

Abstract
We have recently discovered an allosteric switch in Ras, bringing an additional level of complexity to this GTPase whose mutants are involved in nearly 30% of cancers. Upon activation of the allosteric switch, there is a shift in helix 3/loop 7 associated with a disorder to order transition in the active site. Here, we use a combination of multiple solvent crystal structures and computational solvent mapping (FTMap) to determine binding site hot spots in the “off” and “on” allosteric states of the GTP-bound form of H-Ras. Thirteen sites are revealed, expanding possible target sites for ligand binding well beyond the active site. Comparison of FTMaps for the H and K isoforms reveals essentially identical hot spots. Furthermore, using NMR measurements of spin relaxation, we determined that K-Ras exhibits global conformational dynamics very similar to those we previously reported for H-Ras. We thus hypothesize that the global conformational rearrangement serves as a mechanism for allosteric coupling between the effector interface and remote hot spots in all Ras isoforms. At least with respect to the binding sites involving the G domain, H-Ras is an excellent model for K-Ras and probably N-Ras as well. Ras has so far been elusive as a target for drug design. The present work identifies various unexplored hot spots throughout the entire surface of Ras, extending the focus from the disordered active site to well-ordered locations that should be easier to target.
Keywords: Ras isoforms, drug target, binding site hot spots, Ras dynamics, allosteric switch
Introduction
Ras proteins are monomeric GTPases that function as “molecular switches” in signal transduction pathways in the cell, where the inactive state is associated with Ras-GDP, and the active state, with Ras-GTP.1 Ras interacts with a number of protein binding partners, such as guanine nucleotide exchange factors2 and GTPase-activating proteins (GAPs),3 that regulate its nucleotide-bound state and effector proteins that interact with Ras-GTP to propagate signals through cascades of protein/protein interactions.4 Due to its involvement in the control of cell proliferation, apoptosis and other critical functions,5,6 defects in Ras that result in impairment of GTP hydrolysis are associated with many human cancers, and thus, Ras is a major therapeutic target.7 However, even though the structure and function of Ras have been studied intensively for nearly 30 years, it has remained elusive as a drug target for the treatment of cancer. The focus on the active site may be a problem due to its inherent flexibility, yet protein/protein interactions in Ras extend to surfaces that have been unexplored. In general, interfaces between protein binding partners have energetically important regions called hot spots that can potentially also interact with small molecules.8,9 Here, we use both experimental and computational approaches to locate putative hot spot regions that can provide insight into potential molecular interactions beyond the active site.
The canonical view of the “molecular switch” is based on conformational changes that occur primarily in the switch I (residues 30–40) and switch II (residues 60–76) regions upon hydrolysis of GTP to GDP.10 This results in loss of affinity for downstream effector proteins and termination of the upstream signal.11 While this overall picture is certainly correct, it overlooks structural nuances associated with an allosteric switch mechanism that modulates the equilibrium between two distinct conformational states in GTP-bound Ras.12 One state (denoted here as the “off” state) has an “empty” allosteric site and is characterized by a disordered active site, particularly switch II, which contains the catalytic residue Q61. We proposed previously that this is the dominant form present under solution conditions used for in vitro hydrolysis rate measurements, explaining the slow rates measured for Ras.13 The other conformational state (the “on” state), which, in our crystals, has a bound calcium acetate in the allosteric site, shows a shift in helix 3/loop 7 toward helix 4 and an ordered active site, with Q61 placed near the catalytic center.12 We propose that this is the catalytically active state and that intrinsic hydrolysis is promoted in some instances by an allosteric modulator in the cell that is mimicked by calcium acetate in the crystal.12 The shift of helix 3 toward helix 4 with an ordered switch II is also found in the complex with RasGAP, which promotes GTP hydrolysis, although the details of the active site differ from those of intrinsic hydrolysis due to the insertion of the arginine finger from RasGAP.3 An equilibrium between the two conformational states in Ras-GppNHp may provide an explanation to the global conformation dynamics that we previously observed for H-Ras-GppNHp.14
The complexity of the Ras system is complicated even further by the fact that Ras is tethered to the membrane through posttranslational modifications at its C-terminal hypervariable region15,16 and that the nature of the bound nucleotide profoundly affects the Ras/membrane interface.17,18 The three isoforms of the human Ras proteins, H-Ras, K-Ras and N-Ras, differ primarily in the sequence of the hypervariable region and in the types of posttranslational modifications that characterize each one.16 The catalytic domains, or G domains, of the three Ras proteins are highly conserved, with no variation in the N-terminal lobe 1 (residues 1–86) and 90% identity in the C-terminal lobe 2 (residues 87–171).19 Lobe 1 includes the catalytic machinery containing the active site with switch I, switch II and the P-loop (residues 10–17), as well as most of the nucleotide binding pocket. We call this the effector lobe, as it contains the protein/protein interaction sites with effectors. Lobe 2 contains the membrane-interacting portions of Ras, including the allosteric site with residues R97, D107 and Y137 and the allosteric switch components involving helix 3/loop 7, as well as helix 4 that has been shown to form salt bridges with membrane phospholipids in Ras-GTP.20 We call this the allosteric lobe. The allosteric site is connected to the active site in H-Ras through helix 3 at one edge of the interlobal region and switch II at the other.
The conformational complexity of Ras proteins and the many modes by which it can be modulated may be at the heart of the difficulty to design inhibitors that effectively interfere with its function. To date, there has been little attention given to the fact that distinct conformational states of Ras-GTP may be directly connected to catalytic competency and that remote binding sites on the protein surface could have a dramatic effect in determining the predominant form. Thus, the active site has been the primary target region for inhibitors, and the structural viewpoint has been biased by the canonical crystal form in which Ras was first crystallized.21–23
In the present article, we use a combination of multiple solvent crystal structures (MSCS)24,25 and computational solvent mapping (FTMap)8,26 to identify hot spots of protein/protein interactions for H-Ras-GppNHp based on groups of crystal structures associated with distinct conformational states. Due to the sensitivity of conformational states to the solvent environment, only the “off” state of the allosteric switch is accessible experimentally by MSCS, and we use FTMap to study two closely related forms of the “on” state. The result is a series of hot spots obtained by partially overlapping and complementary results from MSCS and FTMap, spread through a large area on the surface of Ras-GTP. Each approach emphasizes different properties of the binding regions, and apart from the two strongest hot spots found by both methods, they identify distinct binding pockets on Ras. In essence, the strengths in each of the methods compensate for limitations in the other, resulting in a more complete binding site analysis than if either one were used alone. As we discovered previously,14 H-Ras-GppNHp experiences global conformational rearrangement on a millisecond timescale. We propose that this serves as a mechanism for allosteric coupling of the remote hot spots uncovered by solvent mapping with the effector interface near the active site. Furthermore, we establish that similar conformational exchange exists in the K isoform of Ras, supporting the idea that allosteric coupling in the G domain is present in all three isoforms of Ras.
