

Abstract
AIM: To investigate the role of hepatic peroxisome proliferator-activated receptor-γ (PPAR-γ) in increased susceptibility to endotoxin-induced toxicity in rats with bile duct ligation during endotoxemia.
METHODS: Male Sprague-Dawley rats were subjected to bile duct ligation (BDL). Sham-operated animals served as controls. DNA binding were determined by polymerase chain reaction, Western blotting analysis, and electrophoretic mobility shift assay, respectively. BDL and sham-operated rats received a non-lethal dose of intraperitoneal lipopolysaccharide (LPS) injection (3 mg/kg, i.p.). Additionally, the potential beneficial effects of the PPAR-γ agonist rosiglitazone were determined in BDL and sham-operated rats treated with a non-lethal dose of LPS. Survival was assessed in BDL rats treated with a non-lethal dose of LPS and in sham-operated rats treated at a lethal dose of LPS (6 mg/kg, i.p.).
RESULTS: PPAR-γ activity in rats undergoing BDL was significantly lower than in the sham-controls. Hepatic PPAR-γ gene expression was downregulated at both the mRNA and protein levels. In a parallel group, serum levels of pro-inflammatory cytokines were nearly undetectable in the sham-operated rats. When challenged with a non-lethal dose of LPS (3 mg/kg), the BDL rats had approximately a 2.4-fold increase in serum IL-6, a 2.7 fold increase in serum TNF-α, 2.2-fold increase in serum IL-1 and 4.2-fold increase in serum ALT. The survival rate was significantly lower as compared with that in sham-operated group. Additionally, rosiglitazone significantly reduced the concentration of TNF-α, IL-1β, IL-6 and ALT in sham-operated rats, but not in BDL rats, in response to LPS (3 mg/kg). Also, the survival was improved by rosiglitazone in sham-operated rats challenged with a lethal dose of LPS, but not in BDL rats, even with a non-lethal dose of LPS (3 mg/kg).
CONCLUSION: Obstructive jaundice downregulates hepatic PPAR-γ expression, which in turn may contribute to hypersensitivity towards endotoxin.
Keywords: Obstructive jaundice, Endotoxemia, Liver, Peroxisome proliferator-activated receptor-γ, Rosiglitazone
INTRODUCTION
The high incidence of perioperative septic complications in patients with obstructive jaundice is well documented. This may be related to the high frequency of endotoxemia during cholestasis. It has been reported that up to 50%-70% of patients with obstructive jaundice, even without identifiable source of infection in many cases, have endotoxemia [1-3]. Increased bacterial translocation and absorption of endotoxins, caused by changes in plasma lipoproteins that bind endotoxin, loss of intestinal mucosal integrity, and lack of bile salts in the intestinal lumen, may result in endotoxemia[4-7]. On the other hand, enhanced susceptibility toward endotoxin-induced toxicity, characterized by exaggerated release of several proinflammatory cytokines, more severe organ damage, and reduced survival after non-lethal endotoxemia, is also believed to be responsible for the high morbidity and mortality of patients with obstructive jaundice[8-11]. Previous studies have suggested that prolonged stimulation or over-activation of the Kupffer cells (KCs), the largest numbers of resident macrophages in the liver, is a critical factor in the exaggerated inflammatory response to endotoxin[12-14].
Peroxisome proliferator-activated receptor-γ (PPAR-γ) is a ligand activated transcription factor that belongs to the nuclear receptor family. It is a key regulator of adipocyte differentiation, lipid metabolism, glucose homeostasis, and cell proliferation. Activation of PPARγ could produce anti-inflammatory effects by repressing the expression of inflammatory cytokines in activated macrophages and monocytes[15,16]. In endotoxemia and sepsis, it has been reported that hepatic PPAR-γ expression in KCs is downregulated, possibly by increased plasma tumor necrosis factor-α (TNF-α). Decreased PPAR-γ expression, in turn, increased the production of proinflammatory cytokines and tissue injury[17]. During obstructive jaundice, portal or systemic endotoxemia is frequently observed. Also, circulating proinflammatory cytokines (e.g., TNF-α and interleukin-6) are increased even in the absence of exogenous stimuli under cholestatic conditions[18-21]. We therefore speculated that the PPAR-γ expression and function in the liver are decreased by endotoxemia and/or increased proinflammatory cytokines during obstructive jaundice.
Here we examined whether hepatic PPAR-γ expression and function are decreased in a rat model of obstructive jaundice, to seek for a possible correlation between alteration of PPAR-γ expression and increased susceptibility to endotoxin.
MATERIALS AND METHODS
Animals
Male Sprague-Dawley rats (250-300 g) were purchased from the Animal Center of Shanghai Jiao Tong University School of Medicine (Shanghai, China) and were housed in an air-filtered room at 22-25 °C on a 12-h light/dark cycle, with unlimited access to water and standard rat chow. All experimental procedures were in accordance with the institutional animal care guidelines and approved by the local ethic committees.
Bile duct ligation
Rats were anesthetized with sodium pentobarbital (50 mg/kg, i.p.). The common bile duct was ligated and divided after laparotomy. Sham-operated rats underwent the same procedure without bile duct ligation.
