

Abstract
There is a recent emergence of interest in the genes involved in gametic recognition as drivers of reproductive isolation. The recent population genomic sequencing of two species of sexually primitive yeasts (Liti G, Carter DM, Moses AM, Warringer J, Parts L, James SA, Davey RP, Roberts IN, Burt A, Koufopanou V et al. [23 co-authors]. 2009. Population genomics of domestic and wild yeasts. Nature 458:337–341.) has provided data for systematic study of the roles these genes play in the early evolution of sex and speciation. Here, we discovered that among genes encoding cell surface proteins, the sexual adhesin genes have evolved significantly more rapidly than others, both within and between Saccharomyces cerevisiae and its closest relative S. paradoxus. This result was supported by analyses using the PAML pairwise model, a modified McDonald–Kreitman test, and the PAML branch model. Moreover, using a combination of a new statistic of neutrality, an information theory–based measure of evolutionary variability, and functional characterization of amino acid changes, we found that a higher proportion of amino acid changes are fixed in the sexual adhesins than in other proteins and a greater proportion of the fixed amino acid changes either between the two species or the two subgroups of S. paradoxus are functionally dissimilar or radically different. These results suggest that the accelerated evolution of sexual adhesin genes may facilitate speciation, or incipient speciation, and promote sexual selection in general.
Keywords: sex genes, cell surface proteins, yeast, modified McDonald–Kreitman test, evolutionary variability, adaptive evolution
Introduction
Genes encoding cell surface proteins are highly variable, and it has been argued that their diversification has produced the great diversity in cellular interactions and environmental response (Verstrepen and Fink 2009; Baldo et al. 2010). Among cell surface proteins, those involved in gametic recognition are particularly intriguing. Although the evolution of sex-related genes has been extensively studied in animals including Drosophila species, mammals, and birds, it has rarely been examined in yeasts, one of the sexually most primitive organisms. As opisthokonts, yeasts share a common ancestor with animals and possess sexual reproductive mechanism very similar to that in metazoans. First, sexually reproducing yeasts often contain two mating types, similar to the male and female sexes in animals. Secondly, during mating the two mating types of yeasts need to first recognize each other, followed by cell fusion and nuclear fusion, and ending with a diploid zygote (Lee et al. 2010), which is very similar to the fertilization process in animals. Lastly, even the proteins involved in sexual reproduction in yeast and animals are very similar in 3D structure of their essential domains (Swanson et al. 2011). Thus, the study of sexual reproduction in yeast could shed new lights on the origin and evolution of sex.
Studies in animals, including Drosophila, abalone, and human, show accelerated evolution of the proteins localized on the cell surface of gametes and involved in egg–sperm interaction (Aagaard et al. 2006, 2010; Gasper and Swanson 2006; Panhuis et al. 2006; Jagadeeshan and Singh 2007). Similarly, sexual adhesin proteins have been found in yeasts and well characterized particularly in the baker's yeast, Saccharomyces cerevisiae (Dranginis et al. 2007). However, how these proteins have evolved and how their evolution has contributed to speciation in this group of organisms remain open questions.
Saccharomyces species have two mating types a or α, determined by the presence a sex-specific allele at the mating type locus MAT on chromosome III, which regulates the secretion of sex pheromone peptides, their receptors, and mating-specific cell adhesion proteins. Sexual adhesion of a- and α cells in S. cerevisiae depends on the binding among four proteins on the cell surface, encoded by AGA1, AGA2, SAG1, and FIG2 genes, respectively (Terrance and Lipke 1987; Lipke and Kurjan 1992; Dranginis et al. 2007; Huang et al. 2009; Xie and Lipke 2010). Mating-type specific binding is mediated mainly by two sexual agglutinins, a-agglutinin and α-agglutinin. a-Agglutinin consists of subunits Aga1p and the 68-residue glycopeptide Aga2p, the latter of which is expressed only on a-type cells. Aga1p is anchored in cell wall at one end and cross-linked to Aga2p through two disulfide bonds at the other end (Roy et al. 1991; Cappellaro et al. 1994). During mating, Aga2p is bound to α-agglutinin, that is, Sag1p, which is only expressed on the surface of α-type cells and anchored to cell wall polysaccharide (Lipke et al. 1987, 1989; Chen et al. 1995; de Nobel et al. 1996; Zhao et al. 2001). In addition, mating-specific binding is strengthened by interactions between Aga1p and Fig2p, with each being expressed on both mating types (Huang et al. 2009).
Besides sexual adhesin proteins, yeasts also express other cell surface proteins. Some of these proteins, like mucin-like Muc1p (Flo11p), mediate cell aggregation and biofilm formation in yeast (Dranginis et al. 2007). Another group consists of glycosyl enzymes that are involved in the glycosylation of cell surface proteins, including glycosidases, glycosyl transferases, and transglycosylases. Whether these proteins and their coding genes have evolved differently from sexual adhesin genes/proteins is still a question to be answered (Xie and Lipke 2010). Recently, multiple strains of both S. cerevisiae and its closest relative, S. paradoxus, have been genome-sequenced (Liti et al. 2009). Here we present our study of the evolution of 73 genes encoding cell surface proteins in S. cerevisiae and S. paradoxus. We found that sexual adhesin genes have evolved much faster than any other cell surface–related genes both between and within the two yeast species, which have important implications to speciation and sexual selection.
Materials and Methods
Sequence Extraction, Quality Control, and Alignment
The genomic sequences for 38 strains of S. cerevisiae and 36 strains of S. paradoxus were downloaded from the ftp site of Sanger Institute (ftp://ftp.sanger.ac.uk/pub/dmc/yeast/latest/, last accessed February 25, 2009), which were originally generated by the Saccharomyces Genome Resequencing Project (Liti et al. 2009). The standard sequences for a selected set of 73 cell surface–related genes (Coronado et al. 2007), including the 4 sexual adhesin genes, were obtained from the Saccharomyces Genome Database (http://www.yeastgenome.org, last accessed March 3, 2011). For each of the 73 genes (supplementary table 1, Supplementary Material online), both NUCMER and BlastN programs were used to find the best matching sequences in the genomes of all the strains in both species. A series of custom scripts were used to distinguish orthologs from paralogs, based on a combination of chromosome number, chromosomal positions, and match quality (bit-scores for Blastn or alignment length and percent identity for NUCMER), and to extract the orthologous sequences.
