

Abstract
Agricultural and household organophosphorus (OP) pesticides inhibit acetylcholinesterase (AchE), resulting in increased acetylcholine (Ach) in the central nervous system. In adults, acute and prolonged exposure to high doses of AchE inhibitors causes severe, clinically apparent symptoms, followed by lasting memory impairments and cognitive dysfunction. The neurotoxicity of repeated environmental exposure to lower, subclinical doses of OP pesticides in adults is not as well studied. However, repeated exposure to acetylcholinesterase inhibitors, such as chlorpyrifos (CPF), pyridostigmine, and sarin nerve agent, has been epidemiologically linked to delayed onset symptoms in Gulf War Illness and may be relevant to environmental exposure in farm workers among others. We treated adult mice with a subclinical dose (5 mg/kg) of CPF for 5 consecutive days and investigated hippocampal synaptic transmission and spine density early (2–7 days) and late (3 months) after CPF administration. No signs of cholinergic toxicity were observed at any time during or after treatment. At 2–7 days after the last injection, we found increased synaptic transmission in the CA3-CA1 region of the hippocampus of CPF-treated mice compared with controls. In contrast, at 3 months after CPF administration, we observed a 50% reduction in synaptic transmission likely due to a corresponding 50% decrease in CA1 pyramidal neuron synaptic spine density. This study is the first to identify a biphasic progression of synaptic abnormalities following repeated OP exposure and suggests that even in the absence of acute cholinergic toxicity, repeated exposure to CPF causes delayed persistent damage to the adult brain in vivo.
Keywords: chlorpyrifos, Gulf War Illness, Gulf War syndrome, synapse, acetylcholinesterase
Organophosphorus (OP) pesticides have broad household, agricultural, and military applications worldwide. These compounds inhibit acetylcholinesterase (AchE) and can be acutely toxic to humans at high doses (Dassanayake et al., 2008; Eaton et al., 2008; Jurewicz and Hanke, 2008; Kaplan et al., 1993). OP poisoning results in a biphasic progression of symptoms beginning with nausea, headache, fatigue, and seizures (Aardema et al., 2008; Brown and Brix, 1998). Exposure to moderate and high levels of OP pesticides is often followed by persistent delayed cognitive deficits (Dassanayake et al., 2008; Kaplan et al., 1993) and sensory (Kaplan et al., 1993; Murata et al., 1997) and motor neuropathies (Lotti and Moretto, 2005). Specifically, the OP pesticide chlorpyrifos (CPF) has been shown to cause impairments in short-term memory and cognition in adults (Kaplan et al., 1993) and in mature rodents (Bushnell et al., 1994; Cañadas et al., 2005; Cohn and MacPhail, 1997; Sánchez-Santed et al., 2004).
Although exposure to low levels of OP pesticides does not inhibit AchE to the same extent or result in the same types of severe, acute clinical symptoms as exposure to high doses, epidemiologic evidence suggests that persistent subclinical exposure to OP pesticides also can lead to long-term neurological impairment in adults (Jamal et al., 2002a,b) and in children (Jurewicz and Hanke, 2008; Rauh et al., 2006, 2011). Long-term low-dose OP exposure has been implicated as a risk factor for Parkinson’s disease (Manthripragada et al., 2010) and for amyotrophic lateral sclerosis (ALS) (Morahan et al., 2007). Cognitive (Dassanayake et al., 2009) and sensory (Jamal et al., 2002b; Pilkington et al., 2001) deficits have also been reported following occupational exposure to OP pesticides. Furthermore, combined exposure of U.S. troops to subclinical levels of OP pesticides and to more potent irreversible OP nerve agents, such as sarin and soman, is hypothesized to underlie many neurological symptoms and susceptibilities of Gulf War Illness (GWI) (Golomb, 2008; Haley, 2003a; Haley and Kurt, 1997; Jamal, 1998; Mahoney, 2001).
Environmental exposure to CPF is also a concern for the general population. In 1995, it was estimated that up to 82% of adults in the United States contained detectable levels of CPF metabolites in their urine (Hill et al., 1995). Though the United States restricted CPF from home use in 2002 due to its neurotoxic effects on childhood development (U.S. Environmental Protection Agency Administrator Announcement, 2000), it remains a ubiquitous environmental neurotoxin and is still widely used in golf course maintenance and in agriculture (Eaton et al., 2008).
Although the peripheral, sensory, and autonomic mechanisms of acute high-dose OP poisoning are well studied in adults, the physiological mechanisms of CNS dysfunction arising from repeated exposure to subclinical doses of OP in vivo are not. Behavioral studies in rodents have identified hippocampus-dependent learning and memory as a target for the neurotoxic effects of repeated subclinical CPF exposure (Prendergast et al., 1998; Terry et al., 2003), but the underlying synaptic mechanisms are unclear. The purpose of this study is to identify the early and delayed effects of repeated subclinical OP exposure on the adult mouse hippocampus, including changes in synaptic transmission, synaptic spines, and neuronal numbers, which may cause deficits in hippocampus-dependent cognitive behaviors.
MATERIALS AND METHODS
Animals and drugs.
Treatment of mice with CPF was modified from Cowan et al. (2001). Adult male mice 10–12 weeks old (C57Bl/6J, 25–30 g; Jackson Laboratories, Bar Harbor, ME) were injected (sc) with either 5 mg/kg CPF dissolved in dimethyl sulfoxide (DMSO) or with DMSO alone (vehicle control) for 5 consecutive days at a concentration of 1 mg/ml. This dose was chosen because it has been previously shown to cause < 10% whole-brain AchE inhibition following a single dose (Cowan et al., 2001) and is within range of the threshold (2 mg/kg) for erythrocyte AchE inhibition in humans (Kisicki et al., 1999). For studies investigating the early effects of CPF exposure, testing began 2 days following the last injection; for studies investigating the delayed effects of CPF exposure, testing began 3 months after the last injection. All experimental procedures were approved by the Institutional Animal Care and Use Committee of the UT Southwestern Medical Center and are in accordance with the National Institutes of Health policy on the care and use of laboratory animals.
AchE activity and protein quantification assays.
Mice were euthanized with CO2 gas, and their brains harvested at 3 and 6 h following the first injection and at 3 and 24 h following the last injection. Brains were isolated, bisected, and snap frozen on dry ice then stored at −80°C. The hippocampus was isolated from one hemisphere of each brain, diluted 1:40 (wt/vol) in 0.1M PBS, and homogenized using an ultrasonic processor (Cole Parmer, Vernon Hills, IL). Lysates were centrifuged and the supernatant tested for AchE activity within 24 h of tissue harvesting using the DACE-100 QuantiChrom Acetylcholinesterase Assay Kit (BioAssay Systems, Hayward, CA) according to the manufacturer’s protocol. AchE activity was then normalized to total protein concentration for each sample using the DC Protein Assay kit (Bio-Rad, Hercules, CA). AchE and protein assays were carried out in triplicate in 96-well plates using a Synergy HT Multi-Mode Microplate Reader (Bio-Tek, Winooski, VT).
Electrophysiology.
Mice were briefly anesthetized with the inhalation anesthetic isoflurane (Baxter Healthcare Corporation, Deerfield, IL) and rapidly decapitated for extracellular recordings. For whole-cell patch-clamp recordings, mice were deeply anesthetized with 400 mg/kg (ip) chloral hydrate (Sigma, St Louis, MO) then vascularly perfused through the heart with ice-cold artificial cerebrospinal fluid (ACSF). Brains were quickly removed and placed in ice-cold modified ACSF. Transverse hippocampal slices (300–350 μm for whole-cell recordings and 350–400 μm for extracellular recordings) were made using a vibrating microtome (Vibratome, Bannockburn, IL). Slices were allowed to recover at 33°C for 30 min in normal ACSF and slowly cooled to room temperature over a 45-min period prior to recording. Modified ACSF contained (in millimolar) 75 sucrose, 87 NaCl, 3 KCl, 1.25 NaH2PO4, 7 MgSO4, 26 NaHCO3, 20 dextrose, and 0.5 CaCl2. ACSF contained (in millimolar) 126 NaCl, 3 KCl, 1.25 NaH2PO4, 1 MgSO4, 26 NaHCO3, 10 dextrose, and 2 CaCl2. All solutions were pH 7.4 and saturated with 95% O2/5% CO2.