Experimental protocol
Three sets of experiments were carried out. In the first experiment, rats were sacrificed at day 7 after the surgery to analyze PPAR-γ expression and activation in the liver. In the second experiment, endotoxemia was established by a non-lethal dose of lipopolysaccharide (LPS) (Escherichia coli 0111:B4; Sigma, 3 mg/kg, i.p.) in the BDL or sham-operated rats. Blood was collected for analysis of pro-inflammatory cytokines (e.g., TNF-α, IL-6 and IL-1β) and alanine aminotransferase (ALT) 2 h after LPS challenge. Survival was also monitored in another group. In the third experiment, the potential beneficial effects of the PPAR-γ agonist rosiglitazone were examined in BDL and sham-operated rats treated with a non-lethal dose of LPS (3 mg/kg, i.p.). Survival was examined in BDL rats treated with a non-lethal dose of LPS and in sham-operated rats treated at a lethal dose of LPS (6 mg/kg, i.p.). Rosiglitazone (3 mg/kg, i.p.) or vehicle (10% dimethyl sulfoxide) was administered intraperitoneally as a bolus 15 min prior to LPS injection.
RNA extraction and peroxisome proliferator-activated receptor-γ gene expression
Total RNA was extracted from hepatic tissues using TRIzol reagent (Invitrogen, United States) according to the manufacturer’s protocol. Primers were obtained from Shanghai Sangon Biologic Engineering and Technology and Service (Shanghai, China). The following primer sequences were used: for PPAR-γ mRNA (forward: 5’-ACACCATGCTGGCCTCCCTGA-3’; reverse: 5’-AAGCCTGGGCGGTCTCCACT-3’; size 220 bp), and for β-actin (forward: 5'-CCACACCCGCCACCAGTTCG-3’; reverse: 5’-CTTGCTCTGGGCCTCGTCGC-3’; size 205 bp). Amplification and detection were performed with an ABI PRISM 7300 real-time polymerase chain reaction (PCR) System (Applied Biosystems, Foster City, California, United States) as follows: 30 s at 95 °C, and 40 cycles at 95 °C for 5 s and at 60 °C for 31 s. The DNA-binding dye SYBER Green I for the detection of PCR products was used. The reaction mixture (RT-PCR kit, Code DRR063A, Takara) contained 25 μL Premix Ex Taq, 1 μL forward and reverse primers, 1 μL ROX reference dye, 4 μL cDNA (equivalent to 20 ng total RNA) in a final volume of 50 μL. The amount of gene transcript was measured using a comparative (2-[DELTA][DELTA]CT) method by Applied Biosystems. The reference gene β-actin was used for normalization of the expression data.
Western blotting analysis of peroxisome proliferator-activated receptor-γ proteins
The nuclear extracts were prepared from liver tissues using a nuclear extract kit (Active Motif, Carlsbad, CA), following the manufacturer’s protocol. Protein concentration was measured by the bicinchoninic acid assay method (Pierce). Liver nuclear extracts containing equal amounts of protein were separated in a 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (PAGE). Separated protein bands were transferred onto a nitrocellulose filter. The filter was blocked with 5% non-fat milk, primary rabbit-anti-mouse PPAR-γ antibody (1:200 dilution, Santa Cruz Technologies, Santa Cruz, CA) and β-actin monoclonal antibody (1:4000 dilution, Sigma-Aldrich) were used. After incubation with a horseradish peroxidase-conjugated secondary antibody, chemoluminescence agent A was mixed with B at equal volumes and evenly smeared onto the filter. The chemiluminescence signals were quantified using a Chemi-Smart 3000 (Vilber Lourmat; Marne-la-Vallée, France).
Electrophoretic mobility shift assay of peroxisome proliferator-activated receptor-γ activation
Electrophoretic mobility shift assay (EMSA) was used to detect specific binding of the transcription factor PPAR-γ to its specific DNA consensus sequence. Nuclear protein/DNA binding reactions were conducted for 20 min at room temperature in a 20-μL reaction volume containing 2 μL 10 × binding buffer, 1 μL polydI-dC, 1 μL 50% glycerol, 1 μL 1% NP-40, 1 μL of 1 mol/L KCl, 1 μL 100 mmol/L MgCl2, 1 μL 200 mmol/L EDTA (all the reagents are included in the LightShift Chemiluminescent EMSA kit; Pierce), 20 fmol biotin-labeled probe (PPAR-γ consensus sequence 5’-GGGGTCAGTAAGTCAGAGGCCAGGGA-3’), and 2 μL nuclear extract. Binding reactions were analyzed using 8% PAGE. After blotting to a nylon membrane, the labeled oligonucleotides were detected with the LightShift Chemiluminescent EMSA kit (Pierce). The relative intensity of the bands was analyzed using an LAS-1000 luminoimage analyzer (Fuji Film, Tokyo, Japan).
Measurement of serum cytokines and alanine aminotransferase
Serum concentrations of TNF-α, IL-1β, or IL-6 were quantified using enzyme-linked immunosorbent assay (R&D Systems). Serum ALT levels were determined with an autoanalyzer (Model 7600, Hitachi Co., Tokyo, Japan).