The extracted sequences for each gene were aligned using both ClustalW (Larkin et al. 2007) and MUSCLE (Edgar 2004) programs as implemented in SeaView (Gouy et al. 2010), with manual adjustment for obviously less than optimal alignments particularly in the low-complexity regions. The sequences were then carefully checked against the National Center for Biotechnology Information (NCBI) trace archive whenever needed and possible, with some corrections made based on the trace files submitted to NCBI by Sanger Institute. Given the approach used by Sanger to fill in genomic sequencing gaps, we extracted just unique haplotypes within each species for subsequent analyses. All sequences used for analysis are in coding regions, with none of the genes containing any intron.
Pairwise Substitution Rates and Modified McDonald–Kreitman Test
McDonald–Kreitman (MK) test (McDonald and Kreitman 1991) is often used to examine whether the amino acid sequence changes between species is significantly different from that within species, as an indicator of whether there is “adaptive” evolution (positive selection) between species. However, the traditional MK test uses only the total numbers of segregating sites both within and between species, regardless of the frequencies of each allele, and thus could be significantly skewed by rare alleles. To correct this problem, we designed a modified MK test by using the averaged pairwise synonymous and nonsynonymous changes both within and between species.
For each gene, the synonymous and nonsynonymous substitution rates between any pair of sequences from either the same or different species were first obtained using PAML pairwise model with the maximum likelihood method. The resultant files containing pairwise synonymous (dS) and nonsynonymous (dN) substitution rates were parsed using a custom Perl script and plotted in R.
The numbers of synonymous (S) and nonsynonymous (N) substitutions for each pair of sequences were then calculated by parsing the mlc file from PAML analysis using another custom script. These values were then used to calculate the average numbers of synonymous and nonsynonymous nucleotide changes both within and between the two species, and the net between-species substitutions were calculated by subtracting the average numbers of within-species substitutions from the gross estimate of between-species substitutions following Nei (1987, Equation 8.20). These numbers in turn were used in a MK-like test to compare the rate of evolution between species with that within species.
New Statistic to Test Evolutionary Neutrality
The ratio of nonsynonymous to synonymous substitutions, N/S, is another commonly used method to detect selection. However, the N/S ratio could not be defined or is meaninglessly large when S (the number of synonymous changes) is, or close to, zero. Recently, the fraction of nonsynonymous changes among all evolutionary changes, N/(N + S), has been independently suggested as an improvement over the simple N/S ratio (Stoletzki and Eyre-Walker 2011), making it applicable to all situations, even when the number of synonymous changes is zero. Here we present a further improvement by using the rates, instead of the numbers, of synonymous and nonsynonymous substitutions, producing a new measure called the fraction of nonsynonymous substitution rate:
where dS and dN were calculated as the average synonymous and nonsynonymous substitution rate, respectively, based on all the pairwise sequence comparisons using maximum likelihood methods. Compared with N/(N + S) ratio, this modification standardizes the number of synonymous and nonsynonymous substitutions by the number of potential synonymous and nonsynonymous substitution sites, respectively, and at the same time corrects codon bias and the unequal probability of generating synonymous versus nonsynonymous substitutions from DNA mutations by using maximum likelihood calculation. fN between species was then plotted against that within species for each gene in R, and the comparison was then used to infer whether the gene has evolved neutrally or not. The difference in fN between species compared with that within species could be further quantified as:
where DiS denotes the difference in selection regime between within-species and between-species. This is analogous to the statistics of α (the proportion of adaptive amino acid substitution, Smith and Eyre-Walker 2002) or DoS (direction of selection, Stoletzki and Eyre-Walker 2011), but its calculation is based on average synonymous and nonsynonymous substitution rates, rather than simple counts of synonymous and nonsynonymous substitution sites. The new statistic, termed “Difference in Selection,” does not depend on the assumption that within-species evolution is always neutral, an assumption of α and DoS.
PAML Analyses Using Branch and Site Models
To further test whether the four sexual adhesin genes have evolved at different rates between the two yeast species compared with that within the two species, we first performed PAML analysis using the branch models (Yang 2007) with trees reconstructed using the Neighbor-Joining method. Two models, one assuming a single dN/dS ratio (ω) on all branches and the other setting the branch between the two species to have a different dN/dS ratio from that within species, were statistically tested for significant difference using the log-likelihood ratio test. We then used PAML site models in attempt to identify specific codon sites that might have been under positive selection in each of the four sexual adhesin genes, with the same neighbor-joining tree being used for each gene. The M1a model assumes there are only two types of sites in the gene sequence of interest (one neutral with ω = 1 and the other nearly neutral with ω < 1), whereas the M2a model assumes there is a third class of positively selected sites with ω > 1. These two models were statistically tested using the log-likelihood ratio test to see if one explained the data significantly better than the other.
Site-Specific Characterization of Evolutionary Variability and Biochemical Similarity
We used information theory to quantify sequence variability at individual alignment positions (Schneider 1999). Based on the information theory, we used mutual information (MI) to quantify sequence variations associated with species divergence. MI measures the association of an amino acid residue (aa) with a species (sp) at each site:
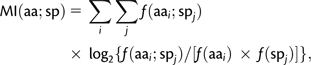 |
where f(aai;spj) is the frequency of residue i occurring in species j and f(aai) and f(spj) are marginal frequencies of residue i and species j, respectively. Furthermore, we normalized MI by total uncertainty to obtain:
where H is the Shannon entropy (Witten and Frank 2000). This normalized MI (U) measures the proportion of variability associated with species relative to the total amount of sequence and species diversity at an alignment site. The larger the U value, the higher is the proportion of sequence variability associated with different species. In our study, sites with U = 0 are invariant across the two Saccharomyces species whereas those with U = 1 demonstrate as fixed differences between the two species. To reduce statistical uncertainties, we calculated U scores only for gene that have at least five alleles present in either species.
We further characterized the amino acid changes at individual sites using the amino acid similarity/dissimilarity matrix included in BLOSUM62 (Henikoff S and Henikoff JG 1992). The distribution of U and BLOSUM62 scores for segregating sites in sexual adhesins were then statistically compared with that in proteins of other functional groups.
Results
Accelerated Rate of Amino Acid Substitutions in Sexual Adhesins
For each of 73 cell surface protein–related genes, we calculated synonymous and nonsynonymous substitution rates between each pair of sequences using both Nei and Gojobori (1986) and maximum likelihood methods, as implemented in PAML. The results from the two methods were quite similar, and we use the maximum likelihood measures for further analysis because this method has taken into consideration of codon bias and mutation rate difference between transitions and transversions.
