

Abstract
Adaptive evolution frequently occurs in episodic bursts, localized to a few sites in a gene, and to a small number of lineages in a phylogenetic tree. A popular class of “branch-site” evolutionary models provides a statistical framework to search for evidence of such episodic selection. For computational tractability, current branch-site models unrealistically assume that all branches in the tree can be partitioned a priori into two rigid classes—“foreground” branches that are allowed to undergo diversifying selective bursts and “background” branches that are negatively selected or neutral. We demonstrate that this assumption leads to unacceptably high rates of false positives or false negatives when the evolutionary process along background branches strongly deviates from modeling assumptions. To address this problem, we extend Felsenstein's pruning algorithm to allow efficient likelihood computations for models in which variation over branches (and not just sites) is described in the random effects likelihood framework. This enables us to model the process at every branch-site combination as a mixture of three Markov substitution models—our model treats the selective class of every branch at a particular site as an unobserved state that is chosen independently of that at any other branch. When benchmarked on a previously published set of simulated sequences, our method consistently matched or outperformed existing branch-site tests in terms of power and error rates. Using three empirical data sets, previously analyzed for episodic selection, we discuss how modeling assumptions can influence inference in practical situations.
Keywords: episodic selection, random effects model, evolutionary model, branch-site model
Introduction
The inference of selection from molecular data, both along a sequence (Nielsen and Yang 1998, Suzuki and Gojobori 1999, Yang et al. 2000) and over the evolutionary tree (Yang and Nielsen 2002, Kosakovsky Pond and Frost 2005a), has been an area of active research and unrelenting debate (Suzuki and Nei 2004, Wong et al. 2004, Nozawa et al. 2009). Selective pressures can vary over both sites and time, resulting in bursts of selection localized to a subset of sites and a small number of lineages, for example, Messier and Stewart (1997).
A class of methods, termed “branch-site” tests (Yang and Nielsen 2002), was the first to offer a model-based phylogenetic hypothesis testing framework for deciding whether or not a lineage (or lineages) of interest had undergone adaptive change. Branch-site tests measure selective pressure by ω, the ratio of nonsynonymous (β) to synonymous (α) substitution rates, and if a proportion of sites in the sequence provides statistically significant support for ω > 1 along the lineages of interest, then episodic positive selection is inferred. The original formulation of the method suffered from high rates of false positives when the model assumptions were violated (Zhang 2004) because the model could misidentify relaxed selective constraints as evidence of diversifying selection and was subsequently revised to address that shortcoming (Zhang et al. 2005). Typically, the lineages to be tested (“foreground” lineages) were specified a priori, until a recent extension outlined and benchmarked a sequential testing approach to examine whether any single lineage was under selection (Anisimova and Yang 2007). These branch-site methods have been used extensively, with well over 1,000 citations to date, highlighting the interest of the evolutionary community in being able to identify instances of episodic selection. Alternative approaches to capturing variable selective pressures include the covarion models of Guindon et al. (2004) and a full Bayesian treatment in the framework of Rodrigue et al. (2010).
In the context of codon evolutionary models, the selective profile of site Ds in a multiple sequence alignment can be characterized by the collection of branch-specific ω values, (ω1,…,ωB), denoted Ωs, where B equals the total number of branches in the phylogeny. Existing branch-site models use three alignment-wide (i.e., shared by all sites) ratios ω− < ωN = 1 ≤ ω+ to model strong conservation, neutral evolution, and diversifying selection, respectively. Assuming these three ω ratios (fig. 1) with no further restrictions, each site can follow one of 3B possible selective profiles—the number of different ways to assign the B branches to the three different selection rate bins. However, it is unclear how to determine which of these selective profiles or, equivalently, assignments of branches to selection rate bins is the most appropriate at a given site.
FIG. 1.

An illustration of episodic selection profiles at a single site with three possible regimes: negative, neutral (or nearly neutral), and diversifying selection along a branch. Panel (A) depicts the phylogeny used for discussion in the text and to carry out robustness simulations; Branch 5 is designated as foreground (FG), and the remaining four branches as background (BG). Panel (B) illustrates the four a priori selective profiles allowed by the model of Zhang et al. (2005). Panel (C) shows 2 of 239 possible selective profiles not modeled by current branch-site models; these profiles are used in robustness simulations (see Methods).
One approach (Yang and Nielsen 2002) is to model each site using only four predefined profiles regardless of the size of the phylogeny. More specifically, 1) every branch belongs either to the a priori known foreground class, which is allowed to experience diversifying selection, or the “background” class, which evolves under purifying selection or neutrally and 2) at a given site, there is no variation in selection strength (ω) among background branches, with all foreground branches either sharing the selection strength of the background or being under shared diversifying selection (fig. 1B). Clearly, these options are not exhaustive: For example, neither variable strength of selection among background or foreground nor positive selection along background branches is allowed. We refer to this approach as the restricted branch-site (rBS) model because the number of selective profiles is limited to the four a priori defined scenarios. Given a 4-taxon tree (fig. 1), and three selection parameters (as in fig. 1B), there are 35 = 243 possible selection configurations, only four of which are accounted for by the branch-site model. The number of ω configurations grows as KB, where K is the number of rate classes, thus making it unlikely that any four selection profiles chosen a priori are going to be sufficiently representative. Because there are no compelling biological reasons to expect that any two branches in the phylogenetic tree will have the same ω at any given site, we do not expect these four predefined selective profiles to provide an adequate description of complex biological data. This model was likely motivated by the need to avoid overfitting in the case of small sample sizes; however, we argue that if branches with differing selective pressures are incorrectly assigned to the same class, likelihood ratio test (LRT)-based branch-site methods can be positively misleading. In this manuscript, we present one case where they falsely identify positive selection on a neutrally evolving lineage (Type I or false positive error), and another where they fail to detect positive selection on a lineage with ω > 1 (Type II or false negative error). In addition, if several branches are claimed to be under positive selection by setting the foreground to one branch at a time, as is done by the sequential testing procedure of Anisimova and Yang (2007), this creates a logical inconsistency—when a branch is found to be under selection, the model under which this was established implies that no other branch could be under positive selection.
We introduce a new class of models in which substitution rates may vary from branch to branch and from site to site. We incorporate this variation via “random effects”—unobserved strengths of selection at sites and branches are incorporated using a discrete or a discretized parametric probability distribution. Parameters defining the distribution are estimated jointly from all sites using maximum likelihood. Random effects likelihood (REL) and complementary fixed effects likelihood (FEL) models are standard tools in statistical modeling. Both types of model have been used to allow sitewise rate variation in phylogenetic models—see Yang:1994kx, where rates over sites in a nucleotide alignment followed a discretized unit-mean gamma distribution (the now ubiquitous + Γ4 model). Nielsen and Yang (1998) and Yang et al. (2000) applied REL models to codon data in order to identify signatures of natural selection, whereas Kosakovsky Pond and Frost (2005b) and Massingham and Goldman (2005) used FEL models for the same purpose. For all these models, likelihoods of individual sites are computed by Felsenstein's pruning algorithm (Felsenstein 1981). However, as we show later, the direct application of the pruning algorithm is intractable for REL models with branchwise as well as sitewise rate variation. It is presumably for this reason that, to date, branch models (Yang 1998, Kosakovsky Pond and Frost 2005a) have only been implemented in the FEL framework and branch-site models only as a four-category sitewise REL model. Our solution involves a simple extension of the pruning algorithm which makes it feasible to implement not only the model proposed here but also several other branch-site REL models.