All recordings were performed at 33°C ± 0.5°C, and all data were collected using Clampex (pClamp software suite version 10.2; Molecular Devices, Sunnyvale, CA). Whole-cell and extracellular recordings were filtered at 1–3 kHz and digitized at 10 kHz. CA3-CA1 synapses were stimulated by a bipolar nickel dichromate stimulating electrode, and field excitatory postsynaptic potentials (fEPSPs) were recorded using glass pipette electrodes (3–5 MΩ) filled with ACSF and placed 400–500 μm apart, laterally, in the stratum radiatum. The distance between the recording electrode and the stimulating electrode was kept constant within these bounds using a SZX7 dissecting microscope (Olympus, Center Valley, PA) at ×5 magnification. Sample size represents number of mice tested with only one slice per mouse included. Response size was determined by fitting a straight line to the initial slope (10–40%) of the fEPSP using automated analysis in Clampfit (pClamp software suite version 10.2; Molecular Devices). For studies of long-term potentiation (LTP), stimulus intensity was set to generate approximately 50% of the maximum fEPSP, as determined by the input/output (I/O) curve. I/O curves were performed in each slice immediately preceding each LTP experiment, and stimulus intensity remained unchanged for the duration of the LTP experiment.
Whole-cell patch-clamp recordings were performed at 33°C ± 0.5°C in the presence of 2μM tetrodotoxin (TTX) to block evoked transmission and 100μM picrotoxin to block fast inhibitory transmission. Neurons were visualized under differential interference contrast microscopy using an AxioExaminerD1 microscope (Zeiss, Thornwood, NY). Spontaneous miniature excitatory postsynaptic currents (mEPSCs) were recorded from a holding potential of −65 mV for 2–3 min intervals starting 3–5 min after break-in using glass pipette electrodes 4–6 MΩ. Internal solution contained (in millimolar) 117 CsMeSO3, 15 CsCl, 8 NaCl, 10 TEA, 3 QX-314, 0.2 EGTA, 10 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 2 ATP, 0.3 GTP, and adjusted to pH 7.3 and 290 mOsm. Only experiments with a high seal resistance (> 3 GΩ) were analyzed and were rejected if the series or input resistance changed by more than 25% during the recording. Sample size represents no more than one neuron per slice and no more than four slices per mouse. Clampfit was used to measure peak amplitude and rise time (10–90%) of mEPSCs using automated analysis. The decay constant (τ) of mEPSCs was calculated from a single-exponential fit using the Levenberg-Marquardt least squares algorithm. Sample size represents one neuron per slice, though more than one slice was used from each mouse. All statistically significant differences observed using the number of neurons recorded as the “N” were also statistically significant when analyzed with averaged data per mouse as the N value.
Histochemistry and histology.
For all spine density experiments, there were 18 neurons from three mice per treatment group (i.e., CPF treated or vehicle treated). Mouse brains were processed for Golgi-Cox staining with the FD Rapid GolgiStain Kit (FD NeuroTechnologies, Ellicott City, MD) according to the manufacturer’s protocol. Mice were deeply anesthetized with isoflurane and rapidly decapitated. Brains were quickly removed, rinsed in double-distilled water, then immersed into impregnation solution (Solution A + B), and then stored for 2 weeks in the dark at room temperature (22°C–25°C). Brains were then transferred to Solution C and shipped to FD NeuroTechnologies within 48 h. Serial cryostat sections (100 μm) were cut coronally through the cerebrum containing the hippocampus and mounted three slices per slide.
CA1 neurons from dorsal hippocampus of Golgi-stained brain slices were selected if apical and basilar dendrites did not have any interfering overlap with neighboring stained neurons. Chosen neurons were in the same region and orientation as those selected for electrophysiological recordings. Neurons were traced using NeuroLucida 3D neuron tracing software (MicroBrightField Bioscience, Williston, VT) at ×100 magnification by an investigator blind to the treatment. Reconstructions were then partitioned with concentric circles of increasing radii beginning at 30 μm, and each circle separated by 30 μm. Dendritic segments were selected for spine counting in both the apical and the basilar dendritic arbors within defined distances from the cell body (30–90, 90–150, 150–210, 210–270, and 270–330 μm from cell body). Dendritic segments were chosen based on the following criteria: (1) segment must be at least 30 μm in length, (2) segment must cross the midpoint of the defined region, (3) segment cannot branch along the measured length, and (4) no interfering crossing of other branches that would interfere with spine counting. Two segments were chosen from each distance in both the apical and the basilar dendritic arbors. Segments from the same region in the same neuron were averaged together prior to statistical analysis.
Stereology was performed on four mice per treatment group. Mice were transcardially perfused with 0.1M PBS and 4% paraformaldehyde. Brains were then cryoprotected in 30% sucrose in PBS before collecting 30 μm coronal sections in 12 series using a freezing sliding microtome. In order to quantify the number of neurons in CA1 and CA3, we employed a rigorous stereological approach using the optical fractionator method using StereoInvestigator software (MicroBrightField Bioscience). Neurons were identified by staining sections using 4′,6-Diamidino-2-phenylindole dihydrochloride (DAPI; Sigma D9542). For counting both CA1 and CA3 neurons, every 12th section (in other words, a section sampling fraction of 1/12) was examined through the entire rostrocaudal extent of the hippocampus using a random series start. For area CA1, the step size across sections was 100 × 100 μm, and the dissector dimensions were 15 × 15 μm, with a height of 12 μm. For CA3, the step size was 140 × 140 μm, and the dissector dimensions were 20 × 20 μm, with a height of 12 μm. These sampling parameters yielded a coefficient of error of less than 10%. All slides were coded and counted by an observer blind to the treatment condition.
Statistics.
All statistical analysis was performed using SigmaPlot (version 11.0; Systat Software, Chicago, IL) and Statistica (version 5.5; StatSoft, Inc., Tulsa, OK) software packages. Statistics were performed using either a Student’s t-test, two-way ANOVA, or two-way repeated measures ANOVA, as appropriate. For the ANOVAs, pairwise comparisons were made using Tukey’s post hoc analysis, and treatment (CPF vs. vehicle) was always used as the between-subjects factor. For mouse weights and AchE activity assays during CPF treatment, treatment day was used as the within-subjects factor, and either intensity (for I/O curves) or interval (for paired-pulse ratio [PPR]) was used as within-subjects factor in electrophysiology experiments. An unpaired, two-sample Student’s t-test was performed to determine significant differences between treatment groups for the following: LTP magnitude, average mEPSC amplitude, mEPSC rise time, mEPSC decay, spine density, and neuron number. A Kolmogorov-Smirnov test was used to determine significance between cumulative frequency curves for mEPSC amplitude. All values are expressed as mean ± SEM.
Compounds.
CPF (diethoxy-sulfanylidene-(3,5,6-trichloropyridin-2-yl)oxy-λ5-phosphane) was obtained from Chem Service (West Chester, PA), and DMSO was obtained from Amresco (Solon, OH). QX-314 (N-(2,6-Dimethylphenylcarbamoylmethyl)triethylammonium bromide) and TTX (Octahydro-12-(hydroxymethyl)-2-imino-5,9:7,10a-dimethano-10aH-[1,3]dioxocino[6,5-d]pyrimidine-4,7,10,11,12-pentol) were obtained from Tocris Bioscience (Ellisville, MO), and picrotoxin was obtained from Enzo Life Sciences (Plymouth Meeting, PA). All other reagents were obtained from Sigma-Aldrich (St Louis, MO).
RESULTS
Five-Day Treatment with 5 mg/kg CPF Does Not Induce Acute Cholinergic Toxicity
Prolonged and acute exposure to high doses of OP compounds, such as CPF, elicits well-defined signs of acute cholinergic toxicity primarily due to overactivation of acetylcholine receptors (AchRs) in the peripheral and autonomic nervous systems (Aardema et al., 2008; Moser, 1995). The present study focuses on the long-term effects of repeated exposure to a subclinical dose of CPF that does not elicit any signs of overt cholinergic toxicity (Cowan et al., 2001). To ensure that mice received subclinical exposure to CPF, signs of OP poisoning were qualitatively observed throughout the treatment period, including weight loss, which is associated with CPF poisoning (Moser, 1995). No signs of cholinergic toxicity were observed in healthy mice (25–30 g), and no change in weight (Fig. 1A) was observed between days 1 and 5 of treatment in either CPF or control groups (two-way repeated measures ANOVA; treatment: p = 0.912; day: p < 0.001; treatment × day interaction: p = 0.572; N = 20 per group).
FIG. 1.

Daily treatment with 5 mg/kg CPF results in AchE inhibition but does not affect body weight. (A) Body weight of control- and CPF-treated mice as measured each day during the treatment period prior to receiving injections. (B) AchE activity measured as percentage control at four different time points during the 5-day CPF treatment period. Data represent means ± SEM. *p < 0.05 compared with vehicle.
