

Abstract
Occlusive transplant vasculopathy (TV) is the major cause for chronic graft rejection. Since endothelial cells (EC) are the first graft cells encountered by activated host lymphocytes, it is important to delineate the molecular mechanisms that coordinate the interaction of EC with activated T cells. Here, the interaction of CD8+ T cells with Ag-presenting EC in vivo was examined using a transgenic heart transplantation model with β-galactosidase (β-gal) expression exclusively in EC (Tie2-LacZ hearts). We found that priming with β-gal peptide-loaded DC failed to generate a strong systemic IFN-γ response, but elicited pronounced TV in both IFN-γ receptor (IFNGR)-competent, and ifngr−/− Tie2-LacZ hearts. In contrast, stimulation of EC-specific CD8+ T cells with β-gal-recombinant mouse cytomegalovirus (MCMV-LacZ) in recipients of ifngr+/+ Tie2-LacZ hearts did not precipitate significant TV. However, MCMV-LacZ infection of recipients of ifngr−/− Tie2-LacZ hearts led to massive activation of β-gal-specific CD8 T cells, and led to development of fulminant TV. Further analyses revealed that the strong systemic IFN-γ “storm” associated with MCMV infection induced upregulation of programmed death-1 ligand 1 (PD-L1) on EC, and subsequent attenuation of programmed death-1 (PD-1)-expressing EC-specific CD8+ T cells. Thus, IFNGR signaling in ECs activates a potent peripheral negative feedback circuit that protects vascularized grafts from occlusive TV.
Keywords: Chronic rejection, CTL, IFN-γ-receptor, PD-L1, Transplantation, Vascular endothelial cells
Introduction
Despite advances in immunosuppressive therapies for acute allograft rejection, successful long-term outcome of transplanted solid organs is still hampered by late graft failure. Chronic graft rejection is caused, in large part, by host-anti-graft immune responses that target cells of the graft vasculature and precipitate transplant vasculopathy (TV) [1]. TV is the main cause of long-term allograft dysfunction and late graft loss, e.g. in heart [2] and kidney transplantation [3]. The most prominent feature of TV is the remodeling of graft vessels caused by neointima formation and progressive luminal narrowing [4]. Since the progressive TV-associated narrowing of vascular lumen affects mainly conduit arteries, this disease entity can also be referred to as “graft arteriosclerosis” [5]. Endothelial cells (EC) are the first graft cells encountered by the host immune system and it is thus most likely that a complex series of immune-mediated EC injuries initiate and drive the process of chronic vascular rejection [6]. Furthermore, ECs are preserved in long-term allografts [7] and are thus able to act long-term as targets of anti-graft immune responses. It is therefore important to study the initial EC injury mechanistically and to dissect the molecular and cellular interactions leading to the precipitation of TV.
Early studies of Russell and colleagues indicated that cardiac TV in a MHC-disparate setting is largely driven by T cells [8], while humoral alloresponses may augment the chronic inflammatory process [9, 10]. IFN-γ appears to be a particularly important immune effector molecule in the pathogenesis of TV [5]. This notion is not only supported by the fact that IFN-γ transcription in endomyocardial biopsies of human heart grafts is upregulated prior to the development of TV [11], but also by several experimental studies showing that IFN-γ is instrumental for the development of TV [5, 12, 13]. Furthermore, IFN-γ has been implicated in the chronic arterial lumen narrowing in genetically hyperlipidemic mice [14, 15]. IFN-γ is mainly produced by activated CD4+ T cells, CD8+ T cells, and NK cells [16]. It leads to the induction of MHC class II expression and up-regulation of MHC class I molecules on vascular EC, hence potentially enhancing their ability to present Ag [5]. A pronounced direct pro-arteriosclerotic effect of IFN-γ exposure on the human vasculature has been demonstrated in a study using human arteries transplanted into T-cell-deficient mice [17]. However, in addition to its clear pro-inflammatory potential, IFN-γ was also found to limit immune responses, e.g. in the regulation of CD8+ T-cell expansion and contraction during bacterial infection [18], in the development of experimental autoimmune encephalomyelitis [19], or in graft-versus-host disease [20, 21].
The major aim of the present study was to determine the role of IFN-γ receptor (IFNGR) signaling in ECs during CD8+ T-cell-mediated vascular rejection. To this end, we employed a transgenic mouse model with EC-specific β-galactosidase (β-gal) expression under the control of the Tie2 promoter (Tie2-LacZ mice) [22]. We have shown that β-gal expressing EC in heterotopically transplanted Tie2-LacZ hearts or orthotopically transplanted Tie2-LacZ livers remain immunologically ignored by naive CTL, even when high numbers of β-gal-specific T-cell receptor transgenic CD8+ T cells are present [23]. To address the effect of IFNGR signaling, ifngr−/−, or ifngr+/+ Tie2-LacZ hearts were transplanted into naïve recipients and EC-specific CD8+ effector T cells were induced by priming with β-gal peptide-pulsed DC or infection with β-galrecombinant mouse cytomegalovirus (MCMV-LacZ). Repetitive priming with β-gal peptide-pulsed DC elicited severe vascular inflammation in transplanted Tie2-LacZ hearts with neointima formation and vascular occlusion, whereas activation of EC-specific CD8+ T cells during infection with MCMV-LacZ caused only mild vascular inflammation. Importantly, transplant arteriosclerosis in ifngr−/− Tie2-LacZ hearts was aggravated, particularly when EC-specific CD8+ T cells were stimulated through MCMV-LacZ infection. Disruption of the IFN-γ-driven cross-talk between endothelial programmed death-1 ligand 1 (PD-L1) and CD8+ T-cell-expressed PD-1 by Ab blockade of PD-L1, or adoptive transfer of PD-1-deficient, EC-specific TCR transgenic CD8+ T cells, respectively, ameliorated TV. This shows that the IFNGR expressed on EC exerts a potent negative feedback on EC-specific CD8+ T cells. Furthermore, this study indicates that important systemic pro-inflammatory stimuli can be translated locally into anti-inflammatory signals for the benefit of the grafted organ.
Results
DC-induced TV
Tie2-LacZ organs can be transplanted into naive C57BL/6 recipients and the EC-associated minor histocompatibility Ag (mhAg) remains immunologically ignored. Transplanted Tie2-LacZ hearts remained fully functional, did not show signs of spontaneous rejection, and, most importantly, retained β-gal-expression in coronary arteries [23]. CD8+ T-cell reactivity is not affected by the presence of the β-gal mhAg in EC. This is shown by the fact that recipients of C57BL/6 (B6→B6) and Tie2-LacZ hearts (T2→B6) displayed comparable levels of β-gal-specific CD8+ T cells both in blood and spleens following priming with DC pulsed with the immunodominant, H2-Kb-binding β-gal497 peptide (Fig. 1).
Figure 1.