The extended pruning algorithm computes the likelihood of each site, treating the selection site profile Ωs as an unobserved variable, under the assumption that the probability of observing a substitution rate at a branch is independent of all other branches. Computationally, our algorithm is equivalent to replacing the standard Markov evolutionary model at a single phylogenetic branch with a mixture of three Markov models (one each for ω−,ωN,andω+), where the mixing coefficients and ω rates are inferred for each branch along with branch lengths, nucleotide substitution biases, and other alignment-wide parameters. Just like existing branch-site methods (Anisimova and Yang 2007), we use sequential likelihood ratio testing to identify which branches support a model with episodic diversifying selection. Unlike existing methods, however, our approach is unrestricted and considers every possible site profile, thus avoiding some of the prominent issues posed by model misspecification and further allows ω rates to vary independently from branch to branch and site to site.
Using an extensive collection of simulated sequences from Anisimova and Yang (2007), we perform a direct comparison of the unrestricted branch-site (uBS) model with the existing, restricted, approach (rBS) to evaluate Type I error and power. We also reinvestigate three empirical data sets that had been previously analyzed with the standard or sequential branch-site method and discover that many, but not all, of the original inferences are supported by our mixture model. Lastly, we report selective episodes not previously detected.
Methods
Codon Model Specification
To facilitate our presentation of episodic selection methods, we first briefly review maximum likelihood codon phylogenetic models (although see Delport et al. 2009 and Anisimova and Kosiol 2009 for detailed reviews). These models assume that substitutions along a branch of a phylogenetic tree can be described by an appropriately parameterized continuous-time stationary Markov process, defined by its instantaneous rate matrix, Q, with elements that describe the rate of substitution of codon i with codon j:
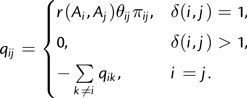 |
(1) |
Here, δ(i,j) is the number of nucleotide differences between codons i and j, πij denote the equilibrium frequency parameters (e.g., πAAA,AAC = qC3,πACC,AAC = qA2), θij are the nucleotide mutational biases, and r(Ai,Aj) = r(Aj,Ai) are the relative substitution rates between amino acids encoded by codons i and j. In the most general model, each of these r(Ai,Aj)'s can be independently estimated (see Delport et al. 2010), but here we follow the common approach of allowing only two rates: α for synonymous (Ai = Aj) and β for nonsynonymous (Ai≠Aj) substitutions. Their ratio, β/α, is the familiar selection parameter, ω. The equilibrium frequency parameters may be estimated empirically either as the product of position-specific nucleotide frequencies (Goldman and Yang 1994) or as the position-specific frequency of the target nucleotide (Muse and Gaut 1994). Because we have previously identified biases using such empirical approaches (Kosakovsky Pond et al. 2010), we use corrected estimates (CF3×4) of nucleotide frequency parameters. Given a phylogenetic tree T (fig. 1), with B branches and branch lengths ti,i = 1,…,B, the likelihood of changing from state i to j at a site along branch b in time tb is given by the (i,j) element of the transition matrix PQ(tb) = eQtb. Subsequently, the likelihood of observing the alignment is evaluated as the product of site-likelihoods (with sites ranging from 1 to the number S of sites in the alignment), each of which is calculated using the standard pruning algorithm (Felsenstein 1981) given the data, a phylogenetic tree, T, and instantaneous rate matrix, Q.
Sitewise REL Models
Before extending Felsenstein's pruning algorithm, we first summarize how it is used in the context of the commonly used class of sitewise REL models. We pick our notation to allow extension to other types of REL models in the sections that follow. Throughout, we consider only the case of a finite number of discrete categories; extension to continuous-valued unobserved variables is straightforward, but computationally impractical, at least in the standard frequentist phylogenetic framework.
In a sitewise REL model, we think of each site as belonging to a site category, with the possible site categories ranging from 1 to K. For notational convenience, we present the special case where the categories differ only in terms of their ω values—allowing us to denote the category for site s by ωs. Considering all sites simultaneously, the configuration of categories over all sites is a vector Ω∀b = (ω1,…,ωS), where the subscript makes it explicit that this configuration is shared by all branches. We model the joint probability of the configuration as the product of independent factors:
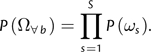 |
(2) |
The individual category probabilities P(ωs) are shared across all sites. Although the independence of sites is a standard assumption in the literature and allows for a particularly efficient likelihood calculation, it is not necessary. For example, P(Ω∀b) has been modeled as a Hidden Markov process to permit spatial correlations among site categories (Felsenstein and Churchill 1996).
Another alternative to the model assumption of equation (2) would have been to allow only a small number of configurations. For example, we could imagine a model where sites are divided a priori into “buried” and “exposed” residues (e.g., Yang and Swanson 2002) and propose the following four configurations: 1) all sites conserved; 2) all sites evolving neutrally; 3) buried sites conserved and exposed sites under positive selection; and 4) buried sites evolving neutrally and exposed sites under positive selection. One could calculate the alignment-wide likelihood under each configuration and infer which of the configurations fits the data best. We mention this not because we think it is a good model (surely, it would not be biologically realistic to assume such a limited number of possible configurations) but because it is directly analogous to the existing branch-site model of Zhang et al. (2005). Our contribution in this manuscript is to upgrade from a branch-site model with four prechosen configurations such as these to one that is analogous to a REL model where the categories of different sites are independent.
Returning to standard sitewise REL models, the likelihood of the data Ds observed at site s (conditioned implicitly on non-ω model parameters) is
| (3) |
| (4) |
where the first sum is over all site categories, A denotes a vector of ancestral node states, and the sum over A is taken over all possible such vectors. Labeling each nonroot node with the number of its parental branch, and the root node as 0, we can write this out more fully using
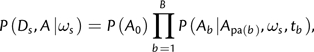 |
(5) |
where Ab denotes the state at node b and p a(b) is the parent node of b. The task of Felsenstein's pruning algorithm is to calculate the sum
| (6) |
which, because each of the terms P(Ab|A p a(b),ωs,tb) in equation (5) depends only on a local part of the tree (a child and parent node and the branch connecting them), can be factorized efficiently and calculated by means of a postorder tree traversal. In what follows, we retain this property so that the same tree traversal remains an efficient way to calculate the desired likelihood.
