

Abstract
AIM: To evaluate the safety and clinical efficacy of a new immunotherapy using both α-Gal epitope-pulsed dendritic cells (DCs) and cytokine-induced killer cells.
METHODS: Freshly collected hepatocellular carcinoma (HCC) tumor tissues were incubated with a mixture of neuraminidase and recombinant α1,3-galactosyltransferase (α1,3GT) to synthesize α-Gal epitopes on carbohydrate chains of the glycoproteins of tumor membranes. The subsequent incubation of the processed membranes in the presence of human natural anti-Gal IgG resulted in the effective phagocytosis to the tumor membrane by DCs. Eighteen patients aged 38-78 years with stage III primary HCC were randomLy chosen for the study; 9 patients served as controls, and 9 patients were enrolled in the study group.
RESULTS: The evaluation demonstrated that the procedure was safe; no serious side effects or autoimmune diseases were observed. The therapy significantly prolonged the survival of treated patients as compared with the controls (17.1 ± 2.01 mo vs 10.1 ± 4.5 mo, P = 0.00121). After treatment, all patients in the study group had positive delayed hypersensitivity and robust systemic cytotoxicity in response to tumor lysate as measured by interferon-γ-expression in peripheral blood mononuclear cells using enzyme-linked immunosorbent spot assay. They also displayed increased numbers of CD8-, CD45RO- and CD56-positive cells in the peripheral blood and decreased α-fetoprotein level in the serum.
CONCLUSION: This new tumor-specific immunotherapy is safe, effective and has a great potential for the treatment of tumors.
Keywords: Hepatocellular carcinoma, α-Gal epitope, Dendritic cell, Tumor-associated antigen, Dendritic cell-activated cytokine-induced killer cell
INTRODUCTION
Primary hepatocellular carcinoma (HCC), the most common neoplasm of the liver, is generally resistant to conventional treatment. The prognosis of the patients is poor, especially at the late stage, with a mean survival of less than ten months[1-3]. This poor prognosis has led to the interest in various alternative therapies. Immunotherapy is particularly appealing because of its specificity against tumor cells, absence of serious side effects and its potential to eradicate residual tumors after conventional treatment.
One area of active research is immunotherapy with cytokine-induced killer cell (CIK)[1-3]; unfortunately, its efficiency is limited because of low specificity to tumor cells. Another approach is tumor-associated antigen (TAA)-pulsed dendritic cell (DC) therapy, but the outcome is still not satisfactory because of the low immunogenicity of TAA[4-6].
DCs are the most effective antigen-presenting cells (APCs) responsible for initiating an immune response[7]. Upon exposure to tumor cells, isolated tumor antigen, and even to tumor mRNA, DCs are capable of presenting endogenous and exogenous antigens to naïve T cells in a human leukocyte antigen (HLA)-restricted manner and expressing several adhesive and costimulatory surface molecules. However, the poor immunogenicity of TAA may be the reason why tumor cells fail to adequately stimulate DCs for effective presentation to immune cells[8]. A possible method for increasing the uptake of TAAs by DCs is to complex them with an IgG antibody. These immune complexes would bind to Fcγ receptors (Fcγ-Rs) on DCs and induce phagocytosis of TAAs. The augmentation of antigen processing, presentation by immune complex and subsequent activation of immune responders has been demonstrated in several studies. For example, targeting tetanus toxoid (TT) to APC Fcγ-Rs by complexing it with anti-TT IgG results in a 10- to 1000-fold increase in Th-cell activation[9,10]. Similarly, the targeting of autologous tumor cell vaccines by DC Fcγ-Rs uptake would also lead to an effective immune response against the tumor cells[11]. Such targeting was achieved by complexing the tumor cell membranes in situ with naturally existing antibodies against the α-Galactosyl epitope (Gal-α1, 3Gal-β1, 4-GlcNAc-R, α-Gal)[12]. These anti-Gal antibodies have been shown to be detrimental in xenotransplantation[13] and to destroy retroviral vectors used for gene therapy[14]. In humans, the α-Gal epitope on the cell membrane is absent, but the natural anti-Gal antibody is abundant in serum[15]. Expression of the α-Gal epitope on tumor cells could result in in situ binding of the patient’s natural anti-Gal IgG. The binding complex can then opsonize DC phagocytosis and enhance TAA presentation to naïve T or CIK cells, which are then activated and attack the remaining tumor cells in vivo. However, patients with malignancies usually have very low immunity, especially in cellular immune responses. The cause of the low immunity is not quite clear. One possible explanation is that the patient’s T cells have been anergized by tumor antigens in the absence of APCs and cannot be activated again with the same antigen in vivo.
To maximize APC phagocytosis and the activation of tumor-specific T/CIK cells, we used newly differentiated T/CIKs from bone marrow stem cells instead of circulating T cells. Here, we present a new therapy for HCC using α-Gal epitope-expressing tumor cell-pulsed DCs to activate tumor-specific T/CIKs in vitro. The activated immune responders were then expanded in the presence of a high concentration of cytokines ex vivo.