Results
The hot spots identified by either MSCS or FTMap are listed in Table 1. The “off” state, accessible experimentally by MSCS and computationally by FTMap, shows two sites in common, six that appear in MSCS but not in FTMap and four that are present in the FTMap but not in the MSCS results. In addition, there is a single site that appears uniquely in the FTMap of one of two “on” states. Thus, taken together, there is a total of 13 sites analyzed in this work.
Table 1.
Summary of hot spots on the surface of the GTP-bound form of Ras
| Cluster | Solvent molecules | Protein contacts | Allosteric state | Location | Binding partner |
|---|---|---|---|---|---|
| MSCSa and FTMapb | |||||
| 1 | MSCS HEX, DMF, ETF, HEZ FTMap Off state: 52 probes On state 1: 75 probes |
R68, Q95, Y96, Q99, D92 E62, R68, D92, Q95, Y96, Q99, R102 |
MSCS: Off state FTMap: Off state On state 1 |
Interlobal region, between switch II and helix 3 | Importin-β binds Ran at this site |
| 2 | MSCS HEX, DMF, GOL, ETF FTMap Off state: 61 probes On state 1: 58 probes On state 2: 31 probes |
H94, L133, S136, Y137+symm (see Table 3) F90, E91, I93, H94, L133, Y137 |
MSCS: Off state FTMap: Off state On state 1 On state 2 |
Allosteric lobe, between helix 3 and helix 4 | Near membrane |
| MSCS | |||||
| 3 | GOL, DMF, RSF, GOL (second molecule) | S17, I21, Q25, H27, V29, D33, T35, D38, Y40+symm (see Table 3) | Off state | Effector lobe, opposite switch I relative to GppNHp | Raf-RBD |
| 4 | RSF, RSG | F28, D30, K147 | Off state | Interlobal region | |
| 5 | RSF, GOL | A11, G12, N86, K88, S89, D92 | Off state | Interlobal region, between P-loop and N-terminus of helix 3 | RasGAP |
| 6 | RSF, YEG | D30, E31, Tyr32, GppNHp | Off state | Effector lobe near N-terminus of switch I | RasGAP |
| 7 | RSF, GOL | L23, N26, K42, Val44, V45, R149, E153, Y157 | Off state | Interlobal region C-terminus of helix 1 | Raf-CRD |
| 8 | RSF, RSG | G13, Y32, N86, K117, GppNHp | Off state | Interlobal region between P-loop and switch I | RasGAP |
| FTMap | |||||
| R128 | Off state: 64 probes On state 1: 58 probes On state 2: 40 probes |
R123, S127, Q131, Y141, I142, Q143 | Off state On state 1 On state 2 |
Allosteric lobe between helix 4 and helix 5 | Membrane |
| R135 | Off state: 35 probes On state 2: 42 probes |
Q131, R135, G138, I139, P140, Y141 | Off state On state 2 |
Allosteric lobe between helix 4 and helix 5 | Membrane |
| Y71 | Off state: 40 probes On state 2: 81 probes |
K5, V7, E37, S39, D54, L56, Y71, T74 | Off state On state 2 |
Effector lobe near switch II | |
| Allosteric | Off state: 83 probes On state 1: 55 probes On state 2: 45 probes |
R97, K101, V109, E108, P110, M111, I139, E162, I163, Q165, H166 | Off state On state 1 On state 2 |
Allosteric lobe near helix 3 and loop 7 | Near membrane |
| Loop 7 | On state 2: 65 probes | E76, K104, V109, P110, I163, H166 | On state 2 | Allosteric lobe near loop 7 opposite side from allosteric site | Near membrane |
The abbreviations for the organic solvent names are the respective residue names used in the coordinate files submitted to the PDB.
The number of probes reported for FTMap is the sum of probes at that site in the four structures in each set for the off state, “on” state 1 and “on” state 2.
Multiple solvent crystal structures
MSCS is a powerful experimental method in determining hot spots of protein/protein or protein/ligand interactions on the surfaces of proteins.24,25 It yields a set of many structures of the same protein, each in a different solvent environment. The clustering of organic solvent molecules at a particular site on the superimposed structures has been empirically observed to be indicative of a hot spot for molecular interaction.25 For each structure, protein crystals are cross-linked in glutaraldehyde and soaked in a high concentration of organic solvent for data collection. The crystals used for the MSCS of Ras-GppNHp had symmetry of the space group R32, with helix 3/loop 7 representing the “off” state of the allosteric switch and a disordered switch II [as in Protein Data Bank (PDB) ID: 2RGE].27 Structures were obtained for crystals soaked in 50% 2,2,2-trifluoroethanol (ETF), 50% isopropanol (IPA), 70% glycerol (GOL), 90% R, S,R-bisfuranol (RSF), 20% S,R,S-bisfuranol (RSG), 60% 1,6-hexanediol (HEZ), neat cyclopentanol (YEG), neat hexane (HEX) and 55% dimethylformamide (DMF). Note that the RSF and RSG molecules are enantiomers and that the Ras crystals are highly tolerant of one but not the other, resulting in the vastly different concentrations used for soaks in the two solvents (90% RSF and 20% RSG). The structure of the cross-linked crystal in aqueous solution (XLINK) was solved as a control. Data collection and refinement statistics for the 10 crystal structures in the MSCS set are presented in Table 2, and representative electron density for the organic solvents is shown in Fig. 1. The refined structures were superimposed based on the nucleotide for MSCS analysis. The distinct organic solvent environments do not have a significant effect on the overall conformation of the protein, although some of the solvents tend to promote some degree of order in switch II as previously reported.28 The overall main-chain root-mean-square deviation (RMSD) within the MSCS set of structures is 0.27 Å. For comparison, the overall main-chain RMSD between representative structures in the “on” state (PDB ID: 3K8Y) and in the “off” state (PDB ID: 2RGD) of the allosteric switch is 0.48 Å.