We also measured AchE activity in the hippocampus at four time points during and after the CPF treatment period (Fig. 1B) and found a main effect of treatment on AchE activity (Two-way ANOVA treatment: p < 0.001; day: p = 0.613; treatment × day interaction p = 0.131; N = 12 per group). We found no significant inhibition of AchE with CPF at 3 h (2.97% compared with vehicle; p = 0.822) or at 6 h (18.34% compared with vehicle; p = 0.162) following the first injection in CPF-treated mice compared with vehicle controls, confirming that 5 mg/kg is a low dose of CPF, and does not cause significant inhibition of AchE activity after one injection. However, repeated administration of this low dose of CPF does result in “buildup” of AchE inhibition. We found significant inhibition of AchE activity with CPF treatment at 3 h (30.86% compared with vehicle; p < 0.05) and 24 h (38.58% compared with vehicle; p < 0.05) following the last injection of the 5-day treatment period. This additive effect of AchE inhibition with repeated CPF exposure in mice is consistent with a previous report of progressive buildup of CPF metabolites and AchE inhibition following 10-day exposure to 3 and 10 mg/kg CPF sc over a 10-day treatment period in rats (Ellison et al., 2011). Taken together, these results indicate that repeated low-dose CPF exposure does result in accumulation of AchE inhibition even in the absence of overt clinical signs of cholinergic toxicity and that our 5 mg/kg dose is appropriate for studying effects of repeated subclinical OP exposure.
Hippocampal Synaptic Transmission Is Enhanced at 1 Week Following CPF Treatment
Repeated systemic administration of subclinical CPF causes an early increase in basal synaptic transmission (Fig. 2A), as evidenced by an increase in the I/O relationship (I/O curve) and in the maximum slope of the fEPSP at stimulus intensities above 3.0 mA (two-way repeated measures ANOVA; treatment: p = 0.107; intensity: p < 0.001; treatment × intensity interaction: p < 0.001, N = 8 mice per group). At the maximum stimulus intensity applied, the mean fEPSP slope was 44% greater in CPF-treated mice compared with vehicle-treated mice (−0.33 ± 0.04 mV/ms for vehicle vs. −0.47 ± 0.04 mV/ms for CPF; Student’s t-test, p < 0.036).
FIG. 2.

Hippocampal synaptic transmission is enhanced in CPF-treated mice at 1 week following treatment. (A) fEPSP slope was measured at stimulus intensities of 0–5 mA. Maximal fEPSP slope is greater in slices from CPF-treated mice than vehicle-treated mice. *p < 0.05 compared with vehicle. Inset: Average of 10 consecutive traces from vehicle-treated (left, dark) and CPF-treated (right, light) mice at 0, 1.5, 3.0, and 4.5 mA stimulus intensities. Scale bar: 0.4 mV, 5 ms. (B) PPR is similar in vehicle-treated and CPF-treated slices at interstimulus intervals of 50–500 ms. (C) LTP is not affected by CPF treatment relative to vehicle controls. Arrow indicates high-frequency stimulation (4 1s, 100 Hz trains at 0.1 Hz). Inset: Average of 10 consecutive traces immediately preceding (solid) and 60 min following (dashed) LTP induction protocol in vehicle-treated (left, dark) or CPF-treated (right, light) mice. Scale bar: Veh: 0.2 mV, CPF: 0.25 mV; 10 ms. For all panels, data represent means ± SEM.
To determine whether this increase in synaptic transmission might be mediated by alterations in presynaptic release mechanisms, we looked for changes in the PPR of fEPSPs as a measure of presynaptic function. In this experiment, two consecutive pulses of equal stimulus intensity, determined at 50–60% of the stimulus intensity needed to elicit maximal fEPSP magnitude, are applied at specific intervals ranging from 50 to 500 ms. As a general principle, the ratio of the second pulse to that of the first is inversely related to the initial release probability of the presynaptic neuron (Debanne et al., 1996; Dobrunz, 2002; Dobrunz and Stevens, 1997; Manabe et al., 1993). No difference in PPR (Fig. 2B) was observed between CPF-treated and vehicle-treated mice at any interstimulus interval (two-way repeated measures ANOVA; treatment: p = 0.117; interstimulus interval: p < 0.001; treatment × interval interaction: p = 0.318; veh N = 8, CPF N = 9). These results indicate that the immediate increase in excitatory transmission with CPF treatment does not affect short-term plasticity and is not likely mediated by a change in initial release probability.
Early effects of CPF treatment on LTP were also analyzed in the first week following the last injection using a strong conditioning protocol (4, 1 s, 100 Hz trains given at 0.1 Hz; Fig. 2C). Following a 20-min baseline, fEPSP slope was measured at 0.1 Hz for 60 min post-tetanus. Both groups demonstrated reliable LTP, and when the magnitude of LTP was measured between 50 and 60 min post-tetanus, no difference in fEPSP slope was found between CPF-treated and control mice (163.93 ± 11.61 mV/ms for vehicle vs. 176.84 ± 18.67 mV/ms for CPF; Student’s t-test, p = 0.577; veh N = 8, CPF N = 9). Thus, despite an increase in excitatory synaptic transmission, expression and maintenance of LTP were unaffected.
We also used Golgi staining and histology of 18 neurons from three mice per treatment group to determine whether changes in spine density could account for the early increase in hippocampal synaptic transmission in CPF-treated mice (Fig. 3). CA1 neurons were chosen in Golgi-stained hippocampal sections from either vehicle control (Fig. 3B) or CPF-treated (Fig. 3C) mice, with one neuron chosen per section. Average spine density of CA1 neurons was similar between CPF and control mice (Fig. 3A; 21.51 ± 0.29 spines/10 μm for vehicle vs. 21.31 ± 0.44 spines/10 μm for CPF; Student’s t-test, p = 0.700). Next, we investigated distance-dependent changes in spine density along CA1 dendrites in CPF-treated and vehicle-treated mice. No difference in spine density was observed between CPF-treated and control mice with regard to branch order on apical segments (Fig. 3D; Student’s t-test, p > 0.05) or on basal segments (Fig. 3F; Student’s t-test, p > 0.05). At apical dendrites 270–310 μm from the soma, there was a small but significant decrease in spine number in CPF-treated mice compared with vehicle controls (Fig. 3E; 25.77 ± 0.518 spines/10 μm for vehicle vs. 21.97 ± 1.13 spines/10 μm for CPF; Student’s t-test, p = 0.022), but at distances < 210 μm from the soma, no difference in spine number was observed (Fig. 3E; Student’s t-test, p > 0.05). In addition, no change in spine density, measured in 60 μm increments beginning 30 μm from the soma, was found on basal dendrites between mice treated with CPF and those treated with vehicle (Fig. 3G; Student’s t-test, p > 0.05). Therefore, CA1 spine number is not greatly affected at 1 week following CPF treatment.
FIG. 3.

CPF treatment has no major effect on spine number on apical CA1 dendrites in the first week following treatment. (A) Total synaptic spine density is unaffected by CPF exposure at 1 week following treatment. Magnification of dendritic spines (×100) in Golgi-stained CA1 pyramidal neurons from vehicle-treated (B) and CPF-treated mice (C). Scale bars: 10 μm. Changes in spine density between vehicle-treated and CPF-treated mice are not dependent on dendritic branch order for apical (D) or basilar segments (F). Legend in panel D also applies to panels E, F, and G. Spine density is slightly decreased in CPF-treated mice at apical segments 270–310 μm from the soma (E) but not at basilar segments (G). *p < 0.05 compared with vehicle. For all panels, data represent means ± SEM.
Hippocampal Synaptic Transmission Is Severely Impaired 3 Months Following CPF Treatment
To determine the delayed effects of prolonged low-dose exposure to CPF on hippocampal function, we compared I/O relationships of stimulus intensity with fEPSP slope in CPF-treated and control- and vehicle-treated mice (Fig. 4A). We found a large decrease in fEPSP slope at almost all stimulus intensities compared with control mice, indicating a delayed decrease in synaptic transmission following CPF treatment (treatment: p < 0.001; intensity: p < 0.001; treatment × stimulus intensity interaction: p < 0.001; veh N = 12, CPF N = 11). At the maximum stimulus intensity, mice receiving CPF injections exhibited a 52% reduction in mean fEPSP slope compared with mice receiving vehicle injections (−1.35 ± 0.10 mV/ms for vehicle vs. −0.70 ± 0.08 mV/ms for CPF; Student’s t-test, p < 0.001). Thus, in striking contrast to our findings during the first week following treatment in which the maximal fEPSP slope was increased in CPF-treated mice (Fig. 2A), we find a delayed, significant reduction in hippocampal synaptic transmission at 3 months following CPF treatment (Fig. 4A).