MATERIALS AND METHODS
Patients
Written informed consent was obtained from all the patients who participated in the study before treatment according to the guidelines of the Ethics Committee of the Armed Police General Hospital, which approved this study. Inclusion criteria were a Karnofsky score of ≥ 60[16], the lowest possible maintenance dose of glucocorticoid therapy (prednisone < 5mg/d) and normal baseline hematological parameters before treatment. These parameters included: hemoglobin ≥ 10.0 g/dL; total granulocyte count ≥ 4000/μL; platelet count ≥ 100 000/μL; blood urea nitrogen ≤ 30 mg/dL; creatinine ≤ 2 mg/dL; alkaline phosphatase and aspartate aminotransferase levels less than 2 times the normal upper limit; prothrombin and activated partial thromboplastin not more than 1.4 times higher than the control; and total bilirubin < 35 μmol/L. Exclusion criteria included cachexia, severe jaundice, a large amount of ascites, human immunodeficiency virus positive status, drug addiction, pregnancies, severe pulmonary and cardiac diseases, treatment with a large dose of steroids and a history of an autoimmune disorder or prior history of other malignancies.
Nine patients with clinical stage III, histologically-proven primary HCC were enrolled in the study group. There were 2 women and 7 men ranging in age from 38 to 78 years (56.8 ± 13.9 years, Table 1). After surgical removal of the tumor masses, the patients received radio-, chemo- or interventional therapies. They were monitored after the treatment by computed tomography (CT). None of the patients was receiving steroids at the time of treatment.
Table 1.
Outcomes of immunotherapy in the study group
| Patient no. | Sex | Age (yr) | Pre-treatment KPS | No. of injection | DTH positive ≥ 5 mm | Adverse events | Tumor markers AFP | Imaging response | Survival months till now |
| 1 | F | 38 | 70 | 4 | 60 mm | Fever, rash | Decrease | PD | 14.4D |
| 2 | M | 46 | 100 | 5 | 70 mm | Fever, rash, erythema | Decrease | PD | 19.6A |
| 3 | M | 55 | 75 | 6 | 45 mm | Fever, multiple erythema | Decrease | PR | 18.8A |
| 4 | M | 65 | 90 | 2 | 60 mm | Fever | Decrease | NC | 16.8D |
| 5 | M | 72 | 70 | 5 | 25 mm | Fever, rash | Decrease | NC | 15.2D |
| 6 | M | 78 | 90 | 6 | 65 mm | Fatigue, fever | Decrease | PR | 16.0D |
| 7 | M | 40 | 80 | 4 | 60 mm | Fever, rash | Decrease | NC | 15.6D |
| 8 | M | 54 | 70 | 6 | 5 mm | Fever | Increase | PD | 17.6D |
| 9 | F | 63 | 95 | 7 | 50 mm | Rash | Decrease | PR | 20.0A |
AFP: α-fetoprotein; PR: Partial response; PD: Progress disease, NC: No change; A: Still alive; D: Dead; KPS: Karnofsky performance status; DTH: Delayed-type hypersensitivity.
There were 9 patients with clinical stage III, histologically-proven HCC in the control group. This group consisted of one woman and 8 men, aged 45-66 years. They all underwent similar conventional treatment as those of the study group.
Preparation of primary hepatocellular carcinoma cell suspension
The surgically removed tumor samples were immediately placed in ice-cold phosphate buffered saline (PBS) containing 3 mmol ethylenediaminetetraacetic acid (EDTA). A fine scalpel was used to remove as much adjacent non-tumor tissues as possible. The tumor tissue was then dispersed using a curved needle to create a cell suspension, which was filtered using a 70 μm cell strainer (BD Biosciences, United Kingdom). The cells were resuspended (2 × 108 cells/mL) in ice-cold PBS containing 3 mmol EDTA. The cells were irradiated five times at a total dose of 100 Gy, with a 5-min break during which the cells were placed on ice. They were then washed twice with ice-cold PBS.
Synthesis of α-Gal epitopes on intact tumor cells
Tumor cells prepared as described above were first resuspended at a concentration of 2 × 108 cells/mL in saline containing 0.1 mol Tris-HCl pH 7.0, 15 mmol MnCl2, and 1 mmol UDP-Gal (Sigma-Aldrich, United Kingdom) and then incubated with 1 mU/mL neuraminidase (Sigma-Aldrich, United Kingdom) and 50 μg/mL recombinant bovine α1,3-galactosyltransferase (α1,3GT, produced in our lab) at 37 °C for 1 h with constant rotation. The cells were washed with RPMI-1640 medium, resuspended in RPMI-1640 medium (Gibco, Beijing) supplemented with 10% AB serum and incubated on ice for 1 h to promote maximum binding of the natural anti-Gal antibody to newly synthesized α-Gal epitope on the tumor cell surface. The cells (2 × 108 cells/mL) were washed and sonicated in ice water using a Sonicator-3000 (Misonix, Inc., United States) to destroy all tumor cells and centrifuged briefly to remove the viscous DNA. Finally, the tumor cell lysate was aliquoted and stored at -80 °C for future use.
Synthesis of the α-Gal epitope on tumor cells was analyzed by flow cytometry with 10 μg/mL purified human natural anti-Gal antibody[17], followed by staining with fluorescein isothiocyanate (FITC)-conjugated mouse anti-human immunoglobulin antibody (Dako, Denmark). Intact tumor cells incubated with neuraminidase only were used as a negative control because they lack the α-Gal epitope.