Branch-Site REL Models
To define a branch-site REL model, we replace our sitewise category variable ωs with a branch-site category variable ωbs. Each branch-site combination is considered to belong to one of our K categories. We still aim to calculate the likelihood for a single site s, so we consider the configuration Ωs = (ω1s,…,ωBs) of branch categories. Our new approach is based on the observation that if the branch categories are independent, so that
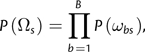 |
(7) |
then the likelihood at a site can be computed efficiently without the need to apply the pruning algorithm for every possible value of Ωs. By definition,
| (8) |
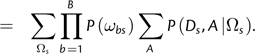 |
(9) |
Changing the order of summations, this can be written as follows:
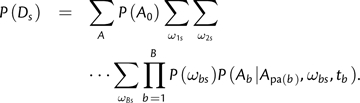 |
(10) |
This is identical to the quantity calculated by Felsenstein's algorithm except for the presence of the P(ωbs) terms and the summations over ω values. Thinking algorithmically, and as indicated in equation (10), the entire space of KB values of Ωs can be traversed by B nested loops, where the outermost loop iterates over ω1s, the second loop over ω2s etc. Note that each product term P(ωbs)P(Ab|A p a(b),ωbs,tb) depends on only one branch. Hence, the sum computed by B nested loops (O(KB) operations) is equivalent to a product of B sums (O(KB) operations):
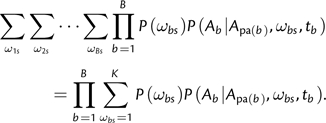 |
Consequently, we can rewrite equation (10):
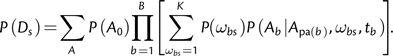 |
(11) |
The summation in parentheses can be viewed as the transition probability matrix of a mixture of K Markov substitution models, with P(Ab|A p a(b),ωbs,tb) being the model-specific likelihoods at branch b, and P(ωb) being the mixing proportions. If Qωbs is the rate matrix associated with ωbs (as in equation (1)), then this transition probability matrix can be computed as
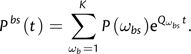 |
(12) |
The sum over A in equation (11) can be carried out efficiently using Felsenstein's pruning algorithm, with the transition matrices along each branch defined as K-process mixtures as above. In other words, in order to compute the likelihood of an alignment site, we first assume that the probability of a particular selective regime at a branch is independent of that at any other branch, and apply the pruning algorithm as usual, except that the substitution model along each branch is given as the mixture of equation (12).
Depending on how the mixing coefficients and the transition matrices in equation (12) are parameterized, we can obtain different types of branch-site models. In principle, for every branch-site combination (b,s), there could be K independently estimated mixing proportions P(ωbs) and selection parameters ωbs. However, this approach will yield a model with considerably more parameters than observations. Three simpler model types appear promising.
Nonspecific Branch-Site REL.
ωbs and P(ωbs) for each category K are shared by all branches and sites. There are K alignment-wide ω parameters (Ωk), and the probability that P(ωbs = Ωk) = qk is described by an alignment-wide frequency parameter qk,∑kqk = 1. This is a simple model with 2K − 1 parameters estimated from the entire alignment but may not incorporate enough biological realism. We used it as the first step of the optimization process for our more complex model to obtain initial parameter estimates.
Site-Specific Branch-Site REL.
P(ωbs) is a function of s, that is, every site (or more precisely site pattern) has its own set of mixing coefficients, shared across all branches. ωbs are shared by all sites and branches. This model has KS + K − S parameters: KΩk parameters estimated jointly from the alignment and S sets of qsk mixing parameters, with ∑kqsk = 1,∀s = 1,…,S, so that P(ωbs = Ωk) = qsk. Because the number of parameters grows with the size of the alignment, the model will be asymptotically ill behaved. However, for fixed length alignments with many sequences, it may be possible to learn site-specific mixing parameters reliably.
Branch-Specific Branch-Site REL.
ωbs and P(ωbs) are functions of b, that is, every branch has its own set of model parameters (ωbk) and mixing coefficients (qbk,∑kqbk = 1), but they are estimated jointly from all sites. This model has (2K − 1)B parameters and is investigated in the present manuscript. It has the attractive property that the model parameters we learn include, for every branch, the proportion of sites belonging to every selection category.
A New Test for Episodic Selection
We define and fit a branch-specific branch-site REL model (termed unrestricted branch site or uBS). For consistency with several existing REL models, we restrict ω at every branch to take on one of K = 3 values ωb− ≤ ωbN ≤ 1 ≤ ωb+, representative of strong and weak conservation and positive diversifying selection. In our experience (e.g., see Kosakovsky Pond et al. 2010), models that permit multiple classes of sites with ω < 1 fit protein-coding sequence alignments much better than those with one of the ω values fixed at 1. We denote their mixing proportions qb−, qbN, and qb+ (subject to qb− + qbN + qb+ = 1), respectively. All model parameters are estimated by maximum likelihood. Next, we fit B models (one for each branch), where model b = 1,…,B differs from the unrestricted model by the additional constraint of ωb+ = 1. Each of these models, therefore, disallows diversifying selection along a single branch while leaving all other background branches unrestricted. Compare this with the requirement that all background branches have uniform neutral or negative selection regimes in the standard branch-site model (Zhang et al. 2005). As described most recently in Anisimova and Yang (2007), the evidence for positive selection along branch b can be evaluated by a LRT using the asymptotic distribution of the LR statistic defined by (χ12 + χ02)/2 (Self and Liang 1987). If B branches are tested in sequence, it is necessary to correct the nominal significance level for each individual test to control the cumulative (or family wise) error rate of the tests. Anisimova and Yang (2007) compared multiple such corrections in the context of branch-site methods and reported that their performance was broadly similar. With that in mind, we settled on the correction procedure due to Holm (1979), which is more powerful and as easy to compute as the simple Bonferroni correction. Briefly, if the desired Type I error for the event “any of the B tests is a false positive under the null model” is α, then the testing procedure first ranks p values for each individual test in increasing order p(1) ≤ p(2) ≤ ⋯ ≤ p(B) and rejects first k hypotheses if p(i) ≤ α/(B − i + 1) for i = 1,…,k and p(k + 1) > α/(B − k). Our testing procedure uses a single alternative hypothesis and requires that B + 1 model fits be performed, whereas the testing procedure of Anisimova and Yang (2007) demands the fitting of 2B models because a different null and alternative pair must be evaluated for each branch.
Evaluating the Robustness of the rBS Model
We simulated data according to two selection scenarios along a 4-taxon tree (fig. 1A) using the codon substitution model defined above, with equal codon equilibrium frequencies (π = 1/61) and the HKY85 (Hasegawa et al. 1985) nucleotide substitution biases (i.e., θac = θat = θcg = θgt = 2;θag = θct = 1). This choice of base frequencies and nucleotide substitution biases will deemphasize the differences in how frequency parameters and nucleotide substitution biases are modeled in rBS and uBS.