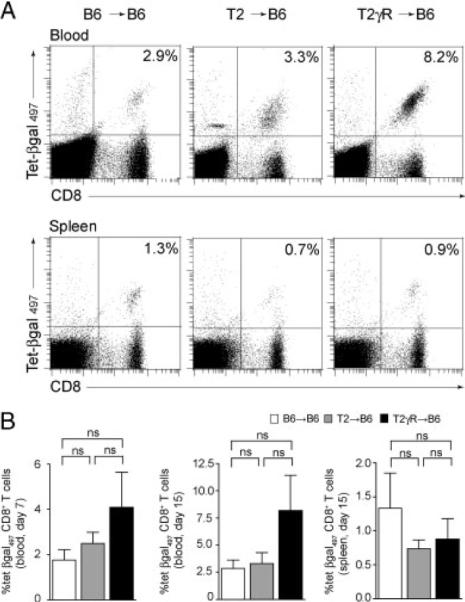
Flow cytometric analysis of CD8+ T cells responses in B6 recipients of B6, T2, or T2γR hearts following immunization with β-gal peptide-pulsed DC. (A) Representative dot-plot histograms showing H2-Kb/β-gal497 tetramer staining of CD8+ T cells from blood (upper row) and spleen (lower row) on day 15. Values in the upper right panel indicate percent H2-Kb/β-gal497 tetramer-positive cells in the CD8+ T-cell compartment. (B) Mean percentage of H2-Kb/β-gal497 tetramer-positive cells in the CD8 compartment from blood on days 7 and 15, and spleens on day 15 post immunization. Values indicate percent H2-Kb/β-gal497 tetramer-positive cells in the CD8+ T-cell compartment+SEM. Pooled data from four independent experiments (blood day 7: B6→B6, n = 9; T2→B6, n = 20; T2γR→B6, n = 9; blood day 15: B6→B6, n = 8; T2→B6, n = 14; T2γR→B6, n = 5; spleen day 15: B6→B6, n = 8, T2→B6, n = 14; T2γR→B6, n = 5). Statistical analysis was performed using one-way ANOVA with Bonferroni post test (ns, not significant).
Based on our previous experience with DC-based immunization against tumors [24, 25] and the requirement of prolonged DC-mediated Ag presentation to precipitate autoimmune disease [26, 27], immunization of transplant recipients with β-gal497 peptide-pulsed DC was performed repeatedly, and hearts were evaluated for signs of vascular rejection on day 15 following the first DC application. Histological analysis revealed typical signs of chronic vascular rejection including intimal thickening and vascular occlusion. Moreover, perivascular infiltration with inflammatory cells (mainly CD8+ T cells), and perivascular fibrosis was observed only in T2→B6 hearts (Fig. 2A), but not in B6→B6 hearts (Fig. 2A). Using a scoring system based on criteria published by Russell et al. [12] and by Hirozane et al. [28], we found that T2→B6 hearts from DC-immunized recipients showed significantly more perivascular infiltration, intimal thickening, and perivascular fibrosis compared to B6→B6 hearts (Fig. 2B). Thus, once β-gal-specific CD8+ T cells were induced in T2→B6 recipients, EC presenting the mhAg became target cells for the activated CD8+T cells and the process of TV development was initiated.
Figure 2.

Transplant vasculopathy induced by DC immunization. (A) Representative sections of transplanted B6→B6, T2→B6, and T2γR→B6 hearts on day 15 after DC immunization (H&E, EVG, and immunostaining for CD8). Original magnification ×400; scale bar, 100 μm. (B) Detailed analysis of immunopathological vascular alterations on day 15 post immunization. Individual scores for perivascular infiltration, intimal thickening, and perivascular fibrosis were determined, and the cumulative rejection score was calculated as the sum of the three individual parameters. Pooled data from four independent experiments (mean+SEM; B6→B6, n = 10; T2→B6, n = 18; T2γR→B6, n = 5). Statistical analysis was performed using one-way ANOVA with Bonferroni post test (***, p<0.001; **, p<0.01; ns, not significant).
Next, we assessed the role of IFNGR signaling in DC-induced TV. To this end, Tie2-LacZ hearts lacking the IFN-γ-receptor (T2γR) were transplanted into C57BL/6 recipients (T2γR→B6), and mice were challenged with β-gal497 peptide-loaded DC as above. In comparison to T2→B6 hearts, T2γR→B6 hearts displayed slightly more perivascular infiltration, intimal thickening, and perivascular fibrosis (Fig. 2A and B). In accordance with the mild aggravation of TV due to the lack of the IFNGR in T2γR hearts, we observed mildly increased levels of β-gal-specific CD8+ T cells in the blood of T2γR→B6 recipients on days 7 and 15 (Fig. 1A and B). These data suggested that IFN-γ might to some extent shield the graft from CD8+ T-cell-mediated attack. Since treatment with recombinant IFN-γ can alter the course of allograft rejection [29, 30], we transplanted T2 hearts into B6 recipients and treated these mice with recombinant IFN-γ. To ensure comparable initial priming of CD8+ T cells, IFN-γ was applied daily from days 6–12. Indeed, this treatment did not impact on the generation of DC-induced CD8+ T cells on days 7 or 12. However, the ensuing relative decrease in βgal-specific CD8+ T cells on day 15 in IFN-γ-treated T2 heart recipients indicates that chronic systemic application of rIFN-γ can down-tune CD8+ T-cell responses (Supporting Information Fig. 1A). Nevertheless, the altered CD8+ T-cell response did not result in an attenuation of TV (Supporting Information Fig. 1B) suggesting that systemic application of IFN-γ is not suitable to examine the role of the IFNGR in this experimental system. The lack of a protective effect of daily application of 9 μg rIFN-γ in the DC-mediated vascular rejection of Tie2-LacZ hearts (Supporting Information Fig. 1) might reflect particular–yet unresolved–dose-requirements in this setting.
Transplant arteriosclerosis after MCMV-LacZ infection
Viral infections generally activate CD8+ T cells to produce high amounts of IFN-γ [31]. We, therefore, used infection with MCMV-LacZ [32] to elicit a fulminant IFN-γ response in addition to CD8+ T-cell-mediated attack of ECs. IFN-γ-levels in virus-infected mice were significantly higher compared to IFN-γ levels in DC-immunized mice (Fig. 3A and B). Bg1 TCR transgenic T cells recognizing the subdominant H2-Kb-binding β-gal96 peptide [23] were used to augment the effect CD8+ T cells on ECs. IFN-γ levels were comparable in MCMV-LacZ infected mice receiving TCR transgenic β-gal-specific Bg1 CD8+ T cells and those without adoptive transfer (Fig. 3A and B).
Figure 3.
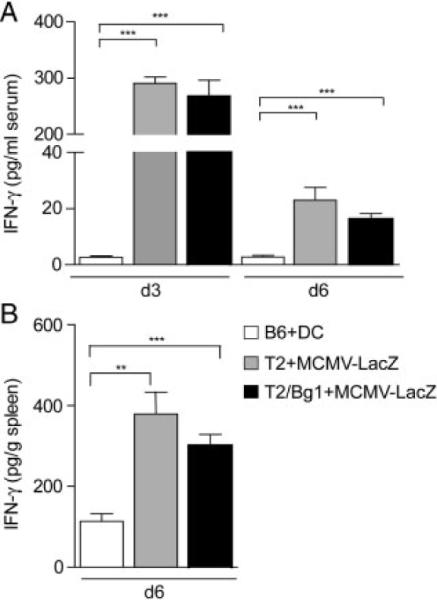
IFNγ-concentration in serum (A) and spleen homogenates (B) of DC-immunized, MCMV-LacZ infected B6, and Bg1 TCR transgenic T-cell-transfused B6 mice. ELISA results are expressed as the mean1 SEM of six mice per group. Statistical analysis was performed using the Student's t-test (**p<0.001; **p<0.01).