Generation of dendritic cells from peripheral blood mononuclear cells
A concentrated 50-mL leukocyte fraction was generated through restricted peripheral blood leukapheresis. The peripheral blood mononuclear cells (PBMCs) were then purified using Ficoll-Hypaque (Sigma-Aldrich, United Kingdom) density gradient centrifugation. The cells were resuspended in RPMI-1640 medium supplemented with 10% human AB group serum, plated at a concentration of 5 × 106 cells/mL and allowed to adhere to tissue culture plates for DC preparation. The adherent cells were further cultured at 37 °C for 5 d in PRMI-1640 in the presence of 1000 U/mL recombinant human granulocyte-macrophage colony-stimulating factor (GM-CSF) (Amoytop Biotech, Xiamen, China), 500 U/mL recombinant human IL-4 (Amoytop Biotech, China), and 1% penicillin/streptomycin (Sino-American, Beijing). They were next grown for an additional 2 d in the presence of 1000 U/mL tumor necrosis factor-α (TNF-α) (R&D Systems, Inc., Shanghai). The semi-adherent and non-adherent cells were harvested by pipetting and used as DCs for pulsing with the tumor cell lysate prepared as described above[18].
The DCs were incubated with mouse anti-human CD3, CD4, CD8, CD14, CD16, CD19, CD40, CD86, CD80, CD83, CD56, CD45RO, MHC I and HLA-DR monoclonal antibodies (BD Biosciences, Shanghai) for 30 min at 4 °C. Species- and isotype-matched monoclonal antibodies were used as negative controls. Subsequently, the cells were washed and incubated with FITC-conjugated rabbit anti-mouse antibody (Dako, Denmark) and analyzed using FACSCalibur (Becton Dickinson, United States).
Pulsation of autologous dendritic cells by α-Gal expressing tumor cell lysate
The abovementioned DCs (1 × 107 - 4 × 107/mL) were co-cultured with the α-Gal-expressing HCC cell lysate (equal to 2 × 108 cells) in PRMI-1640 medium supplemented with 10% human AB group serum and incubated in 5% CO2 at 37 °C overnight. The co-incubated DCs were washed twice and resuspended in PRMI-1640 medium at a concentration of 1 × 107 cells/mL in preparation for the activation of tumor-specific T/CIKs.
Generation of tumor-specific T/cytokine-induced killer cells
The non-adherent mononuclear cells obtained from patient bone marrow were resuspended in PRMI-1640 supplemented with 10% human AB group serum at a concentration of 1 × 107 cells/mL. IFN-γ (Amoytop Biotech, Xiamen, China; 1000 U/mL) was added on day 1; Muromonab CD-3(OKT3) monoclonal antibody (Sunbio, China; 50 ng/mL), IL2 (Canerotc, China; 500 U/mL) and rIL-1α (Amoytop Biotech, Xiamen, China; 100 U/mL) were added on day 2. Fresh complete medium containing IL2 was added every two or three days to expand the CIKs.
The CIKs were collected on day 12 and co-cultured with the tumor cell lysate-pulsed DCs in PRMI-1640 (a ratio of DC:CIK, 1:30) supplemented with 10% AB serum for an additional 72 h in the presence of IFN-γ, OKT3, and IL2. The co-cultured cells were harvested in 2% human albumin saline for injection.
Clinical study design
The patients of the study group received the first injection on the third day after completing the radio- or chemotherapy, and the subsequent doses were administered every week. The first single dose was initiated with 2 × 109 T/CIK cells and increased to 20 × 109 cells per injection.
Patients were monitored for immediate and delayed toxicities. The survival in months was calculated from the time of hospitalization to the time of death or to the present time. Treatment responses were evaluated by clinical observations, laboratory tests and radiological findings. CT scan was performed every 3 mo to evaluate the lesions after the injections. Tumor size was estimated by direct measurement of the volume of the abnormal enhancement region on the CT. The responses were classified into the following categories: (1) disappearance of the entire tumor, classified as complete response; (2) a reduction in tumor size by 25% or more, as partial response (PR); (3) either a decrease in tumor size by less than 25% or an increase in tumor size by less than 25%, classified as no change (NC); and (4) an increase in tumor size by 25% or more, classified as progressive disease (PD).
Delayed-type hypersensitivity reaction
To test the delayed-type cytotoxicity response, 1 μg of autologous tumor cell lysate with no α-Gal epitopes was administered intradermally into the forearm before and after the injections. A positive DTH reaction was recorded as ≥ 5 mm in diameter of rash or any size of blister at the injection site after 48 h.