First (robustness simulation 1, RS1), we designated branch 5 (fig. 1 A) as a neutrally evolving foreground, that is, the one to be tested for episodic diversifying selection by the models), branch (ω = 1), whereas background branches 1 and 3 were simulated under strong diversifying selection (ω = 10), and background branches 2 and 4—under strong purifying selection (ω = 0.1). This scenario was crafted to include variable selection along background branches which is not handled by any of the four classes of the branch-site model, and hence the standard branch-site test of selection along branch 5 will be fitting the data using two incorrect models. Second (RS2), we designated branch 5 as a positively selected foreground branch (ω = 2), whereas background branches 1 and 2 are under strong diversifying selection (ω = 10) and background branches 3 and 4 are under strong purifying selection (ω = 0.05). These two scenarios are designed to explore the asymptotic behavior of the tests and use sequences longer than most genes. A test with poor asymptotic properties when a specific model assumption is violated may appear to behave acceptably on smaller samples due to, for example, lack of power. If test errors increase with sample size, this may point to fundamental issues with the approach.
Evaluating the Performance of the Unrestricted Branch-Site Model
Anisimova and Yang (2007) generated several thousand alignments under seven selective regimes, three of which included no positive selection (to test for Type I error or false positives) and four included varying extents of diversifying selective pressure (to assess Type II error or power). These simulation alignments were kindly provided by the authors, and we reanalyzed the data for a direct comparison with our approach. For complete details on these simulations, we refer the reader to table 2 and text in Anisimova and Yang (2007). Briefly, either 4 or 8 taxon balanced trees were used for simulations, with 1,000 (4 taxa) or 200 (8 taxa) 300-codon long replicates/scenario.
Table 2.
uBS on the CD2 Data Set
| Branch | Mean ω | ω– | q– | ωN | qN | ω+ | q+ | LRT | p | Corrected p |
| Pig | 1.341 | 0.000 | 0.443 | 0.919 | 0.000 | 2.811 | 0.557 | 3.276 | 0.035 | 0.352 |
| Cow | 1.914 | 0.000 | 0.025 | 0.000 | 0.513 | 10.732 | 0.462 | 23.465 | 0.000 | 0.000 |
| 3 | 1.480 | 0.000 | 0.328 | 0.000 | 0.370 | 7.824 | 0.303 | 5.989 | 0.007 | 0.079 |
| Horse | 1.244 | 0.000 | 0.001 | 0.000 | 0.569 | 5.190 | 0.430 | 11.463 | 0.000 | 0.005 |
| Cat | 1.598 | 0.252 | 0.463 | 1.000 | 0.137 | 6.544 | 0.400 | 13.309 | 0.000 | 0.002 |
| 6 | 0.664 | 0.000 | 0.906 | 0.118 | 0.000 | 37.328 | 0.094 | 7.432 | 0.003 | 0.038 |
| RHmonkey | 22.503 | 1.000 | 0.007 | 1.000 | 0.316 | 113.398 | 0.677 | 1.196 | 0.137 | 0.822 |
| Baboon | 0.000 | 0.000 | 0.550 | 0.000 | 0.336 | 0.000 | 0.113 | 0.000 | 1.000 | 1.000 |
| 9 | 0.400 | 0.047 | 0.000 | 0.443 | 1.000 | 0.009 | 0.000 | 0.000 | 1.000 | 1.000 |
| Human | 0.002 | 0.126 | 0.468 | 0.215 | 0.384 | 2.963 | 0.148 | 0.000 | 0.500 | 1.000 |
| Chimpanzee | 24.634 | 0.313 | 0.000 | 0.812 | 0.000 | 47.512 | 1.000 | 0.630 | 0.214 | 1.000 |
| 12 | 0.368 | 0.000 | 0.149 | 0.000 | 0.803 | 12.624 | 0.048 | 1.393 | 0.119 | 0.952 |
| 13 | 1.915 | 1.000 | 0.020 | 1.000 | 0.888 | 39.772 | 0.092 | 8.823 | 0.001 | 0.019 |
| 14 | 0.432 | 0.156 | 0.039 | 0.162 | 0.730 | 2.581 | 0.232 | 1.315 | 0.126 | 0.880 |
| Rat | 1.093 | 0.000 | 0.552 | 0.002 | 0.000 | 2.998 | 0.448 | 0.367 | 0.272 | 1.000 |
| Mouse | 0.524 | 0.400 | 0.947 | 0.799 | 0.000 | 22.217 | 0.053 | 2.240 | 0.067 | 0.605 |
Mean ω is estimated under the free-ratio MG94 × REV model (no site-to-site rate variation). ω and q values reflect the branch-level mixture of negative, (nearly) neutral, and positive selection models. LRT: likelihood ratio test statistic, p: uncorrected p-value obtained using the mixture of χ20 and χ21 distributions; corrected p: after an application of Holm's multiple testing correction. Internal branches are numbered concordantly with figure 2. Branches found by uBS to be under positive diversifying selection are shown in italic.
In addition, we test our approach in a high information content setting, using sequences with 1,000 codons simulated along a 16-taxon balanced tree (supplementary fig. S3, Supplementary Material online). We subdivide the length of the sequence into three partitions, such that a site is simulated under one of three potential selection models. The first two models are homogeneous with respect to the tree and encompass purifying selection (ω = 0.1) and neutrality (ω = 1) with proportions, p1 = 0.8 and p2 = 0.05, respectively. Finally, the third model, with proportion p3 = 0.15, is heterogeneous with respect to the tree, comprising neutral evolution (ω = 1) at all branches, except a set of three branches at which strong diversifying selection is simulated (ω = 5). We considered two modifications of this scenario: a lower proportion of selected sites (p2 = 0.15,p3 = 0.05) or weaker selection (ω = 2 in the third model).
Finally, we reexamine three empirical alignments previously analyzed for evidence of episodic selection: a data set consisting of 19 lysozyme c sequences (S = 130 codons) from primates, initially analyzed by Messier and Stewart (1997); CD2 gene sequences (S = 187 codons) coding for a cell adhesion molecule located on the surface of certain type of lymphocyte, isolated from 10 mammalian species and originally analyzed by Lynn et al. (2005); and 10 mammalian sequences (S = 1,162 codons) of the tumor suppressor gene BRCA1 (Zhang et al. 2005).
Implementation
The model is implemented as a collection of HyPhy (Kosakovsky Pond et al. 2005) Batch Language scripts and is distributed as a part of HyPhy v2.0020110306 or later as BranchSiteREL.bf file in the Positive Selection rubrik of standard analyses.
Results
Test Performance on Simulated Data
We applied our uBS sequential selection test to parametric replicates generated under seven different selection profiles previously used by Anisimova and Yang (2007) to evaluate the original sequential branch-site test for detecting episodic selection (Zhang et al. 2005) and to two additional sets robustness simulations. Details of simulation results are collated in table 1.
When sequences are simulated under rBS assumptions (fig. 1), that is, those which conform to the null or the alternative model of Zhang et al. (2005), both uBS and rBS perform comparably (NC1, NC2, and SC in table 1), with similar family wise error rates (FWER) and power. It is encouraging that our unrestricted method does not appear to be strongly underpowered compared with rBS, even when the data are simulated to favor the former (38% vs. 44% power on SC with one sequence). The same holds for data generated under models which deviate from rBS assumptions but not too strongly (NI, SI1 in table 1).