There was a wide range of variation among the 73 genes in their synonymous and nonsynonymous divergence rates between the two species (fig. 1). Genes encoding cell wall structural proteins (light green in fig. 1, same below) and the enzymes involved in modifications of cell wall proteins (green) varied broadly in synonymous substitution rates between the two yeast species, but their nonsynonymous substitution rates seemed to be very limited, a pattern consistent with high levels of functional constraints on these proteins. The results were similar for genes encoding transmembrane proteins, cytoplasmic proteins, and mating pheromone and receptors. The genes coding for sexual adhesins (red) showed relatively low to moderate levels of synonymous substitution rates, but their nonsynonymous substitution rates were significantly higher than those for the genes in all the other groups, both between species (fig. 1) and within species (supplementary figs. 1A and 1B, Supplementary Material online). The between-species patterns of divergence in these different groups of genes were also reflected in fN, the fraction of nonsynonymous substitution rate, which was much higher for the four sexual adhesin genes, though none of them had a ratio greater than 0.5, which is equivalent to a dN/dS ratio of 1 (fig. 2, supplementary fig. 2, Supplementary Material online).
FIG. 1.

The between-species dN and dS for genes in different groups. All the 73 genes under study are grouped based on their functions and cellular locations and shown in different colors. The sexual adhesin genes, shown in red, clearly have higher nonsynonymous substitution rates whereas their synonymous substitution rates are within the range of the other genes. The line indicates dN/dS = 1.
FIG. 2.

The fraction of nonsynonymous substitution rate (fN) between species for different gene groups. The gene grouping scheme is the same as those in figure 1. The variation in fN for each gene group is shown in box plot, with each circle representing an outlier pair of sequences from the two different species.
Interestingly, the sexual adhesins have higher rates of between-species divergence than other sex-related genes. For example, the sex pheromone and receptor genes vary broadly in the synonymous rates (dS), but show very similar low nonsynonymous rates (dN) (figs. 1 and 2). Four of these genes are among the most conserved as measured by fN (fig. 2 and supplementary fig. 2, Supplementary Material online), whereas the other two genes are still far below those of the sexual adhesin genes. Sexual adhesins are also distinct from other extracellular proteins, including FLO10, the only FLO gene for which enough reliable sequences were obtained. The genes coding these extracellular proteins are quite similar to sexual adhesins in synonymous substitution rates, their nonsynonymous substitution rates vary to some degree but are still distinctly lower than those of the sexual adhesins (fig. 1). Consequently, the fN measures for these extracellular proteins are much lower than those for sexual adhesins (fig. 2 and supplementary fig. 2, Supplementary Material online).
Between- and Within-Species Comparison and Test of Neutrality
To test whether the rate of amino acid sequence changes between species is significantly different from that within species for the four sexual adhesin genes, we carried out a modified MK test (see Materials and Methods). The results showed that although the four sexual adhesin genes had much higher nonsynonymous substitution changes between the two species compared with other groups of genes, the rate of nonsynonymous changes between species is not significantly higher than that within species for any of the four sexual adhesin genes (P = 0.40, 0.90, 0.49, and 0.88 for AGA1, AGA2, FIG2, SAG1, respectively, table 1), which differs from the results of the traditional MK test, particularly for AGA1 (supplementary table 2, Supplementary Material online).
Table 1.
Modified MK Test and Neutrality Indices for the Evolution of Four Sexual Adhesin Genes.
| Gene | S-Within | S-Between | N-Within | N-Between | P Value | DiS |
| AGA1 | 40 | 190 | 30 | 184 | 0.40 | 0.0634 |
| AGA2 | 6 | 25 | 7 | 26 | 0.90 | –0.0287 |
| FIG2 | 48 | 561 | 65 | 649 | 0.49 | –0.0389 |
| SAG1 | 21 | 173 | 26 | 195 | 0.88 | –0.0233 |
NOTE.—Columns 2–3 indicate the number of synonymous changes within and between the two yeast species, respectively. Similarly, columns 4–5 represent the number of nonsynonymous changes either within or between the two species. Column 6 shows the P value for the modified MK test.
Similarly, the modified MK test was carried out on all the other 69 genes and then compared with the results from the traditional test (supplementary table 3, Supplementary Material online). For genes shown to be significantly different for their within- and between-species evolutionary patterns using the traditional test, the modified test tends to reduce the significance of that difference, though not always. Nineteen genes are shown to be significantly more constrained in nonsynonymous changes between the two yeast species in the traditional MK test, but the significance levels are reduced in the modified test for 17 of them (supplementary table 3, Supplementary Material online). For those genes not shown to be significant in the traditional MK test, the change in direction and degree of P value varies case by case. Furthermore, none of these genes show both statistical significance for within- and between-species difference (indicated by the P value) and a positive value of α or DiS, the combination of which is a signature of positive selection for amino acid changes between species.
As an independent test for selection, we carried out PAML branch model analysis for the four sexual adhesins to examine whether the branch between the two species has evolved at different rates than that within the two species because they showed accelerated amino acid substitution rates both between and within species compared with other groups (figs. 1 and 2, supplementary figs. 1 and 2, Supplementary Material online). The results show that there is no significant difference in the model assigning the branch between the two species a different ω and that assuming all branches have evolved at the same rate (supplementary table 4, Supplementary Material online). The above results together suggest that these four sexual adhesin genes have evolved at similarly high rates both within and between species.
We further quantified the fraction of nonsynonymous substitution rates (fN) both between and within species (fig. 3). There is a linear relationship between the average fN-between and fN-within for most of the genes, and the best-fit regression line for the data was fN-between = –0.01322 + 0.52943 × fN-within (fig. 3). So the nonsynonymous substitution rate between the two species is only roughly half of that within the species, which suggests that purifying selection is much stronger between the two yeast species than that within the species for most of these genes. However, the four sexual adhesin genes differed from all the other genes in having higher nonsynonymous substitution rates both within and between species but still a higher ratio between the two than most of the other genes. These results suggest the sexual adhesin genes might have evolved under either weaker functional constraint or stronger positive selection, particularly between the two yeast species, than other genes.
FIG. 3.

Comparing the fraction of nonsynonymous substitution rate (fN) between species versus that within species. The best-fit linear regression line represents fN-between = –0.01322 + 0.52943 × fN-within. MFA1 gene is another major outlier (to the upper left in purple) due to lack of statistical power, as it contains no nonsynonymous substitution within species and very few segregating sites between species.