Interferon-γ enzyme-linked immunospot assay
Human interferon-γ secretion by effectors was assessed by enzyme-linked immunosorbent spot assay (ELISPOT). Multiplescreen 96-well assay plates (Dakewe, Shenzhen, China) were precoated overnight at 4 °C with anti-IFN-γ antibody according to the manufacturer’s instructions. After washed with PBS and 0.05% Tween-20, the plates were blocked for 1 h at 37 °C with 100 μL of RPMI-1640 supplemented with 1% bovine serum albumin (Sigma-Aldrich, Beijing). Mononuclear cells from the patients were plated in triplicate wells at a density of 1 × 104 cells/100 μL of PRMI-1640 medium supplemented with 10% AB serum, cultured overnight, washed and incubated with anti-IFN-γ mAb-biotin (Dakewe, Shenzhen, China). After washing, goat anti-biotin antibodies (Dakewe, Shenzhen, China) were added and incubated for 1 h at 37 °C, and then 30 μL of activator solution (Dakewe, Shenzhen, China) was used to develop the spots. The number of spots in each well was counted using the Bioreader 4000-PRO-X (Bio-Sys, Germany). The cutoff criterion for positive spots was defined as a spot size more than 3 times the SD greater than the mean value of the spot diameter obtained in the absence of the DCs.
RESULTS
Dendritic cells phenotype
Peripheral mononuclear cells (1.5 × 108 - 6.6 × 108) were induced to differentiate into DCs in the presence of GM-CSF, IL-4 and TNF-α. The final yield of DCs was 1 × 107 - 4 × 107 cells after 7 days of incubation. The phenotype of the purified DCs was 100% HLA class I positive, > 90% CD1a, CD80, and HLA-DR positive, > 70% CD83 and CD86 positive, and CD3, CD4, CD8, CD14, CD19 and CD56 negative.
Synthesis of α-Gal epitopes on intact HCC cells
Intact tumor cells from the 9 patients were isolated for the synthesis of α-Gal epitopes (Figure 1). The tumor cells expressed abundant α-Gal epitopes on their surface after incubation with both neuraminidase and recombinant bovine α1, 3GT as indicated by the extensive binding of human purified natural anti-Gal antibody. However, the cells incubated with recombinant α1,3GT in the absence of neuraminidase weakly expressed the α-Gal epitope at a level that was 10-50 folds lower than the cells incubated with both neuraminidase and recombinant α1,3GT. The reason for this result is that most of the N-acetyllactosamine units on HCC cells were capped by sialic acid and could be exposed by neuraminidase.
Figure 1.
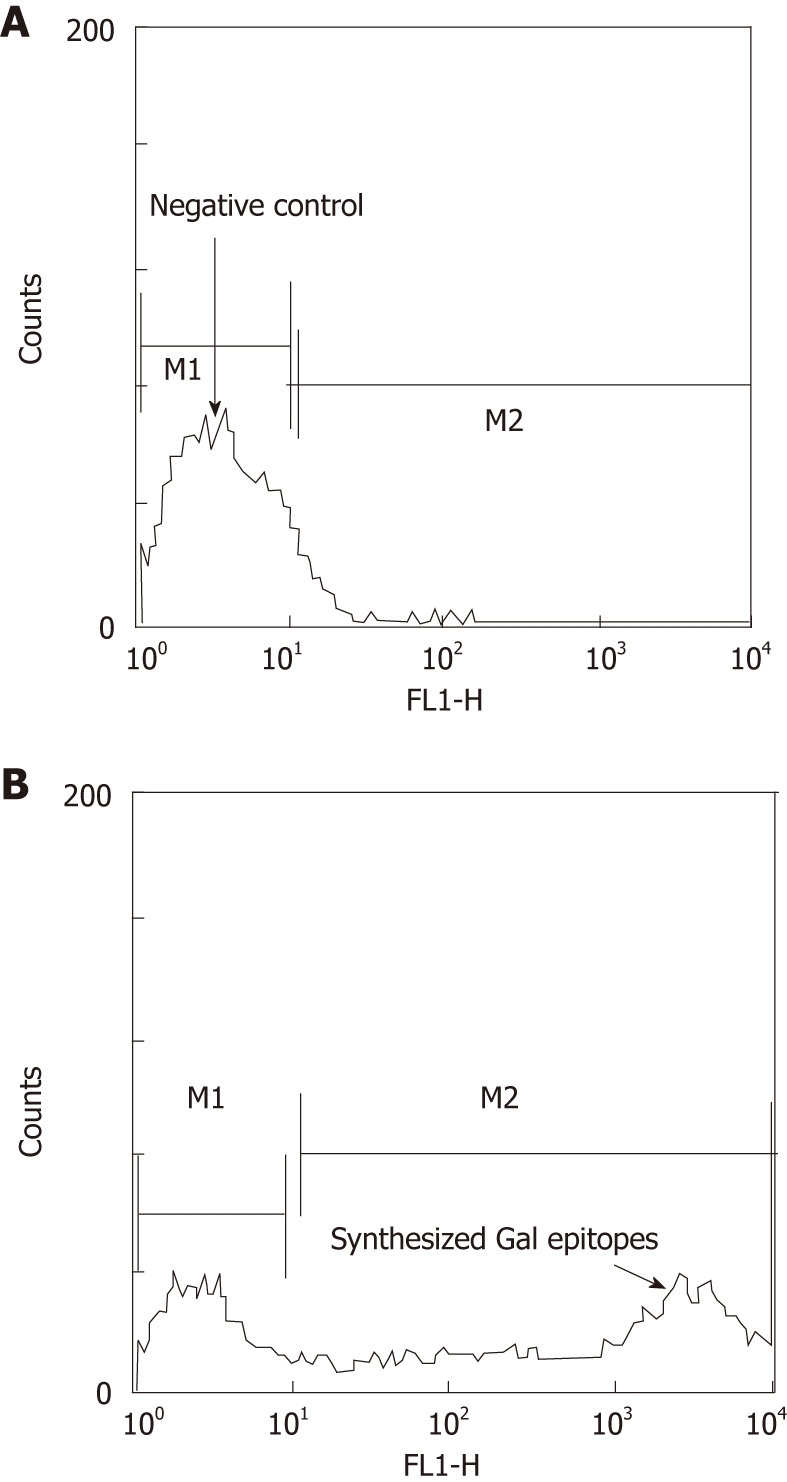
Synthesis of α-Gal epitopes on tumor cell membranes. A: Negative control, tumor cells treated with neuraminidase only, cells with no synthesized epitopes; B: Tumor cells treated with both neuraminidase and α1,3GT, > 60% tumor cells with synthesized α-Gal epitopes.