Table 2.
Diffraction data collection and structure refinement statistics
| XLINK | ETF (50%) | IPA (50%) | GOL (70%) | RSF (90%) | RSG (20%) | HEZ (60%) | YEG (neat) | HEX (neat) | DMF (55%) | |
|---|---|---|---|---|---|---|---|---|---|---|
| Data | ||||||||||
| Space group | R32 | R32 | R32 | R32 | R32 | R32 | R32 | R32 | R32 | R32 |
| Unit cell a, b, c (Å) | 88.73, 88.73, 135.05 | 88.32, 88.32, 133.91 | 89.40, 89.40, 135.25 | 89.28, 89.28, 134.70 | 89.14, 89.14, 133.09 | 88.50, 88.50, 134.09 | 88.79, 88.79, 135.09 | 87.63, 87.63, 134.24 | 88.33, 88.33, 135.13 | 88.53, 88.53, 134.95 |
| No. of unique reflections | 25,152 (2450) | 17,122 (769) | 23,053 (2300) | 27,479 (2689) | 22,501 (2213) | 26,262 (2586) | 22,719 (2235) | 38,553 (3680) | 31,339 (3049) | 23,412 (2320) |
| Resolution (Å) | 30–1.6 (1.66–1.60) | 50–1.8 (1.89–1.82) | 50–1.7 (1.76–1.70) | 30–1.6 (1.66–1.60) | 50–1.7 (1.76–1.70) | 50–1.6 (1.66–1.60) | 50–1.7 (1.76–1.70) | 50–1.4 (1.45–1.40) | 20–1.5 (1.57–1.52) | 50–1.7 (1.74–1.68) |
| Rsym or Rmerge | 0.12 (0.49) | 0.06 (0.54) | 0.04 (0.24) | 0.08 (0.43) | 0.04 (0.52) | 0.04 (0.24) | 0.03 (0. 18) | 0.06 (0.48) | 0.07 (0.53) | 0.03 (0.12) |
| I/σI | 13 (1.8) | 30 (1.4) | 53 (10.4) | 39 (3.3) | 44 (2.9) | 33 (3.7) | 62 (10.7) | 31 (2.7) | 20 (1.7) | 64 (16.9) |
| Completeness (%) | 91.7 (78.7) | 89.5 (37.6) | 95.5 (91.6) | 97.5 (91.0) | 95.8 (87.6) | 97.4 (93.2) | 99.4 (100.0) | 94.9 (83.3) | 94.8 (80.4) | 99.1 (98.2) |
| Redundancy | 5.5 (4.2) | 10.9 (1.9) | 10.5 (10.5) | 9.6 (8.0) | 8.7 (8.0) | 3.8 (3.6) | 10.5 (10.5) | 6.9 (4.8) | 4.3 (2.7) | 11 (10) |
| Refinement | ||||||||||
| Resolution (Å) | 26.7–1.6 | 22.1–1.8 | 20.5–1.7 | 26.8–1.6 | 33.4–1.7 | 33.3–1.6 | 22.0–1.7 | 33.0–1.4 | 19.8–1.5 | 22.2–1.7 |
| No. of reflections used Rwork/Rfree | 22,527/2467 | 14,841/1658 | 20,044/2198 | 24,249/2690 | 21,819/2168 | 23,637/2625 | 20,148/2220 | 33,652/3743 | 27,262/3026 | 21,023/2297 |
| Rwork/Rfree | 18.5/20.1 | 17.6/20.4 | 18.1/20.8 | 18.3/20.5 | 19.7/22.0 | 17.7/19.9 | 17.2/19.8 | 16.7/18.5 | 18.8/20.7 | 16.2/18.2 |
| No. of atoms | ||||||||||
| Protein | 1331 | 1335 | 1312 | 1324 | 1247 | 1328 | 1324 | 1340 | 1335 | 1312 |
| GppNHp | 32 | 32 | 32 | 32 | 32 | 32 | 32 | 32 | 32 | 32 |
| Organic no. of atoms/no. of molecules | 0/0 | 12/2 | 0/0 | 48/8 | 117/13 | 18/2 | 8/1 | 12/2 | 12/2 | 20/4 |
| Ca2+/Mg2+ | 2/2 | 2/2 | 2/1 | 2/2 | 3/1 | 2/1 | 2/2 | 2/2 | 3/1 | 2/2 |
| Water | 124 | 116 | 138 | 88 | 48 | 124 | 108 | 147 | 71 | 127 |
| B-factors (average) | ||||||||||
| Protein | 29.5 | 27.9 | 21.9 | 27.5 | 28.1 | 22.7 | 22.0 | 16.8 | 20.2 | 17.1 |
| GppNHp | 22.7 | 21.5 | 15.1 | 20.0 | 20.6 | 15.8 | 15.3 | 11.3 | 14.4 | 11.1 |
| Organic | N/A | 47.2 | N/A | 41.9 | 47.1 | 44.5 | 42.1 | 36.4 | 31.6 | 28.5 |
| Ca2+/Mg2+ | 23.5/31.3 | 30.0/32.0 | 26.7/15.2 | 29.4/24.8 | 31.3/22.8 | 21.6/16.0 | 24.1/18.8 | 15.7/14.2 | 20.6/14.5 | 22.5/9.6 |
| Water | 35.6 | 35.1 | 29.6 | 36.8 | 35.3 | 30.8 | 30.6 | 26.7 | 25.5 | 26.1 |
| RMSD bonds | ||||||||||
| Length (Å) | 0.007 | 0.007 | 0.008 | 0.007 | 0.008 | 0.007 | 0.008 | 0.007 | 0.007 | 0.008 |
| Angles (°) | 1.14 | 1.12 | 1.08 | 1.13 | 1.20 | 1.08 | 1.10 | 1.17 | 1.18 | 1.17 |
| Ramachandran profile (%)a, favored/outlier | 95.2/0.6 | 97.0/1.2 | 98.8/0.0 | 97.6/0.6 | 97.4/0.7 | 97.6/0.0 | 96.9/0.6 | 97.0/0.0 | 97.0/0.0 | 96.3/0.0 |
| PDB IDs | 3RRY | 3RS2 | 3RS7 | 3RRZ | 3RSL | 3RSO | 3RS4 | 3RS0 | 3RS3 | 3RS5 |
The abbreviations for the organic solvent names are the respective residue names used in the coordinate files submitted to the PDB. XLINK, cross-linked in aqueous solution. A single crystal was used for each structure. The values in parentheses are for the highest-resolution shell.