Recipients of B6 and T2 hearts displayed only low levels of β-gal-specific CD8+ T cells following infection with 2×105 pfu MCMV-LacZ, both in blood and spleens (Fig. 4A and B). Adoptive transfer of Bg1 T cells led only to slightly increased β-gal96 peptide-specific CD8+ T-cell numbers in blood (Fig. 4B) Still, the slightly elevated frequency of EC-specific CD8+ T-cell precursors in T2→-B6/Bg1 mice was sufficient to enhance vascular inflammation as compared to T2→B6 recipients, which had only the endogenous repertoire of β-gal-specific CD8+ T cells (Fig. 5A and B). Neither B6→B6 nor B6→B6/Bg1 recipients developed signs of TV following MCMV-LacZ infection (Fig. 5A and B). However, we observed a pronounced expansion of both the immunodominant β-gal497− and subdominant β-gal96-specific CD8+ T-cell populations when we infected B6/Bg1 recipients of T2γR hearts (T2γR→B6/Bg1) with MCMV-LacZ (Fig. 4). T2γR→B6 hearts developed severe transplant arteriosclerosis that even led to damage of the myocardium (Fig. 5A and B). Therefore, it appears that the lack of the IFNGR exclusively on the transplanted hearts augmented the clonal burst of EC-specific CD8+ T cells and thereby exacerbated TV.
Figure 4.
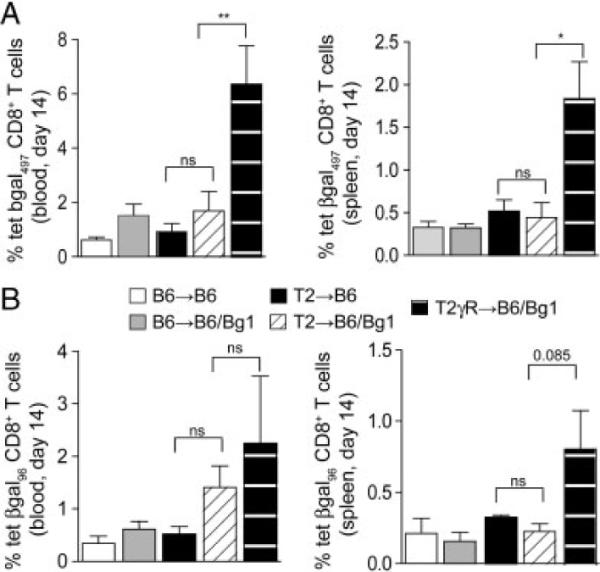
CD8+ T-cell responses in MCMV-LacZ infected heart recipients. Recipients of B6, T2, and T2γR hearts were i.v. infected with 2 × 105 pfu MCMV-LacZ and β-gal-specific CD8+ T-cell responses were measured in blood and spleens on day 14 post infection using H2-Kb/β-gal497 (A) and H2-Kb/β-gal96 (B) tetramers. Elevation of β-galspecific CD8+ T-cell frequencies in B6→B6/Bg1, T2→B6/Bg1, and T2γR→B6/Bg1 mice was achieved by adoptive transfer of 2 × 105 transgenic Bg1 CD8+ T cells on the day of infection. Mean percentages 1SEM of tetramer-positive cells in the CD8 compartment are indicated. Pooled data from two independent experiments (B6→B6, n = 7; B6→B6/Bg1, n = 7; T2→B6, n = 5; T2→B6/Bg1, n = 6; T2γR→B6/Bg1, n = 8). Statistical analysis was performed using one-way ANOVA with Bonferroni post test (**p<0.01; *p<0.05; ns, not significant).
Figure 5.
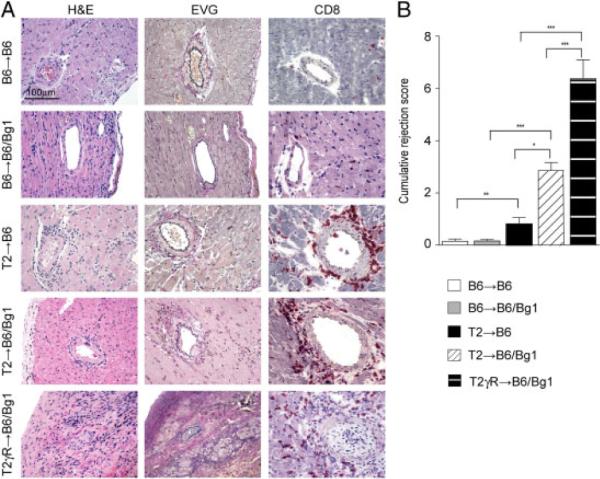
In situ analysis of hearts from MCMV-LacZ-infected graft recipients. B6→B6/Bg1, T2→B6/Bg1, and T2γR→B6/Bg1 mice received 2 × 105 transgenic Bg1 CD8+ T cells by adoptive transfer on the day of infection. (A) Representative sections of transplanted B6, T2, and T2γR hearts on day 14 after MCMV-LacZ infection (H&E, EVG, and immunostaining for CD8). Original magnification ×400; scale bar, 100 μm. (B) Analysis of immunopathological vascular alterations on day 14 post infection. The cumulative rejection score (mean±SEM) was calculated as the sum of three individual parameters including perivascular infiltration, intimal thickening, and perivascular fibrosis. Data from four independent experiments were pooled; (B6→B6, n = 7; B6→B6/Bg1, n = 13; T2→B6, n = 5; T2→B6/Bg1, n = 10; T2γR→B6/Bg1, n = 8). Statistical analysis was performed using one-way ANOVA with Bonferroni post test (***p<0.001; **p<0.01; *p<0.05).
IFN-γ-activated negative feedback circuit
Next, we wanted to study how the IFNGR-deficiency of the graft alone may control the population of EC-specific CD8+ T cells. We hypothesized that the expression of IFNGR-dependent inhibitory receptors by the graft might contribute to the down-tuning of EC-specific CD8+ T cells. Examples of such receptors are PD-L1 and the herpes virus entry mediator (HVEM), which have been shown to impinge on the course of transplant rejection [33, 34]. Since the PD-1-pathway is particularly important in the attenuation of antiviral CD8+ T-cell responses [35, 36], we assessed first whether the PD-1 molecule is expressed on CD8+ T cells in heart recipients following MCMV-LacZ infection. Indeed, compared to the low expression of PD-1 in naive B6 mice, CD8+ T cells in recipients of B6, T2, or T2γR hearts showed a pronounced upregulation of PD-1 expression following MCMV-LacZ infection (Fig. 6A). Furthermore, expression of both PD-1 ligands, and the IFN-γ-induced chemokine CXCL10 was induced in transplanted hearts on day 14 following MCMV-LacZ infection but only if the β-gal transgene was expressed in the transplanted hearts (Fig. 6B). While PD-L2, a co-inhibitory molecule mainly expressed by bone marrow-derived APC, was upregulated in severely inflamed T2γR hearts, up-regulation of PD-L1 was completely dependent on the presence of the IFNGR in the transplanted heart (Fig. 6B and C). These data suggested that the aggravated TV in T2γR might be a consequence of decreased endothelial PD-L1 expression in this situation.
Figure 6.