Safety of α-Gal-pulsed dendritic cells and tumor-specific cytokine-induced killer cells
The DCs pulsed with tumor cell membrane expressing synthesized epitopes were co-cultured with autologous CIKs to induce tumor-specific CIKs. The cell mixture was administered to the patients for 2-7 times with a mean of 5.
The patients did not develop any grade III or IV adverse effects associated with the treatment. These effects are defined by the National Cancer Institute Common Toxicity Criteria. No other serious side effects were observed other than death from tumor progression. There were no substantial changes in blood tests, such as anti-dsDNA, anti-ANA antibodies, etc. All patients developed a mild fever, which lasted 1-3 d after the second and third injections. Five patients showed mild to median rash or multiple erythema, suggesting the presence of delayed-type DTH. All symptoms vanished within 1-2 wk (Table 1).
Delayed-type hypersensitivity reaction
To test the cell-mediated immune responses, tumor lysate with no α-Gal epitopes was administered intradermally into the forearm before and after treatment. A positive DTH skin test reaction was defined as ≥ 5 mm in diameter after 48 h arbitrarily. As a result, the reaction was less than 5 mm in diameter in all studied patients before immunotherapy. However, after treatment, only one patient demonstrated a negative response and all other patients showed a positive reaction; patients 1, 2, 4, 6, 7 and 9 displayed a reaction of more than 50 mm in diameter (Table 1).
Clinical responses
There were no statistically significant differences between the study and control groups in age (56.8 ± 13.9 years vs 49.7 ± 19.7 years; P = 0.39). The overall survival in months was determined from the date of hospitalization to the date of death or present time. The response to the treatment was evaluated by clinical observations and laboratory findings.
The quality of life in the study group improved significantly, e.g., the Karnofsky performance status score increased to 90 in 8 patients after treatment. The median survival time for the study and control groups was 17.1 mo and 10.1 mo, respectively. The Kaplan Meier test showed that the survival curves were significantly different between the two groups (P = 0.00121) (Figure 2). Three patients in the study group were still alive and in stable condition at the time the paper was written, and they have survived for more than 2 years (Table 1); and three patients showed a partial response in the size of the tumor as shown on CT. The lab test indicated that the AFP in the serum decreased in 8 patients after three injections (Table 1).
Figure 2.

Kaplan Meier survival curves of the patients with stage III hepatocellular carcinoma. Study group, n = 9; control group, n = 9; P = 0.00121.
Increase of tumor-specific T cells in patients’ peripheral blood
The phenotype (CD3, CD4, CD8, CD16, CD19, CD45RO and CD56) of the patients’ peripheral blood lymphocytes was analyzed using flow cytometry before and after treatment. A moderate increase in the number of CD3- and CD4-positive cells was observed after immunotherapy; the increase in number of CD45RO+, CD8+ and CD56 cells was significant (Table 2).
Table 2.
Ratios of post- and pre-immunotherapy in cell populations and interferon-γ expression levels
| Patient no. | CD8+ post vs pre | CD45RO+ post vs pre | CD56+ post vs pre | IFN- γ fold increasepost vs pre |
| 1 | 5.09 | 6.78 | 4.81 | 15.32 |
| 2 | 5.22 | 4.97 | 5.66 | 12.25 |
| 3 | 6.33 | 3.78 | 4.77 | 12.45 |
| 4 | 4.81 | 1.42 | 1.39 | 11.11 |
| 5 | 2.15 | 2.43 | 2.17 | 3.88 |
| 6 | 3.67 | 3.66 | 5.86 | 7.94 |
| 7 | 3.78 | 3.94 | 2.81 | 8.31 |
| 8 | 0.55 | 2.02 | 0.65 | 1.03 |
| 9 | 5.23 | 6.67 | 3.55 | 12.73 |
IFN: Interferon; CD: Cluster of differentiation..
The tumor-specific cytotoxicity of PBMCs was assessed by IFN-γ ELISPOT assay. PBMCs were isolated and stimulated with α-Gal-expressing tumor cell lysate. Untreated PBMCs were used as a negative control. The tumor antigens were found to generate strong tumor-specific T cell responses by virtue of their ability to induce increased frequencies of IFN-γ-producing T cells after immunotherapy (Table 2).
