

Abstract
Chlamydiae are evolutionarily well-separated bacteria that live exclusively within eukaryotic host cells. They include important human pathogens such as Chlamydia trachomatis as well as symbionts of protozoa. As these bacteria are experimentally challenging and genetically intractable, our knowledge about them is still limited. In this study, we obtained the genome sequences of Simkania negevensis Z, Waddlia chondrophila 2032/99, and Parachlamydia acanthamoebae UV-7. This enabled us to perform the first comprehensive comparative and phylogenomic analysis of representative members of four major families of the Chlamydiae, including the Chlamydiaceae. We identified a surprisingly large core gene set present in all genomes and a high number of diverse accessory genes in those Chlamydiae that do not primarily infect humans or animals, including a chemosensory system in P. acanthamoebae and a type IV secretion system. In S. negevensis, the type IV secretion system is encoded on a large conjugative plasmid (pSn, 132 kb). Phylogenetic analyses suggested that a plasmid similar to the S. negevensis plasmid was originally acquired by the last common ancestor of all four families and that it was subsequently reduced, integrated into the chromosome, or lost during diversification, ultimately giving rise to the extant virulence-associated plasmid of pathogenic chlamydiae. Other virulence factors, including a type III secretion system, are conserved among the Chlamydiae to variable degrees and together with differences in the composition of the cell wall reflect adaptation to different host cells including convergent evolution among the four chlamydial families. Phylogenomic analysis focusing on chlamydial proteins with homology to plant proteins provided evidence for the acquisition of 53 chlamydial genes by a plant progenitor, lending further support for the hypothesis of an early interaction between a chlamydial ancestor and the primary photosynthetic eukaryote.
Keywords: bacterial pathogens, sexually transmitted disease, symbiosis, intracellular bacteria, protozoa, pathogen–host interaction
Introduction
Chlamydiae (Chlamydiales) have traditionally been viewed as a small group of highly successful bacterial pathogens of humans and animals. Two members of the family Chlamydiaceae, Chlamydia trachomatis and Chlamydia (aka Chlamydophila) pneumoniae, are among the best known bacterial pathogens. Chlamydia trachomatis is responsible for more than 90 million trachoma cases each year worldwide and also represents the most frequently sexually transmitted bacterial pathogen (World Health Organization 2001, 2008). Chlamydia pneumoniae is the cause of up to 10% of community-acquired pneumonia cases and is putatively associated with a number of chronic diseases such as arteriosclerosis (Mahony et al. 2003). Chlamydiae have a remarkably broad host range and diversity. The phylum Chlamydiae is evolutionarily well separated from all other bacteria and consists of at least eight families. In addition, there is evidence for an even greater, previously unseen diversity of the Chlamydiae (Corsaro et al. 2003; Horn 2008).
All Chlamydiae infect eukaryotic host cells, which are required for replication, and there is no close free-living relative known. This obligate intracellular lifestyle is facilitated by a characteristic developmental cycle consisting of two morphologically and physiologically distinct stages. The elementary body (EB) functions mainly to survive the extracellular environment and to infect new host cells. Upon infection, the EB differentiates to a reticulate body (RB) that multiplies inside a host-derived vacuole. The developmental cycle is completed when the RBs convert back to EBs, which are set free by host cell lysis or exocytosis in order to start a new round of infection.
The discovery of Chlamydiae that are not primarily pathogens of humans but also occur as symbionts or pathogens in other hosts opened up new possibilities to investigate the evolution of this unique group of bacteria, their intracellular lifestyle, and their adaptation to different eukaryotic hosts (Horn 2008). In this study, we sequenced and compared the genomes of representatives of three major families within the Chlamydiae: Simkaniaceae, Parachlamydiaceae, and Waddliaceae. Simkania negevensis Z was originally discovered as a contaminant of a human cell culture and was later proposed to be a new emerging pathogen causing respiratory infections (Kahane et al. 1993; Friedman et al. 2003). Parachlamydia acanthamoebae UV-7 primarily lives in amoebae but may also be associated with disease in humans (Greub and Raoult 2002; Collingro et al. 2005). Waddlia chondrophila 2032/99 was isolated from an aborted bovine fetus and subsequently implicated in abortion in humans (Henning et al. 2002; Baud et al. 2008). In contrast to the Chlamydiaceae, Simkania, Parachlamydia, and Waddlia all grow well in amoebae, which might therefore represent their natural hosts and reservoir.
The availability of these genome sequences and the comparison with other available chlamydial genome sequences allowed a comprehensive overview of genomic diversity among the Chlamydiae. Here we describe and discuss aspects of their pan-genome. We used these data to trace the phylogenetic relationships among the Chlamydiae and to track down evolutionary ancient traits of this important phylum. We show that despite many evolutionarily well-conserved features, members of the Chlamydiae show both divergent and convergent adaptations to their intracellular life in different eukaryotic hosts.
Materials and Methods
Preparation of Genomic DNA
Simkania negevensis Z was grown on Vero cells, and EBs were purified on Renografin gradients as described previously (Kahane et al. 1999, 2001). For preparation of high molecular weight DNA, EBs were suspended in TE buffer with 50 mM dithiothreitol and incubated at 37 °C for 1 h and then further incubated at 37 °C for 1 h in the presence of 1% NP-40 and DNase-free RNase at 0.5 mg/ml. Proteinase K was added to a final concentration of 0.2 mg/ml and incubated overnight at 37 °C. The suspension was extracted with phenol saturated in TE buffer and then with phenol–chloroform–isoamyl alcohol (24:24:1 by volume) followed by dialysis at 4 °C against 2xSSC buffer (0.15 M NaCl, 15 mM sodium citrate, pH 7.0).
Parachlamydia acanthamoebae UV-7 was cultured in Acanthamoebae sp. UWC1 in trypticase soy yeast extract medium (Collingro et al. 2005). EBs were harvested and purified by step gradient density centrifugation including repeated DNase (20 mg/ml) and RNase (20 mg/ml) treatments at 37 °C for 45–90 min (Heinz, Pichler, et al. 2010). High molecular weight genomic DNA was isolated from purified EBs by UNSET lysis, phenol–chloroform extraction, and ethanol precipitation as described previously (Hugo et al. 1992; Horn et al. 1999).
Waddlia chondrophila 2032/99 (Henning et al. 2002) was cultured in Buffalo Green Monkey kidney cells in Nephros LP medium (BioWhittaker Europe S.P.R.L.) at 37 °C and 5% CO2 in a fully humidified cabinet for 3 days. Cells were harvested using a cell scraper, and the suspension was centrifuged at 2,000 × g and 4 °C for 15 min to remove host cell debris. Subsequently, the supernatant was centrifuged at 20,000 × g and 4 °C for 40 min. The pellet was resuspended in phosphate buffered saline and subjected to DNA extraction using the High Pure PCR Template Preparation Kit (Roche Diagnostics).
Genome Sequencing
The complete genome sequences of S. negevensis Z and P. acanthamoebae UV-7 and a draft genome sequence of W. chondrophila 2032/99 were determined using the whole-genome shotgun method (Myers et al. 2007). For S. negevensis and P. acanthamoebae, physical and sequencing gaps were closed using a combination of primer walking, generation, and sequencing of transposon-tagged libraries of large-insert clones and multiplex PCR (Tettelin et al. 2001). The coverage of the sequenced genomes was 8–16×.
Genome Annotation and Analysis
The PEDANT software system was used for genome sequence analysis (Frishman et al. 2001). Prediction of coding sequences (CDSs) was performed with GeneMarkS (Besemer et al. 2001) and a minimal length of predicted CDSs of 42 nucleotides. CDSs were assigned to the National Center for Biotechnology Information (NCBI) clusters of orthologous group (COG) catalogue (Tatusov et al. 2003) by PEDANT. Biochemical pathway prediction and reconstruction was performed using KEGG (Wixon and Kell 2000). Transfer RNA (tRNA) genes were identified with tRNAscan-SE (Lowe and Eddy 1997). Noncoding RNAs were identified by search against Rfam (Gardner et al. 2009) and inspected manually. Eukaryotic domains in predicted proteins were identified using the precalculated lists of eukaryotic-like domains at effectors.org (Jehl et al. 2011) and the search for InterPro signatures (Hunter et al. 2009) as provided by the PEDANT program.
Identification of Putative Inc Proteins
A hydrophilicity profile using the Kyte–Doolittle method with a window size of seven was generated in the program MacVector (Rastogi 2000) for all proteins with hypothetical function and without a signal peptide determined by SignalP prediction (Bendtsen et al. 2004). Hydrophilicity plots were analyzed manually, and all proteins with a dominant bilobed hydrophobic domain (Bannantine et al. 2000) were designated candidate Incs. Coiled-coil regions were determined by the program MARCOIL (http://www.isrec.isb-sib.ch/webmarcoil/webmarcoilC1.html) (Delorenzi and Speed 2002) and the program COILS (http://www.ch.embnet.org/software/COILS_form.html) (Lupas et al. 1991). Cluster analysis was performed with the program PRIMER 5.0 (Primer-e Ltd) based on a presence/absence Bray–Curtis similarity matrix by complete linkage (Bray and Curtis 1957).
Clusters of Orthologues
The chlamydial pan-genome was analyzed by calculation of clusters of orthologous proteins from 19 complete chlamydial genomes (Chlamydia muridarum Nigg [NC_002620], C. trachomatis A/HAR-13 [NC_007429], C. trachomatis 434/Bu [NC_010287], C. trachomatis L2b/UCH-1/proctitis [NC_010280], C. trachomatis D/UW-3/CX [NC_000117], C. trachomatis B/TZ1A828/OT [NC_012687], C. trachomatis Jali20 [NC_012686], Chlamydia abortus S26/3 [NC_004552], Chlamydia caviae GPIC [NC_003361], C. pneumoniae AR39 [NC_002179], C. pneumoniae CWL029 [NC_000922], C. pneumoniae J138 [NC_002491], C. pneumoniae TW-183 [NC_005043], Chlamydia felis Fe/C-56 [NC_007899], C. pneumoniae LPCoLN [CP001713], W. chondrophila WSU 86-1044 [NC_014225], Protochlamydia amoebophila UWE25 [NC_005861], P. acanthamoebae UV-7 [to be added], and S. negevensis Z [to be added]). Clusters of orthologues were determined using SIMAP and bidirectional best basic local alignment search tool (BLAST) hits (BBH) with a cutoff value of 1 × 10−10 (Rattei et al. 2010). The clustering process was performed using an in-house software from the SIMAP project. The clustering process is equivalent to the procedure described for the NCBI COGs (Tatusov et al. 2003), with the only difference that fused proteins were not split but formed individual COGs. The obtained clusters of orthologues were classified with respect to the presence of proteins from each of the included chlamydial proteomes, sorted using an in-house program, and subsequently inspected manually. The affiliation of chlamydial proteins to the NCBI COG catalogue was used for functional classification. Proteins specific to the Chlamydiae were identified by homology search against the SIMAP database (Rattei et al. 2010) including all cellular organisms using a maximum E-value of 1.0 × 10−04, a minimal identity of 20%, and a minimal length ratio of both proteins of 0.5 (i.e., 50% of the lengths of both proteins required to be part of the alignments). The proteins were inspected manually after extraction using BLAST against the GenBank database.
Genome-Based Phylogenetic Analysis
Thirty-seven conserved proteins (supplementary table S1, Supplementary Material online) were used for phylogenetic analyses. Protein sequences were extracted from GenBank using genomic BLAST (Cummings et al. 2002) and BLAST (Altschul et al. 1990) searches against the genome databases of S. negevensis Z, W. chondrophila 2032/99, and P. acanthamoebae UV-7. Alignments were performed using MAFFT version 6 (Katoh et al. 2009). Concatenation of multiple alignments was performed using an in-house program. A phylogenetic tree considering 5,831 amino acid positions was constructed using RAxML version 7.0.0 and 1,000 bootstrap samples (parameters -m PROTGAMMAJTT -x 12345 -N 1000 -f a) (Stamatakis 2006). In addition, the concatenated alignment was imported into the ARB program (Ludwig et al. 2004). Trees obtained with the neighbor joining and maximum likelihood treeing methods including Tree-Puzzle (Schmidt et al. 2002) implemented in ARB were consistent with the RAxML-inferred tree topology.