FIG. 4.

Evoked hippocampal synaptic transmission is decreased in CPF-treated mice at 3 months following treatment. (A) fEPSP slope was measured at stimulus intensities of 0–5 mA. Maximal fEPSP slope is greatly decreased in slices from CPF-treated mice than vehicle-treated mice. *p < 0.001 compared with vehicle. Inset: Average of 10 consecutive traces from vehicle-treated (left, dark) and CPF-treated (right, light) mice at 0, 1.5, 3.0, and 4.5 mA stimulus intensities. Scale bar: 1.0 mV, 5 ms. (B) PPR is similar in vehicle-treated and CPF-treated slices at interstimulus intervals of 50–500 ms. (C) LTP is not affected by CPF treatment relative to vehicle controls. Inset: Average of 10 consecutive traces immediately preceding (solid) or 60 min following (dashed) LTP induction protocol in vehicle- (left, dark) or CPF- (right, light) treated mice. Arrow indicates tetanic stimulation (4, 1 s, 100 Hz trains at 0.1 Hz). Scale bar: 1.0 mV; 10 ms. For all panels, data represent means ± SEM.
Such a dramatic decrease in synaptic transmission could be expected to impair hippocampal synaptic plasticity. Therefore, delayed effects of CPF treatment on short- and long-term plasticity were investigated. When PPR was compared between CPF-treated and vehicle-treated mice, we found no significant difference at any interstimulus interval 50–500 ms (Fig. 4B; two-way repeated measures ANOVA; treatment: p = 0.107; interstimulus interval: p < 0.001; treatment × interval interaction: p = 0.306; N = 11 per group), suggesting that at our dose, CPF does not affect short-term plasticity and, by extrapolation, does not affect presynaptic function at CA3-CA1 synapses. Similarly, when we compared magnitude of LTP at 50–60 min following tetanic stimulation, no significant difference was observed between CPF-treated and vehicle-treated mice (Fig. 4C; 140.92 ± 0.87 mV/ms for vehicle vs. 140.21 ± 0.48 mV/ms for CPF; Student’s t-test, p = 0.475). The lack of deficits in short-term synaptic plasticity with CPF treatment suggests that synaptic changes in presynaptic function may not be responsible for the large reduction in excitatory transmission in mice receiving CPF injections.
Another potential cause for a decrease in fEPSP slope with CPF treatment is a change in the number of synapses or neurons in the CA3-CA1 region of treated mice. We first tested this hypothesis by measuring amplitude and frequency of spontaneous miniature excitatory currents (mEPSCs) from CA1 pyramidal neurons (Fig. 5). Whole-cell patch-clamp recordings (veh N = 28 neurons from eight mice, CPF N = 21 neurons from nine mice, no more than one neuron per slice) were made from CA1 cell bodies in the stratum pyramidale (s.p.) in the presence of the Na+ channel blocker TTX (2μM) to block activity-dependent synaptic transmission and the GABAA receptor antagonist picrotoxin (100μM) to block fast inhibitory transmission (Fig. 5A). Mean mEPSC frequency was dramatically reduced 3 months following CPF treatment compared with vehicle controls (Fig. 5B, 1.41 ± 0.21 Hz for vehicle vs. 0.61 ± 0.06 Hz for CPF; Student’s t-test, p < 0.01), suggesting a reduction in synapse number or neuronal number as potential mechanisms underlying the decrease in synaptic transmission with CPF treatment (taking into account the lack of altered PPR as a measure of presynaptic function). Average mEPSC amplitude (Fig. 5C; −6.62 ± 0.31 pA for vehicle vs. −7.73 ± 0.66 pA for CPF; Student’s t-test, p = 0.159) and the cumulative distribution of amplitudes were unaffected (Fig. 5D; two-sample Kolmogorov-Smirnov test, p > 0.100). We also observed decreased input resistance in CPF-treated mice compared with controls (Fig. 5E; 180.68 ± 17.92 MΩ for vehicle vs. 126.81 ± 15.19 MΩ for CPF) that slowed both the rise time (Fig. 5F; 2.70 ± 0.16 ms for vehicle vs. 4.66 ± 0.50 ms for CPF; Student’s t-test, p < 0.001) and the decay constant (Fig. 5G; 9.30 ± 0.52 ms for vehicle vs. 13.74 ± 1.36 ms for CPF; Student’s t-test, p = 0.001) of mEPSCs. These results suggest that the decrease in basal synaptic transmission in CPF-treated mice is primarily due to a decrease in either the number of CA3 synapses onto CA1 pyramidal neurons or to a decrease in CA3 or CA1 neuron number, though decreased input resistance may further impede the ability of CA1 neurons to respond to incoming stimuli in CPF-treated mice.
FIG. 5.

Spontaneous miniature hippocampal synaptic transmission frequency is decreased in CPF-treated mice at 3 months following treatment. (A) Fifteen seconds raw mEPSC recordings from vehicle-treated (top) and CPF-treated (bottom) mice at 3 months following the last injection. Scale bar: 10 pA, 150 ms. (B) mEPSC frequency is decreased in CPF-treated mice compared with vehicle-treated controls, and mEPSC amplitude (C) is unchanged. The cumulative distribution of mEPSC amplitudes (D) remains unchanged following CPF treatment compared with controls. Input resistance is decreased with CPF treatment (E), and rise time (F) and decay constant (G) of mEPSCs are prolonged in CPF-treated mice. Data represent means ± SEM. *p < 0.05 and **p < 0.001 compared with vehicle.
Spine Number But Not Neuron Number Is Greatly Decreased at 3 Months Following CPF Treatment
Prolonged direct exposure of organotypic hippocampal slice cultures to CPF at 100μM, but not 10μM, has been shown to cause CA1 neuron loss (Terry et al., 2003), and in utero repeated exposure of rats to low-dose CPF causes neuron loss in the cerebellum that persists into adulthood (Abou-Donia et al., 2006). To determine whether repeated exposure of adult mice to low doses of CPF in vivo results in neuronal loss sufficient to explain our observed decrease in synaptic transmission, we measured neuron number in the CA1 and CA3 regions of the entire hippocampus of four mice per treatment group using unbiased stereology. No difference in mean CA1 neuron number across the entire hippocampus was observed between CPF-treated and vehicle-treated mice (Fig. 6A, 145,297 ± 5040 neurons/mouse for vehicle vs. 145,938 ± 8719 neurons/mouse for CPF; Student’s t-test, p = 0.951) nor was there a difference in CA3 neuron number (Fig. 6B, 121,252 ± 12,627 neurons/mouse for vehicle vs. 131,698 ± 16,443 neurons/mouse for CPF; Student’s t-test, p = 0.632), indicating that low-dose CPF treatment does not cause a significant lasting loss of neurons in the CA1 (Fig. 6C, vehicle treated; Fig. 6E, CPF treated) or CA3 regions (Fig. 6D, vehicle treated; Fig. 6F, CPF treated).
FIG. 6.
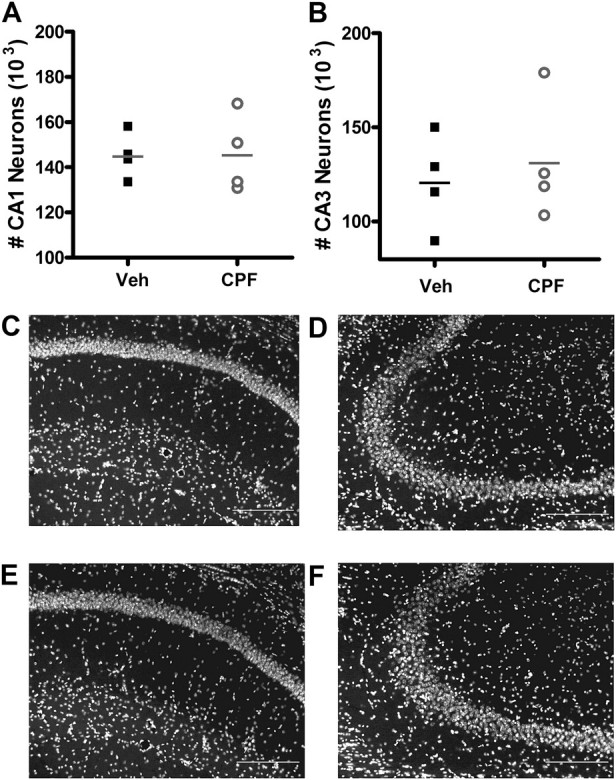
Neuron number is not affected at 3 months following CPF treatment. (A) CA1 and (B) CA3 neuron numbers are not affected by CPF treatment. Data represent single neuron counts from each mouse, with horizontal line indicating mean neuron count per treatment group ± SEM. Legend in panel A also applies to panel B. DAPI nuclear-stained sections at ×20 magnification from CA1 pyramidal neurons from vehicle-treated (C) and CPF-treated (E) mice and from CA3 pyramidal neurons from vehicle-treated (D) and CPF-treated (F) mice showing no loss of neurons at 3 months following treatment. Scale bars: 20 μm.