The advantages of uBS over rBS become apparent when the assumptions of the latter are inappropriate for the data (SI2 and SI3). Already, in the SI2 scenario, where two branches are experiencing episodic diversifying selection, uBS provides a considerable boost in power for 8-taxon trees (63% vs. 48.5%). The greatest difference between our approach and rBS is revealed in the SI3 simulation scenario, when four background branches in a 4-taxon tree were simulated under episodic selection, whereas the single foreground branch was evolved neutrally or under purifying selection. The intent of SI3 in Anisimova and Yang (2007) was to violate the assumptions of the rBS model as much as conceivably possible and investigate how this would reflect on Type I errors. Although the rBS model controlled the rates of false positives (FWER 1.7%), it suffered a severe loss of power—the cumulative power was reported at only 35.3%, despite pervasive episodic selection in this case. In contrast, uBS achieved 92.5% power while maintaining FWER of 6.0%.
Given sufficient deviations from modeling assumptions (RS1, RS2 in table 1), rBS tests for selection on foreground branches can be severely misleading. For RS1, the null model (ω2 = 1) is rejected in favor the alternative model (ω2 ≥ 1), implying positive selection along the neutral lineage five with frequencies much higher than the nominal error rate of the tests, and a very skewed distribution of the p-values (supplementary fig. S1, Supplementary Material online). The null hypothesis rejection rate increases as the length (S codons) of the alignment is increased. For example, at test p = 0.05, the null model was rejected 12/100 times for S = 1,000, 31/100 times for S = 2,000, 74/100 times for S = 5,000, and in 97/100 cases for S = 10,000. Nominal p-values are commonly interpreted as the acceptable rate of false positives of the test, hence p = 0.05 should result in about 5/100 false rejections of the null. Lowering p = 10 − 4 still yields 34/100 false positives for S = 10,000, suggesting that the rate of false positives is difficult to control. The estimate of ω along lineage 5 is biased, with mean and variance inversely proportional to sample size. On the same data, uBS had well-controlled rates of false positives, which did not correlate with the length of the alignments. For RS2, the rBS test now performs as if the null model (ω = 1 on branch 5) were correct—the rate of rejections is similar to the rate expected under the null model and the ω2 estimate is now biased downward to ω2≈1.0 and very low power (2–5%) to detect selection along branch 5 (table 1). We observed shrinking estimator variances for larger sample sizes (fig. S2), showing that the lack of power is not due to insufficient sample sizes. In contrast, uBS showed very low rates of false positives on the negatively selected branches (0%) and power ranging from 44% (S = 1,000) to 100% (S = 10,000) on the interior branch of the tree simulated to be under diversifying selection.
Table 1.
uBS Performance on Simulated Data
| Simulation Scenario | Sequences/Codons | Branch 1 | Branch 2 | Branch 3 | Branch 4 | Branch 5 | FWER |
Power |
||
| rBS | uBS | rBS | uBS | |||||||
| NC1 | 4 | 0.008 | 0.006 | 0.01 | 0.007 | 0.005 | 0.043 | 0.036 | — | — |
| 8 | 0.005 | 0.005 | 0.005 | 0.015 | 0.00 | 0.044 | 0.03 | — | — | |
| NC2 | 4 | 0.014 | 0.01 | 0.016 | 0.007 | 0.07 | 0.053 | 0.053 | — | — |
| 8 | 0.005 | 0.015 | 0.01 | 0.005 | 0.000 | 0.045 | 0.035 | — | — | |
| NI | 4 | 0.006 | 0.012 | 0.009 | 0.001 | 0.005 | 0.051 | 0.033 | — | — |
| 8 | 0.03 | 0.025 | 0.01 | 0.005 | 0.005 | 0.08 | 0.07 | — | — | |
| SC | 4 | 0.005 | 0.008 | 0.004 | 0.004 | 0.101 | 0.026 | 0.02 | 0.084 | 0.101 |
| 8 | 0.015 | 0.015 | 0.000 | 0.005 | 0.38 | 0.045 | 0.035 | 0.44 | 0.38 | |
| SI1 | 4 | 0.007 | 0.007 | 0.005 | 0.007 | 0.103 | 0.033 | 0.025 | 0.082 | 0.103 |
| 8 | 0.00 | 0.015 | 0.005 | 0.015 | 0.435 | 0.06 | 0.035 | 0.495 | 0.435 | |
| SI2 | 4 | 0.116 | 0.004 | 0.008 | 0.009 | 0.07 | 0.033 | 0.021 | 0.166 | 0.176 |
| 8 | 0.53 | 0.01 | 0.01 | 0.00 | 0.195 | 0.02 | 0.02 | 0.485 | 0.630 | |
| SI3 | 4 | 0.295 | 0.484 | 0.599 | 0.667 | 0.06 | 0.017 | 0.06 | 0.353 | 0.925 |
| RS1 | 1,000 | 1 | 0.01 | 1 | 0.00 | 0.00 | 0.12 | 0.01 | 1.00 | 1.00 |
| RS1 | 2,000 | 1 | 0.00 | 1 | 0.00 | 0.08 | 0.31 | 0.08 | 1.00 | 1.00 |
| RS1 | 5,000 | 1 | 0.01 | 1 | 0.00 | 0.03 | 0.74 | 0.03 | 1.00 | 1.00 |
| RS1 | 10,000 | 1 | 0.00 | 1 | 0.01 | 0.03 | 0.97 | 0.03 | 1.00 | 1.00 |
| RS2 | 1,000 | 1 | 1 | 0.00 | 0.00 | 0.44/0.03* | 0.00 | 0.00 | 1.00 | 1.00 |
| RS2 | 2,000 | 1 | 1 | 0.00 | 0.00 | 0.83/0.02* | 0.00 | 0.00 | 1.00 | 1.00 |
| RS2 | 5,000 | 1 | 1 | 0.00 | 0.00 | 0.98/0.03* | 0.00 | 0.00 | 1.00 | 1.00 |
| RS2 | 10,000 | 1 | 1 | 0.00 | 0.00 | 1.00/0.05* | 0.00 | 0.00 | 1.00 | 1.00 |
RS1 and RS2 are described in the text and figure 1. Simulations NC1, NC2, NI, SC, SI1, SI2, and SI3 are taken from Anisimova and Yang (2007) (see table 2 therein for complete details of simulation parameters). The first three simulations (NC1, NC2, and NI) do not include any lineages under positive selection, whereas the last four include one or more lineages under selection at some sites in the alignment. Branches that experience positive selection are typeset in italic. Entries for Branch 1–Branch 5 columns show the proportion of replicates where any branch from this class was found to be under positive selection at p ≤ 0.05. FWER is the proportion of replicates where at least one branch was falsely classified as undergoing positive selection. The Power column lists the proportion of replicates for which at least one branch under positive selection was correctly classified as such. *: the second number reports the proportion of replicates where Branch 5 was reported under positive selection by rBS.
Test Performance as a Function the Strength and Extent of Episodic Selection
For the 16-taxon tree and 1,000-codon long sequences with lineages A, B, and AB (supplementary fig. S3, Supplementary Material online) are under positive diversifying selection, we observed the following test performance.