Phylogeny of Proteins of the Type III Secretion Apparatus
Protein sequences of the T3S apparatus were obtained using genomic BLAST searches, aligned with MAFFT, and two conserved proteins from each genomic island (SctVU, SctJT, and SctNQ) were concatenated. Phylogenetic analyses were performed with MrBayes 3.1 (Ronquist and Huelsenbeck 2003) using the mixed amino acid model and standard settings. For SctVU, 1,500,000 generations with a final standard deviation of 0.0061, for SctJT 800,000 generations with a final standard deviation of 0.0096, and for SctNQ 1,000,000 generations with a final standard deviation of 0.0028 were performed.
Phylogeny of Plasmid Proteins
For phylogenetic analysis of T4S apparatus proteins TraUNF, protein sequences were obtained using genomic BLAST searches and the respective proteins conserved in S. negevensis, P. amoebophila, and P. acanthamoebae UV-7. Proteins were aligned with MAFFT and concatenated. A filter using only amino acid positions conserved in at least 15% of all used sequences was generated with the ARB program package (Ludwig et al. 2004). MrBayes 3.1 (Ronquist and Huelsenbeck 2003) applying the mixed amino acid model and standard settings was used for phylogenetic calculations (200,000 generations, standard deviation 0.0013). Phylogenetic analysis of other plasmid-encoded proteins (pGP1-D, pGP5-D, and pGP6-D) was performed using MAFFT alignments and PhyML (Guindon and Gascuel 2003) available at the Mobyle portal of the Institut Pasteur (http://mobyle.pasteur.fr/cgi-bin/portal.py?form=phyml) with standard settings and 1,000 bootstraps.
Phylogeny of MOMP and MOMP-Like Proteins
Phylogenetic analysis of major outer membrane proteins (MOMPs) and MOMP-like proteins was performed using a MAFFT alignment and MrBayes 3.1 (Ronquist and Huelsenbeck 2003) applying the mixed amino acid model and standard settings (1,800,000 generations with a final standard deviation of 0.098).
Chlamydial Orthologues in Plants
To identify chlamydial proteins sharing a common evolutionary origin with plant proteins, we calculated phylogenetic trees for each protein encoded in the genomes of S. negevensis Z, W. chondrophila 2032/99, P. acanthamoebae UV-7, and P. amoebophila UWE25 using the fully automated software PhyloGenie (Frickey and Lupas 2004). The reference database for PhyloGenie was generated from all bacterial and archaeal genomes available in the NCBI RefSeq database in September 2009 (Pruitt et al. 2007) and from additional 41 genome sequences of eukaryotes available in SIMAP (supplementary table S9, Supplementary Material online). Taxon names in the reference database and the NCBI taxonomy database were edited to remove characters that control the structure of tree files in the Newick format. The PhyloGenie software was executed for each query protein using default parameters with modifications -blammerparams=- taxid f. For the BLAST calculations in PhyloGenie, NCBI BLAST (version 2.2.19) was used. Protein phylogenies were calculated with RAxML using the gamma JTT model with rapid bootstrapping and 100 bootstraps (parameters -m PROTGAMMAJTT -N 100 -f a) (Stamatakis et al. 2005) on the basis of full or partial automatic alignments produced by the BLAMMER program included in PhyloGenie. The obtained phylogenetic trees were postprocessed by an in-house script, which sorted all operational taxonomic units according to their distances in the tree to the query protein. All trees showing lowest distance values for Chlamydiae and Plantae (Viridiplantae and Rhodophyta) proteins were inspected manually, and only trees with bootstrap support values above 70% for the monophyly of Chlamydiae and Plantae were considered. Subcellular locations of plant homologs were predicted with WoLF PSORT (Horton et al. 2007), TargetP 1.1, and ChloroP 1.1 (Emanuelsson et al. 2007).
Data Deposition
The genome sequences were deposited at EMBL/GenBank/DDBJ (accession numbers FR872580, FR872581, FR872582, and FR872583–FR872668), the PEDANT database (http://pedant.gsf.de), and chlamydiaeDB (http://www.chlamydiaedb.org/).
Results and Discussion
Genome Features
The main features of the genomes analyzed here including selected known chlamydial genomes are summarized in table 1 (Horn et al. 2004; Greub et al. 2009; Bertelli et al. 2010). The closed genome sequences of Simkania and Parachlamydia contain 2,519 and 2,788 predicted CDSs. The draft genome sequence of Waddlia contains 2,028 predicted CDSs (on 86 contigs). The most noticeable feature of the Simkania, Parachlamydia, and Waddlia genomes is the 2- to 3-fold larger genome size compared with the Chlamydiaceae (fig. 1; Stephens et al. 1998; Kalman et al. 1999; Read et al. 2000, 2003; Carlson et al. 2005). As typical intracellular bacteria, the genome sizes of the Chlamydiaceae are small (1 Mb) and highly reduced in gene content. Although genome reduction generally correlates with a decrease in the genomic G + C content (Sällström and Andersson 2005; Gómez-Valero et al. 2007; Moran et al. 2008), the G + C content of the Chlamydiae is approximately 40% regardless of genome size (fig. 1).
Table 1.
General Features of the Genomes of Representative Members of Four Chlamydial Families
| Chlamydia trachomatis D/UW-3/CXa,b | Protochlamydia amoebophila UWE25c | Parachlamydia acanthamoebae Hall's Coccusd | P. acanthamoebae UV-7e | Simkania negevensis Ze | Waddlia chondrophila 2032/99e,f | W. chondrophila WSU 86-1044g | |
| Chromosome size (nt) | 1,042,519 | 2,417,793 | 2,971,261 | 3,072,383 | 2,496,337 | 2,141,577 | 2,116,312 |
| Plasmid size (nt) | 7,493 | — | — | — | 132,038 | — | 15,593 |
| Contigs | 2 | 1 | 95 | 1 | 2 | 86 | 2 |
| Predicted CDSs | 895 | 1,986 | 2,809 | 2,788 | 2,519 | 2,028 | 1,934 |
| G + C content (%) | 41 | 35 | 38 | 39 | 38 | 43 | 43 |
| Coding regions (%) | 89 | 82 | 89 | 90 | 91 | 93 | 93 |
| Average CDSs length (bp) | 1,050 | 1,003 | 930 | 988 | 951 | 978 | nd |
| Hypothetical CDSs | 368 (13%) | 450 (18%) | 406 (20%) | ||||
| CDSs unknown functionh | 1,580 (57%) | 1,518 (60%) | 1,050 (52%) | ||||
| tRNAs | 37 | 37 | 35 | 40 | 35 | 34 | 37 |
| RRNA genes | 6 | 9 | 10 | 3 | 6f | 6 | |
| sRNAs | 5 | nd | nd | 3 | 4 | nd | nd |
This study.
Although the genome is a draft sequence, two rRNA operons, 44 ribosomal proteins, some complete metabolic pathways, and a complete T3SS are present.
CDSs assigned to the poorly characterized NCBI COG classes R and S and CDSs without NCBI COG assignment, nd, not determined.
FIG. 1.

Genome size reduction in the Chlamydiae. In contrast to the highly reduced genomes of the Chlamydiaceae (gray squares), the genomes of Simkania, Waddlia, and the Parachlamydiaceae are significantly larger (black squares). The reduction of genome size within the Chlamydiae does not correlate with a decrease in genomic G + C content, an effect observed in other bacterial groups, for example, the Gammaproteobacteria (light gray circles).
Genome-Based Phylogeny of the Chlamydiae
Although relationships among the Chlamydiae are traditionally inferred based on phylogenetic analysis of 16S ribosomal RNA (rRNA) sequences, this has become increasingly difficult with accumulating sequence data. Currently, using 16S rRNA phylogenetic analysis alone, it is not possible to reliably determine the branching order of the diverse chlamydial families. Therefore, we used a set of 37 ribosomal and other phylogenetic marker proteins (supplementary table S1, Supplementary Material online) comprising in total 5,831 amino acid positions to trace the evolutionary history of the Chlamydiae. Based on this analysis, the closest relatives of the Chlamydiae are the Verrucomicrobia and the Lentisphaera (Wagner and Horn 2006; Kamneva et al. 2010), comprising mainly free-living microorganisms (fig. 2). Within the Chlamydiae, the Chlamydiaceae branch most deeply, that is, they split first and evolved independently from the other chlamydial families. The next split occurs between the Simkaniaceae and the common ancestor of the Waddliaceae and the Parachlamydiaceae, which subsequently split into the two families. This scenario is also largely supported in the phylogeny of the chlamydial type III and type IV secretion systems (see below).
FIG. 2.

Phylogeny of the Chlamydiae based on 37 conserved proteins. Maximum likelihood–based phylogenetic (RAxML) analysis of a concatenated set of 32 ribosomal proteins, RpoBC, GyrB, RecA, and ET-Tu (TufA) (5831 amino acid positions) from complete genome sequences was used. If not indicated, bootstrap (1,000 iterations) support is 100%. The Verrucomicrobia (gray box) are represented by Verrucumicrobium spinosum, Opitutus terrae, and Akkermansia muciniphila. Bar indicates 10% estimated sequence divergence.
Members of the Chlamydiaceae are notable for their degree of genome conservation with high genomic synteny (Stephens et al. 1998; Kalman et al. 1999; Read et al. 2000, 2003; Carlson et al. 2005). Little to no synteny is observed within the Parachlamydiaceae and between the Simkaniaceae, Parachlamydiaceae, and Waddliaceae when compared with the Chlamydiaceae (supplementary fig. S1, Supplementary Material online). This lack of synteny reflects the evolutionary distance between these families and their different evolutionary trajectories as symbionts of protozoa and pathogens of animals.
The Pan-Genome of the Chlamydiae
Given the low degree of synteny and in order to determine the similarities and differences of the Chlamydiae on a genomic level, we performed a clustering analysis of orthologous groups of proteins based on BBH (using a cutoff E-value of 1.0 × 10−10) using representatives of all available complete chlamydial genome sequences. This analysis provided an overview of the chlamydial pan-genome, which describes the full complement of their genes. It served to determine features conserved among all Chlamydiae or specific to individual evolutionary lineages. Despite the family-level divergence, a surprisingly large number (n = 560) of genes are shared by all Chlamydiae representing the core gene set (fig. 3). This corresponds to approximately half of the genes encoded in the genomes of the Chlamydiaceae. On the other hand, this core genome is expanded by a high number of genus- and family-specific genes, reflecting the differences in genome size and illustrating the genomic variability of the Chlamydiae.
FIG. 3.

The chlamydial pan-genome. The Venn diagram illustrates the number of shared and specific (accessory) genes among the Chlamydiae based on clusters of orthologs. Only complete genome sequences were used (Chlamydiaceae n = 15).
The Chlamydial Core Gene Set
The set of 560 core genes of the Chlamydiae comprises mainly housekeeping genes. It also contains members of all 100 COGs (according to the NCBI COG catalogue; Tatusov et al. 2003), which are conserved in 99% among all intracellular bacteria (Merhej et al. 2009). Many (122) of the 560 core genes cannot be assigned to an NCBI COG or their function is unknown (NCBI COG codes R and S). Using a maximum E-value of 1.0 × 10−04, a minimal identity of 20%, and a minimal length ratio of both proteins of 0.5, only 27 core genes are specific for the Chlamydiae and are not present in any other cellular organism sequenced so far (supplementary table S2, Supplementary Material online).
A pan-genome typically describes the full complement of genes in a bacterial species, incorporating all known strains. Here we were able to expand the pan-genome concept to a higher taxonomic grouping, the phylum Chlamydiae (order Chlamydiales), and still found more than 500 conserved core genes. This could be explained by evolutionary “bottlenecks” related to the largely conserved chlamydial developmental cycle and the intracellular lifestyle (Stephens et al. 2009). Genes presumably important for these hallmark features are likely to be present within this core gene set, and many will be found among the 122 clusters containing hypothetical genes pending further characterization.
