O1 | 0.0544 (6) | 0.0586 (7) | 0.0499 (6) | 0.0023 (4) | 0.0334 (5) | −0.0022 (4) |
| N1 | 0.0447 (6) | 0.0544 (7) | 0.0417 (6) | 0.0019 (5) | 0.0228 (5) | −0.0006 (5) |
| N2 | 0.0490 (7) | 0.0566 (7) | 0.0446 (7) | 0.0073 (5) | 0.0296 (5) | 0.0048 (5) |
| N3 | 0.0569 (7) | 0.0541 (7) | 0.0457 (7) | −0.0004 (5) | 0.0299 (6) | 0.0032 (5) |
| C1 | 0.0563 (9) | 0.0936 (13) | 0.0502 (9) | 0.0081 (8) | 0.0304 (7) | 0.0076 (8) |
| C2 | 0.0662 (10) | 0.0974 (15) | 0.0620 (10) | −0.0021 (9) | 0.0438 (9) | −0.0068 (10) |
| C3 | 0.0558 (10) | 0.0863 (14) | 0.0938 (15) | 0.0098 (8) | 0.0457 (10) | −0.0057 (11) |
| C4 | 0.0695 (12) | 0.1019 (16) | 0.0965 (17) | 0.0321 (11) | 0.0423 (12) | 0.0232 (13) |
| C5 | 0.0631 (10) | 0.0820 (12) | 0.0656 (11) | 0.0196 (8) | 0.0353 (8) | 0.0179 (9) |
| C6 | 0.0457 (7) | 0.0643 (9) | 0.0448 (7) | 0.0019 (6) | 0.0244 (6) | −0.0001 (6) |
| C7 | 0.0482 (8) | 0.0673 (9) | 0.0425 (8) | 0.0090 (6) | 0.0245 (6) | 0.0085 (6) |
| C8 | 0.0461 (7) | 0.0452 (7) | 0.0361 (6) | −0.0067 (5) | 0.0215 (5) | −0.0085 (5) |
Geometric parameters (Å, °)
| F1—C3 | 1.3568 (19) | C1—H1A | 0.9300 |
| O1—C8 | 1.2393 (16) | C2—C3 | 1.355 (3) |
| N1—C7 | 1.2661 (18) | C2—H2A | 0.9300 |
| N1—N2 | 1.3714 (16) | C3—C4 | 1.365 (3) |
| N2—C8 | 1.3634 (17) | C4—C5 | 1.376 (2) |
| N2—H1N2 | 0.90 (2) | C4—H4A | 0.9300 |
| N3—C8 | 1.3333 (18) | C5—C6 | 1.379 (2) |
| N3—H1N3 | 0.87 (2) | C5—H5A | 0.9300 |
| N3—H2N3 | 0.91 (2) | C6—C7 | 1.4621 (19) |
| C1—C2 | 1.390 (2) | C7—H7A | 0.9300 |
| C1—C6 | 1.389 (2) | ||
| C7—N1—N2 | 115.71 (12) | C3—C4—C5 | 118.7 (2) |
| C8—N2—N1 | 119.96 (12) | C3—C4—H4A | 120.6 |
| C8—N2—H1N2 | 118.4 (12) | C5—C4—H4A | 120.6 |
| N1—N2—H1N2 | 120.3 (12) | C4—C5—C6 | 120.79 (17) |
| C8—N3—H1N3 | 115.9 (13) | C4—C5—H5A | 119.6 |
| C8—N3—H2N3 | 120.3 (14) | C6—C5—H5A | 119.6 |
| H1N3—N3—H2N3 | 120 (2) | C5—C6—C1 | 118.85 (14) |
| C2—C1—C6 | 120.56 (17) | C5—C6—C7 | 122.08 (14) |
| C2—C1—H1A | 119.7 | C1—C6—C7 | 119.06 (15) |
| C6—C1—H1A | 119.7 | N1—C7—C6 | 121.99 (14) |
| C3—C2—C1 | 118.24 (16) | N1—C7—H7A | 119.0 |
| C3—C2—H2A | 120.9 | C6—C7—H7A | 119.0 |
| C1—C2—H2A | 120.9 | O1—C8—N3 | 123.66 (12) |
| F1—C3—C2 | 118.27 (19) | O1—C8—N2 | 119.18 (13) |
| F1—C3—C4 | 118.9 (2) | N3—C8—N2 | 117.15 (12) |
| C2—C3—C4 | 122.86 (16) | ||
| C7—N1—N2—C8 | −170.19 (13) | C4—C5—C6—C7 | 178.57 (19) |
| C6—C1—C2—C3 | −0.3 (3) | C2—C1—C6—C5 | 0.3 (3) |
| C1—C2—C3—F1 | −179.4 (2) | C2—C1—C6—C7 | −178.40 (17) |
| C1—C2—C3—C4 | 0.0 (4) | N2—N1—C7—C6 | −177.87 (13) |
| F1—C3—C4—C5 | 179.6 (2) | C5—C6—C7—N1 | 8.7 (3) |
| C2—C3—C4—C5 | 0.2 (4) | C1—C6—C7—N1 | −172.65 (16) |
| C3—C4—C5—C6 | −0.1 (4) | N1—N2—C8—O1 | 178.19 (12) |
| C4—C5—C6—C1 | −0.1 (3) | N1—N2—C8—N3 | −3.3 (2) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N3—H1N3···O1i | 0.88 (2) | 2.07 (2) | 2.8954 (19) | 158 (2) |
| N2—H1N2···O1ii | 0.92 (2) | 2.00 (2) | 2.9155 (19) | 179 (2) |
Symmetry codes: (i) −x+2, y+1/2, −z+3/2; (ii) −x+2, −y+1, −z+2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: HB6436).
References
- Allen, F. H., Kennard, O., Watson, D. G., Brammer, L., Orpen, A. G. & Taylor, R. (1987). J. Chem. Soc. Perkin Trans. 2, pp. S1–19.
- Bruker (2009). SADABS, APEX2 and SAINT Bruker AXS Inc., Madison, Wisconsin, USA.
- Dogan, H. N., Duran, A. & Yemni, E. (1999). Drug Metab. Drug Interact. 15, 187–195. [DOI] [PubMed]
- Fun, H.-K., Goh, J. H., Padaki, M., Malladi, S. & Isloor, A. M. (2009a). Acta Cryst. E65, o1591–o1592. [DOI] [PMC free article] [PubMed]
- Fun, H.-K., Yeap, C. S., Padaki, M., Malladi, S. & Isloor, A. M. (2009b). Acta Cryst. E65, o1619–o1620. [DOI] [PMC free article] [PubMed]
- Furniss, B. S., Hannaford, A. J., Rogers, V., Smith, P. W. G. & Tatchell, A. R. (1978). Vogel’s Textbook of Practical Organic Chemistry, 4th ed., p. 1112. London: ELBS.
- Pandeya, S. N. & Dimmock, J. R. (1993). Pharmazie, 48, 659–666. [PubMed]
- Pandeya, S. N., Misra, V., Singh, P. N. & Rupainwar, D. C. (1998). Pharmacology, 37, 17–22. [DOI] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
- Sriram, D., Yogeeswari, P. & Thirumurugan, R. S. (2004). Bioorg. Med. Chem. Lett. 14, 3923–3924. [DOI] [PubMed]
- Yogeeswari, P., Sriram, D., Pandeya, S. N. & Stables, J. P. (2004). Farmaco, 59, 609–613. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S1600536811040797/hb6436sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536811040797/hb6436Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536811040797/hb6436Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report