15% of Sites under Selection with ω=5.
uBS achieved 100% power and FWER of 2%, demonstrating that larger and more informative alignments allow the test to be more discriminative and accurate, as expected. For the same data set, rBS was surprisingly conservative with 0% FWER, but only 6% power.
5% of Sites under Selection with ω=5.
uBS achieved only 9% power at FWER of 2%, demonstrating that if too few sites are under selection, the ability of the test to detect episodic selection is severely impacted.
15% of Sites under Selection with ω = 2.
uBS attained 8% power at FWER of 3%, indicating that a weak selection signal is considerably more difficult to identify.
Empirical Data Applications
First, we analyzed CD2 gene sequences coding for a cell adhesion molecule located on the surface of certain types of lymphocytes. These sequences were isolated from ten mammalian species and were previously analyzed by Lynn et al. (2005) using a branch (no site-to-site variation) method (Yang 1998) and more recently by Anisimova and Yang (2007) with a branch-site method. Lynn and colleagues found that lineages leading to pig, cow, horse, cat, the (pig and cow) ancestor (lineage 3 in fig. 2A), and the primate clade ancestral lineage (13) were under positive selection because the mean point estimate of ω at those branches exceeded one and the branch heterogeneity test (Yang 1998) rejected the hypothesis that all lineages were under the same selective pressure. Anisimova and Yang (2007) identified positive selection along lineages leading to cow, cat, and the ancestor of (pig, cow, horse, and cat) clade using a sequential rBS test; and pointed out that comparing the value of point estimate of ω to 1 was only suitable for exploratory analyses and did not constitute a valid statistical test. Our uBS model confirms (at p ≤ 0.05) episodic selection along the same three lineages reported by Anisimova and Yang (2007) but also identifies two additional lineages—the horse lineage and the most recent common ancestor of the primate clade. Neither of these lineages approached significance in the analysis of Anisimova and Yang, but because CD2 appears to have undergone extensive episodic selection at multiple lineages, the assumptions of the rBS test are likely to be violated in these data, for example, leading to loss of power by rBS (as was shown in SI3 simulations). The patterns of episodic selection were complex (fig. 2A and table 2), with marked differences in the extent (proportion) and strength (ω+) of selection along different lineages. Interestingly, Branches 6 (not reported by Lynn et al. 2005) and 13 (not reported by Anisimova and Yang 2007) appear to experience very strong selective forces (ω6+ = 37.2,ω13+ = 39.7) on a small percentage of sites (q6+ = 0.094,q13+ = 0.092), whereas the other three selected branches (cow, horse, and cat) each have approximately 40% of sites under relatively weaker positive selection (ω = 5.2–10.7).
FIG. 2.
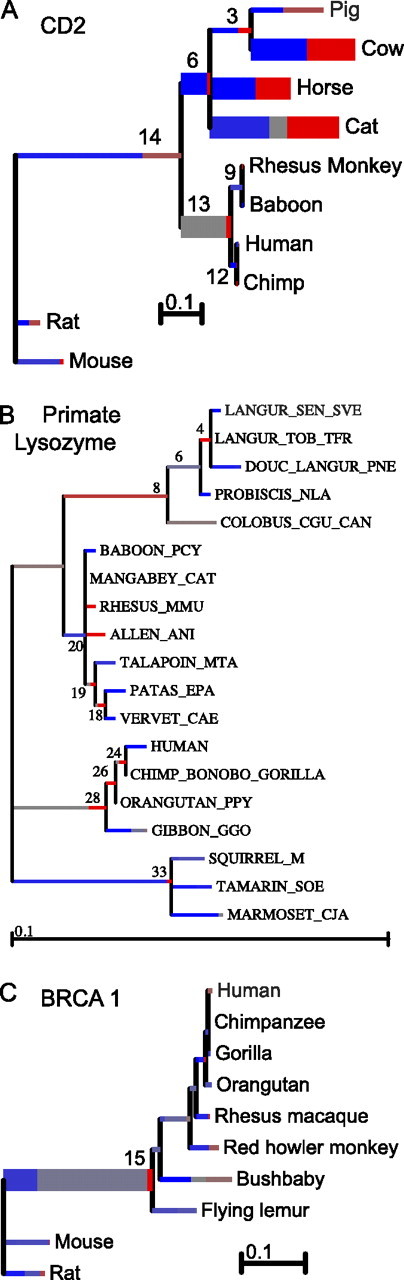
Empirical data sets analyzed for episodic selection. Each tree is scaled on the expected number of substitutions/nucleotide. The hue of each color indicates strength of selection, with primary red corresponding to ω > 5, primary blue to ω = 0, and grey to ω = 1. The width of each color component represents the proportion of sites in the corresponding class. Thicker branches have been classified as undergoing episodic diversifying selection by the sequential test at p ≤ 0.05.
Next, we reexamined a data set consisting of 19 lysozyme c sequences from primates initially analyzed by Messier and Stewart (1997) and more recently by Zhang et al. (2005). The authors suspected positive selection along the lineage leading to the colobine monkeys and hominoids for which the lysozyme protein may have acquired a different digestive function that allows them to lyse symbiotic bacteria. Yang (1998) confirmed positive selection along the hominoid lineage (and elevated ω compared with background on the colobine lineage) using codon models that permitted no site-to-site rate variation. Indeed, it appears that if one assumes negative or neutral selection elsewhere on the phylogeny, the “average” strength of selection along the lineages of interest exceeds or approaches one. It was therefore somewhat unexpected that more sensitive rBS models did not find evidence of episodic diversifying selection along the two lineages (Zhang et al. 2005). uBS reached the same conclusion—no single lineage had sufficient statistical support for episodic diversifying selection under a sequential (branch at a time) test. The inferred selective mixture for the hominoid ancestral lineages (28 in fig. 2B) showed 18.2% of sites under very strong selection ω > 100 and an uncorrected p-value of 0.008, that is, were we to test only for selection only along this lineage based on a priori information, we would find episodic diversifying selection at p < 0.05. For the colobine ancestral lineage (8 in fig. 2B), 100% of sites were allocated to the positive selection regime (ω = 3.4), yet the test p-value was only 0.10.
The last data set we analyzed contains ten mammalian sequences of the tumor suppressor gene BRCA1. Zhang et al. (2005) previously analyzed eight of these sequences as the chimpanzee and human lineages are suspected to be under positive selection but found no evidence of positive selection along any lineages. Our sequential analysis found evidence of episodic diversifying selection on the lineage ancestral to primates and lemurs (Branch 15 in fig. 2C) with 3.3% of sites in the ω+ = 17.3 class. The human lineage shows borderline (uncorrected) significance with p = 0.076 (all sites under weaker positive selection, ω = 2.26), whereas the chimpanzee lineage is not significant (uncorrected p = 0.16). These findings are in qualitative agreement with previous analyses (Zhang et al. 2005).