Orthologs Shared by Simkania, Waddlia, and Parachlamydiaceae
Given the large genome size of Simkania, Waddlia, and Parachlamydiaceae (represented by the amoeba symbionts P. amoebophila and P. acanthamoebae) compared with the Chlamydiaceae (fig. 1, table 1), we were initially interested in those proteins that are shared by Simkania, Waddlia, and Parachlamydiaceae but are absent in the Chlamydiaceae, as well as the pool of proteins specific to each. There are just 171 clusters of orthologous proteins shared by Simkania, Waddlia, and Parachlamydiaceae (fig. 3, supplementary table S3, Supplementary Material online). About half (83) include hypothetical proteins, with either no assignment to NCBI COGs or with unknown function. Other shared proteins are involved in central metabolic processes enabling greater biosynthetic diversity and versatility in responding to changing environmental conditions compared with the Chlamydiaceae. This observation agrees well with amoebae being natural reservoirs for Simkania, Waddlia, and the Parachlamydiaceae and providing a far less homeostatic environment than multicellular organisms. For example, a glucokinase allows phosphorylation of glucose enabling independence from host-derived phosphorylated glucose. The tricarboxylic acid cycle enzymes aconitate hydratase (AcnB), isocitrate dehydrogenase (Icd), and citrate synthase (GltA) are present, enabling regeneration of nicotinamide adenine dinucleotide (phosphate). In addition, several amino acid synthesis pathways and the folate biosynthesis pathway are more complete than in the Chlamydiaceae. There is also an entire gene set for production of menaquinone (menA-F), which can be used in the quinone pool during oxidative phosphorylation instead of host-derived ubiquinone. A possible reason for the observed more complete metabolic networks in Simkania, Waddlia, and Parachlamydiaceae might also be a less strict or more recent obligate association with the host compared with the Chlamydiaceae. In view of the evidence for an ancient obligate intracellular lifestyle of the chlamydial progenitor, this alternative explanation might, however, be less likely than the requirement to cope with changing environmental conditions and different host cells.
Simkania, Waddlia, and the Parachlamydiaceae show high numbers of species-specific proteins (between 595 and 1,340 proteins, respectively; fig. 3, supplementary tables S5–S8, Supplementary Material online). The majority of these proteins (69–82%) are uncharacterized proteins of unknown function. Among the species-specific proteins for which a function can be inferred, many are involved in transport and metabolism, indicating species-specific adaptations with respect to metabolic host dependency. For example, Simkania encodes complete NAD+ and tryptophan synthesis pathways, both of which are fully or partially absent in all other Chlamydiae.
A Chemosensory System in Parachlamydia
Another remarkable set of species-specific proteins can be found in Parachlamydia. In contrast to all other Chlamydiae, Parachlamydia encodes a minimal chemotaxis system similar to the Escherichia coli system (supplementary table S9, Supplementary Material online) (Szurmant and Ordal 2004). The 15 proteins belonging to this system include methyl-accepting chemotaxis proteins (MCPs) as well as coupling proteins, histidine kinases, a response regulator, a methyltransferase, and a methylesterase. The respective functional domains are present and conserved in all these proteins (Alexander and Zhulin 2007; Wuichet et al. 2007). That the genes encoding these proteins are undisrupted by mutation suggests that they are fully functional. Consistent with this notion, we could detect transcription of 12 chemotaxis genes (except mcp1, cheA2, and cheW2; not shown) during growth of P. acanthamoebae UV-7 in its amoeba host, suggesting that they are required during intracellular growth. However, since Parachlamydia does not encode any flagellar motor proteins and as chemotaxis inside its unicellular host is dispensable, the role of this system in Parachlamydia is unclear. Most microorganisms capable of chemotaxis also employ proteins homologous to the chemotaxis system known as chemosensory systems (Kirby 2009). These systems are used for temporal regulation of signal transduction for other purposes, such as prokaryotic cell differentiation in Myxococcus xanthus or exopolysaccharide production in Pseudomonas aeruginosa (Kirby 2009). To our knowledge, the presence of a chemosensory system in the absence of a flagellar motor has not been described so far, and it remains to be determined how widespread this situation is among other prokaryotes. For Parachlamydia, it is not possible to distinguish between an origin of this system from a free-living and motile chlamydial ancestor (and subsequent loss in all other chlamydial lineages) or acquisition by lateral gene transfer (possibly in two independent events from Cyanobacteria and Clostridia as suggested by sequence homology to the respective chemotaxis genes in these bacteria).
The Chlamydial Type III Secretion System
Mechanisms for host cell interaction are conserved to varying degrees among the Chlamydiae. In the following, we discuss the occurrence of important recognized Chlamydiaceae virulence factors in Simkania, Parachlamydia, and Waddlia.
Chlamydiae secrete effector proteins for interaction with their respective host cells via a type III secretion (T3S) system (Peters et al. 2007; Beeckman and Vanrompay 2009). Most structural components of this apparatus are also encoded in the Simkania, Parachlamydia, and Waddlia genomes (fig. 4; supplementary text, Supplementary Material online). In addition to the full set of genes for T3S structural components, the Chlamydiaceae genomes encode some homologs of the flagellar apparatus (FliF, FliI, and FlhA), which might be important for bacterial replication and/or intracellular survival (Peters et al. 2007; Stone et al. 2010). These genes are missing in all other chlamydial genomes.
FIG. 4.

The chlamydial type III secretion system. (A) Genomic organization of the T3S system in representative chlamydial genomes. The genes encoding T3S are located on different genomic regions labeled in shades of green, orange, and red, respectively. Structural components of the secretion machinery are labeled in dark color shades or gray; genes encoding effector proteins are labeled in medium color shades; genes encoding T3S chaperones are labeled in light color shades; genes encoding hypothetical proteins are displayed white. (B–D) Phylogeny of the chlamydial T3S system based on concatenated protein sequences encoded on the three main genomic loci displayed above. (B) SctV and SctU; (C) SctJ and SctT; (D) SctN and SctQ. Scale bar represents 0.1 expected changes per site. If not indicated, Bayesian posterior probabilities are 100%.
In contrast to proteobacterial T3S genes, chlamydial T3S genes are organized in multiple regions distributed around the genome (Horn et al. 2004; Peters et al. 2007; Bertelli et al. 2010). This organization is also observed in the genomes reported here (fig. 4A). Consistent with this distribution and unlike the Proteobacteria, there is no evidence for lateral gene transfer of chlamydial T3S components, suggesting that the T3S system was present already in the last common ancestor of all Chlamydiae (Horn et al. 2004). To further investigate this hypothesis, we performed phylogenetic analyses with two structural T3S proteins from each of three genomic T3S regions. This analysis supported with high confidence a common ancestry of all chlamydial T3S systems (fig. 4B–D). The trees mostly recapitulate and confirm the chlamydial phylogeny determined using rRNA genes and 37 conserved proteins, respectively (fig. 2). Interestingly, the monophyly of the Parachlamydiaceae comprising P. acanthamoebae and P. amoebophila is not well supported in the SctNQ trees (fig. 4D), and in the SctJT-based trees (fig. 4C), Parachlamydia clusters together with Waddlia. This pattern could be explained by an ancient exchange of the genomic T3S region encoding SctJT between ancestors of Waddlia and Parachlamydia. Alternatively, this might indicate a lack of phylogenetic resolution with respect to the T3S system of the Parachlamydiaceae and Waddlia.
Type III Effector Proteins
Many of the proteins secreted by the T3S pathway are proteins targeted to the host-derived membrane encapsulating the chlamydiae inside their host cell termed the inclusion membrane (Rockey et al. 2002). Located directly at the interface between host and bacteria, these inclusion membrane proteins (Incs), which have virtually no homologs in any other microorganism, are likely candidates for manipulation of the eukaryotic cell. Characterized Inc proteins function in the fusion of chlamydial inclusions, the interaction with host proteins, interference with host cell cytokinesis, and inhibition of NFkappaB activation (Hackstadt et al. 1999; Delevoye et al. 2004; Cortes et al. 2007; Alzhanov et al. 2009; Wolf et al. 2009). Although Inc proteins do neither show a common sequence motif nor consistent sequence similarities, most Inc proteins share a characteristic large bilobed hydrophobic domain in hydropathy plots (Rockey et al. 2002). This conserved feature was recently used to identify novel Inc proteins in Protochlamydia (Heinz, Rockey, et al. 2010). By employing this approach, we identified 120 putative Inc proteins in the genomes of Parachlamydia (n = 38), Simkania (n = 41), and Waddlia (n = 41). Only three putative Inc proteins are conserved throughout all Chlamydiae. Cluster analysis of putative Inc proteins demonstrated great differences between the members of the Chlamydiaceae and all other chlamydial families (20% Bray–Curtis similarity). The two members of the Parachlamydiaceae were less than 50% similar in terms of their Inc proteins (supplementary fig. S2, Supplementary Material online). The pronounced differences among the Chlamydiae with respect to Inc proteins indicate that their function as virulence factors is likely to be dependent on the host cell type. Interestingly, Parachlamydia and Waddlia grouped together in this cluster analysis to the exclusion of Protochlamydia. This fits well with the observation that Protochlamydia seems unable to infect mammalian cells, whereas Simkania, Waddlia, and Parachlamydia can enter and multiply in mammalian cells, albeit with varying efficiencies (Kahane et al. 1999; Henning et al. 2002; Greub et al. 2003).
We noted an enrichment of coiled-coil domains among the putative Inc proteins, which contained up to eight coiled-coil domains of more than 200 amino acids. Coiled-coil proteins are very common in eukaryotic organisms and perform a wide variety of functions, including important structural roles in the cytoskeleton as well as in eukaryotic membrane fusion processes. The overrepresentation of this domain in Inc proteins supports a role for many of these proteins in the interaction with the host cell such as manipulation of the cytoskeleton or interactions with the Golgi complex (Carabeo et al. 2003; Delevoye et al. 2008).
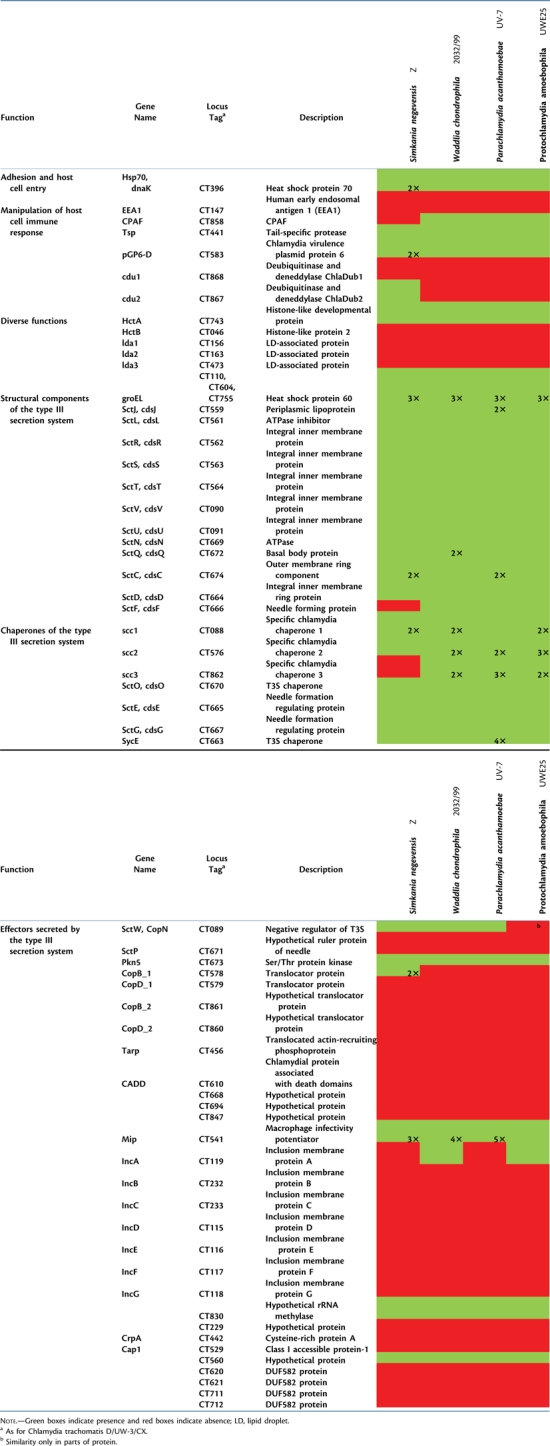
In addition to Inc proteins, several other T3S effectors of the Chlamydiaceae are known to be secreted into the host cell or the chlamydial inclusion (Beeckman and Vanrompay 2009). Some of these proteins are also encoded in the Waddlia, Simkania, Parachlamydia, or Protochlamydia genomes (table 2, supplementary text and table S10, Supplementary Material online). For example, the Ser/Thr kinase PknD, which seems to play a major role in chlamydial growth (Johnson et al. 2009), the Ser/Thr protease Pkn5, and the negative regulator of the T3S system SctW (CopN) are present. Other important effectors like CADD, translocated actin-recruiting phosphoprotein (Tarp), Cap1, and CrpA are missing in all Chlamydiae other than the Chlamydiaceae. Thus, although the structural components of the chlamydial T3S pathway are highly conserved, there is considerable variability among secreted effector proteins (table 2). This diversity of secreted effectors reflects the diversity of chlamydial host cells and indicates that they are important for niche differentiation.