DISCUSSION
Previous immunotherapy treatment for primary HCC used passive[19], adoptive[20], and non-specific strategies that yielded limited benefits[21]. There are several possible reasons for the weak and inefficient immune responses against tumor cells. The first and perhaps the most important reason is that the antigenicity of TAA is not strong enough to stimulate the host immune system and cannot be recognized as “foreign”[22]; thus, TAA cannot generate strong tumor-specific immune responses. Another explanation for the inability of TAAs to induce an efficient tumor-specific immune response is the lack of costimulatory molecules on tumor cells[23,24]. The activation of naïve T cells requires recognition by the T-cell receptors (TCRs) of TAA peptides in association with the major histocompatibility complex (MHC) (Signal 1), as well as a costimulatory signal (Signal 2) that is not antigen-specific. The interaction of TCRs on naive T cells with TAAs on tumors without the delivery of signal 2 is thought to result in T cell anergy; this anergy leads to the tolerance of TAAs[23,24], which is similar to the tolerance of normal self-antigens during development. In contrast, if the antigenicity of TAA has been artificially amplified, the uptake of tumor cell membranes by APCs would result in proteolytic degradation of the TAAs, followed by presentation of the TAA peptides in association with MHC class I and class II molecules. These molecules together with costimulatory molecules on APCs would activate naive T helper (Th) cells[25].
The second reason is that T cell function is impaired in most patients with malignant tumors[26]. The immunity in tumor patients is generally quite low, and it may be difficult for the circulating T cells to be activated due to anergy to tumor antigens. To overcome these problems, an ideal method is to enhance the TAA, pulse patient DCs with TAA-enriched tumor cells and use the newly differentiated naïve T/CIK cells from bone marrow stem cells. The DCs then activate the patients’ naïve T/CIK cells to produce tumor-specific immune responders ex vivo.
This new approach consists of four steps to fulfill the ideal requirements for immunotherapy described above. The first step was to increase the antigenicity of TAA based on the bio-synthesis of α-Gal epitopes on the tumor membrane. In the second step, the adhesive cells from PBMCs were induced to differentiate into DCs. At the same time, the isolated non-adhesive mononuclear cells from bone marrow were induced to naïve T/CIKs. In the third step, α-Gal epitope TAAs pulsed DCs. In the final step, the TAA-pulsed DCs and the T/CIKs were co-cultured for a short time to promote TAA delivery to naïve T/CIKs.
In this study, the naïve T/CIKs activated by TAA-pulsed DCs elicited significant cytotoxic responses against tumor cells in 8 of the 9 patients as determined by ELISPOT in the restimulated PBMCs. IFN-γ increased over 10 folds after three injections in patients 1, 2, 3, 4 and 9. While determining whether the injection of DCs and CIKs could increase the T cell reaction against a tumor antigen, we found that the numbers of CD3-, CD4-, CD8-, CD4RO- and CD56-positive cells increased after treatment. Another important finding is that patients treated with the tumor-specific immune cells had a prolonged survival than the control patients. These findings indicate the therapeutic relevance of the observed enhancement of tumor-specific cytotoxicity to HCC.
However, the immunotherapy increased the tumor marker AFP in the serum after the first treatment. We hypothesize that the injection of the immune responders destroyed tumor cells and gradually released the AFP. The specific binding of α-Gal epitope and its natural anti-Gal antibody promotes DC delivery of TAA to T lymphocytes and CIKs in vitro. The ex vivo-activated, tumor-specific immunoresponse cells may account for the robust increase in the survival of late-stage HCC patients in this study as compared with the control group. This study used α-Gal epitopes added to the tumor cell surface to increase tumor antigenicity, TAA-pulsed DCs and ex vivo-activated CIKs rather than TAA-pulsed DCs alone or CIKs alone, which represented a divergence from previously reported immunotherapeutic trials.
In summary, this new therapeutic technique can significantly increase the tumor-specific immune responders in circulation and the survival of advanced HCC patients with no serious side effects. At the same time, clinical studies in other tumors, such as pancreatic carcinoma, lung cancer and lymphoma, are being conducted to reassess the role that the therapy may play in prolonging the survival of the patients.
ACKNOWLEDGMENTS
We thank Dr. Dong XB, King’s College, London, United Kingdom for his reviewing the first draft.
COMMENTS
Background
Hepatocellular carcinoma (HCC), the most common primary neoplasm of the liver, is generally resistant to conventional treatment. This led to the interest in studies of alternative approaches. Immunotherapy is particularly appealing because of the potential specificity of the immune response in eradicating residual tumor cells after conventional treatment. However, the biggest obstacles of tumor immunotherapy are the low antigenicity of tumor cells and the weak immunity of the patients.
Research frontiers
Previous reports have suggested that synthesis of the immunostimulatory xenoantigen α-Gal on malignant cells could be used as a means to enhance tumor-associated antigen (TAA) immunogenicity and that treatment with cytokine-induced killer cells (CIKs) may benefit patients with various types of tumors. Although the α-Gal epitope can increase the antigenicity of tumor cells and CIKs can kill tumor cells, the combination of α-Gal epitope-capped TAAs pulsed dendritic cells (DCs) with the newly differentiated naive T/CIKs to treat tumors has never been reported. In this study, the authors demonstrate that the novel technique has a great potential for tumor treatment.