Next, we performed histological analysis on Golgi-stained hippocampal sections from vehicle-treated (Fig. 7B) and CPF-treated (Fig. 7C) mice to determine if the late decrease in hippocampal synaptic transmission was due to a decrease in synaptic spine number. As described for early effects of CPF treatment, 18 neurons were analyzed per treatment group with one neuron analyzed per section and six neurons analyzed per mouse. Interestingly, total spine density was reduced by 40% in CPF-treated mice compared with vehicle controls (Fig. 7A, 20.97 ± 0.74 spines/10 μm for vehicle vs. 12.57 ± 0.77 spines/10 μm for CPF; Student’s t-test, p < 0.001), indicating loss of synaptic spines as the likely mechanism of the large reduction in CA3-CA1 synaptic transmission. The delayed decrease in spine density with CPF treatment was observed throughout the length of the apical dendrite, as measured in 60 μm increments beginning at 30 μm from the soma (Fig. 7D; Student’s t-test, p < 0.005). Similarly, the decrease in spine density was also significant along the entire basal dendrite, also measured in 60 μm increments beginning at 30 μm from the soma (Fig. 7F; Student’s t-test, p < 0.001). With regard to branch order, CPF treatment caused a decrease in spine density at first, second, and third order dendritic branches of apical dendrites (Fig. 7E; Student’s t-test, p < 0.05) and at all branch points (1–5) of basal dendrites (Fig. 7G; Student’s t-test, p < 0.05).
FIG. 7.

Spine density is dramatically decreased in CPF-treated mice compared with controls at 3 months following treatment. (A) Total spine density is decreased in CPF-treated mice compared with vehicle controls (*p < 0.001 compared with vehicle). Magnification of dendritic spines (×100) in Golgi-stained CA1 pyramidal neurons from vehicle-treated (B) and CPF-treated mice (C). Scale bars: 10 μm. The decrease in spine density of (D) apical dendrites (*p < 0.01 compared with vehicle) and (F) basilar dendrites in CPF-treated mice is not dependent on distance from soma (*p < 0.001 compared with vehicle). (E) The decrease in spine density of apical dendrites is significantly different from controls up to the third branch point (*p < 0.05 compared with vehicle). (G) The decrease in spine density of basilar dendrites in CPF-treated mice is not dependent on number branch points. For panels (D–G), data represent means ± SEM.
DISCUSSION
Early versus Delayed Effects of Repeated Subclinical CPF Treatment
This study is the first to report that repeated subclinical doses of CPF result in a biphasic progression of hippocampal synaptic dysfunction in adult mice, with early effects being strikingly different than delayed effects. CA3-CA1 synaptic transmission was enhanced in CPF-treated mice compared with vehicle-treated mice at the 1-week time point. These results are consistent with studies on therapeutic AchE inhibitors showing improved hippocampal synaptic function and learning and memory in patients with Alzheimer’s disease (Birks, 2006).
Conversely, at 3 months following the last injection, we have identified a dramatic decrease in the slope of fEPSPs in response to extracellular stimulation in CPF-treated mice compared with vehicle-treated controls. This is accompanied by a drastic decrease in the frequency of mEPSCs, slowing of mEPSC kinetics, and decreased input resistance of CA1 neurons in CPF-treated mice. Although our results are in line with reports of CPF-induced delayed cytotoxicity in vitro (Rush et al., 2010; Tan et al., 2009; Terry et al., 2003), we did not observe any change in neuron number in either the CA3 or CA1 regions with treatment in vivo. However, we did observe a large novel decrease in synaptic spine density across both basilar and apical dendrites of CA1 neurons, indicating that synaptic spine loss rather than neuron loss is responsible for the decrease in basal hippocampal synaptic transmission at 3 months following CPF treatment.
Effects of Route and Dose on AchE Inhibition
The effects of CPF have been shown to be dependent on route of exposure (Carr and Nail, 2008; Marty et al., 2007; Smith et al., 2009), length of exposure (Marty et al., 2007), dose (Baireddy et al., 2011), and vehicle (Carr and Nail, 2008; Marty et al., 2007; Smith et al., 2009). The 5 mg/kg dose of CPF used in this study does not cause significant inhibition in the adult mouse brain following a single injection and may be considered low dose. However, when given systemically over a 5-day period, we see considerable buildup of AchE inhibition following the last CPF injection.
No signs of cholinergic toxicity were observed at any time during treatment, consistent with Ellison et al. (2011) who saw a similar lack of clinical symptoms in adult rats following a 10-day treatment period with 3 and 10 mg/kg CPF sc. Similarly, Middlemore-Risher et al. (2010) reported that no signs of cholinergic toxicity were elicited by 14-day treatment of adult rats with 18 mg/kg CPF despite a 63.3% reduction in AchE activity in hippocampus. Therefore, this dose is higher than is likely to be found through dietary or environmental exposure, though still low enough not to elicit signs of cholinergic toxicity. It remains unclear whether this exposure is in any way comparable to that experienced by veterans suffering from GWI (Golomb, 2008; Haley and Kurt, 1997) or by patients suffering from chronic organophosphate–induced neuropsychiatric disorder (COPIND) (Jamal et al., 2002a).
Cholinergic Modulation of Glutamatergic Hippocampal Circuits
Ach positively modulates glutamatergic transmission in the hippocampus (reviewed in Cobb and Davies, 2005; Drever et al., 2010) as well as learning and memory (reviewed in Hasselmo, 2006) primarily through the septohippocampal pathway. The primary role of AchE in the CNS is to maintain ambient levels of AchE in the extracellular space and when inhibited leads to increased activation of nicotinic (nAchR) and muscarinic (mAchRs) Ach receptors. Low-dose nicotine increases cell excitability in hippocampus (Penton et al., 2011; Szabo et al., 2008) and has been shown to lower the threshold for LTP induction at CA3-CA1 synapses (Fujii et al., 1999, 2000a,b), as does septal stimulation (Gu and Yakel, 2011). mAchRs are slower acting second messenger-coupled receptors that enhance hippocampal excitatory transmission through modulation of Ca2+, K+, and mixed ion currents (reviewed in Cobb and Davies, 2005) that are also capable of modulating glutamatergic CA3-CA1 synaptic plasticity (Gu and Yakel, 2011). Given that our 5-day treatment protocol causes a buildup of AchE inhibition, the early increase in excitatory transmission in CPF-treated mice is likely due to increased activation of AchRs by moderate AchE inhibition. The delayed effects of AchE inhibition on synaptic spines and synaptic transmission remain a mystery that will require multiple future experimental approaches to delineate.
Noncholinergic Effects of Repeated CPF Treatment
CPF has additional effects on the CNS arising independently or indirectly from AchE inhibition (Pope, 1999), including alterations in gene expression (Ray et al., 2010), endocannabinoid receptors (Baireddy et al., 2011), CREB phosphorylation (Schuh et al., 2002), cAMP signaling (Song et al., 1997), protein kinase C signaling (Slotkin and Seidler, 2009), and axonal transport (Terry et al., 2003, 2007). In the developing nervous system, CPF has been shown to impair axon growth in vitro (Howard et al., 2005; Yang et al., 2008, 2011) while also stimulating dendritic growth (Howard et al., 2005) through nonenzymatic mechanisms. Transient upregulation of neurotrophins, such as brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF), has also been identified in young rats treated with subclinical doses of CPF (Betancourt et al., 2007) and in primary hippocampal neurons in vitro (Tan et al., 2009). However, the role of CPF on axonal and dendritic length as well as neurotrophin signaling in mature rodents following CPF treatment in vivo is unknown.