Regulation of PD-1 and PD-L1 during MCMV-LacZ infection. (A) PD-1 expression on CD8+ T cells in naive B6 and B6→B6/Bg1, T2→B6/Bg1, and T2γR→B6/Bg1 mice on day 6 after MCMV-LacZ infection. Mean percentages of PD-1+ cells in the CD8 compartment are indicated (B6 naive, n = 3; B6→B6/Bg1, n = 3; T2→B6/Bg1, n = 6; T2γR→B6/Bg1, n = 6). (B) Expression of IFN-γ, CXCL10, PD-L1, and PD-L2 mRNA in the indicated heart transplants on day 14 after MCMV-LacZ infection as determined by real-time RT-PCR. Pooled data from two independent experiments (B6 naive, n = 8; B6→B6/Bg1, n = 4; T→B6/Bg1, n = 6; T2γR→B6/Bg1, n = 6). Statistical analysis was performed using one-way ANOVA with Bonferroni post test (***p<0.001; **p<0.01; *p<0.05; ns, not significant). (C) In situ analysis for PD-L1 expression in heart sections on day 14 post MCMV-LacZ infection (original magnification ×400; scale bar, 100 μm).
In a next set of transplantation experiments, we therefore determined whether a global blockade of PD-L1 or an abrogation of CD8+ T-cell-expressed PD-1 would contribute to the aggravation of TV during MCMV-LacZ infection. To this end, T2 or T2γR hearts were transplanted into B6 mice, and recipient mice received CD8+ Bg1 T cells. One group of T2→B6 mice received PD-1-deficient Bg1 cells (T2→B6/Bg1PD-1−/−), and a second group of T2→B6/Bg1 mice received blocking anti-PD-L1 Ab on days 1, 2. 5, 8, and 11 post infection (T2→B6/Bg11αPD-L1). Immunohistochemical analysis of heart sections on day 14 after infection revealed that both the deficiency of PD-1 on EC-specific CD8+ T cells, and the global blockade of PD-L1 led to an exacerbation of arterial inflammation (Fig. 7A and B). These data demonstrate that IFN-γ-induced graft protection is – to a large extent – dependent on the interaction of PD-1 expressed by EC-specific CD8+ T cells and IFN-γ-induced expression of PD-L1 on ECs.
Figure 7.
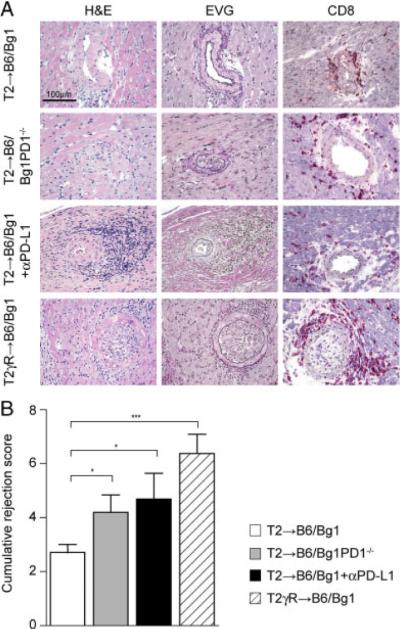
Impact of PD-L1 blockade and PD-1-deficiency on transplant vasculopathy. Recipients of the indicated hearts were infected with MCMV-LacZ. Blockade of PD-L1 was achieved by repeated treatment with anti-PD-L1 Ab (T2→B6/αPD-L1) and action of PD-1 on EC was partially abrogated by adoptive transfer of PD-1-deficient Bg1 cells (T2→B6/Bg1PD-1−/−). (A) Representative sections of transplanted T2, and T2gR hearts on day 14 after MCMV-LacZ infection (H&E, EVG, and immunostaining for CD8). Original magnification ×400; scale bar, 100 μm. (B) Analysis of immunopathological vascular alterations on day 14 post infection. The cumulative rejection score (mean+SEM) is indicated. Data from two independent experiments were pooled (T2→B6/Bg1, n = 10; T2→B6/Bg1PD-1−/−, n = 5; T2→B6/Bg1+αPD-L1, n = 8; T2γR→B6/Bg1; n = 8). Statistical analysis was performed using the Student's t-test (***p<0.001; *p<0.05).
Discussion
Evidence from a large body of literature indicates that the signature Th type 1 (Th1) cytokine IFN-γ and molecules involved in the Th1 pathway, such as IL-12, are crucial and necessary for chronic arterial injury (summarized in Ref. [5]). However, our data support the notion that the pleiotropic cytokine IFN-γ acts in a context-dependent fashion: immunostimulatory effects of IFN-γ are needed to eliminate pathogens from the vasculature [37, 38], but are counteracted by particularly vulnerable cells to protect them from immunopathological damage. Our results indicate that IFNGR signaling in EC provides a potent negative feedback on EC-specific CD8+ T cells, hence limiting vascular pathology. This mechanism may not only operate in TV, but may also limit endothelial destruction and, thus fatal parenchymal damage during viral infection [39]. Here, the degree of attenuation of EC-specific CD8+ T cells clearly correlated with the levels of systemic IFN-γ. Provoking vascular rejection via repetitive priming with DC elicited only low levels of systemic IFN-γ, and hence almost no impact of IFNGR signaling on the amount of EC-specific CD8+ T cells and vascular pathology was observed. In contrast, the MCMV-LacZ infection-induced “IFN-γ storm” was associated with a significant protection against arterial inflammation. Notably, the lack of the IFNGR solely on cells of the grafted hearts was sufficient to exert a pronounced PD-1/PD-L1-mediated negative feedback on the EC-specific CD8+ population. This indicates that graft-reactive T cells are fine-tuned within the graft itself.
Donor EC persist in grafted organs and can thus contribute to chronic immune stimulation. Under particular circumstances, EC may even initiate CTL responses that mediate allograft rejection [40], and EC can be recognized by CD8+ T cells through the indirect Ag presentation pathway involving cross-presentation of exogenous Ag [41–43]. Priming of naive recipients of T2 or T2γR hearts, as done here by either vaccination with β-gal peptide-pulsed DC or infection with MCMV-LacZ, allowed us to address the role of IFNGR signaling in the interaction of EC with effector CD8+ T cells, excluding an involvement of EC in the initial activation of the T cells. Our study confirmed that IFN-γ can act on grafted tissue to promote its survival [44–46] and extended the current knowledge by showing that the efficacy of CD8+ down-tuning strongly correlates with the levels of systemic IFN-γ. IFNGR signaling within secondary lymphoid organs is critical for the generation of T-cell responses, e.g. due to stimulation of Ag presentation via MHC class I and class II, or effects on T-cell proliferation. For example, the IFNGR is necessary for generation of antiviral CD4+ and CD8+ effector T cells, and to promote optimal memory T-cell responses [47–49]. Thus, there appears to be a pronounced dichotomy of IFN-γ action on target cells within secondary lymphoid organs (APC and T cells) versus target cells within peripheral organs such as ECs. Such clear-cut distinction between lymphoid versus peripheral action of IFN-γ would explain how this cytokine can protect epithelial cells from graft-versus-host disease while – at the same time – it can exert potent antitumor functions against leukemic cells within secondary lymphoid organs [21].
Protection of EC from CD8+ T-cell-mediated injury can be achieved by IFN-γ regulated expression of PD-L1, as shown in this study. Likewise, other peripheral cells such as pancreatic islet cells [50], or cardiomyocytes [51] can be protected through the PD-1/PD-L1 pathway from T-cell-mediated damage. Other tissue-specific co-inhibitory molecules such as HVEM can contribute to the attenuation of effector T-cell responses [33]. The partial exacerbation of the CD8+ T-cell-mediated TV observed in our model by disruption of the PD-1/PD-L1 pathway suggests that this feedback circuit is most likely only one component of the potent protective capacity of IFN-γ in the periphery.