Table 2.
Presence/Absence of Selected Virulence-Associated Proteins of the Chlamydiaceae in Simkania, Waddlia, and the Parachlamydiaceae.
 |
Other Known Chlamydiaceae Virulence Genes
A number of proteins associated with virulence in the Chlamydiaceae are also present in Simkania, Parachlamydia, and Waddlia (table 2, supplementary table S10, Supplementary Material online). These comprise homologs of the general stress response proteins DnaK, GroEL, and GroES as well as the chromosome condensation protein HctA. Present in all chlamydial genomes are a temperature-activated serine protease specific for unfolded proteins (HtrA) (Huston et al. 2007), the tail-specific protease (Tsp), which degrades p65 protein during host cell immune response (Lad et al. 2007), and the Chlamydia plasmid protein pGP6-D that stimulates the host's T-cell response (Olsen et al. 2007).
Interestingly in Simkania, a homolog of the Chlamydia protease-like activity factor (CPAF) is missing, whereas a homolog is present in Waddlia and the Parachlamydiaceae. This protein is unique and was considered ubiquitous to the Chlamydiae. It was identified as a key virulence factor of the Chlamydiaceae and interferes with major histocompatibility complex expression, the apoptosis pathway, and proinflammatory signaling of the host cell (Christian et al. 2010; Huston 2010). Even a search for remote homology using hidden Markov models generated using known CPAF sequences did not reveal any similar sequence (or even remnants) of CPAF in Simkania. However, two proteins showing homology to Tsp and containing the S41 peptidase domain present in all CPAF proteins are encoded in the Simkania genome; it is an open question whether these would be able to compensate the central functions of CPAF (supplementary table S10, Supplementary Material online).
Simkania encodes some virulence factors in common with the Chlamydiaceae that are absent in Waddlia and the Parachlamydiaceae, including a protein that shows similarity to ChlaDub2, a deubiquitinating and deneddylating cysteine protease (Misaghi et al. 2006), and two homologs of the major outer membrane protein (table 2).
Among known factors important for pathogenicity of the Chlamydiaceae and missing in all analyzed genomes are the Tarp that recruits actin of the host cell to modulate entry to nonphagocytic cells (Jewett et al. 2006) (table 2, supplementary table S10, Supplementary Material online). This suggests that the host cell entry mechanisms of Chlamydiaceae and other members of the Chlamydiae are distinct. Also other factors required for evasion of the host cell immune system (Cap1) or modulation of host cell apoptosis (CADD) are missing in Simkania, Waddlia, and the Parachlamydiaceae. Since there is some evidence for a possible pathogenicity of these chlamydiae (Greub and Raoult 2002; Friedman et al. 2003), the lack of these important proteins indicates that they either use alternative mechanisms or that they are not specifically adapted for evasion of the human immune response.
The Chlamydial Outer Membrane
The chlamydial outer membrane is crucial for adhesion and invasion of host cells. The Chlamydiaceae possess a unique outer membrane complex (OMC), whose composition changes during the developmental cycle and plays an important role in chlamydial pathogenicity (Hatch 1999). Many of the proteins identified in the OMC of the Chlamydiaceae such as porin B (PorB), OprB, and most polymorphic outer membrane proteins (Pmps) (Birkelund et al. 2009; Liu et al. 2010) have no homologs in Simkania, Waddlia, and the Parachlamydiaceae. Nevertheless, some proteins of the Chlamydiaceae OMC can be found in all members of the Chlamydiae (supplementary table S11, Supplementary Material online).
The main component of the OMC of the Chlamydiaceae is the major outer membrane protein MOMP (OmpA), which is a major antigen and functions as a large porin (Sun et al. 2007). In Simkania, two MOMP homologs are present. They show highest similarity to MOMPs of veterinary Chlamydia species (26–30% identity). In addition, a large number of more divergent MOMP-like proteins was previously observed in Waddlia (n = 11) (Bertelli et al. 2010), which are related to the major outer membrane proteins of Legionella species and other proteins from free-living bacteria (supplementary fig. S3, Supplementary Material online). Interestingly, Simkania encodes an even higher number of MOMP-like proteins (n = 35), which (with one exception) have evolved independently from MOMP-like proteins of Waddlia. MOMP and MOMP-like proteins are notably absent in Protochlamydia and rare in the Parachlamydia (supplementary table S11, Supplementary Material online), suggesting that proliferation and diversification of MOMP-like proteins occurred mainly in Waddlia and Simkania. In Protochlamydia, MOMP appears to be functionally replaced by a group of putative porins that show structural but no sequence similarity with MOMP and hence are the result of convergent evolution (Heinz et al. 2009; Heinz, Pichler, et al. 2010).
The OMC of the Chlamydiaceae is characterized by cross-linkage via disulfide bridges. These are mainly formed by the two cysteine-rich proteins OmcA (Omp3) and OmcB (Omp2). The large cysteine-rich protein OmcB is present in all Chlamydiae except Simkania. The small cysteine-rich protein OmcA was found in Protochlamydia based on gene neighborhood and compositional analysis and was confirmed experimentally (Horn et al. 2004; Heinz, Pichler, et al. 2010). No OmcA homolog was identified in Simkania, Parachlamydia, and Waddlia.
Polymorphic membrane proteins are an expanded family of outer membrane proteins of the Chlamydiaceae implicated in pathogenesis (Henderson and Lam 2001). Members of this family are surprisingly less abundant in Simkania, Waddlia, and the Parachlamydiaceae, suggesting that the expansion of this family was associated with the diversification of the Chlamydiaceae and the adaptation to human and mammalian hosts. In Simkania, three homologs of PmpB could be identified. They show low sequence similarity to the Chlamydiaceae Pmps but possess leader peptides and C-terminal autotransporter domains and harbor the typical GG[A/L/V/I][I/L/V/Y]…FXXN motifs (Henderson and Lam 2001).
Taken together, the outer membrane of the members of the Chlamydiae shows only few conserved features and is highly divergent with respect to protein composition. This is also supported by recent experimental and in silico analyses of Protochlamydia outer membrane proteins (Heinz et al. 2009; Heinz, Pichler, et al. 2010). In addition, a cluster analysis of all predicted outer membrane proteins further illustrates the distinctness of Simkania, Waddlia, and the Parachlamydiaceae from the Chlamydiaceae (supplementary fig. S4, Supplementary Material online). The compositional diversity of the outer membrane likely reflects the recognized versatility of Simkania, Waddlia, and the Parachlamydiaceae with respect to protozoan hosts compared with the Chlamydiaceae.
Putative Virulence Genes Not Present in the Chlamydiaceae
In contrast to the Chlamydiaceae, the pathogenic potential of Simkania, Waddlia, and Parachlamydiaceae is not as clear based on available clinical and experimental data (Greub and Raoult 2002; Friedman et al. 2003). Whereas there are millions of documented cases for the Chlamydiaceae each year worldwide, data for Simkania, Waddlia, and Parachlamydiaceae as disease agents are limited. In addition, the inconsistent occurrence of Chlamydiaceae virulence-associated proteins in these organisms observed in this study does not allow an unambiguous conclusion to be drawn (table 2). Although Simkania, Waddlia, and the Parachlamydiaceae are lacking a number of virulence-associated proteins of the Chlamydiaceae, they encode other putative virulence factors identified either by similarity to eukaryotic domains or to bacterial proteins with confirmed virulence function.
A remarkable overrepresentation of proteins containing eukaryotic domains was recently observed in the genomes of bacteria that are associated with protozoa (Schmitz-Esser et al. 2010). This trend is also evident in the genomes of Simkania, Waddlia, and Parachlamydia that contain 29, 17, and 48 proteins, respectively, with predicted eukaryotic domains (supplementary table S12, Supplementary Material online). The most abundant eukaryotic domains are F-box and ankyrin domains, which are important for protein–protein interactions and mediate interaction with the host cell in other pathogenic bacteria. For example, for Legionella, it was shown that ankyrin repeat proteins are necessary for intracellular proliferation in protozoan and mammalian hosts (Habyarimana et al. 2008; Price et al. 2010). Other eukaryotic domains include SET, patatin, ERG4_ERG24, SNARE-associated, WD40, TPR_1, and leucine-rich repeats (supplementary table S12, Supplementary Material online). It seems very likely that these proteins are secreted into the host cell to mediate host cell interaction.
Simkania, Waddlia, and the Parachlamydiaceae encode the protein Dps (DNA-binding protein from starved cells). Originally, proteins of the Ferritin/Dps family were implicated only in iron storage and during oxidative stress response. However, the Dps of the amoeba symbiont Legionella jeonii responds to the hydrogen peroxide and phagocytic activity of its host, thereby protecting its DNA during host cell invasion (Park et al. 2006). Dps is also important for resistance to host cell phagocytosis and for establishing a productive infection in human pathogens such as Salmonella enterica (Halsey et al. 2004).
Simkania, Waddlia, and the Parachlamydiaceae encode proteins belonging to the thermolysin family M4. Members of this enzyme family are secreted metallopeptidases that degrade extracellular proteins. Many metallopeptidases play important roles in disease pathogenesis such as λ-toxin of Clostridium perfringens or the M4 thermolysin-like peptidases of Helicobacter pylori, Vibrio cholerae, and Legionella spp. (Adekoya and Sylte 2009).
A Conjugative Plasmid in Simkania
Most members of the Chlamydiaceae bear a 7.5-kb plasmid that is considered a virulence factor as it encodes proteins involved in transcriptional regulation of chromosomal genes important for in vivo infectivity (Olsen et al. 2007; Carlson et al. 2008; O'Connell et al. 2011). Waddlia chondrophila WSU 86-1044 harbors a 15.6-kb plasmid (pWc) encoding 22 predicted proteins from which only two show similarity with proteins encoded by the plasmids of the Chlamydiaceae (Bertelli et al. 2010). Nine of the pWc proteins have homologs in the W. chondrophila 2032/99 genome; however, these are distributed over five contigs suggesting chromosomal integration in this strain.
Whereas a plasmid was not found in Parachlamydia or Protochlamydia, Simkania harbors a 132-kb plasmid that is approximately ten times larger than those of the Chlamydiaceae and Waddlia (table 1). This plasmid, pSn, resembles an F-type conjugative plasmid encoding 138 predicted proteins. Typically F-plasmids carry modules for plasmid stability, replication, adaptation, and propagation (reviewed in Norman et al. 2009), and there is evidence for the presence of all these components on pSn (supplementary text and table S13, Supplementary Material online). Several proteins encoded on pSn play a potential role in host adaptation. Some of these are involved in protection against heavy metals, whereas others are involved in metabolic processes like starch and glycerophospholipid metabolism. Some proteins are present that could play a role in pathogenicity or host cell interaction, including 1 of 3 Mip homologs of Simkania (supplementary text and supplementary table S13, Supplementary Material online).