Statistical analysis
The data were expressed as means ± SE. Data were analyzed by analysis of variance, followed by the Student-Newman-Keuls test. The survival curve was estimated by the Kaplan-Meier method and statistical significance was assessed by log-rank test. P values less than 0.05 were considered as significant.
RESULTS
Peroxisome proliferator-activated receptor-γ activation and expression are decreased upon bile duct ligation-induced cholestasis
We started the investigation by measuring modification of PPAR-γ activation and expression upon BDL and consequent cholestasis. We measured levels of PPAR-γ activation through electrophoretic mobility shift assay of PPAR-γ DNA binding in nuclear extracts from liver of treated and control rats (Figure 1A). PPAR-γ activity in rats undergoing BDL was significantly lower than in the sham-controls (0.62 ± 0.07 vs 1.00 ± 0.08, P = 0.0257). Hepatic PPAR-γ gene expression was downregulated at both the mRNA (1 ± 0.09 vs 0.09 ± 0.03, P = 0.0000002, Figure 1B) and protein levels (0.70 ± 0.02 vs 0.59 ± 0.01, P = 0.0089, Figure 1C).
Figure 1.

Modified measurement of peroxisome proliferator-activated receptor-γ activation and expression upon bile duct ligation and consequent cholestasis. A: Electrophoretic mobility shift assay (top panel) of peroxisome proliferator-activated receptor (PPAR)-γ. DNA binding in nuclear extracts from liver, and relative densitometric analysis (lower panel). Data are expressed as mean ± SE (n = 3). aP < 0.05 for bile duct ligation (BDL) group vs sham group; B: PPAR-γ mRNA expression in the liver. PPAR-γ expression in the liver of BDL rats decreased significantly compared with the sham-group determined by quantitative real-time reverse transcription-polymerase chain reaction. Data are expressed as mean ± SE (n = 6). bP < 0.01 for BDL group vs sham group; C: Western blotting analysis (top panel) of PPAR-γ in nuclear extracts from liver, and relative densitometric analysis (lower panel, n = 3). Data are expressed as mean ± SE (n = 3). bP < 0.01 for BDL group vs sham group.
Enhanced susceptibility toward endotoxin-induced toxicity in rats with pre-existing cholestasis
In a parallel group, serum levels of pro-inflammatory cytokines were nearly undetectable in the sham-operated rats. Biliary obstruction per se resulted in the induction of pro-inflammatory cytokines, such as TNF-α, IL-1β and IL-6 (66.0 ± 6.5 vs 27.9 ± 3.2, P = 0.00036; 56.0 ± 7.6 vs 19.7 ± 2.6, P = 0.0011; 60.2 ± 5.8 vs 30.7 ± 3.5, P = 0.0014, Table 1), and liver injury, as indicated by increased circulating level of ALT (119.7 ± 11.1 vs 41.2 ± 3.2, P = 0.00005, Table 1). When challenged with a non-lethal dose of LPS (3 mg/kg), the BDL rats had approximately 2.75-fold increase in serum TNF-α, 2.21-fold increase in serum IL-1β, 2.41-fold increase in serum IL-6 and 4.17-fold increase in serum ALT (2364.7 ± 196.3 vs 861.1 ± 44.2, P = 0.00006; 2373.0 ± 265.1 vs 1073 ± 97.7, P = 0.00098; 10802.0 ± 853.2 vs 4476.7 ± 430.2, P = 0.00005; 305.0 ± 37.5 vs 73.2 ± 7.7, P = 0.0001, Table 1). The survival rate was significantly lower than in the sham-operated group (20% vs 90%, P = 0.000009, Figure 2). These results indicated that enhanced susceptibility toward endotoxin-induced toxicity is present in BDL rats, which is consistent with previous studies.
Table 1.
Serum alanine aminotransferase, tumor necrosis factor-α, IL-1β and IL-6 levels in rats after administration of lipopolysaccharide (n = 6, mean ± SE)
| Sham | BDL | Sham + LPS | BDL + LPS | |
| ALT (U/L) | 41.2 ± 3.2 | 119.7±11.1a | 73.2 ± 7.7 | 305.0 ± 37.5a |
| TNF-α (pg/mL) | 27.9 ± 3.2 | 66.0 ± 6.5a | 861.1 ± 44.2 | 2364.7 ± 196.3a |
| IL-1β (pg/mL) | 19.7 ± 2.6 | 56.0 ± 7.6a | 1073 ± 97.7 | 2373.0 ± 265.1a |
| IL-6 (pg/mL) | 30.7 ± 3.5 | 60.2 ± 5.8a | 4476.7 ± 430.2 | 10802.0 ± 853.2a |
P < 0.01 for bile duct ligation group vs Sham group. BDL: Bile duct ligation; LPS: Lipopolysaccharide; ALT: Alanine aminotransferase; TNF-α: Tumor necrosis factor-α.
Figure 2.