A number of approaches for intervention in this feedback circuit have been proposed such as ligation of PD-1 or the HVEM-receptor BTLA on activated T cells [33]. A particularly attractive scenario would be the potential exploitation of the cell type-specific signal transduction pathways downstream of the IFNGR in EC [52], which may harbor specific targets that could permit stimulation of the peripheral inhibitory signals while sparing the stimulatory pathway in immune cells. Taken together, pro-inflammatory IFNGR signaling in EC appears to be a central regulatory step in the control of EC-specific CD8+ T-cell responses and hence, in the promotion of shielding the graft from CD8+ T-cell-mediated damage.
Materials and methods
Mice
Male and female C57BL/6 mice were obtained from Charles River (Sulzfeld, Germany). Tie2-LacZ mice expressing the LacZ gene under the control of the EC-specific Tie2 promoter [22] had been backcrossed with C57BL/6 for at least 14 times. Tie2-LacZ mice were further crossed with IFN-γ-R−/− (C57BL/6 background) mice [53] to obtain Tie2-LacZxIFN-γ-R−/− (T2γR) mice. TCR transgenic Bg1 mice that express a TCR recognizing the H2-Kb-restricted β-gal96–103 peptide on more than 95% of their CD8+ T cells have been described previously [23]. Bg1 mice were crossed with C57BL/6 mice expressing the congenic marker Thy 1.1 and transgene expression was monitored by staining of blood cells with anti-Vβ7 by flow cytometry. Thy 1.1+Bg1 mice were further crossed with PD 1−/− mice [54] to obtain Bg1×PD-1−/− mice. The presence of the β-gal transgene in Tie2-LacZ mice as well as the absence of PD-1 and the IFNGR were determined by PCR from genomic DNA. All animals were kept under conventional conditions in individually ventilated cages and fed with normal chow diet. Experiments were carried out with age and sex-matched animals. Experiments were performed in accordance with Swiss Kantonal and Federal legislations (permissions SG07/62, SG97/64, and SG07/65).
Viruses and peptides
Recombinant MCMV expressing the β-gal protein under the transcriptional control of the human CMV ie1/ie2 promoter-enhancer (MCMV-LacZ RM427 [32]) was kindly provided by Professor E. S. Mocarski (Stanford University, San Francisco, CA, USA). MCMV-LacZ was propagated and titrated on NIH 3T3 cells (ECACC, UK) and injected intravenously at a dose of 2 × 105 pfu. β-gal96–103 (DAPIYTNV) [55] and β-gal497–504 (ICPMYARV) [56] peptides were purchased from Neosystem (Strasbourg, France).
Ab and flow cytometry
Anti-CD8-PerCp was obtained from BD PharMingen (Basel, Switzerland). Anti-CD8-FITC, anti-CD8-APC, and anti-PD-1 were obtained from Biolegend (LuBioScience GmbH, Lucerne, Switzerland). For flow cytometry, single cell suspensions were generated from the indicated organs and 1 × 106 cells were incubated with the indicated mAb at 4°C for 20 min. For PBL samples, erythrocytes were lysed with FACS Lysing Solution (BD PharMingen). Cells were analyzed by flow cytometry, gating on viable leukocytes using 7-aminoactinomycin D (Sigma) using a FACScalibur flow cytometer and the CellQuest software (BD Biosciences). For in vivo anti-PD-L1 blockade, anti-PD-L1 mAb (MIH5, rat IgG2a, eBioscience, San Diego, CA, USA) was administered i.p. at 200 μg per mouse on days –1, 2, 5, 8, and 11 post infection with MCMV-LacZ.
Construction of tetrameric MHC class I peptide complexes
MHC class I monomers complexed with β-gal (H-2Kb) were produced as previously described [57] and tetramerized by addition of streptavidin–PE (Molecular Probes, Eugene, OR, USA). At the indicated time points following infection, organs were removed and single cell suspensions were prepared. Aliquots of 1 × 106 cells or 300 μL of blood were stained using 50 μL of a solution containing tetrameric class I-peptide complexes at 37°C for 10 min followed by staining with anti-CD8-FITC, anti-CD8-APC (Biolegend), or anti-CD8-PerCp (BD Pharmingen) at 4°C for 20 min. Absolute cell counts were determined by counting leukocytes in an improved Neubauer chamber.
DC preparation and injection
DC were generated from C57BL/6 bone marrow as described previously [58]. Before injection, cells were loaded with the βgal497–504 peptide at a concentration of 10−5 M for 1 h at 37°C, washed three times with ice-cold BSS, and resuspended in BSS at a concentration of 4 × 105 DC/mL. DC were injected in 500 μL BSS i.v. on days 0, 2, 10, and 12.
Adoptive transfer of TCR transgenic T cells
Single cell suspensions from spleens of Thy1.1+ Bg1 mice were subjected to hypotonic red blood cell lysis and cells were washed twice with ice-cold BSS and resuspended in BSS at a concentration of 2 β 106 splenocytes/mL. Heart-transplanted recipients (B6→B6, Tie2-LacZ→B6, Tie2-LacZxIFN-γR−/−→B6) were injected i.v. with 106 Bg1-Thy 1.1+ splenocytes (corresponding to 2 × 105 CD8+ T cells) in 500 μL BSS on the day of MCMV-LacZ infection.
ELISA
Mouse IFN-γ-concentration in serum and spleen homogenates was measured by enzyme-linked immunosorbent assay (ELISA; BD OptEIA; BD Bioscience) according to the manufacturer's instructions. High-binding 96-well polystyrene plates (Corning, New York, NY, USA) were coated with the monoclonal IFN-γ Ab (clone AN-18; BD Bioscience) in 0.1 M carbonate-bicarbonate buffer, pH 9.5. Plates were incubated overnight at 4°C. Before use, plates were washed five times in PBS (pH 7.2) containing 0.05% Tween-20 (PBS-T) (Sigma-Aldrich). Non-specific binding was blocked with PBS (pH 7.2) containing 10% FBS (Assay diluent) (Lonza, Belgium) for 1 h at RT. After washing, sera were diluted 1:2 in assay diluent and twofold serial dilutions were added to the wells. Plates were incubated for 2 h at RT, followed by five washes with PBS-T. The optimal dilution, 1:250, of the biotinylated anti-mouse IFN-γ Ab and the peroxidase-conjugated streptavidin–horseradish Ab (BD Bioscience) in assay diluents was added, followed by 1 h of incubation at RT and five additional washes with PBS-T. Tetramethylbenzidine (KPL, Gaithersburg, MD, USA) in H2O2 was used as the enzyme substrate. The reaction was stopped with 1.25 M H2SO4 and the optical densities were read at 450 nm using an automatic ELISA plate reader (Tecan).