A putative type IV secretion (T4S) system encoded in the tra region of pSn may be used for plasmid propagation via conjugation (supplementary table S13, Supplementary Material online; Anthony et al. 1999; Lawley et al. 2003; Smillie et al. 2010). The T4S tra region has high synteny to the chromosomal tra regions of Rickettsia bellii (Ogata et al. 2006) and Protochlamydia (Greub et al. 2004; Horn et al. 2004) (fig. 5A). Remnants of the tra region could also be identified in the Parachlamydia genome, whereas Waddlia appears to lack the tra region. Simkania encodes 15 predicted components of the T4S machinery on pSn (supplementary table S13, Supplementary Material online). Among them are the coupling protein (TraDF), components needed for mating pair formation (TraLF, -EF, -KF, -BF, -CF, -WF, -UF, -HF, TrbCF), and proteins needed for mating pair stabilization (TraNF, -GF; fig. 6A). Other functions generally encoded within F-plasmid tra regions are missing in pSn. TraTF and TraSF, which enable surface and entry exclusion and thus prevent redundant plasmid exchange, are not present on pSn. Notably, proteins involved in DNA transport, TraMF, TraJF, and TraYF, which participate in relaxosome formation, and the relaxase TraIF are missing. Instead, similar to R. bellii, pSn encodes two proteins commonly found on the Agrobacterium tumefaciens Ti-plasmid, TraATi and TraDTi (Ogata et al. 2006).
FIG. 5.

The origin of the type IV secretion system in the Chlamydiae. (A) Parachlamydia acanthamaebae UV-7, Protochlamydia amoebophila UWE25, and Simkania negevensis Z contain an F-plasmid–like tra region with high similarity and synteny to the tra region of Rickettsia bellii RML-369C. Genome synteny is shown by trapezes; pairwise protein identity is color coded. (B) Relationship of the chlamydial T4S system to other bacterial T4S systems based on phylogenetic (Bayesian) analysis of concatenated TraUNF proteins. Scale bar represents 0.1 expected changes per site. If not indicated, Bayesian posterior probabilities are 100%.
FIG. 6.
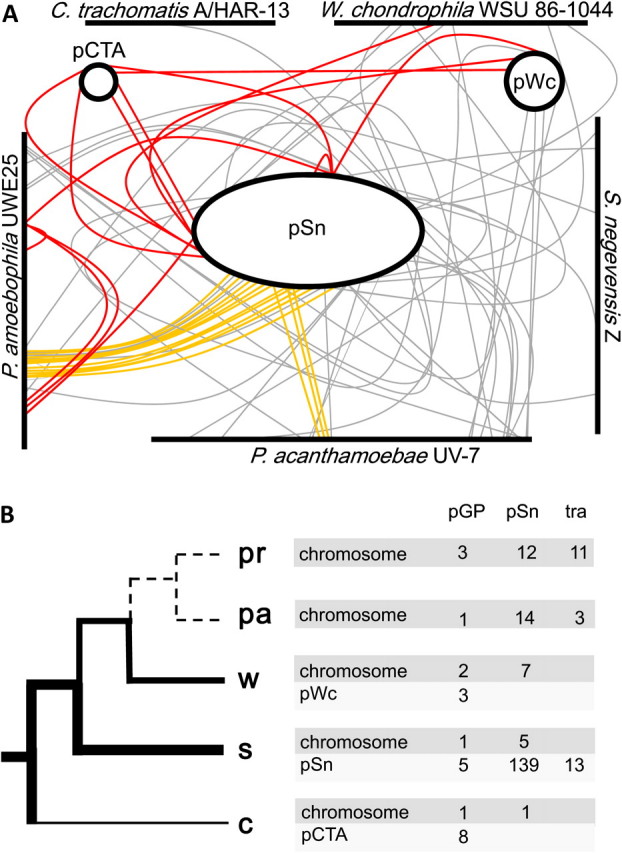
Evolution of the chlamydial plasmid. The presence of plasmid-specific genes in all evolutionary lineages within the Chlamydiae and phylogenetic analysis suggests the evolution of the chlamydial plasmid from a large conjugative plasmid acquired by the chlamydial ancestor. (A) Homologs of chlamydial plasmid-encoded proteins on plasmids and chromosomes of representatives of the Chlamydiaceae, Simkania, Waddlia, and Parachlamydiaceae. Red lines indicate homologs with the virulence plasmid of the Chlamydiaceae. Orange lines indicate genes encoding proteins of the T4S system possibly originally involved in conjugation; gray lines represent all other homologs. Chromosome and plasmid sizes and the positions of the respective genes are approximated to scale. pSn, plasmid of Simkania negevensis Z; pCTA, plasmid of Chlamydia trachomatis A/HAR-13; pWc, plasmid of Waddlia chondrophila WSU 86-1044. (B) Schematic representation of the evolution of the Chlamydiae with the evolution of the chlamydial plasmid superimposed. The thickness of the tree branches indicates plasmid size/reduction or loss of the plasmid (dashed lines) in the respective evolutionary lineages. The number of chromosomal and plasmid homologs with the plasmids of the Chlamydiaceae, Simkania, and T4S system proteins (Tra) are given for each lineage next to the branch tips (including the chromosomal copies of pGP6-D found in all lineages). pr, Protochlamydia; pa, Parachlamydia acanthamoebae UV-7, w, W. chondrophila WSU 86-1044; s, Simkania; c, Chlamydiaceae.
Taken together, pSn meets all criteria of a typical conjugative plasmid with highest similarity to F-type plasmids. pSn is thus the first example of a mobilizable conjugation system in the Chlamydiae. Chlamydiae cannot be readily manipulated genetically, and methods to generate targeted mutants became available only very recently (Heuer et al. 2007; Binet and Maurelli 2009; Kari et al. 2011). A conjugative plasmid might be an alternative, feasible way for genetic manipulation using Simkania as a model organism.
The Evolutionary History of Chlamydial Plasmids
Phylogenetic analysis of three F-plasmid–like conjugative proteins present in Simkania, Parachlamydia, and Protochlamydia (TraUNF) demonstrated that the evolutionary history of the chlamydial T4S system matches the phylogeny inferred from 16S rRNA analysis and concatenated conserved proteins (figs. 2 and 5B). Thus, the tra region has long been present in Simkania and the Parachlamydiaceae. The most parsimonious scenario for its evolutionary history is that it was acquired by a progenitor of the Simkaniaceae, the Parachlamydiaceae, and the Waddliaceae. Although it was lost in the Waddliaceae, a (partial) T4S system is still encoded on the chromosomes of the Parachlamydiaceae and maintained on the Simkania plasmid (fig. 5A). Is it possible that the acquisition of a conjugative plasmid by chlamydiae is even more ancient? Could a single conjugative plasmid represent the source of all chlamydial plasmids, including the plasmids of the Chlamydiaceae? Plasmids of the Chlamydiaceae are known to share a common ancestor (Thomas et al. 1997), and the presence of plasmid-encoded proteins of the Chlamydiaceae on the Simkania plasmid pSn (n = 3) and the W. chondrophila WSU 86-1044 plasmid pWc (n = 2) indeed supports a common evolutionary origin of all chlamydial plasmids (fig. 6A). In addition, homologs of plasmid-encoded proteins of the Chlamydiaceae are found on the chromosomes of Simkania, Waddlia, Protochlamydia, and Parachlamydia.
We speculate that the large Simkania plasmid pSn resembles most closely the conjugative plasmid originally acquired by the chlamydial ancestor. This original plasmid was subsequently reduced in size due to gene loss, and some of its genes moved to a chromosomal location (fig. 6B). In support of this hypothesis, more than 20 homologs of pSn proteins are present in the Parachlamydiaceae and Waddlia (fig. 6A). Similar to the situation in the rickettsiae where most species infecting vertebrates have lost the plasmid genes involved in conjugation (Gillespie et al. 2007, 2010; Weinert et al. 2009), these genes are absent in the genomes of extant Chlamydiaceae.
The detection of traces of the evolutionary history of the chlamydial plasmid is consistent with the organismal phylogeny and suggests a single acquisition although subsequent lineage-specific diversification might be counterintuitive. However, plasmid mobility is generally confined within larger clades (Smillie et al. 2010), and the obligate intracellular lifestyle of the Chlamydiae has likely hampered the exchange of plasmids between phylogenetically different chlamydiae and between chlamydiae and other bacteria.
Clues for the origin of the chlamydial conjugative plasmid are available from phylogenetic analysis of the tra region, which indicates a close relationship of the chlamydial and rickettsial T4S. This is further supported by the mosaic structure of the tra region consisting of genes of F-plasmid and Ti-plasmid origin in Simkania and some rickettsiae (fig. 5). All Rickettsiales T4S systems share a common ancestor, and the acquisition of T4S is considered a key event during the transition of the rickettsiae to an intracellular lifestyle (Gillespie et al. 2010). The chlamydial T4S system is most closely related to that of R. bellii, Rickettsia massiliae, and Orientia tsutsugamushi in TraUNF-based phylogenetic trees (fig. 5B), suggesting that chlamydial and rickettsial plasmids share a common evolutionary history. Although it seems likely that the chlamydiae have acquired a conjugative plasmid from a rickettsial progenitor, phylogenetic analysis of additional T4S proteins failed to provide a clear picture of the direction of transfer.
Chlamydiae and the Evolution of Plants
The comparison of genomic data from all domains of life revealed an unusually large proportion of chlamydial proteins with close relationship to plant proteins, suggesting at least a transient association of these organisms and horizontal gene transfer during their early evolutionary history (Stephens et al. 1998; Brinkman et al. 2002; Horn et al. 2004; Huang and Gogarten 2007; Tyra et al. 2007; Becker et al. 2008; Moustafa et al. 2008; Suzuki and Miyagishima 2010). This surprising finding led to the hypothesis that an ancient endosymbiotic Protochlamydia-like bacterium resided in the progenitor of extant plant cells (Huang and Gogarten 2007; Moustafa et al. 2008). In this scenario, the Protochlamydia-like bacterium facilitated the establishment of the cyanobacteria-derived plastid in the primary photosynthetic lineage by contribution of genes (and gene products), which were transferred eventually to the nucleus of the plant ancestor. We further investigated this hypothesis and the occurrence of horizontal gene transfer between chlamydiae and plants by applying a comprehensive phylogenomic approach in the light of an extended data set including additional Plantae (Viridiplantae and Rhodophyta) (Adl et al. 2005) as well as Simkania, Waddlia, and the Parachlamydiaceae (supplementary table S14, Supplementary Material online).
In this analysis, 53 chlamydial proteins show a commonorigin with Plantae proteins in maximum likelihood–based phylogenies with bootstrap support values above 70% (table 3, supplementary table S15, Supplementary Material online). This number is comparable to the number of chlamydial proteins in plant genomes identified earlier, but remarkably, these sets of proteins only overlap partly (n = 29) (Stephens et al. 1998; Brinkman et al. 2002; Horn et al. 2004; Huang and Gogarten 2007; Tyra et al. 2007; Becker et al. 2008; Moustafa et al. 2008; Suzuki and Miyagishima 2010). There are two reasons why several plant proteins with a reported close relationship to Chlamydiae could not be confirmed in this study. First, we used a significantly larger data set than previous studies, and in our analyses, several proteins showed closer relationships to proteins of other organisms whose genomes were not available earlier. Second, a number of plant proteins with similarity to chlamydial proteins were not supported by our more stringent approach.
Table 3.
Phylogenetic Scenarios Observed for Chlamydial Proteins Encoded in Plant Genomes.
| Evolutionary Scenario | Branching Pattern in Trees | Number of Treesa |
| I | (Chlamydiae, Plantae) | 9 |
| II | (Chlamydiaeb, Plantae) | 8 |
| III | (Chlamydiaec, Plantae) | 6 |
| IV | (Chlamydiaceae (other Chlamydiae, Plantae)) | 5 |
| V | (Chlamydiaec, Rhodophyta) | 1 |
| VI | (Chlamydiaec, Chlorophyta) | 2 |
| VII | (Rhodophyta (Chlamydiae, Viridiplantae)) | 3 |
| VIII | (Chlorophyta (Chlamydiae, Streptophyta)) | 4 |
| IX | Protein present in Plantae and Chlamydiaeb only | 9 |
| X | Other | 3 |
NOTE.—Details and trees are available in supplementary table S15 and the Supplementary Material online.
Orthologs were considered only once.
Protein trees do not reflect the 16S rRNA-based tree topology within the Chlamydiae.