Functionally Correlated Selection Regimes at Different Sites in Sexual Adhesins
As expected, different sites within a gene/protein sequence might have evolved under different evolutionary pressures, often in correlation with functional differences among sites. We further examined the evolution of the sexual adhesin genes in detail by characterizing the evolutionary variability using the information theory–based measure U and the chemical properties of amino acid changes. The index U defines the pattern of occurrence for each position in a protein sequence as a number between 0 (conserved in all sequences) and 1 (a fixed difference between species). Figures 4 and 5 show that most positions contain either low variability or fixed differences between species. Similar to the fixed amino acid differences between the two yeast species, some sites in the four sexual adhesin genes also possess fixed amino acid changes between two groups of S. paradoxus, though the number of such sites is much smaller than that of fixed differences between the two species. Relatively few positions show the intermediate variability characteristic of relaxed selection.
FIG. 4.
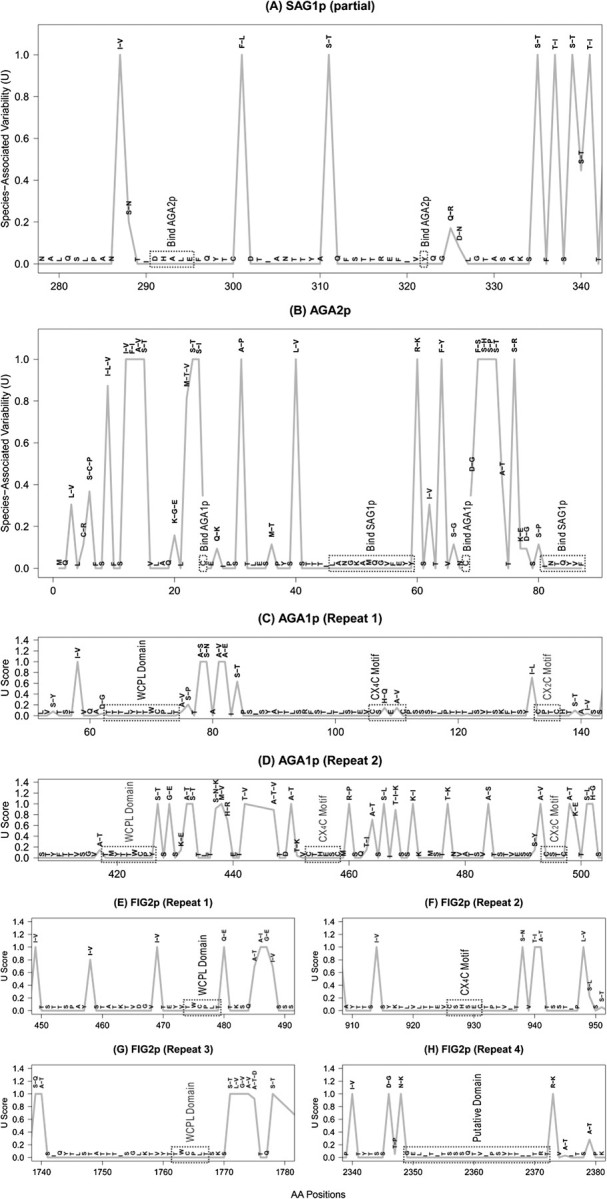
Evolutionary variability of amino acid residues in the sexual adhesins. The variability index (U) is a measure of the proportion (0 to 1) of amino acid variability associated with specific taxa and is calculated based on information theory (see Materials and Methods). The amino acids at aligned positions are shown, with a single amino acid state representing constant sites and multiple states variable sites. (A) Sag1p (partial sequence), (B) Aga2p, (C,D) Aga1p (Repeats 1 and 2, respectively), and (E–H) Fig2p (Repeats 1–4, respectively).
FIG. 5.

Evolutionary variability and functional characterization of amino acid residues in sexual adhesins. To improve statistical reliability, the analysis included only genes that have at least five unique sequences in either species. BLOSUM62 scores were obtained for all two-state variable sites. Plots were based on a total of 884 two-state sites in the 4 sexual-adhesin families (22% of aligned positions) and 2,139 two-state sites in 64 proteins in other groups (7.1% of aligned positions). Substitutions with negative BLOSUM scores were considered nonconservative and those with zero or positive BLOSUM scores conservative. (A) Categorization of amino acid substitutions based on U and BLOSUM62 matrix for sexual adhesins and other proteins. We used a cutoff value of U ≥ 0.8 to delineate substitutions fixed either between the two yeast species or between the two subspecies of S. paradoxus. From top left clockwise, the numbers corresponding to each quadrant show counts and fraction (in parenthesis) of sites belonging to each of the four categories: fixed nonconservative substitutions, fixed conservative substitutions, conservative polymorphisms, and nonconservative polymorphisms. (B) Histogram of the distribution of amino acid changes, as measured by BLOSUM62 scores, in sexual adhesins and other proteins. Error bars show two standard deviations above and below the mean based on the binomial sampling variance.
The Conservation of Known Binding Sites in Sexual Adhesins
Several motifs and residues in Aga1p, Aga2p, Sag1p, and Fig2p are known to be involved in the interactions among these sexual adhesins. We examined how these sites have evolved both within and between the two yeast species. In general, we found high conservation of sites known to be involved in binding between the adhesins though there are many species-specific differences in the sequences near these sites (fig. 4).
In α-agglutinin, Sag1p, all cysteine residues are conserved (U = 0), as well as the residues proposed to interact with its ligand Aga2p. The Sag1p Asp291-His-Ala-Leu-Glu295 (DHALE) motif in the third immunoglobulin region (Cappellaro et al. 1991) and other residues (including Asp216Tyr) shown to be important in the binding to a-agglutinin (de Nobel et al. 1996) are all conserved both within and between the two species (U = 0, fig. 4A).
The a-agglutinin subunit Aga2p has two regions involved in its binding with α-agglutinin, Sag1p (fig. 4B). The first region, which consists of 15 contiguous amino acids at positions 47–61, is highly conserved both within and between the two species, except for only one fixed amino acid difference between the species. However, the amino acids involved, lysine in S. cerevisiae and arginine in S. paradoxus, are both long-chain basic residues, and the substitution may have only a very minor effect on activity. At the C-terminal region of Aga2p, the sequence INTQYVF is necessary for tight binding (Shen et al. 2001) and is completely conserved (U = 0) both within and between the two species.