Quantitative real-time PCR
Hearts were homogenized in Trizol (Sigma) using a MagNA Lyser instrument (Roche Diagnostics). RNA was isolated by isopropanol precipitation, washed with ethanol (70%), and resuspended in DEPC-water. RNA (10 μg) was subjected to RT-PCR analysis. For RT-PCR the high capacity cDNA archive Kit from Applied Biosystem (ABI PRISM, Warrington, UK) was used according to the specifications of the manufacturer to generate cDNA from RNA samples. Quantitative real-time PCR was performed using a LightCycler (Roche Diagnostics) and the LightCycler FastStart DNA MasterPLUS Sybr Green I reaction mix (Roche Diagnostics) following the manufacturer's protocol. Data analysis was performed with LightCycler Software 3 (Roche Diagnostics). Oligonucleotides were purchased from Microsynth (Balgach, Switzerland). The following oligonucleotides were used as primers for quantitative real-time PCR IFN-γ: 5′-AACGCTACACACTGCATCTTGG-3′ and 5′-GCCGTGGCAGTAACAGCC-3′, CCL10: 5′-CCTATCCTGCCCACGTGTTGAG-3′ and 5′-CGCACCTCCACATAGCTTACAG-3′ PD-L1: 5′-TGGACAAACAGTGACCACC AA-3′ and 5′-CCCCTCTGTCCGGGAAGT-3′, PD-L2: 5′-GTACCGTTGCCTGGTCATCT-3′ and 5′-GCCAGGACACTTCTGCTAGG-3′, TBP: 5′-CCTTCACCAATGACTCCTATGAC-3′ and 5′-CAAGTTTACAGCCAAGATTCAC-3′. Thermal cycling started with HotStarTaq activation for 15 min at 95°C. Thereafter, 50 cycles of amplification were run consisting of 10 s at 95°C, 10 s 60°C, and 20 s of 72°C. The relative expression of target genes was measured in triplicates and normalized to the expression amount of TBP. A negative control, containing reagents only and cDNA-standard dilutions, to generate a standard curve, were included in each run.
Immunohistology
Organs were fixed in 4% formalin and embedded in paraffin. Sections were stained with H&E, or resorcin-fuchsin for the Elastic van Gieson staining (EVG). For immunohisto-chemistry, freshly removed organs were immersed in HBSS and snap-frozen in liquid nitrogen. Frozen tissue sections were cut in a cryostat and fixed in acetone for 10 min. Sections were incubated with Ab against CD8 (clone YTS169.4.2), PD-L1 (eBioscience) followed by goat anti-rat Ig (Caltag Labs) and alkaline phosphatase-labeled donkey anti-goat Ig (Jackson ImmunoResearch Labs). Alkaline phosphatase was visualized by using AS-BI phosphate/New Fuchsin, and sections were counterstained with hemalum, and images were acquired using a Leica DM R microscope equipped with a Leica DC300 FX camera. Digital images were processed using Adobe Photoshop.
Histological scoring of chronic vascular transplant rejection
Based on histological changes observed in chronic allograft rejection, we established a modified scoring system to analyze different degrees of vascular transplant rejection based on criteria previously published [12, 28] using a scale from 0 to 3 for the different pathological alterations. The degrees for intimal thickening of coronary arteries (0 = <10%; 1 = 10 to<50%; 2 = 50 to<90%; 3 = 90–100% luminal occlusion in at least one artery/section), perivascular fibrosis (0 = no changes; 1 = minor fibrotic changes around 1–3 arteries/section; 2 = vast fibrotic changes around 1–3 arteries/section; 3 = vast fibrotic changes around more than three arteries/section), and perivascular and vascular inflammatory infiltrates (0 = no inflammatory infiltrates; 1 = few inflammatory infiltrates around 1–3 arteries/section; 2 = vast inflammatory infiltrations around 1–3 arteries/section; 3 = vast inflammatory infiltrates in more than three arteries/section) were determined. Six to eight sections from each heart were evaluated by two independent observers blinded for the tested specimen.
Heterotopic heart transplantation
Heterotopic vascularized cardiac transplantation was performed according to the method described by Corry et al. [59]. Donor hearts were explanted from either male Tie2-LacZ or male C57BL/6 mice. The donor heart was removed from the chest after intracaval injection of 1 mL of heparin (100 U/mL), rinsed with NaCl (0.9%), and placed on ice. After isolation of the recipient's abdominal aorta and inferior vena cava, the donor ascending aorta and pulmonary artery were joined end-to-side to the recipient's aorta and vena cava, respectively, using 10–0 nylon running suture. The abdomen was closed with individual running sutures to musculofascial layer and skins. The recipient mouse was then warmed for a few hours during recovery from anesthesia and had free access to water and food. The function of the transplanted heart was assessed daily by abdominal palpation.
Statistical analysis
To evaluate statistically significant differences, the unpaired two-tailed Student's test or one-way ANOVA with Bonferroni post test was used. p values smaller than 0.05 were considered statistically significant. Statistical data analysis was performed using Graph-Pad Prism version 4.03 for Windows (GraphPad Software, San Diego, CA, USA).
Acknowledgements
We would like to thank Silvia Behnke, Andre Fitsche, and Karin Eugster for help with immunohistochemistry, and Luisa Cervantes-Barragan and Tobias Junt for critical reading of the manuscript. The project received support from the Swiss National Science Foundation (to B. L.), the Velux Foundation (to B. L.), and the Kanton of St. Gallen (to B. L.).
Abbreviations
- β-gal
β-galactosidase
- EC
endothelial cells
- EVG
Elastic van Gieson
- HVEM
herpes virus entry mediator
- IFNGR
IFN-γ receptor
- MCMV-LacZ
β-galactosidase-recombinant murine cytomegalovirus
- mhAg
minor histocompatibility antigen
- PD-1
programmed death-1
- PD-L1
programmed death ligand 1
- TV
transplant vasculopathy
Footnotes
Supporting Information available online
Conflict of interest: The authors declare no financial or commercial conflict of interest.
References
- 1.Libby P, Pober JS. Chronic rejection. Immunity. 2001;14:387–397. doi: 10.1016/s1074-7613(01)00119-4. [DOI] [PubMed] [Google Scholar]
- 2.Schmauss D, Weis M. Cardiac allograft vasculopathy: recent developments. Circulation. 2008;117:2131–2141. doi: 10.1161/CIRCULATIONAHA.107.711911. [DOI] [PubMed] [Google Scholar]
- 3.Nankivell BJ, Borrows RJ, Fung CL, O'Connell PJ, Allen RD, Chapman JR. The natural history of chronic allograft nephropathy. N. Engl. J. Med. 2003;349:2326–2333. doi: 10.1056/NEJMoa020009. [DOI] [PubMed] [Google Scholar]
- 4.Mitchell RN, Libby P. Vascular remodeling in transplant vasculopathy. Circ. Res. 2007;100:967–978. doi: 10.1161/01.RES.0000261982.76892.09. [DOI] [PubMed] [Google Scholar]
- 5.Tellides G, Pober JS. Interferon-gamma axis in graft arteriosclerosis. Circ. Res. 2007;100:622–632. doi: 10.1161/01.RES.0000258861.72279.29. [DOI] [PubMed] [Google Scholar]
- 6.Valantine HA. Cardiac allograft vasculopathy: Central role of endothelial injury leading to transplant “atheroma.”. Transplantation. 2003;76:891–899. doi: 10.1097/01.TP.0000080981.90718.EB. [DOI] [PubMed] [Google Scholar]