Neurotrophin signaling may also contribute to the long-term deficits in hippocampal synaptic transmission and the decrease in spine number observed 3 months after repeated CPF administration. Transient upregulation of extracellular signal-regulated kinase (ERK) signaling observed by Tan et al. (2009) was followed by a delayed cytotoxic decrease in ERK phosphorylation after treatment. This may indicate a delayed downregulation of neurotrophin or glutamatergic NMDA receptor (NMDAR) signaling in CPF-treated mice. BDNF also modulates the frequency and late component of mEPSCs in hippocampal neurons through NMDARs (Madara and Levine, 2008), offering another potential mechanism by which synapse number and synaptic transmission are impaired at CA3-CA1 synapses at 3 months following CPF administration in vivo.
Clearly, the connection between CPF exposure and delayed decreases in synaptic spine density and synaptic transmission is ripe for future studies. Understanding the biochemical mechanisms of spine formation and maintenance and how these mechanisms are altered following CPF exposure will be critical to identifying potentially novel therapeutic targets for delayed effects of such exposures in humans.
CPF Susceptibility, Prevention, and Treatment
The results of this study have potentially serious implications for public health, as the consequences of repeated subclinical exposure to OP pesticides have been shown to cause lasting deficits in cognitive performance in humans (Kaplan et al., 1993) and in rodents (Cañadas et al., 2005; Cohn and MacPhail, 1997; Sánchez-Santed et al., 2004). In addition, repeated exposure to low doses of CPF renders the hippocampus susceptible to subsequent “second hit” neurological and environmental insults, including additional doses of CPF and related compounds. In support of this, impairments in working memory were found to be more profound in mice receiving two injections of toxic high-dose CPF 22 weeks apart than with a single toxic dose of CPF (Cañadas et al., 2005; Sánchez-Santed et al., 2004), and heritable mutations in the paraoxonase1 (PON1) gene that is responsible for OP metabolism have been shown to further increase susceptibility to OP pesticides (Costa et al., 2003).
In humans, occupational exposure is one potential source of second hit toxicity. For example, agricultural workers and their families are exposed to higher doses of CPF and other OP compounds than the general population and retain higher concentrations of metabolites in their urine (Coronado et al., 2006). This prolonged daily exposure does not elicit acute symptoms of OP toxicity, yet is believed to be the cause of delayed COPIND (Jamal et al., 2002a).
Military personnel are at greater risk of CPF- and OP-induced neurotoxicity because they may be exposed to subclinical levels of OP pesticides, irreversible OP nerve agents, and pyridostigmine bromide. Combined exposure to low levels of these compounds is thought to be a contributing factor in GWI (Golomb, 2008; Haley, 2003a; Haley and Kurt, 1997; Jamal, 1998; Mahoney, 2001). Furthermore, the delayed reduction in synaptic transmission may lead to reduced hippocampal “reserve.” With already weakened synaptic transmission, these veterans may experience earlier symptoms in neurodegenerative disorders such as Alzheimer’s dementia or in response to traumatic brain injury. Similar delayed neurotoxic effects of CPF on other central neurons may underlie increased incidence of ALS (Haley, 2003b; Saeed et al., 2006) and Parkinson’s disease (Kamel and Hoppin, 2004) in Gulf War veterans exposed to AchE inhibitors. In future studies, it will be of great interest to determine if substantia nigra or motor neurons are similarly affected by low-dose CPF exposure.
Our finding that synaptic spine loss, rather than neuron loss, is the cause of CPF-induced synaptic impairments suggests that the delayed effects of CPF exposure may be treatable. Thus, the results of this study warrant further research into the biochemical mechanisms of delayed CPF-induced synaptic spine loss in an effort to identify novel therapeutic targets for the detrimental long-term effects of repeated subclinical CPF exposure on the hippocampus.
FUNDING
Department of Veterans Administration Affairs (contract number VA549-P-0027) awarded and administered by the Dallas, TX VA Medical Center to C.M.P. The content of this publication does not necessarily reflect the position or the policy of the U.S. government, and no official endorsement should be inferred.
Acknowledgments
We thank Ajay Rao for technical assistance in the drug treatment of mice and Drs Robert W. Haley, James Bibb, and Christopher Sinton for helpful advice and comments on this manuscript.
References
- Aardema H, Meertens JH, Ligtenberg JJ, Peters-Polman OM, Tulleken JE, Zijlstra JG. Organophosphorus pesticide poisoning: Cases and developments. Neth. J. Med. 2008;66:149–153. [PubMed] [Google Scholar]
- Abou-Donia MB, Khan WA, Dechkovskaia AM, Goldstein LB, Bullman SL, Abdel-Rahman A. In utero exposure to nicotine and chlorpyrifos alone, and in combination produces persistent sensorimotor deficits and Purkinje neuron loss in the cerebellum of adult offspring rats. Arch. Toxicol. 2006;80:620–631. doi: 10.1007/s00204-006-0077-1. [DOI] [PubMed] [Google Scholar]
- Baireddy P, Liu J, Hinsdale M, Pope C. Comparative effects of chlorpyrifos in wild-type and cannabinoid Cb1 receptor knockout mice. Toxicol. Appl. Pharmacol. 2011;256:324–329. doi: 10.1016/j.taap.2011.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betancourt AM, Filipov NM, Carr RL. Alteration of neurotrophins in the hippocampus and cerebral cortex of young rats exposed to chlorpyrifos and methyl parathion. Toxicol. Sci. 2007;100:445–455. doi: 10.1093/toxsci/kfm248. [DOI] [PubMed] [Google Scholar]
- Birks J. Cholinesterase inhibitors for Alzheimer's disease. Cochrane Database Syst. Rev. 2006 doi: 10.1002/14651858.CD005593. 1, CD005593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MA, Brix KA. Review of health consequences from high-, intermediate- and low-level exposure to organophosphorus nerve agents. J. Appl. Toxicol. 1998;18:393–408. doi: 10.1002/(sici)1099-1263(199811/12)18:6<393::aid-jat528>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Bushnell PJ, Kelly KL, Ward TR. Repeated inhibition of cholinesterase by chlorpyrifos in rats: Behavioral, neurochemical and pharmacological indices of tolerance. J. Pharmacol. Exp. Ther. 1994;270:15–25. [PubMed] [Google Scholar]
- Cañadas F, Cardona D, Davila E, Sánchez-Santed F. Long-term neurotoxicity of chlorpyrifos: Spatial learning impairment on repeated acquisition in a water maze. Toxicol. Sci. 2005;85:944–951. doi: 10.1093/toxsci/kfi143. [DOI] [PubMed] [Google Scholar]
- Carr RL, Nail CA. Effect of different administration paradigms on cholinesterase inhibition following repeated chlorpyrifos exposure in late preweaning rats. Toxicol. Sci. 2008;106:186–192. doi: 10.1093/toxsci/kfn164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb SR, Davies CH. Cholinergic modulation of hippocampal cells and circuits. J. Physiol. 2005;562:81–88. doi: 10.1113/jphysiol.2004.076539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn J, MacPhail RC. Chlorpyrifos produces selective learning deficits in rats working under a schedule of repeated acquisition and performance. J. Pharmacol. Exp. Ther. 1997;283:312–320. [PubMed] [Google Scholar]
- Coronado GD, Vigoren EM, Thompson B, Griffith WC, Faustman EM. Organophosphate pesticide exposure and work in pome fruit: Evidence for the take-home pesticide pathway. Environ. Health Perspect. 2006;114:999–1006. doi: 10.1289/ehp.8620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa LG, Richter RJ, Li WF, Cole T, Guizzetti M, Furlong CE. Paraoxonase (PON 1) as a biomarker of susceptibility for organophosphate toxicity. Biomarkers. 2003;8:1–12. doi: 10.1080/13547500210148315. [DOI] [PubMed] [Google Scholar]