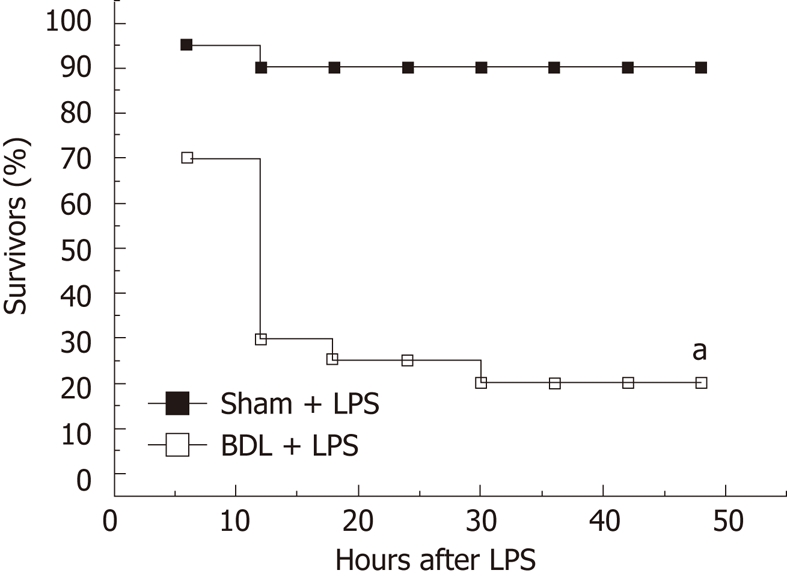
Survival rates of rats after administration of lipopolysaccharide (3 mg/kg, i.p.). The survival curve was estimated by the Kaplan-Meier method and the log-rank method was used to compare differences in survival rates between groups (n = 20). aP < 0.01 for bile duct ligation group vs sham group. BDL: Bile duct ligation; LPS: Lipopolysaccharide.
Peroxisome proliferator-activated receptor-γ agonist rosiglitazone does not protect jaundiced rats from endotoxemia
To further verify a possible correlation between alterations of PPAR-γ function and increased susceptibility to endotoxin, we investigated whether PPAR-γ agonist rosiglitazone had anti-inflammatory effects in BDL rats. Treatment with rosiglitazone significantly reduced the concentration of TNF-α, IL-1β, IL-6, and ALT in sham-operated rats, but not in BDL rats, in response to LPS (3 mg/kg) (Table 2). The survival was improved by rosiglitazone in sham-operated rats challenged with a lethal dose of LPS (6 mg/kg) (50% vs 20%, P = 0.040) but not in BDL rats, even with a non-lethal dose of LPS (35% vs 25%, P = 0.49) (Figure 3). These data indicated that endogenous anti-inflammatory pathway of nuclear receptor PPAR-γ is impaired by obstructive jaundice, which in turn may be at least in part, responsible for the increased susceptibility to endotoxin.
Table 2.
Effect of pretreatment with rosiglitazone on serum alanine aminotransferase, tumor necrosis factor-α, IL-1β and IL-6 levels in rats after administration of lipopolysaccharide (n=6, mean ± SE)
| Sham + vehicle | BDL + vehicle | Sham + ROSI | BDL + ROSI | |
| ALT (U/L) | 78.6 ± 8.1 | 329.4 ± 40.5 | 42.3 ± 5.7a | 303.0 ± 46.6 |
| TNF-α (pg/mL) | 770.5 ± 52.5 | 2017.9 ± 167.5 | 517.0 ± 69.7a | 1984.0 ± 181.1 |
| IL-1β (pg/mL) | 1096.1 ± 101.7 | 2345.2 ± 194.7 | 648.4 ± 111.4a | 2226.7 ± 209.8 |
| IL-6 (pg/mL) | 4479.4 ± 377.4 | 10774.4 ± 675.4 | 2758.5 ± 497.7a | 9847.1 ± 865.1 |
P < 0.05 for vehicle (dimethyl sulfoxide) pretreatment vs rosiglitazone pretreatment group. BDL: Bile duct ligation; ROSI: Rosiglitazone; ALT: Alanine aminotransferase; TNF-α: Tumor necrosis factor-α.
Figure 3.
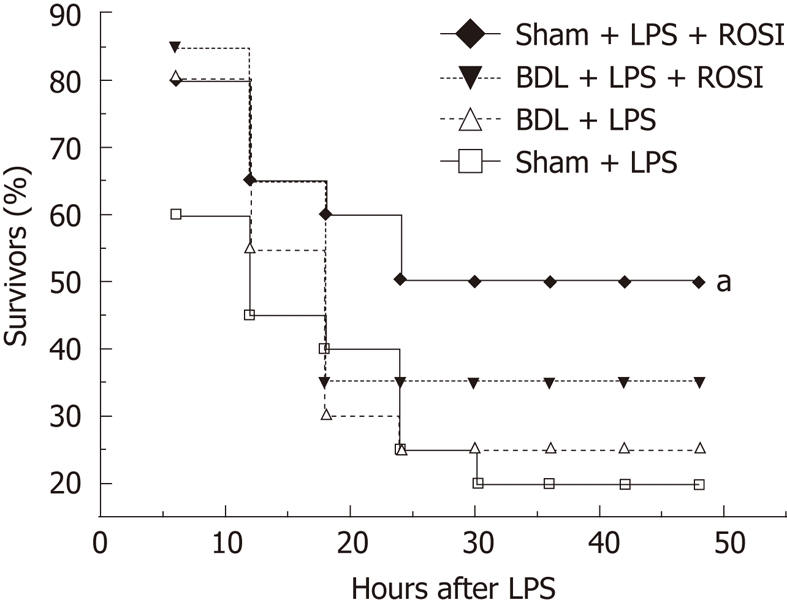
Effects of pretreatment with rosiglitazone (3 mg/kg, i.p.) on survival rates in rats after administration of lipopolysaccharide (6 mg/kg for sham-group and 3 mg/kg for bile duct ligation group, i.p.). The survival curve was estimated by the Kaplan-Meier method and the log-rank method was applied to compare differences in survival rates between groups (n = 20). Rosiglitazone pretreatment significantly improve the survival in sham + lipopolysaccharide (LPS) (6 mg/kg) group but not in bile duct ligation + LPS (3 mg/kg) group, aP < 0.05 vs sham + LPS group. BDL : Bile duct ligation; ROSI: Rosiglitazone.