- 7.Rifle G, Mousson C, Herve P. Endothelial cells in organ transplantation: Friends or foes? Transplantation. 2006;82:S4–S5. doi: 10.1097/01.tp.0000231368.36476.4a. [DOI] [PubMed] [Google Scholar]
- 8.Russell PS, Chase CM, Winn HJ, Colvin RB. Coronary atherosclerosis in transplanted mouse hearts. I. Time course and immunogenetic and immunopathological considerations. Am. J. Pathol. 1994;144:260–274. [PMC free article] [PubMed] [Google Scholar]
- 9.Russell PS, Chase CM, Winn HJ, Colvin RB. Coronary atherosclerosis in transplanted mouse hearts II Importance of humoral immunity. J. Immunol. 1994;152:5135–5141. [PubMed] [Google Scholar]
- 10.Russell PS, Chase CM, Colvin RB. Alloantibody- and T cell-mediated immunity in the pathogenesis of transplant arteriosclerosis: lack of progression to sclerotic lesions in B cell-deficient mice. Transplantation. 1997;64:1531–1536. doi: 10.1097/00007890-199712150-00005. [DOI] [PubMed] [Google Scholar]
- 11.Ueland T, Sikkeland LI, Yndestad A, Eiken HG, Holm T, Guevara C, Haug T, et al. Myocardial gene expression of inflammatory cytokines after heart transplantation in relation to the development of transplant coronary artery disease. Am. J. Cardiol. 2003;92:715–717. doi: 10.1016/s0002-9149(03)00836-1. [DOI] [PubMed] [Google Scholar]
- 12.Russell PS, Chase CM, Winn HJ, Colvin RB. Coronary atherosclerosis in transplanted mouse hearts III Effects of recipient treatment with a monoclonal antibody to interferon-gamma. Transplantation. 1994;57:1367–1371. [PubMed] [Google Scholar]
- 13.Nagano H, Mitchell RN, Taylor MK, Hasegawa S, Tilney NL, Libby P. Interferon-gamma deficiency prevents coronary arteriosclerosis but not myocardial rejection in transplanted mouse hearts. J. Clin. Invest. 1997;100:550–557. doi: 10.1172/JCI119564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gupta S, Pablo AM, Jiang X. c., Wang N, Tall AR, Schindler C. IFN-gamma potentiates atherosclerosis in ApoE knock-out mice. J. Clin. Invest. 1997;99:2752–2761. doi: 10.1172/JCI119465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Whitman SC, Ravisankar P, Elam H, Daugherty A. Exogenous interferon-gamma enhances atherosclerosis in apolipoprotein E−/− mice. Am. J. Pathol. 2000;157:1819–1824. doi: 10.1016/s0002-9440(10)64820-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schoenborn JR, Wilson CB. Regulation of interferon-gamma during innate and adaptive immune responses. Adv. Immunol. 2007;96:41–101. doi: 10.1016/S0065-2776(07)96002-2. 41-101. [DOI] [PubMed] [Google Scholar]
- 17.Tellides G, Tereb DA, Kirkiles-Smith NC, Kim RW, Wilson JH, Schechner JS, Lorber MI, Pober JS. Interferon-gamma elicits arteriosclerosis in the absence of leukocytes. Nature. 2000;403:207–211. doi: 10.1038/35003221. [DOI] [PubMed] [Google Scholar]
- 18.Badovinac VP, Tvinnereim AR, Harty JT. Regulation of antigen-specific CD8(+) T cell homeostasis by perforin and interferon-gamma. Science. 2000;290:1354–1358. doi: 10.1126/science.290.5495.1354. [DOI] [PubMed] [Google Scholar]
- 19.Willenborg DO, Fordham S, Bernard CC, Cowden WB, Ramshaw IA. IFN-gamma plays a critical down-regulatory role in the induction and effector phase of myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. J. Immunol. 1996;157:3223–3227. [PubMed] [Google Scholar]
- 20.Yang YG, Dey BR, Sergio JJ, Pearson DA, Sykes M. Donor-derived interferon gamma is required for inhibition of acute graft-versus-host disease by interleukin 12. J. Clin. Invest. 1998;102:2126–2135. doi: 10.1172/JCI4992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang H, Asavaroengchai W, Yong YB, Wang MG, Wang S, Sykes M, Yang YG. Paradoxical effects of IFN-{gamma} in graft-versus-host disease reflect promotion of lymphohematopoietic graft-versus-host reactions and inhibition of epithelial tissue injury. Blood. 2009;113:3612–3619. doi: 10.1182/blood-2008-07-168419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schlaeger TM, Bartunkova S, Lawitts JA, Teichmann G, Risau W, Deutsch U, Sato TN. Uniform vascular-endothelial-cell-specific gene expression in both embryonic and adult transgenic mice. Proc. Natl. Acad. Sci. USA. 1997;94:3058–3063. doi: 10.1073/pnas.94.7.3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bolinger B, Krebs P, Tian Y, Engeler D, Scandella E, Miller S, Palmer DC, et al. Immunologic ignorance of vascular endothelial cells expressing minor histocompatibility antigen. Blood. 2008;111:4588–4595. doi: 10.1182/blood-2007-09-114769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ochsenbein AF, Klenerman P, Karrer U, Ludewig B, Pericin M, Hengartner H, Zinkernagel RM. Immune surveillance against a solid tumor fails because of immunological ignorance. Proc. Natl. Acad. Sci. USA. 1999;96:2233–2238. doi: 10.1073/pnas.96.5.2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ludewig B, Krebs P, Junt T, Metters H, Ford NJ, Anderson RM, Bocharov G. Determining control parameters for dendritic cell-cytotoxic T lymphocyte interaction. Eur. J. Immunol. 2004;34:2407–2418. doi: 10.1002/eji.200425085. [DOI] [PubMed] [Google Scholar]
- 26.Ludewig B, Odermatt B, Landmann S, Hengartner H, Zinkernagel RM. Dendritic cells induce autoimmune diabetes and maintain disease via de novo formation of local lymphoid tissue. J. Exp. Med. 1998;188:1493–1501. doi: 10.1084/jem.188.8.1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ludewig B, Junt T, Hengartner H, Zinkernagel RM. Dendritic cells in autoimmune diseases. Curr. Opin. Immunol. 2001;13:657–662. doi: 10.1016/s0952-7915(01)00275-8. [DOI] [PubMed] [Google Scholar]
- 28.Hirozane T, Matsumori A, Furukawa Y, Sasayama S. Experimental graft coronary artery disease in a murine heterotopic cardiac transplant model. Circulation. 1995;91:386–392. doi: 10.1161/01.cir.91.2.386. [DOI] [PubMed] [Google Scholar]
- 29.Paineau J, Priestley C, Fabre J, Chevalier S, van der MP, Schellekens H, Jacques Y, Soulillou JP. Effect of recombinant interferon gamma and interleukin-2 and of a monoclonal antibody against interferon gamma on the rat immune response against heart allografts. J. Heart Lung Transplant. 1991;10:424–430. [PubMed] [Google Scholar]
- 30.Halloran PF, Miller LW, Urmson J, Ramassar V, Zhu LF, Kneteman NM, Solez K, Afrouzian M. IFN-gamma alters the pathology of graft rejection: protection from early necrosis. J. Immunol. 2001;166:7072–7081. doi: 10.4049/jimmunol.166.12.7072. [DOI] [PubMed] [Google Scholar]
- 31.Wherry EJ, Ahmed R. Memory CD8 T-cell differentiation during viral infection. J. Virol. 2004;78:5535–5545. doi: 10.1128/JVI.78.11.5535-5545.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Manning WC, Mocarski ES. Insertional mutagenesis of the murine cytomegalovirus genome: one prominent alpha gene (ie2) is dispensable for growth. Virology. 1988;167:477–484. [PubMed] [Google Scholar]