- Cowan J, Sinton CM, Varley AW, Wians FH, Haley RW, Munford RS. Gene therapy to prevent organophosphate intoxication. Toxicol. Appl. Pharmacol. 2001;173:1–6. doi: 10.1006/taap.2001.9169. [DOI] [PubMed] [Google Scholar]
- Dassanayake T, Gawarammana IB, Weerasinghe V, Dissanayake PS, Pragaash S, Dawson A, Senanayake N. Auditory event-related potential changes in chronic occupational exposure to organophosphate pesticides. Clin. Neurophysiol. 2009;120:1693–1698. doi: 10.1016/j.clinph.2009.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dassanayake T, Weerasinghe V, Dangahadeniya U, Kularatne K, Dawson A, Karalliedde L, Senanayake N. Long-term event-related potential changes following organophosphorus insecticide poisoning. Clin. Neurophysiol. 2008;119:144–150. doi: 10.1016/j.clinph.2007.09.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debanne D, Guerineau NC, Gahwiler BH, Thompson SM. Paired-pulse facilitation and depression at unitary synapses in rat hippocampus: Quantal fluctuation affects subsequent release. J. Physiol. 1996;491(Pt 1):163–176. doi: 10.1113/jphysiol.1996.sp021204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrunz LE. Release probability is regulated by the size of the readily releasable vesicle pool at excitatory synapses in hippocampus. Int. J. Dev. Neurosci. 2002;20:225–236. doi: 10.1016/s0736-5748(02)00015-1. [DOI] [PubMed] [Google Scholar]
- Dobrunz LE, Stevens CF. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron. 1997;18:995–1008. doi: 10.1016/s0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- Drever BD, Riedel G, Platt B. The cholinergic system and hippocampal plasticity. Behav. Brain Res. 2010;221:505–514. doi: 10.1016/j.bbr.2010.11.037. [DOI] [PubMed] [Google Scholar]
- Eaton DL, Daroff RB, Autrup H, Bridges J, Buffler P, Costa LG, Coyle J, McKhann G, Mobley WC, Nadel L, et al. Review of the toxicology of chlorpyrifos with an emphasis on human exposure and neurodevelopment. Crit. Rev. Toxicol. 2008;38(Suppl. 2):1–125. doi: 10.1080/10408440802272158. [DOI] [PubMed] [Google Scholar]
- Ellison CA, Smith JN, Lein PJ, Olson JR. Pharmacokinetics and pharmacodynamics of chlorpyrifos in adult male Long-Evans rats following repeated subcutaneous exposure to chlorpyrifos. Toxicology. 2011;287:137–144. doi: 10.1016/j.tox.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii S, Ji Z, Morita N, Sumikawa K. Acute and chronic nicotine exposure differentially facilitate the induction of LTP. Brain Res. 1999;846:137–143. doi: 10.1016/s0006-8993(99)01982-4. [DOI] [PubMed] [Google Scholar]
- Fujii S, Ji Z, Sumikawa K. Inactivation of alpha7 ACh receptors and activation of non-alpha7 ACh receptors both contribute to long term potentiation induction in the hippocampal CA1 region. Neurosci. Lett. 2000a;286:134–138. doi: 10.1016/s0304-3940(00)01076-4. [DOI] [PubMed] [Google Scholar]
- Fujii S, Jia Y, Yang A, Sumikawa K. Nicotine reverses GABAergic inhibition of long-term potentiation induction in the hippocampal CA1 region. Brain Res. 2000b;863:259–265. doi: 10.1016/s0006-8993(00)02119-3. [DOI] [PubMed] [Google Scholar]
- Golomb BA. Acetylcholinesterase inhibitors and Gulf War illnesses. Proc. Natl. Acad. Sci. U. S. A. 2008;105:4295–4300. doi: 10.1073/pnas.0711986105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Yakel JL. Timing-dependent septal cholinergic induction of dynamic hippocampal synaptic plasticity. Neuron. 2011;71:155–165. doi: 10.1016/j.neuron.2011.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haley RW. Gulf war syndrome: Narrowing the possibilities. Lancet Neurol. 2003a;2:272–273. doi: 10.1016/s1474-4422(03)00376-4. [DOI] [PubMed] [Google Scholar]
- Haley RW. Excess incidence of ALS in young Gulf War veterans. Neurology. 2003b;61:750–756. doi: 10.1212/wnl.61.6.750. [DOI] [PubMed] [Google Scholar]
- Haley RW, Kurt TL. Self-reported exposure to neurotoxic chemical combinations in the Gulf War. A cross-sectional epidemiologic study. JAMA. 1997;277:231–237. [PubMed] [Google Scholar]
- Hasselmo ME. The role of acetylcholine in learning and memory. Curr. Opin. Neurobiol. 2006;16:710–715. doi: 10.1016/j.conb.2006.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill RH, Jr, Head SL, Baker S, Gregg M, Shealy DB, Bailey SL, Williams CC, Sampson EJ, Needham LL. Pesticide residues in urine of adults living in the United States: Reference range concentrations. Environ. Res. 1995;71:99–108. doi: 10.1006/enrs.1995.1071. [DOI] [PubMed] [Google Scholar]
- Howard AS, Bucelli R, Jett DA, Bruun D, Yang D, Lein PJ. Chlorpyrifos exerts opposing effects on axonal and dendritic growth in primary neuronal cultures. Toxicol. Appl. Pharmacol. 2005;207:112–124. doi: 10.1016/j.taap.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Jamal GA. Gulf War syndrome—A model for the complexity of biological and environmental interaction with human health. Adverse Drug React. Toxicol. Rev. 1998;17:1–17. [PubMed] [Google Scholar]
- Jamal GA, Hansen S, Julu PO. Low level exposures to organophosphorus esters may cause neurotoxicity. Toxicology. 2002a;181–182:23–33. doi: 10.1016/s0300-483x(02)00447-x. [DOI] [PubMed] [Google Scholar]
- Jamal GA, Hansen S, Pilkington A, Buchanan D, Gillham RA, Abdel-Azis M, Julu PO, Al-Rawas SF, Hurley F, Ballantyne JP. A clinical neurological, neurophysiological, and neuropsychological study of sheep farmers and dippers exposed to organophosphate pesticides. Occup. Environ. Med. 2002b;59:434–441. doi: 10.1136/oem.59.7.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurewicz J, Hanke W. Prenatal and childhood exposure to pesticides and neurobehavioral development: Review of epidemiological studies. Int. J. Occup. Med. Environ. Health. 2008;21:121–132. doi: 10.2478/v10001-008-0014-z. [DOI] [PubMed] [Google Scholar]
- Kamel F, Hoppin JA. Association of pesticide exposure with neurologic dysfunction and disease. Environ. Health Perspect. 2004;112:950–958. doi: 10.1289/ehp.7135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan JG, Kessler J, Rosenberg N, Pack D, Schaumburg HH. Sensory neuropathy associated with Dursban (chlorpyrifos) exposure. Neurology. 1993;43:2193–2196. doi: 10.1212/wnl.43.11.2193. [DOI] [PubMed] [Google Scholar]
- Kisicki JC, Seip CW, Combs ML. A rising dose toxicology study to determine the no-observable-effect levels (NOEL) for erythrocyte acetyl-cholinesterase (AChE) inhibition and cholinergic signs and symptoms of chlorpyrifos at three dose levels. Lincoln, Neb: MDS Harris; April 19, 1999. Project 21438. In Lockwood, A. H. (2004). Human testing of pesticides: ethical and scientific considerations. Am J Public Health94, 1908–1916, 94/11/1908 [pii]. http://www.ncbi.nlm.nih.gov/pubmed/15514226?dopt=Citation. [DOI] [PMC free article] [PubMed]
- Lotti M, Moretto A. Organophosphate-induced delayed polyneuropathy. Toxicol. Rev. 2005;24:37–49. doi: 10.2165/00139709-200524010-00003. [DOI] [PubMed] [Google Scholar]
- Madara JC, Levine ES. Presynaptic and postsynaptic NMDA receptors mediate distinct effects of brain-derived neurotrophic factor on synaptic transmission. J. Neurophysiol. 2008;100:3175–3184. doi: 10.1152/jn.90880.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney DB. A normative construction of Gulf War syndrome. Perspect. Biol. Med. 2001;44:575–583. doi: 10.1353/pbm.2001.0068. [DOI] [PubMed] [Google Scholar]
- Manabe T, Wyllie DJ, Perkel DJ, Nicoll RA. Modulation of synaptic transmission and long-term potentiation: Effects on paired pulse facilitation and EPSC variance in the CA1 region of the hippocampus. J. Neurophysiol. 1993;70:1451–1459. doi: 10.1152/jn.1993.70.4.1451. [DOI] [PubMed] [Google Scholar]