No homologs in the Chlamydiaceae.
Among the chlamydial proteins showing a common origin with Plantae proteins in this study, ten different scenarios for their evolutionary history can be distinguished based on the phylogenetic trees obtained (table 3, supplementary table S15, Supplementary Material online). In 31 cases, the phylogenetic trees suggested the acquisition of chlamydial genes by the ancestor of all Plantae from a chlamydial progenitor (scenarios I–VI). In seven cases, chlamydial proteins grouped together only with a subgroup of the Plantae (scenarios VII and VIII), and for nine cases, the direction of gene transfer is less clear (scenario IX and X).
The largest fraction of functionally characterized plant proteins potentially acquired from a chlamydial ancestor is involved in carbohydrate metabolism and energy production (n = 12, 23%; supplementary table S15, Supplementary Material online), including some important housekeeping genes encoding, for example, malate dehydrogenase, a key enzyme in the tricarboxylic acid cycle, or phosphoglycerate mutase required for glycolysis. Metabolite transporters including nucleotide transporters, which have been proposed to play an essential role during the early interaction of chlamydiae, cyanobacteria, and plants, are not overrepresented in our set of horizontally acquired proteins (Huang and Gogarten 2007; Tyra et al. 2007). Five plant proteins of chlamydial origin found in this study function in lipid metabolism, including the deoxyxylulose 5-phosphate pathway which has been identified earlier as being horizontally acquired from bacteria (Lange et al. 2000) and which is used for biosynthesis of isopentenyl diphosphate (a precursor of carotinoids, chlorophyll side chains, plastoquinone, mono-, and diterpenes). Three out of seven enzymes involved in this pathway are of chlamydial origin, whereas the remaining proteins are derived from other bacteria. Nine (17%) horizontally transferred proteins play a role in translation and include tRNA synthetases and tRNA and rRNA modification enzymes. Thus, most of the plant proteins of chlamydial origin are involved in basic plant metabolism, supporting an ancient contribution of chlamydiae to extant plant genomes.
Taken together, this analysis extends previous studies and provides further evidence for a primordial interaction between the progenitors of modern chlamydiae and plants including the occurrence of horizontal gene transfer from chlamydiae to the ancestral primary photosynthetic eukaryote.
Conclusions
This first analysis of genome sequences of representative members from four families within the phylum Chlamydiae provided novel insights into genetic diversity and genome evolution of this unique group of obligate intracellular bacteria. Using a pan-genome approach, we were able to highlight both conserved features such as the core gene set of the Chlamydiae as well as group-specific characteristics including a chemosensory system in Parachlamydia and the largest plasmid yet seen among the Chlamydiae in Simkania. The definition of a core gene set characteristic to all chlamydiae regardless of their primary hosts will help to identify those genes responsible for the unique chlamydial developmental cycle. Virulence-associated proteins among the Chlamydiae varied widely, perhaps highlighting the extent of varying selection effects due to their largely different eukaryotic host cells, from amoeba to humans. Simkania, which is capable of efficiently infecting both amoeba and humans, may represent a valuable model organism to investigate the adaptation of chlamydiae to human hosts. Analysis of the large conjugative plasmid of this organism suggested a common evolutionary origin of all known chlamydial plasmids; its acquisition might have been involved in the transition to an obligate intracellular lifestyle.
Among all chlamydiae, Parachlamydia shows the least truncated metabolic pathways. A recent study suggested that chlamydiae are metabolically active outside of their host cells for extended time periods (Haider et al. 2010). Similar to the strategy used for cell-free cultivation of the intracellular bacterium Coxiella burnetii (Omsland et al. 2009), it might be possible to use the available genome sequence information reported here to create a culture medium for Parachlamydia. As an organism that remains recalcitrant to experimental investigations, a cell-free culture would open up a new range of possibilities to explore chlamydial biology.
Supplementary Material
Supplementary text, tables S1—S15, and figures S1–S4 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
We would like to acknowledge Ilias Lagkouvardos for help with custom Perl scripts. The searches for remote homologs of CPAF were performed by Maximilian Petrasko and Michael Sonntag. We thank the former Institute for Genomic Research (TIGR) and current Institute for Genome Sciences (IGS) faculty and the IGS Informatics group for expert advice and assistance. This work was funded by grants of the Austrian Science Fund FWF (Y277-B03); University of Vienna (Research Focus Project FS573001). Whole-genome sequencing, assembly, and preliminary annotation of S. negevensis Z, P. acanthamoebae UV-7, and W. chondrophila 2032/99 was accomplished with support from the United States National Institute of Allergy and Infectious Diseases (1R01AI051472). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. M.H., T.D.R., G.S.A.M., R.C.B., and P.M.B. designed the study. A.C., E.H., and T.P. designed and performed experiments. A.C., P.T., T.W., E.H., and T.R. designed and performed bioinformatic and phylogenetic analyses. A.C., G.S.A.M., and M.H. wrote the manuscript. K.S., S.K., and M.G.F. provided Waddlia and Simkania genomic DNA, respectively. All authors discussed the results and commented on the manuscript.
References
- Adekoya OA, Sylte I. The thermolysin family (M4) of enzymes: therapeutic and biotechnological potential. Chem Biol Drug Des. 2009;73:7–16. doi: 10.1111/j.1747-0285.2008.00757.x. [DOI] [PubMed] [Google Scholar]
- Adl SM, Simpson AGB, Farmer MA, et al. (28 co-authors) The new higher level classification of eukaryotes with emphasis on the taxonomy of protists. J Eukaryot Microbiol. 2005;52:399–451. doi: 10.1111/j.1550-7408.2005.00053.x. [DOI] [PubMed] [Google Scholar]
- Alexander RP, Zhulin IB. Evolutionary genomics reveals conserved structural determinants of signaling and adaptation in microbial chemoreceptors. Proc Natl Acad Sci U S A. 2007;104:2885–2890. doi: 10.1073/pnas.0609359104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W. Meyers EW, Lipman DJ. Basic Local Alignment Search Tool. J Mol Biol. 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Alzhanov DT, Weeks SK, Burnett JR, Rockey DD. Cytokinesis is blocked in mammalian cells transfected with Chlamydia trachomatis gene CT223. BMC Microbiol. 2009;9:2. doi: 10.1186/1471-2180-9-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony KG, Klimke WA, Manchak J, Frost LS. Comparison of proteins involved in pilus synthesis and mating pair stabilization from the related plasmids F and R100-1: insights into the mechanism of conjugation. J Bacteriol. 1999;181:5149–5159. doi: 10.1128/jb.181.17.5149-5159.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannantine JP, Griffiths RS, Viratyosin W, Brown WJ, Rockey DD. A secondary structure motif predictive of protein localization to the chlamydial inclusion membrane. Cell Microbiol. 2000;2:35–47. doi: 10.1046/j.1462-5822.2000.00029.x. [DOI] [PubMed] [Google Scholar]
- Baud D, Regan L, Greub G. Emerging role of Chlamydia and Chlamydia-like organisms in adverse pregnancy outcomes. Curr Opin Infect Dis. 2008;21:70–76. doi: 10.1097/QCO.0b013e3282f3e6a5. [DOI] [PubMed] [Google Scholar]
- Becker B, Hoef-Emden K, Melkonian M. Chlamydial genes shed light on the evolution of photoautotrophic eukaryotes. BMC Evol Biol. 2008;8:203. doi: 10.1186/1471-2148-8-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeckman DS, Vanrompay DC. Bacterial secretion systems with an emphasis on the Chlamydial type III secretion system. Curr Issues Mol Biol. 2009;12:17–42. [PubMed] [Google Scholar]
- Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: signalP 3.0. J Mol Biol. 2004;340:783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- Bertelli C, Collyn F, Croxatto A, Ruckert C, Polkinghorne A, Kebbi-Beghdadi C, Goesmann A, Vaughan L, Greub G. The waddlia genome: a window into chlamydial biology. PLoS One. 2010;5:e10890. doi: 10.1371/journal.pone.0010890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besemer J, Lomsadze A, Borodovsky M. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001;29:2607–2618. doi: 10.1093/nar/29.12.2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binet R, Maurelli AT. Transformation and isolation of allelic exchange mutants of Chlamydia psittaci using recombinant DNA introduced by electroporation. Proc Natl Acad Sci U S A. 2009;106:292–297. doi: 10.1073/pnas.0806768106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkelund S, Morgan-Fisher M, Timmerman E, Gevaert K, Shaw AC, Christiansen G. Analysis of proteins in Chlamydia trachomatis L2 outer membrane complex, COMC. FEMS Immunol Med Microbiol. 2009;55:187–195. doi: 10.1111/j.1574-695X.2009.00522.x. [DOI] [PubMed] [Google Scholar]
- Bray JR, Curtis JT. An ordination of the upland forest communities of Southern Wisconsin. Ecol Monogr. 1957;27:325–349. [Google Scholar]
- Brinkman FS, Blanchard JL, Cherkasov A, et al. (14 co-authors) Evidence that plant-like genes in Chlamydia species reflect an ancestral relationship between Chlamydiaceae, cyanobacteria, and the chloroplast. Genome Res. 2002;12:1159–1167. doi: 10.1101/gr.341802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carabeo RA, Mead DJ, Hackstadt T. Golgi-dependent transport of cholesterol to the Chlamydia trachomatis inclusion. Proc Natl Acad Sci U S A. 2003;100:6771–6776. doi: 10.1073/pnas.1131289100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson JH, Porcella SF, McClarty G, Caldwell HD. Comparative genomic analysis of Chlamydia trachomatis oculotropic and genitotropic strains. Infect Immun. 2005;73:6407–6418. doi: 10.1128/IAI.73.10.6407-6418.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson JH, Whitmire WM, Crane DD, et al. (12 co-authors) The Chlamydia trachomatis plasmid is a transcriptional regulator of chromosomal genes and a virulence factor. Infect Immun. 2008;76:2273–2283. doi: 10.1128/IAI.00102-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian JG, Vier J, Paschen SA, Hacker G. Cleavage of the NF-{kappa}B-family protein p65/RelA by the chlamydial protease chlamydial protease-like activity factor (CPAF) impairs pro-inflammatory signalling in cells Infected with chlamydiae. J Biol Chem. 2010;285:41320–41327. doi: 10.1074/jbc.M110.152280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingro A, Poppert S, Heinz E, Schmitz-Esser S, Essig A, Schweikert M, Wagner M, Horn M. Recovery of an environmental chlamydia strain from activated sludge by co-cultivation with Acanthamoeba sp. Microbiology. 2005;151:301–309. doi: 10.1099/mic.0.27406-0. [DOI] [PubMed] [Google Scholar]
- Corsaro D, Valassina M, Venditti D. Increasing diversity within Chlamydiae. Crit Rev Microbiol. 2003;29:37–78. doi: 10.1080/713610404. [DOI] [PubMed] [Google Scholar]