Ramachandran profile calculated with MolProbity.
Fig. 1.

Representative electron density maps contoured at 1 σ showing organic solvent molecules. The cluster numbers at the top of each panel correspond to those in Table 1. (a) The four organic molecules found in cluster 1: upper left, DMF; upper right, HEX; lower left, trifluoroethanol (ETF); lower right, HEZ. (b) The four organic molecules found in cluster 2: upper left, HEX; upper right, DMF; lower left, ETF; lower right, GOL. (c) The four organic molecules found in cluster 3: upper left, DMF; upper right, GOL; lower left, RSF; lower right, a second GOL molecule (GOL). Conserved water molecules 304 and 310 are shown. (d) Organic solvent molecules in the interlobal clusters: upper left, RSF in cluster 4; upper right, GOL in cluster 5; GOL in cluster 7; RSF in cluster 8. In all cases, the protein and organic solvents are shown as sticks. Water molecules are represented as red spheres, and hydrogen bonds are represented as red broken lines.
Figure 2 shows the solvent molecule clusters that result after superposition of the structures. The cluster numbering is related to the number of probe molecules in the cluster (Table 1). Remarkably, most of the clusters are associated with functional sites occupied by one or more of the many protein/protein interaction partners of Ras, although the collective patterns revealed cannot be discerned in the structure of any single complex. The hot spots include two sites on the effector lobe (clusters 3 and 6), one site between helices 3 and 4 in the allosteric lobe (cluster 2) and five sites in the interlobal region where the organic solvents make extensive interactions with residues in both lobes (clusters 1, 4, 5, 7 and 8). The clusters between the effector lobe and the allosteric lobe define an extended solvent-accessible cleft that encircles most of the protein (Fig. 2).
Fig. 2.

MSCS results for H-Ras-GppNHp in the “off” state of the allosteric switch. The effector lobe is shown in green, and the allosteric lobe is shown in gray. MSCS clusters 1 through 8 are shown with red spheres superimposed on the organic solvent molecules, which are in stick representation within each cluster. (b) is rotated by 90° relative to (a) in order to show sites 3 and 7.
With the use of a 5-Å-cutoff distance for determination of crystal contacts, clusters 2 and 3 are in extensive crystal contacts (Table 3). Clusters 6 and 8 each make contact with one symmetry-related protein residue, and clusters 1, 4, 5 and 7 are not involved in crystal contacts. There are 12 sites occupied by one organic solvent molecule each, and as expected, they are mostly found in crystal contacts.25 In general, sites occupied by a single organic solvent molecule in the MSCS set are not considered to be hot spots and are therefore not included in Fig. 2.
Table 3.
Crystal contacts associated with MSCS clusters
| Cluster | Solvent molecule | Crystal contacts (5.0 Å cutoff) |
|---|---|---|
| 2 | DMF, GOL, HEX, ETF | Asn85, Asp119, Leu120, Ala121, Ala122 |
| 3 | DMF, GOL, RSF, GOL(2) | Ile21, Gln24, Gln25, His27, Val29, Glu31, Tyr40 |
| 6 | RSF, YEG | Gln95 |
| 8 | RSF, RSG | Gln95 |
Clusters 1 and 2 are the strongest hot spots
Cluster 1 found in the interlobal region and cluster 2 in the allosteric lobe are two major sites on either side of helix 3, the central structural element in the conformational changes that connect the “on” state and the “off” state of the allosteric switch in Ras-GppNHp. Cluster 1 is near the active site, between helix 3 and switch II. Binding of a ligand in this cluster is accompanied by a disorder to order transition in switch II. The result is a binding pocket to which R68 from the effector lobe and Y96 from the allosteric lobe are the major contributors. The structures resulting from soaks in neat HEX and YEG and those soaked in 50% ETF and 70% GOL have an ordered switch II, while the cross-linked structure and those soaked in the remaining solvents have switch II disordered to various extents. The ordered structure of switch II in the “off” state differs significantly from that found in the “on” state of the allosteric switch (PDB ID: 3K8Y) and superimposes well on the non-catalytic conformation found for the RasQ61L-GppNHp mutant (PDB ID: 2RGD).27 Cluster 1 is at the site where importin-β binds the GTP-bound form of the homologous Ran GTPase, stabilizing an identical non-catalytic conformation of switch II (PDB ID: 1IBR).29 Cluster 2 in the MSCS of the “off” state is found between helix 3 and helix 4 about 15 Å from the allosteric site where calcium acetate binds in the “on” state (PDB ID: 3K8Y). Molecular dynamics simulations and a series of fluorescence resonance energy transfer experiments indicate that this region is near the membrane in the Ras-GTP-bound state.17,18
Cluster 4
Cluster 4 is found in the interlobal region at a binding pocket that includes K147, a recently discovered site of ubiquitination on Ras-GTP that results in enhanced affinity for effectors and promotion of downstream signaling.30 Lysine 147 is in the allosteric lobe and is part of the global correlated dynamics that we observe for Ras-GppNHp.14 Asp30 in the effector lobe, located just outside the active site, is also part of the cluster 4 binding pocket and may contribute to an allosteric network that communicates signals to the active site of Ras.