DISCUSSION
In this study, both the expression and function of PPAR-γ in the liver were significantly depressed in rats with BDL as compared with the sham-operated rats. Concomitant to the decrease in PPAR-γ DNA binding, we observed a markedly increased susceptibility to endotoxin, evidenced by higher degree of liver injury, enhanced proinflammatory cytokine release, and a higher mortality. The PPAR-γ agonist rosiglitazone failed to protect BDL rats against endotoxemia.
Despite the use of broad-spectrum antibiotics and improvement in surgical technique, patients with cholestatic liver disease continue to experience a high incidence of postoperative morbidity and mortality[22,23]. Belghiti et al[24] showed that the mortality of patients with obstructive jaundice is much higher (21%) than that of the patients with normal liver function. Among the multiple complications from obstructive jaundice, gram-negative bacterial sepsis is the most common cause of secondary morbidity and mortality[25]. Animal studies have demonstrated that obstructive jaundice exaggerates the release of several cytokines after endotoxin administration, including TNF-α, IL-1β, and IL-6[8-11].
KCs are the largest macrophage population and form the first line of defense against microorganisms entering the portal circulation. It has been demonstrated that the KCs are the primary source of circulating TNF-α and IL-6 in response to LPS[26]. In the context of appropriate immune response, KC activation plays a protective role during the acute phase of hepatopathies. However, in response to prolonged bacterial or endotoxin challenge, KC over-activation may increase the severity of organ damage and lethality. With regards to obstructive jaundice, previous studies indicate that liver KCs are involved in the exaggerated cytokine secretion after endotoxin challenge[12,13]. In jaundiced animals undergoing endotoxin challenge, blockade of KC function with gadolinium chloride or TNF-α antibody has been shown to improve survival and to suppress the systemic proinflammatory response[14]. Consequently, altered function of KCs during cholestasis is thought to play a decisive role in the increased susceptibility to endotoxin-induced toxicity.
PPARs are ligand-activated transcription factors belonging to the nuclear hormone receptor superfamily. Recently, it has been found that PPAR-γ activation inhibits the expression of several inflammatory response genes in activated macrophages, including the genes encoding inducible nitric oxide synthase, TNF-α, gelatinase B, and COX-2[27,28]. Moreover, activation of PPAR-γ reduces the organ injury/dysfunction caused by endotoxin[29] and by hemorrhage and resuscitation[30], as well as systemic inflammation caused by zymosan[31] and by cecal ligation and puncture[32] in rodents. In this study, the expression and function of hepatic PPAR-γ were decreased during obstructive jaundice. PPAR-γ agonist rosiglitazone failed to protect BDL rats from lethal endotoxemia, suggesting that the downregulation of PPAR-γ in jaundiced rats is of functional significance.
It should be pointed out that hepatic PPAR-γ is expressed in both KCs and hepatocytes (HCs). We did not examine the source of PPAR-γ activity in this study. Nevertheless, previous studies have indicated that PPAR-γ gene expression decreased significantly in KCs at 20 h after sepsis by cecal ligation and puncture, whereas PPAR-γ expression in HCs was not altered. Moreover, when isolated KCs or HCs from normal rats were stimulated with LPS or TNF-α for 20 h, KC PPAR-γ protein levels were all significantly decreased. In contrast, neither LPS nor TNF-α affects PPAR-γ protein levels in HCs either in monoculture or in co-culture with KCs. These findings suggest that the KCs and HCs respond to LPS and TNF-α differentially[17]. Therefore, we believe that the downregulated PPAR-γ expression in the liver during obstructive jaundice is most likely due to decreased PPAR-γ in KCs but not in HCs.
Recent studies have indicated the potential efficacy of PPAR-γ ligands as novel therapeutic approaches in sepsis, inflammation and reperfusion injury. However, our observation that treatment with a PPAR-γ ligand fails to provide protection in cholestatic animals suggests that activation of the endogenous PPAR-γ pathway may need to be tailored to the specific conditions, e.g., obstructive jaundice.