Discussion
This work demonstrates that current branch-site methods can have excessive Type I and Type II errors when the data strongly deviate from model assumptions. These models enforce uniform selective pressure on all background branches, thus biasing the estimate of ω along foreground branches. We have demonstrated this behavior to be positively misleading, with decreasing variance for larger sample sizes. The nature of the bias will depend on the distribution of selective pressures along background branches, nucleotide substitution biases, and branch lengths. More critically, the sequential rBS approach (Anisimova and Yang 2007) to test each branch in a phylogeny for evidence of positive selection, while specifically postulating that no other branches in the phylogeny are subject to positive selection, is likely an oversimplification of biological reality. Furthermore, when one branch is found to be under selection by this method, it automatically implies that no other branch (in the background) can be under selection, hence the sequential testing procedure that finds multiple selected branches by setting the foreground to one branch at a time is logically inconsistent.
We have developed and validated a new random effects branch-site model (uBS) to detect positive selection in protein-coding sequences that do not require partitioning lineages into foreground and background branches. This model considers all possible assignments of three selective regimes to the branches in a phylogeny at a given site. If the selective behavior along a branch is independent of that along other branches, our model can be efficiently evaluated in the standard phylogenetic framework. This is accomplished by replacing the standard substitution model along a branch with a mixture of three Markov models: one for purifying, one for nearly neutral, and one for diversifying selection. To detect episodic diversifying selection, we adopt the familiar hypothesis testing framework (Anisimova and Yang 2007) to identify the lineages in a phylogeny that could have undergone episodic selection, and we measure the strength (ω) and extent (proportion of sites) of such selection independently (but jointly) for each branch. uBS is approximately twice as computationally efficient as the current branch-site approach because it tests a series of nulls (no positive selection on a given branch) versus a universal alternative (no constraints on any branches), whereas the sequential rBS approach constructs a separate null and alternative model for each branch. The new approach is more computationally attractive than the family of codon-based covarion models (Guindon et al. 2004), where the addition of each evolutionary modality incurs an expansion of the character state space and the corresponding quadratic-to-cubic (in terms the number of ω classes) increase in algorithmic complexity. However, some aspects of covarion models are more flexible, for example, the switchpoints in the evolutionary process are not delineated by branches in the tree as they are in uBS, hence the two approaches are complementary.
Because our testing procedure does not limit the number and type of site configurations at a site, we expect it to demonstrate improved performance on data that do not conform to the restrictive assumptions of the rBS model. in IntroductionUsing the same set of simulations as in Anisimova and Yang (2007), we demonstrate that uBS has notably higher power and lower error rates than the sequential rBS method when the assumptions of the latter method are strongly violated (scenarios SI2 and SI3). Encouragingly, on the data that do meet rBS restrictions, our approach delivers comparable performance, suggesting that it is not necessary to make a priori assumptions about the patterns of episodic selection. uBS attains 100% power if sufficient data (e.g., 16 sequences, 1,000 codons, and 15% of sites under selection) are supplied. Our reanalysis of three benchmark biological data sets revealed slight differences from published results and confirmed the lower power of sequential rBS methods to detect short bursts of strong selection in a data set subject to pervasive episodic selection.
Much future work remains, however. First, there is no clear understanding of what extent and strength of selection, data sizes, and divergence levels are necessary for episodic selection tools to be appropriately powered, yet not subject to excessive false positive rates. Even based on our limited 16-taxon simulations, it is apparent that uBS rapidly loses power when the proportion of sites under selection is too small or when selective pressures are relaxed. Second, does the location of lineages under selection in the phylogeny (e.g., tips vs. deep internal branches) influence our ability to infer selection? Simulations in this study suggest that there may be more power to detect recent episodic selection at terminal branches, but a more systematic exploration is necessary. Third, how does one go about automatically pooling branches together to boost the power to detect weaker selection that affects the same set of sites in multiple lineages—a good example would be HIV evolution to independently acquire drug-resistance mutations in lineages that represent patients on treatment (Seoighe et al. 2007). Fourth, much of episodic selection is likely to be directional rather than diversifying, hence models must be adapted to include this type of selection as well (e.g., Delport et al. 2008, Kosakovsky Pond et al. 2008). Fifth, might it be beneficial to relax the assumption of constant synonymous rates (Kosakovsky Pond and Muse 2005)? Sixth, naive, or Bayes empirical Bayes approaches developed for rBS for detecting individual sites subject to episodic diversifying selection (Yang et al. 2005), need to be adapted to and evaluated in the context of uBS.
Based on the results, theoretical considerations and computational feasibility presented in this manuscript, we advocate our mixture approach over current tools for the detection of episodic diversifying selection (Anisimova and Yang 2007). Unlike Nozawa et al. (2009), who propounded a severely underpowered (and difficult to extend) counting method for lineage-specific selection detection and made a number of strong claims recently refuted by Yang and dos Reis (2011), we espouse the view that likelihood model-based approaches are a much more appealing way forward. We are convinced that continued improvements in biological realism of evolutionary models, underpinned by gains in computing power and algorithmic development, will provide evolutionary biologists with the tools to better characterize fundamental adaptive processes. uBS demonstrates the potential for continued extension of classical frequentist and hypothesis testing approaches to parallel recent seminal developments in Bayesian approaches to fitting complex substitution models (e.g., Rodrigue et al. 2010).
Supplementary Material
Supplementary figures S1–S3 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
This research was supported in part by the National Institutes of Health (AI43638, AI47745, and AI57167, GM093939), the University of California Universitywide AIDS Research Program (IS02-SD-701), the Joint DMS/NIGMS Mathematical Biology Initiative through Grant NSF-0714991 and by a University of California, San Diego Center for AIDS Research/National Institute of Allergy and Infectious Diseases Developmental Award to S.D.W.F. and S.L.K.P (AI36214). S.D.W.F. is supported in part by a Royal Society Wolfson Research Merit Award. B.M. is supported by Europeaid grant number Sante/2007/174-790 from the European Commission. The authors are grateful to Dr Maria Anisimova for providing simulated sequence alignments used for benchmarking uBS and rBS.