Clusters 3, 5, 6, 7 and 8 overlap with sites occupied by Raf-RBD-CRD and RasGAP
The two clusters on the effector lobe, clusters 3 and 6 in our numbering scheme, overlap with effector Ras binding domains (RBD) (cluster 3) and RasGAP (clusters 3 and 6). They are located on either side of switch I (Fig. 2). Cluster 3 is the location of a hot spot determined experimentally for the interaction with Raf-RBD.31 As is the case for several of the interactions between Ras and Raf-RBD, many of the organic solvent molecules in cluster 3 interact with the protein indirectly through water molecules, rather than forming direct hydrogen bonds with protein atoms (Fig. 1c). Two water molecules, WAT304 and WAT310, are completely conserved in the experimental MSCS data sets and mediate hydrogen-bonding interactions with ligands. These water molecules are also present in the Raps/Raf-RBD complex (PDB ID: 1GUA). The site of interaction for the second Ras binding domain of Raf, the cysteine-rich domain (Raf-CRD), overlaps with cluster 7 at the interlobal region. Binding experiments have shown that Ras mutants N26G and V45E, two residues in the cluster 7 site, fail to bind Raf-CRD.32
Clusters 3 and 6 comprise two of four clusters that overlap with the RasGAP binding surface. The other two, clusters 5 and 8, are in the interlobal region. The locations of these clusters are consistent with sub-sites within a protein/protein interaction site that encompasses the switch regions and extend communication from there to the allosteric lobe of Ras-GppNHp. Superposition of the Ras/RasGAP complex (PDB ID: 1WQ1) onto the MSCS structures shows that RasGAP residue K949 interacts within the site delineated by cluster 3 in very much the same way as does K84 of Raf-RBD.31 RasGAP residues A790, T785 and T786 all superimpose within 1.0 Å of an organic solvent molecule in clusters 5, 6 and 8, respectively. RasGAP A790 hydrogen bonds to Ras residues G12 and K88 within the cluster 5 binding pocket, bridging the P-loop in the effector lobe to the N-terminal portion of helix 3 in the allosteric lobe. T785 makes a hydrogen bond to one of the ribose oxygen atoms of the nucleotide that forms the cluster 6 pocket, and T786 bridges K117 in the allosteric lobe to the other ribose oxygen atom in the effector lobe within the cluster 8 binding site. These interactions appear to serve as anchor points for placing the arginine finger, RasGAP residue R789, in the catalytic center to accelerate the hydrolysis of GTP to GDP, while positioned to turn “on” the allosteric switch, with helix 3/loop 7 shifting toward helix 4 to make room for proper ordering of switch II for catalysis.
Computational solvent mapping (FTMap)
In order to understand the consequences of the allosteric switch on the binding surface of Ras-GppNHp, we use computational solvent mapping, FTMap, to compare hot spots between structures in the “on” and “off” states.8,33 The method uses small organic molecules containing various functional groups as molecular probes. The probes are placed on a dense grid around the protein surface, and a fast Fourier transform correlation algorithm evaluates the energies for billions of conformations. The low-energy positions are clustered, and the clusters are ranked on the basis of average free energy.8 The positions at which several low-energy clusters overlap are defined as consensus sites. It was shown that the large consensus sites are at hot spots important for molecular interactions. The parameters of the FTMap algorithm were selected to find only the sites that are likely to be druggable, that is, can bind drug-sized ligands with micromolar or higher affinity.34 This implies that FTMap generally does not detect relatively weak sites identified in the MSCS experiments as binding fewer solvent molecules or those binding solvents indirectly through water molecules. In spite of these differences with respect to MSCS, FTMap is ideal for comparing the “on” and “off” states of the allosteric switch in Ras-GppNHp, since the differences in the number of probe clusters that bind to a particular site highlight even very small conformational changes if those affect the size or surface properties of the pocket.26
Three groups of Ras structures bound to GTP analogues are used in our FTMap comparisons (Table 4). The first group represents the “off” state of the allosteric switch. Note the inclusion of K-Ras-GppNHp (PDB ID: 3GFT) in this group. For the “on” state, we have two groups of structures taken from the PDB in which switch I conformations are associated with states 1 and 2 for Ras-GppNHp observed by 31P NMR experiments.35,36 State 1 is associated with a slower intrinsic hydrolysis rate and represents many different switch I conformations where Y32 is away from the nucleotide. This includes the conformation found for Y32 in the Ras/RasGAP complex. State 2 represents the catalytic switch I conformation for intrinsic hydrolysis with Y32 closed over the nucleotide.36 This switch I conformation is found in the Raps/Raf-RBD complex.31 It is important to point out that these conformations of switch I do not necessarily imply the “on” or “off” state of the allosteric switch, which is determined by the position of helix 3/loop 7. For example, our structure of Ras-GppNHp in the presence of calcium chloride (PDB ID: 2RGE) represents state 2 in terms of its switch I conformation but is in the “off” state of the allosteric switch with a disordered switch II. In the presence of calcium acetate (PDB ID: 3K8Y), the conformation of switch I is still in state 2, but now, the allosteric switch is “on” with switch II ordered for catalysis. Conversely, Ras in the RasGAP complex is in state 1, which is unfavorable for intrinsic hydrolysis, yet the allosteric switch is “on”, and switch II is ordered, poised for the GAP-enhanced hydrolysis reaction. We call the two “on” state groups in the present study “on” state 1 and “on” state 2. The “on” state 1 has switch I residue Y32 in an open conformation as found in the complex with RasGAP. The “on” state 2 has switch I residue Y32 closed over the nucleotide as seen in the complex with Raf-RBD.
Table 4.
Coordinates used for FTMap analysis
| Protein | Nucleotide | PDB | Space group | Resolution (Å) |
|---|---|---|---|---|
| Off state | ||||
| H-Ras | GppNHp | ETF | R32 | 1.8 |
| H-Ras | GppNHp | HEX | R32 | 1.5 |
| H-Ras | GppNHp | YEG | R32 | 1.4 |
| K-Ras | GppNHp | 3GFT | P212121 | 2.27 |
| On state 1 | ||||
| H-Ras | GppNHp | 5P21 | P3221 | 1.35 |
| H-Ras | GppNHp | 1CTQ | P3221 | 1.26 |
| H-Ras (G12P) | GppCH2p | 1JAH | P3221 | 1.8 |
| H-Ras/RasGAP | GDP | 1WQ1 | P21 | 2.5 |
| On state 2 | ||||
| H-Ras | GppNHp | 3K8Y | R32 | 1.3 |
| H-Ras | GppCH2p | 6Q21 | P1121 | 1.95 |
| H-Ras (G12D) | GppNHp | 1AGP | C2 | 2.3 |
| H-Ras–PLCE | GTP | 2C5L | P212121 | 1.9 |
FTMap results for the “off” state of the allosteric switch
All four structures in the “off” state group resulted in FTMaps with similar cluster locations, including K-Ras, which behaves similar to H-Ras in terms of hot spots. The major difference is a small shift in cluster 1 due to the presence of histidine in K-Ras in place of glutamine at residue 95 in H-Ras. The similarity between H-Ras and K-Ras in terms of surface hot spots associated with the “off” state supports the idea that the Ras isoforms make similar interactions involving the G domain. Figure 3 shows a representative FTMap of the “off” state superimposed on the MSCS experimental results. There are two significant overlaps between experimental and computational clusters. The first is between helix 3 and switch II, corresponding to MSCS cluster 1 near the active site. The geometric centers of the MSCS and FTMap clusters at this site are 2.1 Å apart, with a significant overlap in the protein residues with which the probe molecules in the two sets interact. The second overlapping site between MSCS and FTMap is between helix 3 and helix 4, MSCS cluster 2. The respective centers of geometries are 4.8 Å apart, but again, the probe molecules in each set interact with some protein residues in common. This cluster in the MSCS set is wedged between crystal contacts, which might partially explain the larger shift seen in the geometric centers of the MSCS versus FTMap clusters.