Evidence suggests that the PPAR-γ expression is a function of the inflammatory response. For instance, in rats subjected to sepsis by cecal ligation and puncture or double-hit hemorrhage and sepsis, PPAR-γ expression is downregulated in the liver, bronchial epithelium, and vascular endothelium[33-35]. In mice subjected to endotoxin administration, PPAR-γ expression is also markedly reduced in adipose tissues, heart and lungs[36-39]. Consistent with these reports, the current study demonstrated that biliary obstructive jaundice significantly increases serum amounts of TNF-α, IL-6 and IL-1β. Thus, increased proinflammatory cytokines (e.g., TNF-α) during cholestasis may result in downregulation of the PPAR-γ in the liver.
In conclusion, biliary obstructive jaundice downregulates hepatic PPAR-γ expression and function, which in turn may contribute to enhanced susceptibility to endotoxin-induced toxicity.
COMMENTS
Background
Biliary obstructive jaundice increases susceptibility to endotoxin-induced toxicity. However, the underlying molecular mechanisms are not fully understood.
Research frontiers
Peroxisome proliferator-activated receptor-γ (PPAR-γ) is a member of the nuclear receptor family of ligand-activated transcription factors. Activation of PPARγ could produce anti-inflammatory effects. In this study, the authors demonstrated that hepatic PPAR-γ is down-regulated upon obstructive jaundice, and provided some evidence suggesting that the down-regulation of PPAR-γ contributes to the hypersensitivity to endotoxin.
Innovations and breakthroughs
The expression and function of hepatic PPAR-γ were significantly decreased in the bile duct ligation rats compared with the control rats. Down-regulation of PPAR-γ was accompanied by exaggerated inflammatory response. Treatment with the PPAR-γ agonist rosiglitazone protected sham-operated, but not BDL rats, from endotoxemia.
Applications
Recent studies have suggested the potential efficacy of PPAR-γ ligands as novel therapeutic approaches in sepsis, inflammation, and reperfusion injury. The treatment with a PPAR-γ ligand fails to provide protection of cholestatic animals suggests that activation of the endogenous PPAR-γ pathway needs to be tailored to the specific conditions, e.g., obstructive jaundice.
Peer review
This is an elegant study from a biological observation to a potential clinical aspect.
Footnotes
Supported by China Postdoctoral Science Foundation, No. 20080440626; National Natural Science Foundation of China, No. 30700788 and No. 81001545; and Shanghai Leading Academic Discipline Project, No. S30203
Peer reviewers: Dr. Prasanna Santhekadur, Human and Molecular Genetics, VCU, PO Box 980035, 1220 East Broad, Room 7055, Human and Molecular Genetics, Richmond 23298, VA, United States; Dr. Yi-Hang Wu, China Jiliang University, 258 Xueyuan Street, Hangzhou 310018, Zhejiang Province, China; Dr. Frank G Schaap, Tytgat Institute for Liver and Intestinal Research, Meibergdreef 69-71, Amsterdam 1105 BK, The Netherlands
S- Editor Lv S L- Editor Ma JY E- Editor Xiong L
References
- 1.Ingoldby CJ, McPherson GA, Blumgart LH. Endotoxemia in human obstructive jaundice. Effect of polymyxin B. Am J Surg. 1984;147:766–771. doi: 10.1016/0002-9610(84)90197-1. [DOI] [PubMed] [Google Scholar]
- 2.Hunt DR, Allison ME, Prentice CR, Blumgart LH. Endotoxemia, disturbance of coagulation, and obstructive jaundice. Am J Surg. 1982;144:325–329. doi: 10.1016/0002-9610(82)90011-3. [DOI] [PubMed] [Google Scholar]
- 3.Pain JA, Bailey ME. Measurement of operative plasma endotoxin levels in jaundiced and non-jaundiced patients. Eur Surg Res. 1987;19:207–216. doi: 10.1159/000128702. [DOI] [PubMed] [Google Scholar]
- 4.Kocsár LT, Bertók L, Várterész V. Effect of bile acids on the intestinal absorption of endotoxin in rats. J Bacteriol. 1969;100:220–223. doi: 10.1128/jb.100.1.220-223.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bailey ME. Endotoxin, bile salts and renal function in obstructive jaundice. Br J Surg. 1976;63:774–778. doi: 10.1002/bjs.1800631011. [DOI] [PubMed] [Google Scholar]
- 6.Deitch EA, Sittig K, Li M, Berg R, Specian RD. Obstructive jaundice promotes bacterial translocation from the gut. Am J Surg. 1990;159:79–84. doi: 10.1016/s0002-9610(05)80610-5. [DOI] [PubMed] [Google Scholar]