Innovations and breakthroughs
The authors described a novel immunotherapy for advanced HCC. DCs were pulsed by co-culturing with patient’s α-Gal epitope expressing tumor cells to promote phagocytosis in vitro. The pulsed DCs were employed to activate newly differentiated T cells from bone marrow stem cells which had not been anergized by the tumor antigen. The highly active, tumor-specific immunoresponders were expanded ex vivo.
Applications
The combination of the α-Gal epitope-expressing tumor cell-pulsed DCs and the newly differentiated naive T/CIKs could increase specific anti-tumor immune responses and prolong the survival of HCC patients with no serious side effects. This study represents a new strategy for therapeutic intervention in the treatment of patients with HCC.
Terminology
α-Gal epitope-pulsed dendritic cells (DCs): DCs are potent antigen presenting cells (APCs) that possess the ability to stimulate naïve T cells. The α-Gal epitope expressing tumor cells are co-cultured with DCs in the presence of natural anti-Gal antibody to promote phagocytosis and presentation of tumor antigens to T lymphocytes. Cytokine-induced killer cells (CIKs): CIKs are a heterogeneous subset of ex-vivo expanded T lymphocytes which present a mixed T phenotype. They have been described as highly efficient cytotoxic effector cells capable of recognizing and lysing tumor cell targets in a non-major histocompatibility complex restricted fashion. CIKs could also be activated and attack tumor cells specifically after contact with tumor specific DCs.
Peer review
This is a study looking for the role of ex vivo activated tumor specific CIK therapy on HCC. This is definitely a well planned and performed and well written study. The authors describe a novel immunotherapy for advanced HCC, and the data is compelling.
Footnotes
Supported by Hong Kong Wang Kuan Cheng Grant; and Inner Mongolia Stem Cell Grant, No. kjk10jhg
Peer reviewers: C Bart Rountree, MD, Assistant Professor of Pediatrics and Pharmacology, Penn State College of Medicine, 500 University Drive, H085, Hershey, PA 17033, United States; Mahir M Ozmen, Professor, Department of Surgery, Hacettepe University, Ankara 06830, Turkey
S- Editor Sun H L- Editor Ma JY E- Editor Xiong L
References
- 1.Yu X, Xia W, Zhang T, Wang H, Xie Y, Yang J, Miao J. Enhanced cytotoxicity of IL-24 gene-modified dendritic cells co-cultured with cytokine-induced killer cells to hepatocellular carcinoma cells. Int J Hematol. 2010;92:276–282. doi: 10.1007/s12185-010-0654-1. [DOI] [PubMed] [Google Scholar]
- 2.Hui D, Qiang L, Jian W, Ti Z, Da-Lu K. A randomized, controlled trial of postoperative adjuvant cytokine-induced killer cells immunotherapy after radical resection of hepatocellular carcinoma. Dig Liver Dis. 2009;41:36–41. doi: 10.1016/j.dld.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 3.Shi M, Zhang B, Tang ZR, Lei ZY, Wang HF, Feng YY, Fan ZP, Xu DP, Wang FS. Autologous cytokine-induced killer cell therapy in clinical trial phase I is safe in patients with primary hepatocellular carcinoma. World J Gastroenterol. 2004;10:1146–1151. doi: 10.3748/wjg.v10.i8.1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palmer DH, Midgley RS, Mirza N, Torr EE, Ahmed F, Steele JC, Steven NM, Kerr DJ, Young LS, Adams DH. A phase II study of adoptive immunotherapy using dendritic cells pulsed with tumor lysate in patients with hepatocellular carcinoma. Hepatology. 2009;49:124–132. doi: 10.1002/hep.22626. [DOI] [PubMed] [Google Scholar]
- 5.Yamaguchi S, Tatsumi T, Takehara T, Sasakawa A, Hikita H, Kohga K, Uemura A, Sakamori R, Ohkawa K, Hayashi N. Dendritic cell-based vaccines suppress metastatic liver tumor via activation of local innate and acquired immunity. Cancer Immunol Immunother. 2008;57:1861–1869. doi: 10.1007/s00262-008-0514-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Butterfield LH. Recent advances in immunotherapy for hepatocellular cancer. Swiss Med Wkly. 2007;137:83–90. doi: 10.4414/smw.2006.11077. [DOI] [PubMed] [Google Scholar]
- 7.Chauvin C, Josien R. Dendritic cells as killers: mechanistic aspects and potential roles. J Immunol. 2008;181:11–16. doi: 10.4049/jimmunol.181.1.11. [DOI] [PubMed] [Google Scholar]
- 8.Baratin M, Kayibanda M, Ziol M, Romieu R, Briand JP, Guiller JG, Viguier M. Amino acid modifications in the wild type sequence p53 232-240 overcome the poor immunogenicity of this self tumour epitope. J Pept Sci. 2002;8:327–334. doi: 10.1002/psc.391. [DOI] [PubMed] [Google Scholar]