Fig. 3.

Comparison of the MSCS and FTMap results for Ras-GppNHp in the “off” state of the allosteric switch. The effector lobe is shown in green, and the allosteric lobe is shown in gray. The MSCS clusters are shown as red spheres as in Fig. 2, and the FTMap clusters are shown in purple. (a) and (b) show two orientations of the molecule so that all clusters are visible.
There are four clusters in the FTMap of Ras-GppNHp in the “off” state that do not appear in the experimental solvent mapping (Table 1 and Fig. 3). The first two are found between helix 4 and helix 5 at positions where there are extensive crystal contacts between adjacent Ras molecules in the crystal form used for the MSCS experiments, making the sites inaccessible for solvent mapping. These sites are likely of biological significance as protein/membrane interaction sites. One includes R128 and the other R135 on helix 4 of the allosteric lobe (referred to as R128 and R135 sites, respectively), both residues shown to interact directly with the membrane in molecular dynamics simulations supported by cell biology experiments.17,18 A third site is located in a pocket where L56 from β-strand 3 and Y71 from switch II provide the major interactions with molecules in the cluster (referred to as the Y71 site). There are no crystal contacts surrounding this site, and thus, it was available in the crystal for solvent binding. However, even though switch II was built into the models used for computational solvent mapping, the electron densities used to build the models show discontinuities associated with disorder, as is typical for this region. Therefore, it is not surprising that organic solvent molecules did not appear at this site in the experimental data set.
The fourth site present in FTMap but not in MSCS is the allosteric site, the region where calcium acetate stabilizes the “on” state of the allosteric switch in our previously published structure with PDB ID: 3K8Y. This is the strongest hot spot in the FTMap calculations of Ras-GppNHp structures with the allosteric switch in the “off” state. Probe molecules in this site hydrogen bond to R97 and are in van der Waals’ contact with P110. This site is well hydrated in the crystal structures and devoid of crystal contacts. It is thus available for binding organic solvent molecules in the MSCS experiments. Experimentally, the situation in this pocket is complex. The side chain of His166 partially overlaps with the FTMap cluster in three out of the ten MSCS structures. However, the three H-Ras structures that were used in the computational work all have His166 out of the way. In the seven MSCS structures where the allosteric site is not occluded, there are three water molecules, two of which completely overlap with the FTMap probes in this site. These two water molecules are displaced by His166 in the structures where the site is occluded. The third water molecule, Wat320, is about 2 Å from the probes and is absolutely conserved not only in the MSCS structures but also in all H-Ras structures solved to better than 2 Å resolution. It is also present in the one structure available for K-Ras, although it was not included in the associated model with PDB ID: 3GFT (Fig. 4). We propose that Wat320 is a structural water molecule that is unlikely to be displaced by ligand, unless its interactions with the protein are mimicked.37 Its absence in the FTMap calculations may have contributed to the discrepancy between experimental and computational solvent mapping results.
Fig. 4.

Conserved water molecule Wat320 near the allosteric site. Protein atoms from Ras-GppNHp soaked in 50% ETF is shown in light gray with Wat320 from that model included as a dark sphere. The published model for K-Ras-GppNHp (PDB ID: 3GFT) is superimposed in dark gray. Wat320 is not present in this model, but electron density from a Fo − Fc map contoured at 3 σ calculated using the 3GFT model and associated structure factors downloaded from the PDB show clearly that Wat320 is indeed present in K-Ras-GppNHp. Wat320 makes good hydrogen bonds (broken lines) with the backbone amide and carbonyl groups of M111 and with the side chain of E162 in both isoforms. His166 is turned away from the hot spots identified by FTMap in both models.
Comparing FTMap results for the “off” and “on” states of the allosteric switch
Comparison of the computational solvent mapping results of Ras-GppNHp in the “off” state with “on” states 1 and 2 is shown in Fig. 5. The numbers of molecular probes found at each site derived from the four models in the three groups are in Table 1. Of the six clusters already discussed for the “off” state, three are present in all groups of FTMaps. The allosteric site, which is the strongest cluster in the “off” state, has fewer probes in both “on” states 1 and 2. The position of loop 7 shifted toward helix 4 in the “on” states contributes to making this a smaller pocket. The R128 site between helix 4 and helix 5 and the site between helix 3 and helix 4 corresponding to MSCS cluster 2 are the second and third sites in common to the three groups of FTMaps. Both have similar numbers of probe molecules in the “off” state and in the “on” state 1, but fewer in “on” state 2.
Fig. 5.

Comparison of the FTMap results for Ras in the three conformational substates associated with the GTP-bound form. The “off” state clusters are shown in purple as in Fig. 3. The “on” state 1 clusters are shown in blue, and the “on” state 2 clusters are shown in cyan. (a) and (b) show two orientations of the molecule so that all clusters are visible.
The remaining three sites found in the “off” state are present in one but not both of the “on” states. The R135 site between helix 4 and helix 5 on the allosteric lobe is absent from the “on” state 1 set. The cluster 1 and Y71 sites are inversely populated in the “on” states 1 and 2. The cluster 1 site is the site with the most number of probes in the “on” state 1. The switch regions in this set of structures are further from the nucleotide giving way to a prominent cluster 1 pocket in a location similar to that observed in the “off” state. The increase in the cluster 1 site is at the expense of the Y71 site on the opposite side of switch II, which is absent in the “on” state 1 set. Conversely, as switch II is closer to the nucleotide and helix 3 in the “on” state 2, the Y71 site is increased significantly relative to its presence in the “off” state and is the most highly populated site in the “on” state 2 set. This is at the expense of the cluster 1 site, which is absent in the “on” state 2 due to the positioning of R68 into the cluster 1 binding pocket.