- 7.Van Bossuyt H, Desmaretz C, Gaeta GB, Wisse E. The role of bile acids in the development of endotoxemia during obstructive jaundice in the rat. J Hepatol. 1990;10:274–279. doi: 10.1016/0168-8278(90)90132-b. [DOI] [PubMed] [Google Scholar]
- 8.Pitt HA, Cameron JL, Postier RG, Gadacz TR. Factors affecting mortality in biliary tract surgery. Am J Surg. 1981;141:66–72. doi: 10.1016/0002-9610(81)90014-3. [DOI] [PubMed] [Google Scholar]
- 9.Greve JW, Gouma DJ, Soeters PB, Buurman WA. Suppression of cellular immunity in obstructive jaundice is caused by endotoxins: a study with germ-free rats. Gastroenterology. 1990;98:478–485. doi: 10.1016/0016-5085(90)90841-n. [DOI] [PubMed] [Google Scholar]
- 10.Sewnath ME, Van Der Poll T, Ten Kate FJ, Van Noorden CJ, Gouma DJ. Interleukin-1 receptor type I gene-deficient bile duct-ligated mice are partially protected against endotoxin. Hepatology. 2002;35:149–158. doi: 10.1053/jhep.2002.30272. [DOI] [PubMed] [Google Scholar]
- 11.Lechner AJ, Velasquez A, Knudsen KR, Johanns CA, Tracy TF, Matuschak GM. Cholestatic liver injury increases circulating TNF-alpha and IL-6 and mortality after Escherichia coli endotoxemia. Am J Respir Crit Care Med. 1998;157:1550–1558. doi: 10.1164/ajrccm.157.5.9709067. [DOI] [PubMed] [Google Scholar]
- 12.Nehéz L, Andersson R. Compromise of immune function in obstructive jaundice. Eur J Surg. 2002;168:315–328. doi: 10.1080/11024150260284815. [DOI] [PubMed] [Google Scholar]
- 13.Kennedy JA, Clements WD, Kirk SJ, McCaigue MD, Campbell GR, Erwin PJ, Halliday MI, Rowlands BJ. Characterization of the Kupffer cell response to exogenous endotoxin in a rodent model of obstructive jaundice. Br J Surg. 1999;86:628–633. doi: 10.1046/j.1365-2168.1999.01114.x. [DOI] [PubMed] [Google Scholar]
- 14.Kennedy JA, Lewis H, Clements WD, Kirk SJ, Campbell G, Halliday MI, Rowlands BJ. Kupffer cell blockade, tumour necrosis factor secretion and survival following endotoxin challenge in experimental biliary obstruction. Br J Surg. 1999;86:1410–1414. doi: 10.1046/j.1365-2168.1999.01269.x. [DOI] [PubMed] [Google Scholar]
- 15.von Knethen A, Soller M, Brüne B. Peroxisome proliferator-activated receptor gamma (PPAR gamma) and sepsis. Arch Immunol Ther Exp (Warsz) 2007;55:19–25. doi: 10.1007/s00005-007-0005-y. [DOI] [PubMed] [Google Scholar]
- 16.Andersen V, Christensen J, Ernst A, Jacobsen BA, Tjønneland A, Krarup HB, Vogel U. Polymorphisms in NF-κB, PXR, LXR, PPARγ and risk of inflammatory bowel disease. World J Gastroenterol. 2011;17:197–206. doi: 10.3748/wjg.v17.i2.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou M, Wu R, Dong W, Jacob A, Wang P. Endotoxin downregulates peroxisome proliferator-activated receptor-gamma via the increase in TNF-alpha release. Am J Physiol Regul Integr Comp Physiol. 2008;294:R84–R92. doi: 10.1152/ajpregu.00340.2007. [DOI] [PubMed] [Google Scholar]
- 18.Papakostas C, Bezirtzoglou E, Pitiakoudis M, Polychronidis A, Simopoulos C. Endotoxinemia in the portal and the systemic circulation in obstructive jaundice. Clin Exp Med. 2003;3:124–128. doi: 10.1007/s10238-003-0015-y. [DOI] [PubMed] [Google Scholar]
- 19.Bemelmans MH, Gouma DJ, Greve JW, Buurman WA. Cytokines tumor necrosis factor and interleukin-6 in experimental biliary obstruction in mice. Hepatology. 1992;15:1132–1136. doi: 10.1002/hep.1840150626. [DOI] [PubMed] [Google Scholar]
- 20.Pain JA. Reticulo-endothelial function in obstructive jaundice. Br J Surg. 1987;74:1091–1094. doi: 10.1002/bjs.1800741207. [DOI] [PubMed] [Google Scholar]
- 21.Bemelmans MH, Greve JW, Gouma DJ, Buurman WA. Increased concentrations of tumour necrosis factor (TNF) and soluble TNF receptors in biliary obstruction in mice; soluble TNF receptors as prognostic factors for mortality. Gut. 1996;38:447–453. doi: 10.1136/gut.38.3.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nomura T, Shirai Y, Hatakeyama K. Impact of bactibilia on the development of postoperative abdominal septic complications in patients with malignant biliary obstruction. Int Surg. 1999;84:204–208. [PubMed] [Google Scholar]
- 23.Thompson JN, Edwards WH, Winearls CG, Blenkharn JI, Benjamin IS, Blumgart LH. Renal impairment following biliary tract surgery. Br J Surg. 1987;74:843–847. doi: 10.1002/bjs.1800740932. [DOI] [PubMed] [Google Scholar]