The a-agglutinin subunit Aga2p is bound through two disulfide bonds to Aga1p, which in turn is cross-linked into the cell wall through a modified glycosyl phosphatidylinositol anchor (Dranginis et al. 2007). In particular, Cys7 and Cys50 in Aga2p are bonded to two cysteines in the first repeat motif of Aga1p (Cappellaro et al. 1994; Shen et al. 2001), and these four sites are perfectly conserved in the two species (U = 0, figs. 4B and 4C).
In the wall-bound subunit Aga1p, all of the 10 Cys residues are conserved (U = 0), including those proposed as participants in disulfide bonding to Aga2p (Shen et al. 2001). Each of the two repeats in Aga1p also contains three known motifs, Trp-Cys-Pro-Leu (WCPL), Cys-(aa)4-Cys (CX4C), and Cys-(aa)2-Cys (CX2C), and some of these motifs are important for the Aga1p-Fig2p binding (Huang et al. 2009). As shown in figures 4C and 4D, these motifs in both repeats are conserved, except for two sites within the first CX4C motif, each being polymorphic in S. paradoxus.
Fig2p contains five WCPL motifs as repeats and at least nine CX4C motifs that are largely separate from the WCPL repeats. Six of the nine CX4C motifs are clustered in the C-terminus in S. cerevisiae, whereas S. paradoxus contains extra CX4C motifs, particularly in one of the subgroups. It has been suggested that Fig2p might interact with Aga1p through disulfide bonding (Huang et al. 2009). Correspondingly, most of the sites in these motifs, particularly WCPL residues, are conserved (figs. 4E–H). Similarly, all the cysteine residues in the nine conserved CX4C motifs have U = 0. We have also found many other sites are either completely or largely conserved in the two species, though their functions are not yet known (figs. 4A–H).
In addition, we checked whether the pattern of binding site conservation held across broader taxonomic distances. A Blast search and multiple alignment showed that the sexual adhesin binding sites in Aga2p and α-agglutinin were by-and-large conserved in closely related S. sensu stricto species, but diverged in S. sensu lato (data not shown), in correlation with the phylogenetic relationship of the two species complexes.
High Level of Polymorphism at Sites with ω > 1
In the first attempt to examine whether any site in the sexual adhesin genes have been positively selected, we followed the traditional method using the PAML site models by statistically comparing one model assuming no site with ω > 1 and the other allowing a class of codon sites to have ω > 1 (for details, see Materials and Methods). The likelihood ratio test revealed that for AGA1 and FIG2, the model containing a group of sites with ω > 1 explains the data significantly better than the model containing no such group. Bayes empirical Bayes analysis, as implemented in PAML, further identified 29 such codon sites in FIG2 and 10 sites in AGA1, but none in AGA2 and SAG1. However, none of the 29 sites in FIG2 are statistically significant, whereas half of the sites in AGA1 demonstrate strong statistical significance (Pr[ω > 1] > 99%, supplementary table 5, Supplementary Material online).
However, further analysis of the pattern of amino acid variation at the five sites with Pr(ω > 1) > 99% in AGA1 shows that they are highly polymorphic within either one or both species (supplementary table 6, Supplementary Material online). Furthermore, these highly polymorphic sites do not seem to be in linkage disequilibrium with any other sites in the gene, an expected signature of positive selection on these sites (data not shown).
Adaptive Fixed Nonconservative Amino Acid Substitutions in Sexual Adhesins
Subsequently, we characterized all amino acid variation in sexual adhesins and tested whether the divergence had a characteristic profile of specific substitutions. The U index summarizes the occurrence of substitutions at one position in the protein sequence in association with a particular species, and we compared its value with the nature of the substitutions themselves. Each amino acid substitution was scored for similarity or dissimilarity by the value of the substitution in the BLOSUM62 sequence comparison matrix (Henikoff S and Henikoff JG 1992). Figure 5A shows that the four sexual adhesins showed a high degree of variation, with 884 substitutions at 3,995 sequence positions (22% of positions in the alignment) compared with 2,214 substitutions at 30,009 positions in 60 other proteins in our data set (7% of positions). In total, 78.7% of the substitutions in the sexual adhesins are fixed either between the two species or between the two subgroups of S. paradoxus (U > 0.8), whereas only 45.4% of the substitutions are fixed for the other genes combined together (fig. 5A).
There is an unusual proportion (45.7%) of radical or nonconservative amino acid substitutions in the sexual adhesins, as indicated by a BLOSUM62 substitution value <0, whereas only 26.8% of the substitutions are nonconservative in the other genes combined. In addition, the proportion of fixed nonconservative amino acid substitutions is 3-fold greater in the sexual adhesins (fig. 5A). A frequency plot of the BLOSUM62 scores shows that within the sexual adhesins, the score distribution is much more negative (fig. 5B). The difference in the distribution of different categories of amino acid changes is statistically significant between the four sexual adhesin genes and all the others (χ2 = 689.17, degrees of freedom = 3, P < 0.0001).
To test whether any other individual group show similar patterns of protein evolution, we compared each of them with sexual adhesins using both U and BLOSUM62 scores as above. The results demonstrate that sexual adhesins are statistically significantly different from all the other individual groups in clearly having much higher proportion of fixed and radical amino acid changes between species (supplementary fig. 3, supplementary table 7, Supplementary Material online).
Therefore, the sexual adhesions are characterized not only by a higher frequency of fixed substitutions between the two species or the two incipient species of S. paradoxus but also by a higher frequency of biochemically nonconservative changes among the fixed substitutions, suggesting these changes in sexual adhesins are adaptive.
Discussion
Our study revealed that yeast sexual adhesin proteins and their coding genes have evolved much faster than other cell surface proteins and genes. To our knowledge, this is the first time that the evolution of sexual adhesions has been described in the two closely related yeast species. Our finding is consistent with the general evolutionary pattern of sex-related genes as previously reported in various animal systems, particularly in the model system for evolutionary genetics study, Drosophila (Mueller et al. 2005; Haerty et al. 2007; Jagadeeshan and Singh 2007), as well as in abalone (Aagaard et al. 2006, 2010; Panhuis et al. 2006) and humans (Wyckoff et al. 2000; Gasper and Swanson 2006).