There is one additional site in the “on” state 2 that is not present in either the “off” state or the “on” state 1. It is found on the opposite side of loop 7 relative to the allosteric site, with I163 forming the base of the pocket (referred to as the loop 7 site) (Fig. 5). As loop 7 is found closer to helix 4 in the “on” state 2, a pocket where steric hindrance would occur in the other two sets is formed. Not surprisingly, the site is not present in our MSCS experiments where we solvent mapped the “off” state.
NMR analysis of conformational dynamics in Ras
The analysis of protein surface described above identified multiple hot spots throughout H-Ras-GppNHp involving the effector and allosteric lobes, as well as the interlobal region that connects them. Using spin-relaxation NMR experiments, we demonstrated previously that a major part of the G domain of H-Ras undergoes correlated motion on the millisecond timescale.14 We hypothesize that the global dynamics is a manifestation of allosteric coupling between the two lobes through the conformational switch mechanism identified by X-ray diffraction.12
Our FTMap calculations identified hot spots on the binding surface of K-Ras-GppNHp, which is a drug target of immense importance.38 These hot spots are the same as those identified for H-Ras-GppNHp in a similar conformational substate, suggesting that the analysis presented here for H-Ras applies to K-Ras as well. To determine whether the global conformational dynamics and possible allosteric linkage between the lobes are also present in the K isoform, we performed 15N spin-relaxation dispersion NMR measurements39 on 15N-labeled K-Ras-GppNHp (residues 1–171). Values of the transverse relaxation rate constant R2 for experiments with different Carr–Purcell–Meiboom–Gill (CPMG) frequencies were determined as described previously.14 Figure 6 demonstrates strong dependence of the R2 values on CPMG frequency observed for amide nitrogen spins in K-Ras-GppNHp, characteristic of an exchange process occurring on a microsecond–millisecond timescale.40 Data for residues K88 and L159 are shown as internal control—representative of residues that do not experience millisecond dynamics. The residues with quantifiable relaxation dispersions (V8, V9, N26, L79, T87, F90, D92, I93, H94, H95, K101, R102, V112 and R149) were fit with the two-state exchange model as described in Materials and Methods (Fig. 7a). Despite the fact that the residues are distributed throughout the entire sequence, the exchange rate constants for a number of residues cluster in a relatively narrow range supporting the hypothesis of a global conformational rearrangement. The Monte Carlo analysis of global fitting results yielded the following exchange parameters shown here as the maximum of the best-fit value distribution and its 2.5 and 97.5 percentiles: kex =1200 s−1 [1100 s−1, 1300 s−1], pa =0.89 [0.5, 0.93], with the chemical shift differences between exchanging conformers ranging from 0.6 to 1.4 ppm. Figure 7b shows the spatial distribution of these dynamic residues in a structural model of the G domain of K-Ras. This is indeed very similar dynamics to those we previously published for H-Ras.14
Fig. 6.
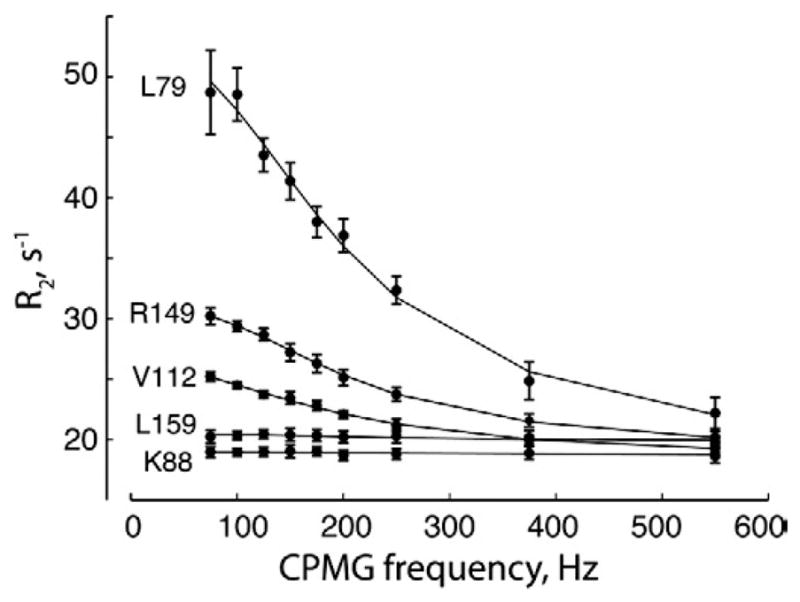
Representative relaxation dispersion profiles for K-Ras-GppNHp. The transverse relaxation rate constant of amide 15N nuclear spins in 1H–15N K-Ras (1–171) in complex with the GTP analogue GppNHp at 20 °C recorded at 14.1 T is plotted as a function of the CPMG pulse train frequency.
Fig. 7.

Conformational exchange dynamics in K-Ras-GppNHp. (a) Best-fit values of the exchange rate constants for K-Ras (1–171)/GppNHp at 20 °C are plotted versus the residue number. The 95% confidence intervals determined by Monte Carlo analysis are shown as asymmetric error bars. The value of the exchange rate constant determined from a global fit is shown as a horizontal continuous line with the corresponding 95% confidence interval given as a shaded area around the line. (b) Mapping of dynamic residues on the molecular model of K-Ras-GppNHp (PDB ID: 3GFT). The GTP analogue GppNHp is shown as green sticks, and the magnesium ion is shown as a yellow sphere. Amide nitrogen atoms of the residues involved in global conformational dynamics are shown as red spheres. Helices and switch regions are labeled. Blue indicates residues, which amide signals were not detected or assigned. The orientation of the molecule is the same as in Fig. 5a.