- Manthripragada AD, Costello S, Cockburn MG, Bronstein JM, Ritz B. Paraoxonase 1, agricultural organophosphate exposure, and Parkinson disease. Epidemiology. 2010;21:87–94. doi: 10.1097/EDE.0b013e3181c15ec6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty MS, Domoradzki JY, Hansen SC, Timchalk C, Bartels MJ, Mattsson JL. The effect of route, vehicle, and divided doses on the pharmacokinetics of chlorpyrifos and its metabolite trichloropyridinol in neonatal Sprague-Dawley rats. Toxicol. Sci. 2007;100:360–373. doi: 10.1093/toxsci/kfm239. [DOI] [PubMed] [Google Scholar]
- Middlemore-Risher ML, Buccafusco JJ, Terry AV., Jr Repeated exposures to low-level chlorpyrifos results in impairments in sustained attention and increased impulsivity in rats. Neurotoxicol. Teratol. 2010;32:415–424. doi: 10.1016/j.ntt.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morahan JM, Yu B, Trent RJ, Pamphlett R. A gene-environment study of the paraoxonase 1 gene and pesticides in amyotrophic lateral sclerosis. Neurotoxicology. 2007;28:532–540. doi: 10.1016/j.neuro.2006.11.007. [DOI] [PubMed] [Google Scholar]
- Moser VC. Comparisons of the acute effects of cholinesterase inhibitors using a neurobehavioral screening battery in rats. Neurotoxicol. Teratol. 1995;17:617–625. doi: 10.1016/0892-0362(95)02002-0. [DOI] [PubMed] [Google Scholar]
- Murata K, Araki S, Yokoyama K, Okumura T, Ishimatsu S, Takasu N, White RF. Asymptomatic sequelae to acute sarin poisoning in the central and autonomic nervous system 6 months after the Tokyo subway attack. J. Neurol. 1997;244:601–606. doi: 10.1007/s004150050153. [DOI] [PubMed] [Google Scholar]
- Penton RE, Quick MW, Lester RA. Short- and long-lasting consequences of in vivo nicotine treatment on hippocampal excitability. J. Neurosci. 2011;31:2584–2594. doi: 10.1523/JNEUROSCI.4362-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilkington A, Buchanan D, Jamal GA, Gillham R, Hansen S, Kidd M, Hurley JF, Soutar CA. An epidemiological study of the relations between exposure to organophosphate pesticides and indices of chronic peripheral neuropathy and neuropsychological abnormalities in sheep farmers and dippers. Occup. Environ. Med. 2001;58:702–710. doi: 10.1136/oem.58.11.702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope CN. Organophosphorus pesticides: Do they all have the same mechanism of toxicity? J. Toxicol. Environ. Health B Crit. Rev. 1999;2:161–181. doi: 10.1080/109374099281205. [DOI] [PubMed] [Google Scholar]
- Prendergast MA, Terry AV, Jr, Buccafusco JJ. Effects of chronic, low-level organophosphate exposure on delayed recall, discrimination, and spatial learning in monkeys and rats. Neurotoxicol. Teratol. 1998;20:115–122. doi: 10.1016/s0892-0362(97)00098-6. [DOI] [PubMed] [Google Scholar]
- Rauh V, Arunajadai S, Horton M, Perera F, Hoepner L, Barr DB, Whyatt R. Seven-year neurodevelopmental scores and prenatal exposure to chlorpyrifos, a common agricultural pesticide. Environ. Health Perspect. 2011;119:1196–1201. doi: 10.1289/ehp.1003160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauh VA, Garfinkel R, Perera FP, Andrews HF, Hoepner L, Barr DB, Whitehead R, Tang D, Whyatt RW. Impact of prenatal chlorpyrifos exposure on neurodevelopment in the first 3 years of life among inner-city children. Pediatrics. 2006;118:e1845–e1859. doi: 10.1542/peds.2006-0338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray A, Liu J, Ayoubi P, Pope C. Dose-related gene expression changes in forebrain following acute, low-level chlorpyrifos exposure in neonatal rats. Toxicol. Appl. Pharmacol. 2010;248:144–155. doi: 10.1016/j.taap.2010.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush T, Liu XQ, Hjelmhaug J, Lobner D. Mechanisms of chlorpyrifos and diazinon induced neurotoxicity in cortical culture. Neuroscience. 2010;166:899–906. doi: 10.1016/j.neuroscience.2010.01.025. [DOI] [PubMed] [Google Scholar]
- Saeed M, Siddique N, Hung WY, Usacheva E, Liu E, Sufit RL, Heller SL, Haines JL, Pericak-Vance M, Siddique T. Paraoxonase cluster polymorphisms are associated with sporadic ALS. Neurology. 2006;67:771–776. doi: 10.1212/01.wnl.0000227187.52002.88. [DOI] [PubMed] [Google Scholar]
- Sánchez-Santed F, Cañadas F, Flores P, López-Grancha M, Cardona D. Long-term functional neurotoxicity of paraoxon and chlorpyrifos: Behavioural and pharmacological evidence. Neurotoxicol. Teratol. 2004;26:305–317. doi: 10.1016/j.ntt.2003.10.008. [DOI] [PubMed] [Google Scholar]
- Schuh RA, Lein PJ, Beckles RA, Jett DA. Noncholinesterase mechanisms of chlorpyrifos neurotoxicity: Altered phosphorylation of Ca2+/cAMP response element binding protein in cultured neurons. Toxicol. Appl. Pharmacol. 2002;182:176–185. doi: 10.1006/taap.2002.9445. [DOI] [PubMed] [Google Scholar]
- Slotkin TA, Seidler FJ. Protein kinase C is a target for diverse developmental neurotoxicants: Transcriptional responses to chlorpyrifos, diazinon, dieldrin and divalent nickel in PC12 cells. Brain Res. 2009;1263:23–32. doi: 10.1016/j.brainres.2009.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JN, Campbell JA, Busby-Hjerpe AL, Lee S, Poet TS, Barr DB, Timchalk C. Comparative chlorpyrifos pharmacokinetics via multiple routes of exposure and vehicles of administration in the adult rat. Toxicology. 2009;261:47–58. doi: 10.1016/j.tox.2009.04.041. [DOI] [PubMed] [Google Scholar]
- Song X, Seidler FJ, Saleh JL, Zhang J, Padilla S, Slotkin TA. Cellular mechanisms for developmental toxicity of chlorpyrifos: Targeting the adenylyl cyclase signaling cascade. Toxicol. Appl. Pharmacol. 1997;145:158–174. doi: 10.1006/taap.1997.8171. [DOI] [PubMed] [Google Scholar]
- Szabo SI, Zelles T, Vizi ES, Lendvai B. The effect of nicotine on spiking activity and Ca2+ dynamics of dendritic spines in rat CA1 pyramidal neurons. Hippocampus. 2008;18:376–385. doi: 10.1002/hipo.20401. [DOI] [PubMed] [Google Scholar]
- Tan DH, Peng SQ, Wu YL, Wang YM, Lu CF, Ding W, Wang QX, Yan CH. Chlorpyrifos induces delayed cytotoxicity after withdrawal in primary hippocampal neurons through extracellular signal-regulated kinase inhibition. Biol. Pharm. Bull. 2009;32:1649–1655. doi: 10.1248/bpb.32.1649. [DOI] [PubMed] [Google Scholar]
- Terry AV, Jr, Gearhart DA, Beck WD, Jr, Truan JN, Middlemore ML, Williamson LN, Bartlett MG, Prendergast MA, Sickles DW, Buccafusco JJ. Chronic, intermittent exposure to chlorpyrifos in rats: Protracted effects on axonal transport, neurotrophin receptors, cholinergic markers, and information processing. J. Pharmacol. Exp. Ther. 2007;322:1117–1128. doi: 10.1124/jpet.107.125625. [DOI] [PubMed] [Google Scholar]
- Terry AV, Jr, Stone JD, Buccafusco JJ, Sickles DW, Sood A, Prendergast MA. Repeated exposures to subthreshold doses of chlorpyrifos in rats: Hippocampal damage, impaired axonal transport, and deficits in spatial learning. J. Pharmacol. Exp. Ther. 2003;305:375–384. doi: 10.1124/jpet.102.041897. [DOI] [PubMed] [Google Scholar]
- U.S. Environmental Protection Agency Administrator Announcement. Prevention Pesticides and Toxic Substances (7506C): Chlorpyrifos Revised Risk Assessment and Agreement with Registrants. Washington, DC: U.S. Environmental Protection Agency; 2000. [Google Scholar]
- Yang D, Howard A, Bruun D, Ajua-Alemanj M, Pickart C, Lein PJ. Chlorpyrifos and chlorpyrifos-oxon inhibit axonal growth by interfering with the morphogenic activity of acetylcholinesterase. Toxicol. Appl. Pharmacol. 2008;228:32–41. doi: 10.1016/j.taap.2007.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Lauridsen H, Buels K, Chi LH, La Du J, Bruun DA, Olson JR, Tanguay RL, Lein PJ. Chlorpyrifos-oxon disrupts zebrafish axonal growth and motor behavior. Toxicol. Sci. 2011;121:146–159. doi: 10.1093/toxsci/kfr028. [DOI] [PMC free article] [PubMed] [Google Scholar]