References
- Anisimova M, Kosiol C. Investigating protein-coding sequence evolution with probabilistic codon substitution models. Mol Biol Evol. 2009;26:255–271. doi: 10.1093/molbev/msn232. [DOI] [PubMed] [Google Scholar]
- Anisimova M, Yang Z. Multiple hypothesis testing to detect lineages under positive selection that affects only a few sites. Mol Biol Evol. 2007;24:1219–1228. doi: 10.1093/molbev/msm042. [DOI] [PubMed] [Google Scholar]
- Delport W, Scheffler K, Botha G, Gravenor MB, Muse SV, Kosakovsky Pond S. Codontest: modeling amino acid substitution preferences in coding sequences. PLoS Comput Biol. 2010;19:e1000885. doi: 10.1371/journal.pcbi.1000885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delport W, Scheffler K, Seoighe C. Frequent toggling between alternative amino acids is driven by selection in HIV-1. PLoS Pathog. 2008;4:e1000242. doi: 10.1371/journal.ppat.1000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delport W, Scheffler K, Seoighe C. Models of coding sequence evolution. Brief Bioinform. 2009;10:97–109. doi: 10.1093/bib/bbn049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. Evolutionary trees from DNA-sequences—a maximum-likelihood approach. J Mol Evol. 1981;17:368–376. doi: 10.1007/BF01734359. [DOI] [PubMed] [Google Scholar]
- Felsenstein J, Churchill GA. A hidden Markov model approach to variation among sites in rate of evolution. Mol Biol Evol. 1996;13:93–104. doi: 10.1093/oxfordjournals.molbev.a025575. [DOI] [PubMed] [Google Scholar]
- Goldman N, Yang Z. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol Biol Evol. 1994;11:725–736. doi: 10.1093/oxfordjournals.molbev.a040153. [DOI] [PubMed] [Google Scholar]
- Guindon S, Rodrigo AG, Dyer KA, Huelsenbeck JP. Modeling the site-specific variation of selection patterns along lineages. Proc Natl Acad Sci U S A. 2004;101:12957–12962. doi: 10.1073/pnas.0402177101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M, Kishino H, Yano TA. Dating of the human ape splitting by a molecular clock of mitochondrial-DNA. J Mol Evol. 1985;22:160–174. doi: 10.1007/BF02101694. [DOI] [PubMed] [Google Scholar]
- Holm S. A simple sequentially rejective multiple test procedure. Scand J Stat. 1979;6:65–70. [Google Scholar]
- Kosakovsky Pond S, Delport W, Muse SV, Scheffler K. Correcting the bias of empirical frequency parameter estimators in codon models. PLoS One 30:e11230. 2010 doi: 10.1371/journal.pone.0011230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Frost SDW. A genetic algorithm approach to detecting lineage-specific variation in selection pressure. Mol Biol Evol. 2005a;22:478–485. doi: 10.1093/molbev/msi031. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Frost SDW. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol Biol Evol. 2005b;22:1208–1222. doi: 10.1093/molbev/msi105. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Frost SDW, Muse SV. Hyphy: hypothesis testing using phylogenies. Bioinformatics. 2005;21:676–679. doi: 10.1093/bioinformatics/bti079. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Muse SV. Site-to-site variation of synonymous substitution rates. Mol Biol Evol. 2005;22:2375–2385. doi: 10.1093/molbev/msi232. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Poon AFY, Leigh Brown AJ, Frost SDW. A maximum likelihood method for detecting directional evolution in protein sequences and its application to influenza A virus. Mol Biol Evol. 2008;25:1809–1824. doi: 10.1093/molbev/msn123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Scheffler K, Gravenor MB, Poon AFY, Frost SDW. Evolutionary fingerprinting of genes. Mol Biol Evol. 2010;27:520–536. doi: 10.1093/molbev/msp260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn DJ, Freeman AR, Murray C, Bradley DG. A genomics approach to the detection of positive selection in cattle: adaptive evolution of the T-cell and natural killer cell-surface protein cd2. Genetics. 2005;170:1189–1196. doi: 10.1534/genetics.104.039040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massingham T, Goldman N. Detecting amino acid sites under positive selection and purifying selection. Genetics. 2005;169:1753–1762. doi: 10.1534/genetics.104.032144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messier W, Stewart CB. Episodic adaptive evolution of primate lysozymes. Nature. 1997;385:151–154. doi: 10.1038/385151a0. [DOI] [PubMed] [Google Scholar]
- Muse SV, Gaut BS. A likelihood approach for comparing synonymous and nonsynonymous nucleotide substitution rates, with application to the chloroplast genome. Mol Biol Evol. 1994;11:715–724. doi: 10.1093/oxfordjournals.molbev.a040152. [DOI] [PubMed] [Google Scholar]
- Nielsen R, Yang ZH. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics. 1998;148:929–936. doi: 10.1093/genetics/148.3.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozawa M, Suzuki Y, Nei M. Reliabilities of identifying positive selection by the branch-site and the site-prediction methods. Proc Natl Acad Sci U S A. 2009;106:6700–6705. doi: 10.1073/pnas.0901855106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigue N, Philippe H, Lartillot N. Mutation-selection models of coding sequence evolution with site-heterogeneous amino acid fitness profiles. Proc Natl Acad Sci U S A. 2010;107:4629–4634. doi: 10.1073/pnas.0910915107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Self SG, Liang KY. Asymptotic properties of maximum likelihood estimators and likelihood ratio tests under nonstandard conditions. J Am Stat Assoc. 1987;82:605–610. [Google Scholar]
- Seoighe C, Ketwaroo F, Pillay V, . (11 co-authors). A model of directional selection applied to the evolution of drug resistance in HIV-1. Mol Biol Evol. 24:1025–1031. [DOI] [PubMed]
- Suzuki Y, Gojobori T. A method for detecting positive selection at single amino acid sites. Mol Biol Evol. 1999;16:1315–1328. doi: 10.1093/oxfordjournals.molbev.a026042. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Nei M. False-positive selection identified by ML-based methods: examples from the Sig1 gene of the diatom Thalassiosira weissflogii and the tax gene of a human T-cell lymphotropic virus. Mol Biol Evol. 2004;21:914–921. doi: 10.1093/molbev/msh098. [DOI] [PubMed] [Google Scholar]
- Wong WSW, Yang Z, Goldman N, Nielsen R. Accuracy and power of statistical methods for detecting adaptive evolution in protein coding sequences and for identifying positively selected sites. Genetics. 2004;168:1041–1051. doi: 10.1534/genetics.104.031153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. J Mol Evol. 1994;39:306–314. doi: 10.1007/BF00160154. [DOI] [PubMed] [Google Scholar]
- Yang Z. Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol Biol Evol. 1998;15:568–573. doi: 10.1093/oxfordjournals.molbev.a025957. [DOI] [PubMed] [Google Scholar]
- Yang Z. dos Reis M. Statistical properties of the branch-site test of positive selection. Mol Biol Evol. 2011;28:1217–1228. doi: 10.1093/molbev/msq303. [DOI] [PubMed] [Google Scholar]
- Yang Z, Nielsen R. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol Biol Evol. 2002;19:908–917. doi: 10.1093/oxfordjournals.molbev.a004148. [DOI] [PubMed] [Google Scholar]
- Yang Z, Wong WSW, Nielsen R. Bayes empirical Bayes inference of amino acid sites under positive selection. Mol Biol Evol. 2005;22:1107–1118. doi: 10.1093/molbev/msi097. [DOI] [PubMed] [Google Scholar]
- Yang ZH, Nielsen R, Goldman N, Pedersen AMK. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics. 2000;155:431–449. doi: 10.1093/genetics/155.1.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ZH, Swanson WJ. Codon-substitution models to detect adaptive evolution that account for heterogeneous selective pressures among site classes. Mol Biol Evol. 2002;19:49–57. doi: 10.1093/oxfordjournals.molbev.a003981. [DOI] [PubMed] [Google Scholar]
- Zhang J. Frequent false detection of positive selection by the likelihood method with branch-site models. Mol Biol Evol. 2004;21:1332–1339. doi: 10.1093/molbev/msh117. [DOI] [PubMed] [Google Scholar]
- Zhang J, Nielsen R, Yang Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol Biol Evol. 2005;22:2472–2479. doi: 10.1093/molbev/msi237. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.