Many previous studies have relied on the traditional MK test (McDonald and Kreitman 1991) to detect adaptive selection between species (Begun et al. 2000; Wyckoff et al. 2000; Lawniczak and Begun 2007). However, because this method uses the total counts of synonymous and nonsynonymous changes within species, regardless of the frequency of each allele, the result can be greatly skewed by low-frequency alleles. The more low-frequency alleles there are within species, the less accurate the result is. The major method that has been proposed to tackle this problem is to remove sites that contain singletons or alleles below an arbitrary frequency (Fay et al. 2001, 2002; Bierne and Eyre-Walker 2004; Zhang and Li 2005; Charlesworth and Eyre-Walker 2006). In contrast, our modified MK test still considers all the alleles in a data set, but weighs all alleles based on their frequencies, and thereby corrects bias caused by singletons or low-frequency alleles. This method does not rely on the assumption that these alleles are necessarily slightly deleterious; in fact, it is independent of any hypothetical fitness effects of these alleles.
The results reported in this study for the modified MK test differ from those of the traditional test in two aspects by using averaged pairwise measures of synonymous and nonsynomous changes, which in turn were calculated based on maximum likelihood method taking into consideration of codon frequency and different substitution rates among bases. As expected, the former has often reduced, in some cases dramatically, the counts of both synonymous and nonsynonymous segregating sites within species, whereas the latter has often increased the counts of fixed differences between species for both synonymous and nonsynonymous changes. However, the overall effect of such changes still varies among genes, though for most of the genes studied here the modified MK test reduces the statistical difference, or suggests more similar evolutionary patterns, between within-species and between-species. It is possible, however, to modify the traditional MK test in a slightly different way, by using the averaged pairwise counts of synonymous and nonsynonymous changes calculated using Nei and Gojobori (1986) method, which though is less preferable than the maximum likelihood method used in this study.
Whether such modifications would make much difference depends on the specific data set being studied, but the modified test would be less sensitive to the cases in which there are many singletons or low-frequency alleles. AGA1 is the only gene for which the traditional MK test shows the rate of nonsynonymous changes between species is significantly higher (supplementary table 2, Supplementary Material online); however, the modified MK test for this gene is not significant (table 1), a result consistent with other analyses reported in this paper. These results suggest adaptive evolution may have been overestimated in some previous studies that did not deal with within-species singletons or low-frequency alleles appropriately, though in other cases it may have been underestimated as recently shown by Charlesworth and Eyre-Walker (2008).
However, the lack of significance for all the four sexual adhesin genes in the modified MK test itself should not be interpreted as lack of adaptive divergence between the two species, as commonly assumed. The result simply means the rate of nonsynonymous changes between species is not significantly higher than that within species. Given the accelerated rates of nonsynonymous substitutions in these genes, they could have evolved adaptively both within and between species, but just at similar rates.
PAML site analysis has often been used to purportedly detect positive selection at individual codon sites by identifying sites with dN/dS (ω) > 1. Such an analysis has indeed identified some sites of such characteristics in two sexual adhesin genes (AGA1 and FIG2) in our study. However, these sites have dN/dS > 1 only because of the high level of within-species polymorphism and do not show any other signatures of selection including linkage disequilibrium between these sites and the neighboring regions. Therefore, these sites with dN/dS > 1 may actually have evolved neutrally, rather than under positive selection, suggesting dN/dS > 1 from PAML site analysis is in itself not sufficient to detect positive selection, at least in some cases.
However, adaptive evolution could be inferred by comparing genes of interests to other genes and by biochemical and functional characterization of amino acid changes. First, the nonsynonymous substitution rate in sexual adhesin genes is very different from that in other sex-related genes. For example, the mating pheromones and their receptors are highly conserved (figs. 1 and 2, supplementary fig. 2), consistent with other studies suggesting that the pheromone and its receptor have coevolved together among different Saccharomyces species, which constrains the evolution of both (McCullough and Herskowitz 1979; Egel-Mitani and Hansen 1987; Marsh 1992; Sen et al. 1997). In contrast, the four sexual adhesin genes have much higher rates of nonsynonymous changes both within and between the two yeast species, which is more remarkable given their relatively low rates of synonymous substitutions.
In further support of a difference between the sexual adhesin genes and genes of other functional groups is the difference in the fraction of nonsynonymous substitution rate fN = dN/(dN + dS). We found for most of the genes in our study, there is strong purifying selection against nonsynonymous changes between the two yeast species, but the four sexual adhesin genes are clearly different from them (fig. 3), consistent with either weaker purifying selection or stronger positive selection between species.
However, our further analysis of evolutionary variability and functional characterization of amino acid changes provide strong evidence that the fixed amino acid changes could likely create functional divergence in these sexual adhesins, both between the two yeast species and between the two subgroups of S. paradoxus. In particular, the fixed substitutions near the known binding sites in the four sexual adhesin proteins (fig. 4) could change the chances and rates of interaction as proteins physically approach each other (Janin and Chothia 1990; van der Merwe et al. 1995). Taken together, the significant differences in nonsynonymous substitution rate, the significantly higher prevalence of non-conservative amino acid replacements, and the biophysical effects of the substitutions are consistent with the idea that the rapid evolution of sexual adhesion genes has been adaptive.
Given the critical roles of sexual adhesins in yeast sexual reproduction, their rapid evolution could have promoted assortative mating and caused reproductive isolation (i.e., speciation) as a result. The divergence of sexual adhesions is correlated with the level of interbreeding between the two yeast species as well as between the two subgroups of S. paradoxus. The deep divergence of the two yeast species at genomic level (Liti et al. 2009) and the severely reduced interbreeding between them suggest reproductive isolation between the two species (Lee et al. 2010), whereas the consistent and substantial divergence between the two subgroups of S. paradoxus at the genomic level indicates the interbreeding between the two has also been relatively rare.
In general, the rapid evolution of gametic recognition proteins and the associated functional changes would limit reproductive compatibility to only certain combinations of gametes of different sexes. In animals, this could promote promiscuous sexual behaviors when individuals cannot tell with which of the mating partners they will be reproductively compatible or most compatible, and the promiscuous mating behaviors in turn would create the phenomenon of “sperm competition.” However, monogamous sexual relationship might be formed when one has learned exactly which type of mating partners is reproductively most compatible with oneself, which could thus maximize its reproductive success rate.
In conclusion, genes involved in sexual adhesion in two yeast species, S. cerevisiae and S. paradoxus, have evolved much faster compared with genes in other functional groups, including other cell surface proteins. Through a modified MK test and PAML branch model analysis, we found these genes have evolved at similarly high rates both within and between the two yeast species. Through a combination of information theory–based analysis of evolutionary variability and functional characterization of amino acid changes, we discovered that the substitutions in sexual adhesins are biased toward dissimilar amino acids relative to other cell surface proteins. The fixed amino acid changes in these sexual adhesins might have created functional divergence either between the two yeast species or between the two subgroups of S. paradoxus, promoting sexual selection and facilitating speciation.