- Cortes C, Rzomp KA, Tvinnereim A, Scidmore MA, Wizel B. Chlamydia pneumoniae inclusion membrane protein Cpn0585 interacts with multiple Rab GTPases. Infect Immun. 2007;75:5586–5596. doi: 10.1128/IAI.01020-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings L, Riley L, Black L, Souvorov A, Resenchuk S, Dondoshansky I, Tatusova T. Genomic BLAST: custom-defined virtual databases for complete and unfinished genomes. FEMS Microbiol Lett. 2002;216:133–138. doi: 10.1111/j.1574-6968.2002.tb11426.x. [DOI] [PubMed] [Google Scholar]
- Delevoye C, Nilges M, Dautry-Varsat A, Subtil A. Conservation of the biochemical properties of IncA from Chlamydia trachomatis and Chlamydia caviae: oligomerization of IncA mediates interaction between facing membranes. J Biol Chem. 2004;279:46896–46906. doi: 10.1074/jbc.M407227200. [DOI] [PubMed] [Google Scholar]
- Delevoye C, Nilges M, Dehoux P, Paumet F, Perrinet S, Dautry-Varsat A, Subtil A. SNARE protein mimicry by an intracellular bacterium. PLOS Pathog. 2008;4:e1000022. doi: 10.1371/journal.ppat.1000022. doi:1000010.1001371/journal.ppat.1000022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delorenzi M, Speed T. An HMM model for coiled-coil domains and a comparison with PSSM-based predictions. Bioinformatics. 2002;18:617–625. doi: 10.1093/bioinformatics/18.4.617. [DOI] [PubMed] [Google Scholar]
- Emanuelsson O, Brunak S, von Heijne G, Nielsen H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protoc. 2007;2:953–971. doi: 10.1038/nprot.2007.131. [DOI] [PubMed] [Google Scholar]
- Frickey T, Lupas AN. PhyloGenie: automated phylome generation and analysis. Nucleic Acids Res. 2004;32:5231–5238. doi: 10.1093/nar/gkh867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman MG, Dvoskin B, Kahane S. Infections with the chlamydia-like microorganism Simkania negevensis, a possible emerging pathogen. Microbes Infect. 2003;5:1013–1021. doi: 10.1016/s1286-4579(03)00188-6. [DOI] [PubMed] [Google Scholar]
- Frishman D, Albermann K, Hani J, Heumann K, Metanomski A, Zollner A, Mewes HW. Functional and structural genomics using PEDANT. Bioinformatics. 2001;17:44–57. doi: 10.1093/bioinformatics/17.1.44. [DOI] [PubMed] [Google Scholar]
- Gardner PP, Daub J, Tate JG, et al. (11 co-authors) Rfam: updates to the RNA families database. Nucleic Acids Res. 2009;37:D136–D140. doi: 10.1093/nar/gkn766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie JJ, Beier MS, Rahman MS, Ammerman NC, Shallom JM, Purkayastha A, Sobral BS, Azad AF. Plasmids and rickettsial evolution: insight from Rickettsia felis. PLoS One. 2007;2:e266. doi: 10.1371/journal.pone.0000266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie JJ, Brayton KA, Williams KP, Diaz MA, Brown WC, Azad AF, Sobral BW. Phylogenomics reveals a diverse Rickettsiales type IV secretion system. Infect Immun. 2010;78:1809–1823. doi: 10.1128/IAI.01384-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Valero L, Silva FJ, Christophe Simon J, Latorre A. Genome reduction of the aphid endosymbiont Buchnera aphidicola in a recent evolutionary time scale. Gene. 2007;389:87–95. doi: 10.1016/j.gene.2006.10.001. [DOI] [PubMed] [Google Scholar]
- Greub G, Collyn F, Guy L, Roten CA. A genomic island present along the bacterial chromosome of the Parachlamydiaceae UWE25, an obligate amoebal endosymbiont, encodes a potentially functional F-like conjugative DNA transfer system. BMC Microbiol. 2004;4:48. doi: 10.1186/1471-2180-4-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greub G, Kebbi-Beghdadi C, Bertelli C, Collyn F, Riederer BM, Yersin C, Croxatto A, Raoult D. High throughput sequencing and proteomics to identify immunogenic proteins of a new pathogen: the dirty genome approach. PLoS One. 2009;4:e8423. doi: 10.1371/journal.pone.0008423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greub G, Mege J-L, Raoult D. Parachlamydia acanthamoebae enters and multiplies within human macrophages and induces their apoptosis. Infect Immun. 2003;71:5979–5985. doi: 10.1128/IAI.71.10.5979-5985.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greub G, Raoult D. Parachlamydiaceae: potential emerging pathogens. Emerg Infect Dis. 2002;8:625–630. doi: 10.3201/eid0806.010210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Habyarimana F, Al-Khodor S, Kalia A, Graham JE, Price CT, Garcia MT, Kwaik YA. Role for the ankyrin eukaryotic-like genes of Legionella pneumophila in parasitism of protozoan hosts and human macrophages. Environ Microbiol. 2008;10:1460–1474. doi: 10.1111/j.1462-2920.2007.01560.x. [DOI] [PubMed] [Google Scholar]
- Hackstadt T, Scidmore-Carlson MA, Shaw EI, Fischer ER. The Chlamydia trachomatis IncA protein is required for homotypic vesicle fusion. Cell Microbiol. 1999;1:119–130. doi: 10.1046/j.1462-5822.1999.00012.x. [DOI] [PubMed] [Google Scholar]
- Haider S, Wagner M, Schmid MC, et al. (13 co-authors) Raman microspectroscopy reveals long-term extracellular activity of Chlamydiae. Mol Microbiol. 2010;77:687–700. doi: 10.1111/j.1365-2958.2010.07241.x. [DOI] [PubMed] [Google Scholar]
- Halsey TA, Vazquez-Torres A, Gravdahl DJ, Fang FC, Libby SJ. The ferritin-like Dps protein is required for Salmonella enterica serovar Typhimurium oxidative stress resistance and virulence. Infect Immun. 2004;72:1155–1158. doi: 10.1128/IAI.72.2.1155-1158.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch TP. Developmental biology. In: Stephens RS, editor. Chlamydia. Washington (DC): American Society for Microbiology; 1999. pp. 29–67. [Google Scholar]
- Heinz E, Pichler P, Heinz C, Op den Camp HJ, Toenshoff ER, Ammerer G, Mechtler K, Wagner M, Horn M. Proteomic analysis of the outer membrane of Protochlamydia amoebophila elementary bodies. Proteomics. 2010 doi: 10.1002/pmic.201000302. [DOI] [PubMed] [Google Scholar]
- Heinz E, Rockey DD, Montanaro J, Aistleitner K, Wagner M, Horn M. Inclusion membrane proteins of Protochlamydia amoebophila UWE25 reveal a conserved mechanism for host cell interaction among the Chlamydiae. J Bacteriol. 2010;192:5093–5102. doi: 10.1128/JB.00605-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz E, Tischler P, Rattei T, Myers G, Wagner M, Horn M. Comprehensive in silico prediction and analysis of chlamydial outer membrane proteins reflects evolution and life style of the Chlamydiae. BMC Genomics. 2009;10:634. doi: 10.1186/1471-2164-10-634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson IR, Lam AC. Polymorphic proteins of Chlamydia spp.—autotransporters beyond the Proteobacteria. Trends Microbiol. 2001;9:573–578. doi: 10.1016/s0966-842x(01)02234-x. [DOI] [PubMed] [Google Scholar]
- Henning K, Schares G, Granzow H, Polster U, Hartmann M, Hotzel H, Sachse K, Peters M, Rauser M. Neospora caninum and Waddlia chondrophila strain 2032/99 in a septic stillborn calf. Vet Microbiol. 2002;85:285–292. doi: 10.1016/s0378-1135(01)00510-7. [DOI] [PubMed] [Google Scholar]
- Heuer D, Kneip C, Maurer AP, Meyer TF. Tackling the intractable—Approaching the genetics of Chlamydiales. Int J Med Microbiol. 2007;297:569–576. doi: 10.1016/j.ijmm.2007.03.011. [DOI] [PubMed] [Google Scholar]
- Horn M. Chlamydiae as symbionts in eukaryotes. Annu Rev Microbiol. 2008;62:113–131. doi: 10.1146/annurev.micro.62.081307.162818. [DOI] [PubMed] [Google Scholar]
- Horn M, Collingro A, Schmitz-Esser S, et al. (13 co-authors) Illuminating the evolutionary history of chlamydiae. Science. 2004;304:728–730. doi: 10.1126/science.1096330. [DOI] [PubMed] [Google Scholar]
- Horn M, Fritsche TR, Gautom RK, Schleifer K-H, Wagner M. Novel bacterial endosymbionts of Acanthamoeba spp. related to the Paramecium caudatum symbiont Caedibacter caryophilus. Environ Microbiol. 1999;1:357–367. doi: 10.1046/j.1462-2920.1999.00045.x. [DOI] [PubMed] [Google Scholar]
- Horton P, Park KJ, Obayashi T, Fujita N, Harada H, Adams-Collier CJ, Nakai K. WoLF PSORT: protein localization predictor. Nucleic Acids Res. 2007;35:W585–W587. doi: 10.1093/nar/gkm259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Gogarten J. Did an ancient chlamydial endosymbiosis facilitate the establishment of primary plastids? Genome Biol. 2007;8:R99. doi: 10.1186/gb-2007-8-6-r99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugo ER, Gast RJ, Byers TJ, Stewart VJ. Purification of amoea mtDNA using the UNSET procedure. In: Lee JJ, Solldo AT, editors. Protocols in protozoology. Lawrence (KN): Allen Press; 1992. pp. D7.1–D7.2. [Google Scholar]
- Hunter S, Apweiler R, Attwood TK, et al. (38 co-authors) InterPro: the integrative protein signature database. Nucleic Acids Res. 2009;37:D211–D215. doi: 10.1093/nar/gkn785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huston WM. Bacterial proteases from the intracellular vacuole niche; protease conservation and adaptation for pathogenic advantage. FEMS Immunol Med Microbiol. 2010;59:1–10. doi: 10.1111/j.1574-695X.2010.00672.x. [DOI] [PubMed] [Google Scholar]
- Huston WM, Swedberg JE, Harris JM, Walsh TP, Mathews SA, Timms P. The temperature activated HtrA protease from pathogen Chlamydia trachomatis acts as both a chaperone and protease at 37 degrees C. FEBS Lett. 2007;581:3382–3386. doi: 10.1016/j.febslet.2007.06.039. [DOI] [PubMed] [Google Scholar]
- Jehl MA, Arnold R, Rattei T. Effective—a database of predicted secreted bacterial proteins. Nucleic Acids Res. 2011;39:D591–D595. doi: 10.1093/nar/gkq1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewett TJ, Fischer ER, Mead DJ, Hackstadt T. Chlamydial TARP is a bacterial nucleator of actin. Proc Natl Acad Sci U S A. 2006;103:15599–15604. doi: 10.1073/pnas.0603044103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DL, Stone CB, Bulir DC, Coombes BK, Mahony JB. A novel inhibitor of Chlamydophila pneumoniae protein kinase D (PknD) inhibits phosphorylation of CdsD and suppresses bacterial replication. BMC Microbiol. 2009;9:218. doi: 10.1186/1471-2180-9-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahane S, Dvoskin B, Mathias M, Friedman MG. Infection of Acanthamoeba polyphaga with Simkania negevensis and S. negevensis survival within amoebal cysts. Appl Environ Microbiol. 2001;67:4789–4795. doi: 10.1128/AEM.67.10.4789-4795.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahane S, Everett KD, Kimmel N, Friedman MG. Simkania negevensis strain ZT: growth, antigenic and genome characteristics. Int J Syst Bacteriol. 1999;49(Pt 2):815–820. doi: 10.1099/00207713-49-2-815. [DOI] [PubMed] [Google Scholar]
- Kahane S, Gonen R, Sayada C, Elion J, Friedman MG. Description and partial characterization of a new chlamydia-like microorganism. FEMS Microbiol Lett. 1993;109:329–334. doi: 10.1016/0378-1097(93)90041-y. [DOI] [PubMed] [Google Scholar]
- Kalman S, Mitchell W, Marathe R, Lammel C, Fan J, Hyman RW, Olinger L, Grimwood J, Davis RW, Stephens RS. Comparative genomes of Chlamydia pneumoniae and C. trachomatis. Nat Genet. 1999;21:385–389. doi: 10.1038/7716. [DOI] [PubMed] [Google Scholar]
- Kamneva OK, Liberles DA, Ward NL. Genome-wide influence of indel substitutions on evolution of bacteria of the PVC superphylum, revealed using a novel computational method. Genome Biol Evol. 2010;2:870–886. doi: 10.1093/gbe/evq071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kari L, Goheen MM, Randall LB, et al. (12 co-authors) Generation of targeted Chlamydia trachomatis null mutants. Proc Natl Acad Sci U S A. 2011;108:7189–7193. doi: 10.1073/pnas.1102229108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Asimenos G, Toh H. Multiple alignment of DNA sequences with MAFFT. Methods Mol Biol. 2009;537:39–64. doi: 10.1007/978-1-59745-251-9_3. [DOI] [PubMed] [Google Scholar]