- 9.Machy P, Serre K, Leserman L. Class I-restricted presentation of exogenous antigen acquired by Fcgamma receptor-mediated endocytosis is regulated in dendritic cells. Eur J Immunol. 2000;30:848–857. doi: 10.1002/1521-4141(200003)30:3<848::AID-IMMU848>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 10.Manca F, Fenoglio D, Li Pira G, Kunkl A, Celada F. Effect of antigen/antibody ratio on macrophage uptake, processing, and presentation to T cells of antigen complexed with polyclonal antibodies. J Exp Med. 1991;173:37–48. doi: 10.1084/jem.173.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galili U, LaTemple DC. Natural anti-Gal antibody as a universal augmenter of autologous tumor vaccine immunogenicity. Immunol Today. 1997;18:281–285. doi: 10.1016/s0167-5699(97)80024-2. [DOI] [PubMed] [Google Scholar]
- 12.Galili U, Chen ZC, DeGeest K. Expression of alpha-gal epitopes on ovarian carcinoma membranes to be used as a novel autologous tumor vaccine. Gynecol Oncol. 2003;90:100–108. doi: 10.1016/s0090-8258(03)00148-3. [DOI] [PubMed] [Google Scholar]
- 13.Poirier N, Blancho G. Recombinant human C1-inhibitor inhibits cytotoxicity induced by allo- and xenoantibodies. Transplant Proc. 2008;40:581–583. doi: 10.1016/j.transproceed.2008.01.045. [DOI] [PubMed] [Google Scholar]
- 14.Rother RP, Fodor WL, Springhorn JP, Birks CW, Setter E, Sandrin MS, Squinto SP, Rollins SA. A novel mechanism of retrovirus inactivation in human serum mediated by anti-alpha-galactosyl natural antibody. J Exp Med. 1995;182:1345–1355. doi: 10.1084/jem.182.5.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buonomano R, Tinguely C, Rieben R, Mohacsi PJ, Nydegger UE. Quantitation and characterization of anti-Galalpha1-3Gal antibodies in sera of 200 healthy persons. Xenotransplantation. 1999;6:173–180. doi: 10.1034/j.1399-3089.1999.00023.x. [DOI] [PubMed] [Google Scholar]
- 16.Lam PT, Leung MW, Tse CY. Identifying prognostic factors for survival in advanced cancer patients: a prospective study. Hong Kong Med J. 2007;13:453–459. [PubMed] [Google Scholar]
- 17.Smorodin EP, Kurtenkov OA, Shevchuk IN, Tanner RH. The isolation and characterization of human natural alphaGal-specific IgG antibodies applicable to the detection of alphaGal-glycosphingolipids. J Immunoassay Immunochem. 2005;26:145–156. doi: 10.1081/ias-200051999. [DOI] [PubMed] [Google Scholar]
- 18.Pajtasz-Piasecka E, Indrová M. Dendritic cell-based vaccines for the therapy of experimental tumors. Immunotherapy. 2010;2:257–268. doi: 10.2217/imt.10.7. [DOI] [PubMed] [Google Scholar]
- 19.Zerbini A, Pilli M, Ferrari C, Missale G. Is there a role for immunotherapy in hepatocellular carcinoma? Dig Liver Dis. 2006;38:221–225. doi: 10.1016/j.dld.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 20.Lamers CH, Sleijfer S, Willemsen RA, Debets R, Kruit WH, Gratama JW, Stoter G. Adoptive immuno-gene therapy of cancer with single chain antibody[scFv(Ig)] gene modified T lymphocytes. J Biol Regul Homeost Agents. 2004;18:134–140. [PubMed] [Google Scholar]
- 21.Cusnir M, Patt YZ. Novel systemic therapy options for hepatocellular carcinoma. Cancer J. 2004;10:97–103. doi: 10.1097/00130404-200403000-00005. [DOI] [PubMed] [Google Scholar]
- 22.Adam JK, Odhav B, Bhoola KD. Immune responses in cancer. Pharmacol Ther. 2003;99:113–132. doi: 10.1016/s0163-7258(03)00056-1. [DOI] [PubMed] [Google Scholar]
- 23.Chamberlain RS, Carroll MW, Bronte V, Hwu P, Warren S, Yang JC, Nishimura M, Moss B, Rosenberg SA, Restifo NP. Costimulation enhances the active immunotherapy effect of recombinant anticancer vaccines. Cancer Res. 1996;56:2832–2836. [PMC free article] [PubMed] [Google Scholar]
- 24.Schultze J, Nadler LM, Gribben JG. B7-mediated costimulation and the immune response. Blood Rev. 1996;10:111–127. doi: 10.1016/s0268-960x(96)90040-5. [DOI] [PubMed] [Google Scholar]
- 25.Feder-Mengus C, Schultz-Thater E, Oertli D, Marti WR, Heberer M, Spagnoli GC, Zajac P. Nonreplicating recombinant vaccinia virus expressing CD40 ligand enhances APC capacity to stimulate specific CD4+ and CD8+ T cell responses. Hum Gene Ther. 2005;16:348–360. doi: 10.1089/hum.2005.16.348. [DOI] [PubMed] [Google Scholar]
- 26.Shirabe K, Motomura T, Muto J, Toshima T, Matono R, Mano Y, Takeishi K, Ijichi H, Harada N, Uchiyama H, et al. Tumor-infiltrating lymphocytes and hepatocellular carcinoma: pathology and clinical management. Int J Clin Oncol. 2010;15:552–558. doi: 10.1007/s10147-010-0131-0. [DOI] [PubMed] [Google Scholar]