Supplementary Material
Supplementary tables and figures are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
We would like to thank Saad Mneimneh for computational consultation, Susan Epstein for discussions, and Lie Di for help with the graphics. This work was supported by the National Institutes of Health (SC1 GM083756 to P.N.L., SC3 GM083722 to W-G.Q., and R03037 to Hunter College, City University of New York) and the National Science Foundation (EF-0905606 to National Evolutionary Synthesis Center).
References
- Aagaard JE, Vacquier VD, MacCoss MJ, Swanson WJ. ZP domain proteins in the abalone egg coat include a paralog of VERL under positive selection that binds lysine and 18-kDa sperm proteins. Mol Biol Evol. 2010;27:193–203. doi: 10.1093/molbev/msp221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aagaard JE, Yi X, MacCoss MJ, Swanson WJ. Rapidly evolving zona pellucida domain proteins are a major component of the vitelline envelope of abalone eggs. Proc Natl Acad Sci U S A. 2006;103:17302–17307. doi: 10.1073/pnas.0603125103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldo L, Desjardins CA, Russell JA, Stahlhut JK, Werren JH. Accelerated microevolution in an outer membrane protein (OMP) of the intracellular bacteria Wolbachia. BMC Evol Biol. 2010;10:48. doi: 10.1186/1471-2148-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begun DJ, Whitley P, Todd BL, Waldrip-Dail HM, Clark AG. Molecular population genetics of male accessory gland proteins in drosophila. Genetics. 2000;156:1879–1888. doi: 10.1093/genetics/156.4.1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierne N, Eyre-Walker A. The genomic rate of adaptive amino acid substitution in drosophila. Mol Biol Evol. 2004;21:1350–1360. doi: 10.1093/molbev/msh134. [DOI] [PubMed] [Google Scholar]
- Cappellaro C, Baldermann C, Rachel R, Tanner W. Mating type-specific cell-cell recognition of Saccharomyces cerevisiae: cell wall attachment and active sites of a- and α-agglutinin. EMBO J. 1994;13:4737–4744. doi: 10.1002/j.1460-2075.1994.tb06799.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappellaro C, Hauser K, Mrsa V, Watzele M, Watzele G, Gruber C, Tanner W. Saccharomyces cerevisiae a- and α-agglutinin: characterization of their molecular interaction. EMBO J. 1991;13:4081–4088. doi: 10.1002/j.1460-2075.1991.tb04984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth J, Eyre-Walker A. The rate of adaptive evolution in enteric bacteria. Mol Biol Evol. 2006;23:1348–1356. doi: 10.1093/molbev/msk025. [DOI] [PubMed] [Google Scholar]
- Charlesworth J, Eyre-Walker A. The McDonald-Kreitman test and slightly deleterious mutations. Mol Biol Evol. 2008;25:1007–1015. doi: 10.1093/molbev/msn005. [DOI] [PubMed] [Google Scholar]
- Chen MH, Shen ZM, Bobin S, Kahn PC, Lipke PN. Structure of Saccharomyces cerevisiae alpha-agglutinin: evidence for a yeast cell wall protein with multiple immunoglobulin-like domains with atypical disulfides. J Biol Chem. 1995;270:26168–26177. doi: 10.1074/jbc.270.44.26168. [DOI] [PubMed] [Google Scholar]
- Coronado JE, Mneimneh S, Epstein SL, Qiu W, Lipke PN. Conserved processes and lineage-specific proteins in fungal cell wall evolution. Eukaryot Cell. 2007;6:2269–2277. doi: 10.1128/EC.00044-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Nobel H, Lipke PN, Kurjan J. Identification of a ligand-binding site in an immunoglobulin fold domain of the Saccharomyces cerevisiae adhesion protein alpha-agglutinin. Mol Biol Cell. 1996;7:143–153. doi: 10.1091/mbc.7.1.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dranginis AM, Rauceo JM, Coronado JE, Lipke PN. A biochemical guide to yeast adhesins: glycoproteins for social and antisocial occasions. Microbiol Mol Biol Rev. 2007;71:282–294. doi: 10.1128/MMBR.00037-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egel-Mitani M, Hansen MT. Nucleotide sequence of the gene encoding the Saccharomyces kluyveri alpha mating pheromone. Nucleic Acids Res. 1987;15:6303. doi: 10.1093/nar/15.15.6303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay JC, Wyckoff GJ, Wu CI. Positive and negative selection on the human genome. Genetics. 2001;158:1227–1234. doi: 10.1093/genetics/158.3.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay JC, Wyckoff GJ, Wu CI. Testing the neutral theory of molecular evolution with genomic data from drosophila. Nature. 2002;415:1024–1026. doi: 10.1038/4151024a. [DOI] [PubMed] [Google Scholar]
- Gasper J, Swanson WJ. Molecular population genetics of the gene encoding the human fertilization protein zonadhesin reveals rapid adaptive evolution. Am J Hum Genet. 2006;79:820–830. doi: 10.1086/508473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouy M, Guindon S, Gascuel O. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. 2010;27:221–224. doi: 10.1093/molbev/msp259. [DOI] [PubMed] [Google Scholar]
- Haerty W, Jagadeeshan S, Kulathinal RJ, et al. (11 co-authors) Evolution in the fast lane: rapidly evolving sex-related genes in drosophila. Genetics. 2007;177:1321–1335. doi: 10.1534/genetics.107.078865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci U S A. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G, Dougherty SD, Erdman SE. Conserved WCPL and CX4C domains mediate several mating adhesin interactions in Saccharomyces cerevisiae. Genetics. 2009;182:173–189. doi: 10.1534/genetics.108.100073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagadeeshan S, Singh RS. Rapid evolution of outer egg membrane proteins in the Drosophila melanogaster subgroup: a case of ecologically driven evolution of female reproductive traits. Mol Biol Evol. 2007;24:929–938. doi: 10.1093/molbev/msm009. [DOI] [PubMed] [Google Scholar]