- Kirby JR. Chemotaxis-like regulatory systems: unique roles in diverse bacteria. Annu Rev Microbiol. 2009;63:45–59. doi: 10.1146/annurev.micro.091208.073221. [DOI] [PubMed] [Google Scholar]
- Lad SP, Yang G, Scott DA, Wang G, Nair P, Mathison J, Reddy VS, Li E. Chlamydial CT441 is a PDZ domain-containing tail-specific protease that interferes with the NF-kappaB pathway of immune response. J Bacteriol. 2007;189:6619–6625. doi: 10.1128/JB.00429-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange BM, Rujan T, Martin W, Croteau R. Isoprenoid biosynthesis: the evolution of two ancient and distinct pathways across genomes. Proc Natl Acad Sci U S A. 2000;97:13172–13177. doi: 10.1073/pnas.240454797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawley TD, Klimke WA, Gubbins MJ, Frost LS. F factor conjugation is a true type IV secretion system. FEMS Microbiol Lett. 2003;224:1–15. doi: 10.1016/S0378-1097(03)00430-0. [DOI] [PubMed] [Google Scholar]
- Liu X, Afrane M, Clemmer DE, Zhong G, Nelson DE. Identification of Chlamydia trachomatis outer membrane complex proteins by differential proteomics. J Bacteriol. 2010;192:2852–2860. doi: 10.1128/JB.01628-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe T, Eddy S. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig W, Strunk O, Westram R, et al. (32 co-authors) ARB: a software environment for sequence data. Nucleic Acids Res. 2004;32:1363–1371. doi: 10.1093/nar/gkh293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupas A, Van Dyke M, Stock J. Predicting coiled coils from protein sequences. Science. 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- Mahony JB, Coombes BK, Chernesky MA. Chlamydia and Chlamydophila. In: Murray PR, Baron EJ, Pfaller MA, Tenover FC, Yolken RH, editors. Manual of clinical microbiology. Washington (DC): ASM Press; 2003. p. 991--1004. [Google Scholar]
- Merhej V, Royer-Carenzi M, Pontarotti P, Raoult D. Massive comparative genomic analysis reveals convergent evolution of specialized bacteria. Biol Direct. 2009;4:13. doi: 10.1186/1745-6150-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misaghi S, Balsara ZR, Catic A, Spooner E, Ploegh HL, Starnbach MN. Chlamydia trachomatis-derived deubiquitinating enzymes in mammalian cells during infection. Mol Microbiol. 2006;61:142–150. doi: 10.1111/j.1365-2958.2006.05199.x. [DOI] [PubMed] [Google Scholar]
- Moran NA, McCutcheon JP, Nakabachi A. Genomics and evolution of heritable bacterial symbionts. Annu Rev Genet. 2008;42:165–190. doi: 10.1146/annurev.genet.41.110306.130119. [DOI] [PubMed] [Google Scholar]
- Moustafa A, Reyes-Prieto A, Bhattacharya D. Chlamydiae has contributed at least 55 genes to Plantae with predominantly plastid functions. PLoS One. 2008;3:e2205. doi: 10.1371/journal.pone.0002205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers GS, Parker D, Al-Hasani K, et al. (25 co-authors) Genome sequence and identification of candidate vaccine antigens from the animal pathogen Dichelobacter nodosus. Nat Biotechnol. 2007;25:569–575. doi: 10.1038/nbt1302. [DOI] [PubMed] [Google Scholar]
- Norman A, Hansen LH, Sorensen SJ. Conjugative plasmids: vessels of the communal gene pool. Philos Trans R Soc Lond B Biol Sci. 2009;364:2275–2289. doi: 10.1098/rstb.2009.0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell CM, Abdelrahman YM, Green E, Darville HK, Saira K, Smith B, Darville T, Scurlock AM, Meyer CR, Belland RJ. TLR2 activation by Chlamydia trachomatis is plasmid-dependent and plasmid-responsive chromosomal loci are coordinately regulated in response to glucose limitation by C. trachomatis but not by C. muridarum. Infect Immun. 2011;3:3. doi: 10.1128/IAI.01118-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata H, La Scola B, Audic S, Renesto P, Blanc G, Robert C, Fournier PE, Claverie JM, Raoult D. Genome sequence of Rickettsia bellii illuminates the role of amoebae in gene exchanges between intracellular pathogens. PLoS Genet. 2006;2:e76. doi: 10.1371/journal.pgen.0020076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen AW, Follmann F, Hojrup P, Leah R, Sand C, Andersen P, Theisen M. Identification of human T cell targets recognized during Chlamydia trachomatis genital infection. J Infect Dis. 2007;196:1546–1552. doi: 10.1086/522524. [DOI] [PubMed] [Google Scholar]
- Omsland A, Cockrell DC, Howe D, Fischer ER, Virtaneva K, Sturdevant DE, Porcella SF, Heinzen RA. Host cell-free growth of the Q fever bacterium Coxiella burnetii. Proc Natl Acad Sci U S A. 2009;106:4430–4434. doi: 10.1073/pnas.0812074106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M, Yun ST, Hwang SY, Chun CI, Ahn TI. The dps gene of symbiotic "Candidatus Legionella jeonii" in Amoeba proteus responds to hydrogen peroxide and phagocytosis. J Bacteriol. 2006;188:7572–7580. doi: 10.1128/JB.00576-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J, Wilson DP, Myers G, Timms P, Bavoil PM. Type III secretion a la Chlamydia. Trends Microbiol. 2007;15:241–251. doi: 10.1016/j.tim.2007.04.005. [DOI] [PubMed] [Google Scholar]
- Price CT, Al-Khodor S, Al-Quadan T, Abu Kwaik Y. Indispensable role for the eukaryotic-like ankyrin domains of the ankyrin B effector of Legionella pneumophila within macrophages and amoebae. Infect Immun. 2010;78:2079–2088. doi: 10.1128/IAI.01450-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, Maglott DR. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35:D61–D65. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rastogi PA. MacVector. Integrated sequence analysis for the Macintosh. Methods Mol Biol. 2000;132:47–69. doi: 10.1385/1-59259-192-2:47. [DOI] [PubMed] [Google Scholar]
- Rattei T, Tischler P, Gotz S, Jehl MA, Hoser J, Arnold R, Conesa A, Mewes HW. SIMAP—a comprehensive database of pre-calculated protein sequence similarities, domains, annotations and clusters. Nucleic Acids Res. 2010;38:D223–D226. doi: 10.1093/nar/gkp949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read TD, Brunham RC, Shen C, et al. (25 co-authors) Genome sequences of Chlamydia trachomatis MoPn and Chlamydia pneumoniae AR39. Nucleic Acids Res. 2000;28:1397–1406. doi: 10.1093/nar/28.6.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read TD, Myers GS, Brunham RC, et al. (21 co-authors) Genome sequence of Chlamydophila caviae (Chlamydia psittaci GPIC): examining the role of niche-specific genes in the evolution of the Chlamydiaceae. Nucleic Acids Res. 2003;31:2134–2147. doi: 10.1093/nar/gkg321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockey DD, Scidmore MA, Bannantine JP, Brown WJ. Proteins in the chlamydial inclusion membrane. Microbes Infect. 2002;4:333–340. doi: 10.1016/s1286-4579(02)01546-0. [DOI] [PubMed] [Google Scholar]
- Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- Sällström B, Andersson SGE. Genome reduction in the Alphaproteobacteria. Curr Opin Microbiol. 2005;8:579–585. doi: 10.1016/j.mib.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Schmidt HA, Strimmer K, Vingron M, von Haeseler A. TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics. 2002;18:502–504. doi: 10.1093/bioinformatics/18.3.502. [DOI] [PubMed] [Google Scholar]
- Schmitz-Esser S, Tischler P, Arnold R, Montanaro J, Wagner M, Rattei T, Horn M. The genome of the amoeba symbiont "Candidatus Amoebophilus asiaticus" reveals common mechanisms for host cell interaction among amoeba-associated bacteria. J Bacteriol. 2010;192:1045–1057. doi: 10.1128/JB.01379-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smillie C, Garcillan-Barcia MP, Francia MV, Rocha EP, de la Cruz F. Mobility of plasmids. Microbiol Mol Biol Rev. 2010;74:434–452. doi: 10.1128/MMBR.00020-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- Stamatakis A, Ludwig T, Meier H. RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics. 2005;21:456–463. doi: 10.1093/bioinformatics/bti191. [DOI] [PubMed] [Google Scholar]
- Stephens RS, Kalman S, Lammel C, et al. (12 co-authors) Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science. 1998;282:754–759. doi: 10.1126/science.282.5389.754. [DOI] [PubMed] [Google Scholar]
- Stephens RS, Myers G, Eppinger M, Bavoil PM. Divergence without difference: phylogenetics and taxonomy of Chlamydia resolved. FEMS Immunol Med Microbiol. 2009;55:115–119. doi: 10.1111/j.1574-695X.2008.00516.x. [DOI] [PubMed] [Google Scholar]
- Stone C, Bulir D, Gilchrist J, Toor R, Mahony J. Interactions between flagellar and type III secretion proteins in Chlamydia pneumoniae. BMC Microbiol. 2010;10:18. doi: 10.1186/1471-2180-10-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun G, Pal S, Sarcon AK, Kim S, Sugawara E, Nikaido H, Cocco MJ, Peterson EM, de la Maza LM. Structural and functional analyses of the major outer membrane protein of Chlamydia trachomatis. J Bacteriol. 2007;189:6222–6235. doi: 10.1128/JB.00552-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Miyagishima SY. Eukaryotic and eubacterial contributions to the establishment of plastid proteome estimated by large-scale phylogenetic analyses. Mol Biol Evol. 2010;27:581–590. doi: 10.1093/molbev/msp273. [DOI] [PubMed] [Google Scholar]
- Szurmant H, Ordal GW. Diversity in chemotaxis mechanisms among the bacteria and archaea. Microbiol Mol Biol Rev. 2004;68:301–319. doi: 10.1128/MMBR.68.2.301-319.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Fedorova ND, Jackson JD, et al. (17 co-authors) The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003;4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tettelin H, Nelson KE, Paulsen IT, et al. (39 co-authors) Complete genome sequence of a virulent isolate of Streptococcus pneumoniae. Science. 2001;293:498–506. doi: 10.1126/science.1061217. [DOI] [PubMed] [Google Scholar]
- Thomas NS, Lusher M, Storey CC, Clarke IN. Plasmid diversity in Chlamydia. Microbiology. 1997;143:1847–1854. doi: 10.1099/00221287-143-6-1847. [DOI] [PubMed] [Google Scholar]
- Tyra HM, Linka M, Weber AP, Bhattacharya D. Host origin of plastid solute transporters in the first photosynthetic eukaryotes. Genome Biol. 2007;8:R212. doi: 10.1186/gb-2007-8-10-r212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner M, Horn M. The Planctomycetes, Verrucomicrobia, Chlamydiae and sister phyla comprise a superphylum with biotechnological and medical relevance. Curr Opin Biotechnol. 2006;17:241–249. doi: 10.1016/j.copbio.2006.05.005. [DOI] [PubMed] [Google Scholar]
- Weinert LA, Werren JH, Aebi A, Stone GN, Jiggins FM. Evolution and diversity of Rickettsia bacteria. BMC Biol. 2009;7:6. doi: 10.1186/1741-7007-7-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization. Global prevalence and incidence of curable STIs. Geneva (Switzerland): World Health Organization; 2001. [Google Scholar]
- World Health Organization. Priority eye diseases. 2008 [Internet]. [cited 2011 Mar 2]. Available from: http://www.who.int/blindness/causes/priority/en/index2.html. [Google Scholar]
- Wixon J, Kell D. The Kyoto encyclopedia of genes and genomes—KEGG. Yeast. 2000;17:48–55. doi: 10.1002/(SICI)1097-0061(200004)17:1<48::AID-YEA2>3.0.CO;2-H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf K, Plano GV, Fields KA. A protein secreted by the respiratory pathogen Chlamydia pneumoniae impairs IL-17 signaling via interaction with human Act1. Cell Microbiol. 2009 doi: 10.1111/j.1462-5822.2009.01290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuichet K, Alexander RP, Zhulin IB. Comparative genomic and protein sequence analyses of a complex system controlling bacterial chemotaxis. Methods Enzymol. 2007;422:1–31. doi: 10.1016/S0076-6879(06)22001-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.