- 24.Belghiti J, Hiramatsu K, Benoist S, Massault P, Sauvanet A, Farges O. Seven hundred forty-seven hepatectomies in the 1990s: an update to evaluate the actual risk of liver resection. J Am Coll Surg. 2000;191:38–46. doi: 10.1016/s1072-7515(00)00261-1. [DOI] [PubMed] [Google Scholar]
- 25.Pain JA, Cahill CJ, Bailey ME. Perioperative complications in obstructive jaundice: therapeutic considerations. Br J Surg. 1985;72:942–945. doi: 10.1002/bjs.1800721203. [DOI] [PubMed] [Google Scholar]
- 26.Hoffmann R, Grewe M, Estler HC, Schulze-Specking A, Decker K. Regulation of tumor necrosis factor-alpha-mRNA synthesis and distribution of tumor necrosis factor-alpha-mRNA synthesizing cells in rat liver during experimental endotoxemia. J Hepatol. 1994;20:122–128. doi: 10.1016/s0168-8278(05)80478-7. [DOI] [PubMed] [Google Scholar]
- 27.Jiang C, Ting AT, Seed B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–86. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- 28.Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- 29.Collin M, Patel NS, Dugo L, Thiemermann C. Role of peroxisome proliferator-activated receptor-gamma in the protection afforded by 15-deoxydelta12,14 prostaglandin J2 against the multiple organ failure caused by endotoxin. Crit Care Med. 2004;32:826–831. doi: 10.1097/01.ccm.0000114821.25573.e7. [DOI] [PubMed] [Google Scholar]
- 30.Abdelrahman M, Collin M, Thiemermann C. The peroxisome proliferator-activated receptor-gamma ligand 15-deoxyDelta12,14 prostaglandin J2 reduces the organ injury in hemorrhagic shock. Shock. 2004;22:555–561. doi: 10.1097/01.shk.0000144132.13900.24. [DOI] [PubMed] [Google Scholar]
- 31.Cuzzocrea S, Pisano B, Dugo L, Ianaro A, Patel NS, Di Paola R, Genovese T, Chatterjee PK, Fulia F, Cuzzocrea E, et al. Rosiglitazone, a ligand of the peroxisome proliferator-activated receptor-gamma, reduces the development of nonseptic shock induced by zymosan in mice. Crit Care Med. 2004;32:457–466. doi: 10.1097/01.CCM.0000109446.38675.61. [DOI] [PubMed] [Google Scholar]
- 32.Zingarelli B, Sheehan M, Hake PW, O’Connor M, Denenberg A, Cook JA. Peroxisome proliferator activator receptor-gamma ligands, 15-deoxy-Delta(12,14)-prostaglandin J2 and ciglitazone, reduce systemic inflammation in polymicrobial sepsis by modulation of signal transduction pathways. J Immunol. 2003;171:6827–6837. doi: 10.4049/jimmunol.171.12.6827. [DOI] [PubMed] [Google Scholar]
- 33.Liu D, Zeng BX, Shang Y. Decreased expression of peroxisome proliferator-activated receptor gamma in endotoxin-induced acute lung injury. Physiol Res. 2006;55:291–299. doi: 10.33549/physiolres.930822. [DOI] [PubMed] [Google Scholar]
- 34.Siddiqui AM, Cui X, Wu R, Dong W, Zhou M, Hu M, Simms HH, Wang P. The anti-inflammatory effect of curcumin in an experimental model of sepsis is mediated by up-regulation of peroxisome proliferator-activated receptor-gamma. Crit Care Med. 2006;34:1874–1882. doi: 10.1097/01.CCM.0000221921.71300.BF. [DOI] [PubMed] [Google Scholar]
- 35.Higuchi S, Wu R, Zhou M, Ravikumar TS, Wang P. Downregulation of hepatic cytochrome P-450 isoforms and PPAR-gamma: their role in hepatic injury and proinflammatory responses in a double-hit model of hemorrhage and sepsis. J Surg Res. 2007;137:46–52. doi: 10.1016/j.jss.2006.04.019. [DOI] [PubMed] [Google Scholar]
- 36.Kaplan JM, Cook JA, Hake PW, O’Connor M, Burroughs TJ, Zingarelli B. 15-Deoxy-delta(12,14)-prostaglandin J(2) (15D-PGJ(2)), a peroxisome proliferator activated receptor gamma ligand, reduces tissue leukosequestration and mortality in endotoxic shock. Shock. 2005;24:59–65. doi: 10.1097/01.shk.0000167108.88376.f2. [DOI] [PubMed] [Google Scholar]
- 37.Vish MG, Mangeshkar P, Piraino G, Denenberg A, Hake PW, O’Connor M, Zingarelli B. Proinsulin c-peptide exerts beneficial effects in endotoxic shock in mice. Crit Care Med. 2007;35:1348–1355. doi: 10.1097/01.CCM.0000260245.61343.B3. [DOI] [PubMed] [Google Scholar]
- 38.Hill MR, Young MD, McCurdy CM, Gimble JM. Decreased expression of murine PPARgamma in adipose tissue during endotoxemia. Endocrinology. 1997;138:3073–3076. doi: 10.1210/endo.138.7.5379. [DOI] [PubMed] [Google Scholar]
- 39.Feingold K, Kim MS, Shigenaga J, Moser A, Grunfeld C. Altered expression of nuclear hormone receptors and coactivators in mouse heart during the acute-phase response. Am J Physiol Endocrinol Metab. 2004;286:E201–E207. doi: 10.1152/ajpendo.00205.2003. [DOI] [PubMed] [Google Scholar]