- Janin J, Chothia C. The structure of protein-protein recognition sites. J Biol Chem. 1990;265:16027–16030. [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, et al. (13 co-authors) Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Lawniczak MK, Begun DJ. Molecular population genetics of female-expressed mating-induced serine proteases in drosophila melanogaster. Mol Biol Evol. 2007;24:1944–1951. doi: 10.1093/molbev/msm122. [DOI] [PubMed] [Google Scholar]
- Lee SC, Ni M, Li W, Shertz C, Heitman J. The evolution of sex: a perspective from the fungal kingdom. Microbiol Mol Biol Rev. 2010;74:298–340. doi: 10.1128/MMBR.00005-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipke PN, Kurjan J. Sexual agglutination in budding yeasts: structure, function, and regulation of adhesion glycoproteins. Microbiol Rev. 1992;56:180–194. doi: 10.1128/mr.56.1.180-194.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipke PN, Terrance K, Wu YS. Interaction of alpha-agglutinin with Saccharomyces cerevisiae a cells. J Bacteriol. 1987;169:483–488. doi: 10.1128/jb.169.2.483-488.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipke PN, Wojciechowicz D, Kurjan J. AG alpha 1 is the structural gene for the Saccharomyces cerevisiae alpha-agglutinin, a cell surface glycoprotein involved in cell-cell interactions during mating. Mol Cell Biol. 1989;9:3155–3165. doi: 10.1128/mcb.9.8.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liti G, Carter DM, Moses AM, et al. Population genomics of domestic and wild yeasts. Nature. 2009;458:337–341. doi: 10.1038/nature07743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh L. Substitutions in the hydrophobic core of the alpha-factor receptor of Saccharomyces cerevisiae permit response to Saccharomyces kluyveri alpha-factor and to antagonist. Mol Cell Biol. 1992;12:3959–3966. doi: 10.1128/mcb.12.9.3959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough J, Herskowitz I. Mating pheromones of Saccharomyces kluyveri: pheromone interactions between Saccharomyces kluyveri and Saccharomyces cerevisiae. J Bacteriol. 1979;138:146–154. doi: 10.1128/jb.138.1.146-154.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JH, Kreitman M. Adaptive protein evolution at the adh locus in drosophila. Nature. 1991;351:652–654. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- Mueller JL, Ravi Ram K, McGraw LA, Bloch Qazi MC, Siggia ED, Clark AG, Aquadro CF, Wolfner MF. Cross-species comparison of drosophila male accessory gland protein genes. Genetics. 2005;171:131–143. doi: 10.1534/genetics.105.043844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M. Molecular evolutionary genetics. New York: Columbia University Press; 1987. [Google Scholar]
- Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- Panhuis TM, Clark NL, Swanson WJ. Rapid evolution of reproductive proteins in abalone and drosophila. Philos Trans R Soc Lond B Biol Sci. 2006;361:261–268. doi: 10.1098/rstb.2005.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Lu CF, Marykwas DL, Lipke PN, Kurjan J. The AGA1 product is involved in cell surface attachment of the Saccharomyces cerevisiae cell adhesion glycoprotein a-agglutinin. Mol Cell Biol. 1991;11:4196–4206. doi: 10.1128/mcb.11.8.4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider TD. Measuring molecular information. J Theor Biol. 1999;7:87–92. doi: 10.1006/jtbi.1999.1012. [DOI] [PubMed] [Google Scholar]
- Sen M, Shah A, Marsh L. Two types of alpha-factor receptor determinants for pheromone specificity in the mating-incompatible yeasts S. cerevisiae and S. kluyveri. Curr Genet. 1997;31:235–240. doi: 10.1007/s002940050200. [DOI] [PubMed] [Google Scholar]
- Shen ZM, Wang L, Pike J, Jue CK, Zhao H, de Nobel H, Kurjan J, Lipke PN. Delineation of functional regions within the subunits of the Saccharomyces cerevisiae cell adhesion molecule a-agglutinin. J Biol Chem. 2001;276:15768–15775. doi: 10.1074/jbc.M010421200. [DOI] [PubMed] [Google Scholar]
- Smith NGC, Eyre-Walker A. Adaptive protein evolution in Drosophila. Nature. 2002;415:1022–1024. doi: 10.1038/4151022a. [DOI] [PubMed] [Google Scholar]
- Stoletzki N, Eyre-Walker A. Estimation of the neutrality index. Mol Biol Evol. 2011;28:63–70. doi: 10.1093/molbev/msq249. [DOI] [PubMed] [Google Scholar]
- Swanson WJ, Aagaard JE, Vacquier VD, Monné M, Al Hosseini HS, Jovine L. The molecular basis of sex: linking yeast to human. Mol Biol Evol. 2011;28:1963–1966. doi: 10.1093/molbev/msr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrance K, Lipke PN. Pheromone induction of agglutination in Saccharomyces cerevisiae a cells. J Bacteriol. 1987;169:4811–4815. doi: 10.1128/jb.169.10.4811-4815.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Merwe PA, McNamee PN, Davies EA, Barclay AN, Davis SJ. Topology of the CD2-CD48 cell-adhesion molecule complex: implications for antigen recognition by T cells. Curr Biol. 1995;5:74–84. doi: 10.1016/s0960-9822(95)00019-4. [DOI] [PubMed] [Google Scholar]
- Verstrepen KJ, Fink GR. Genetic and epigenetic mechanisms underlying cell-surface variability in protozoa and fungi. Annu Rev Genet. 2009;43:1–24. doi: 10.1146/annurev-genet-102108-134156. [DOI] [PubMed] [Google Scholar]
- Witten IH, Frank E. San Francisco (CA): Morgan Kaufmann; 2000. Data mining: practical machine learning tools and techniques with Java implementations. [Google Scholar]
- Wyckoff GJ, Wang W, Wu CI. Rapid evolution of male reproductive genes in the descent of man. Nature. 2000;403:304–309. doi: 10.1038/35002070. [DOI] [PubMed] [Google Scholar]
- Xie X, Lipke PN. On the evolution of fungal and yeast cell walls. Yeast. 2010;27:479–488. doi: 10.1002/yea.1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Zhang L, Li WH. Human SNPs reveal no evidence of frequent positive selection. Mol Biol Evol. 2005;22:2504–2507. doi: 10.1093/molbev/msi240. [DOI] [PubMed] [Google Scholar]
- Zhao H, Shen ZM, Kahn PC, Lipke PN. Interaction of alpha-agglutinin and a-agglutinin, Saccharomyces cerevisiae sexual cell adhesion molecules. J Bacteriol. 2001;183:2874–2880. doi: 10.1128/JB.183.9.2874-2880.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.