- 33.del Rio ML, Buhler L, Gibbons C, Tian J, Rodriguez-Barbosa JI. PD-1/PD-L1, PD-1/PD-L2, and other co-inhibitory signaling pathways in transplantation. Transpl. Int. 2008;21:1015–1028. doi: 10.1111/j.1432-2277.2008.00726.x. [DOI] [PubMed] [Google Scholar]
- 34.Kosuge H, Suzuki J, Kakuta T, Haraguchi G, Koga N, Futamatsu H, Gotoh R, et al. Attenuation of graft arterial disease by manipulation of the LIGHT pathway. Arterioscler. Thromb. Vasc. Biol. 2004;24:1409–1415. doi: 10.1161/01.ATV.0000134645.53285.02. [DOI] [PubMed] [Google Scholar]
- 35.Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, Betts MR, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 2009;10:29–37. doi: 10.1038/ni.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sester U, Presser D, Dirks J, Gartner BC, Kohler H, Sester M. PD-1 expression and IL-2 loss of cytomegalovirus-specific T cells correlates with viremia and reversible functional anergy. Am. J. Transplant. 2008;8:1486–1497. doi: 10.1111/j.1600-6143.2008.02279.x. [DOI] [PubMed] [Google Scholar]
- 37.Dal Canto AJ, Virgin HW, Speck SH. Ongoing viral replication is required for gammaherpesvirus 68-induced vascular damage. J. Virol. 2000;74:11304–11310. doi: 10.1128/jvi.74.23.11304-11310.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dal Canto AJ, Swanson PE, O'Guin AK, Speck SH, Virgin HW. IFN-gamma action in the media of the great elastic arteries, a novel immunoprivileged site. J. Clin. Invest. 2001;107:R15–R22. doi: 10.1172/JCI11540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Iwai Y, Terawaki S, Ikegawa M, Okazaki T, Honjo T. PD-1 inhibits antiviral immunity at the effector phase in the liver. J. Exp. Med. 2003;198:39–50. doi: 10.1084/jem.20022235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kreisel D, Krupnick AS, Gelman AE, Engels FH, Popma SH, Krasinskas AM, Balsara KR, et al. Non-hematopoietic allograft cells directly activate CD8+T cells and trigger acute rejection: an alternative mechanism of allorecognition. Nat. Med. 2002;8:233–239. doi: 10.1038/nm0302-233. [DOI] [PubMed] [Google Scholar]
- 41.Valujskikh A, Lantz O, Celli S, Matzinger P, Heeger PS. Cross-primed CD8(+) T cells mediate graft rejection via a distinct effector pathway. Nat. Immunol. 2002;3:844–851. doi: 10.1038/ni831. [DOI] [PubMed] [Google Scholar]
- 42.Bagai R, Valujskikh A, Canaday DH, Bailey E, Lalli PN, Harding CV, Heeger PS. Mouse endothelial cells cross-present lymphocyte-derived antigen on class I MHC via a TAP1- and proteasome-dependent pathway. J. Immunol. 2005;174:7711–7715. doi: 10.4049/jimmunol.174.12.7711. [DOI] [PubMed] [Google Scholar]
- 43.Limmer A, Ohl J, Kurts C, Ljunggren HG, Reiss Y, Groettrup M, Momburg F, et al. Efficient presentation of exogenous antigen by liver endothelial cells to CD8+T cells results in antigen-specific T-cell tolerance. Nat. Med. 2000;6:1348–1354. doi: 10.1038/82161. [DOI] [PubMed] [Google Scholar]
- 44.Halloran PF, Afrouzian M, Ramassar V, Urmson J, Zhu LF, Helms LM, Solez K, Kneteman NM. Interferon-gamma acts directly on rejecting renal allografts to prevent graft necrosis. Am. J. Pathol. 2001;158:215–226. doi: 10.1016/s0002-9440(10)63960-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mele TS, Kneteman NM, Zhu LF, Ramassar V, Urmson J, Halloran B, Churchill TA, et al. IFN-gamma is an absolute requirement for spontaneous acceptance of liver allografts. Am. J. Transplant. 2003;3:942–951. doi: 10.1034/j.1600-6143.2003.00153.x. [DOI] [PubMed] [Google Scholar]
- 46.Coley SM, Ford ML, Hanna SC, Wagener ME, Kirk AD, Larsen CP. IFN-gamma dictates allograft fate via opposing effects on the graft and on recipient CD8 T cell responses. J. Immunol. 2009;182:225–233. doi: 10.4049/jimmunol.182.1.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Whitmire JK, Tan JT, Whitton JL. Interferon-gamma acts directly on CD8+T cells to increase their abundance during virus infection. J. Exp. Med. 2005;201:1053–1059. doi: 10.1084/jem.20041463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Whitmire JK, Benning N, Whitton JL. Cutting edge: early IFN-gamma signaling directly enhances primary antiviral CD4+T cell responses. J. Immunol. 2005;175:5624–5628. doi: 10.4049/jimmunol.175.9.5624. [DOI] [PubMed] [Google Scholar]
- 49.Whitmire JK, Eam B, Benning N, Whitton JL. Direct interferon-gamma signaling dramatically enhances CD4+ and CD8+T cell memory. J. Immunol. 2007;179:1190–1197. doi: 10.4049/jimmunol.179.2.1190. [DOI] [PubMed] [Google Scholar]
- 50.Keir ME, Liang SC, Guleria I, Latchman YE, Qipo A, Albacker LA, Koulmanda M, et al. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J. Exp. Med. 2006;203:883–895. doi: 10.1084/jem.20051776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grabie N, Gotsman I, DaCosta R, Pang H, Stavrakis G, Butte MJ, Keir ME, et al. Endothelial programmed death-1 ligand 1 (PD-L1) regulates CD8+T-cell mediated injury in the heart. Circulation. 2007;116:2062–2071. doi: 10.1161/CIRCULATIONAHA.107.709360. [DOI] [PubMed] [Google Scholar]
- 52.Miura A, Honma R, Togashi T, Yanagisawa Y, Ito E, Imai J, Isogai T, et al. Differential responses of normal human coronary artery endothelial cells against multiple cytokines comparatively assessed by gene expression profiles. FEBS Lett. 2006;580:6871–6879. doi: 10.1016/j.febslet.2006.11.041. [DOI] [PubMed] [Google Scholar]
- 53.Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 54.Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, Sasayama S, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 2001;291:319–322. doi: 10.1126/science.291.5502.319. [DOI] [PubMed] [Google Scholar]
- 55.Overwijk WW, Surman DR, Tsung K, Restifo NP. Identification of a Kb-restricted CTL epitope of beta-galactosidase: potential use in development of immunization protocols for “self” antigens. Methods. 1997;12:117–123. doi: 10.1006/meth.1997.0461. [DOI] [PubMed] [Google Scholar]
- 56.Oukka M, Cohen-Tannoudji M, Tanaka Y, Babinet C, Kosmatopoulos K. Medullary thymic epithelial cells induce tolerance to intracellular proteins. J. Immunol. 1996;156:968–975. [PubMed] [Google Scholar]
- 57.Altman JD, Moss PAH, Goulder PJR, Barouch DH, McHeyzer-Williams MG, Bell JI, McMichael AJ, Davis MM. Phenotypic analysis of antigen-specific T lymphocytes. Science. 1996;274:94–96. [PubMed] [Google Scholar]
- 58.Ludewig B, Ehl S, Karrer U, Odermatt B, Hengartner H, Zinkernagel RM. Dendritic cells efficiently induce protective antiviral immunity. J. Virol. 1998;72:3812–3818. doi: 10.1128/jvi.72.5.3812-3818.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Corry RJ, Winn HJ, Russell PS. Primarily vascularized allografts of hearts in mice The role of H-2D, H-2 K and non-H-2 antigens in rejection. Transplantation. 1973;16:343–350. doi: 10.1097/00007890-197310000-00010. [DOI] [PubMed] [Google Scholar]