Discussion
Previous structure analysis of Ras-GppNHp has focused primarily on the active site containing switch I, switch II and the P-loop that surround the phosphate groups of the nucleotide. This part of the molecule contains most sites of oncogenic mutations and is within the effector lobe with which proteins interact to activate signal transduction in the cell. The complexes between Ras and effectors such as Raf,31 phosphoinositide 3-kinase41 and RalGDS42 show that they interact through the effector lobe, which, according to results of molecular dynamics simulations of full-length Ras-GTP in the membrane,18 is oriented toward the cytoplasm. Conversely, the Ras-GTP surface that interacts with the membrane, including the allosteric site, is completely within the allosteric lobe. The regulator proteins such as SOS (son of sevenless) and RasGAP, however, interact both at the effector lobe and at the interface between the two lobes. While GAPs interact with Ras-GTP to promote GTP hydrolysis by insertion of the arginine finger into the active site to stabilize the transition state in the hydrolysis reaction as originally reported,3 it also promotes a shift in helix 3 to the “on” state, facilitating the placement of switch II residues in the active site for catalysis. This is accomplished by the interaction of GAP residues within binding pockets at the interlobal region mapped in our MSCS experiments of the “off” state. This latter effect had not previously been noted, primarily because the canonical crystal structure of Ras-GppNHp available at the time, exemplified by PDB ID: 1CTQ, had a similar “on” state conformation as seen in the Ras/RasGAP complex. The “off” state for the wild-type H-Ras-GppNHp was first observed in our more recent structure obtained from a different crystal form, PDB ID: 2RGE. We hypothesize that, in the absence of GAPs or an activator of the allosteric switch, the “off” state of Ras-GTP is the major conformer in the cell. Presumably, the “off” state of the unligated Ras-GTP can recognize the GAP protein, and upon binding, the shift to the “on” state 1 occurs to promote GAP-catalyzed hydrolysis. In the case of intrinsic hydrolysis in the presence of Raf, we propose that Ca2+ and a yet undetermined negatively charged ligand associated with the membrane bind at the allosteric site. This would promote the shift in helix 3/loop 7 that allows placement of switch II residues for catalysis associated with the “on” state 2.12 Thus, in both GAP-catalyzed and intrinsic hydrolyses in the presence of Raf, the helix 3/loop 7 shifts away from the effector lobe, making room for switch II to adopt an ordered conformation for catalysis.
In the present work, we have combined two powerful and complementary methods for identifying binding site hot spots on the surface of Ras in its GTP-bound state. MSCS identified eight clusters, two of which are in the effector lobe (clusters 3 and 6), one in the allosteric lobe (cluster 2) and the remaining five, the majority, are in the interlobal region (clusters 1, 4, 5, 7 and 8). Only clusters 1 and 2, the strongest MSCS hot spots, were also present in the FTMap of the “off” state. Cluster 3 represents a hot spot for Ras binding domains such as the Raf-RBD, but targeting this site would be challenging due to its highly polar nature and the flexibility of the nearby switch I region in solution. Nonetheless, any attempt would benefit from the inclusion of the conserved water molecules as part of the protein template or a requirement that the ligand mimic the conserved water interactions with the protein. FTMap does not identify cluster 3, perhaps due to the fact that conserved water molecules were not included in the calculations. Also, it is in extensive crystal contacts between two Ras molecules in the crystal, whereas the FTMap only considered one molecule in the calculations. However, this is also the case for cluster 2, which FTMap picked up as a significant site.
MSCS clusters 4, 5, 6, 7 and 8 appear to be weaker hot spots with only two bound organic solvent molecules each in the MSCS data set. It is therefore not surprising that they do not appear in the FTMap calculations finding strong hot spots that are likely to be druggable.33 With the exception of cluster 6, these clusters are found in the interlobal region and are sites of interaction with protein binding partners of Ras. Interestingly, several are occupied simultaneously with one of the stronger hot spots (e.g., 3 and 7 in the case of Raf-RBD-CRD and 3, 5, 6 and 8 in the case of RasGAP). These weaker hot spots at the interlobal region provide valuable information because although they are unlikely to be good targets as individual sites, they could be targeted as a group or could be used in conjunction with one of the stronger sites in ligand design. What MSCS provides here is the location of hot spots at protein/protein interfaces, such as that of Ras/RasGAP, which might effectively be targeted by relatively small molecules. The interlobal region is particularly interesting in that the hot spots are close together and that ligand binding to multiple sites might be possible to shift the population equilibrium to the “on” state. This would be expected to facilitate hydrolysis of GTP, even in the context of a complex such as Ras/Raf, as effector binding sites are limited to the effector domain, leaving the interlobal sites largely unoccupied.
The FTMap calculations identified hot spots primarily on the allosteric lobe. Of the seven clusters identified by FTMap, only one is in the effector lobe (the Y71 site), and one is in the interlobal region (overlapping with MSCS cluster 1). These two sites are on either side of the highly flexible switch II, and for this reason, it may be difficult to target the “on” states selectively. For example, although one might reason that ligand binding at the cluster 1 site could promote a GAP-bound-like conformation of the “on” state based on the fact that it is the strongest FTMap cluster in the “on” state 1, our MSCS experiments show that organic solvents binding at this site appear to stabilize the “off” state with switch II in an anti-catalytic conformation.
The other five FTMap clusters are entirely within the allosteric lobe. The R128 and R135 sites between helix 4 and helix 5 have been established experimentally as important sites of protein/membrane interactions and, thus, could be targeted to interfere with signaling through this venue. The site between helix 3 and helix 4 (MSCS cluster 2) is also near the membrane and could be targeted in a similar way. The remaining two clusters identified by FTMap are near the allosteric site, on either side of loop 7. The allosteric site is highly polar and will be challenging to target, but any attempt should take into account the conserved water molecule found at this site. The loop 7 site appears only in the “on” state 2 and might be an interesting site to consider. Other than the loop 7 site, hot spots on the allosteric lobe appear both in the “off” and in at least one, if not both, “on” state. This is perhaps not surprising, given that the Ras interactions with the membrane are dependent on the state of the nucleotide rather than on conformational substates, with extensive interactions in Ras-GTP and less so in Ras-GDP.18
The highly dynamic nature of the effector lobe, particularly switch I and switch II, makes this region the least likely for successful ligand design. The extent of dynamics may be appreciated in Fig. 7b where blue coloring indicates extreme broadening of amide signals from residues in the effector lobe due to extensive conformational exchange. The residues affected less are shown as red spheres—they were amenable to quantitative measurements of spin relaxation. These residues collectively report on a correlated conformational exchange process, most likely, originating from the exchange-broadened effector lobe and penetrating deep into the G domain.
The effector lobe, traditionally, has been the major area of focus for targeting Ras; therefore, it is not surprising that results have been elusive. In this work, we identified novel hot spots in the allosteric lobe, which look very promising as targets for disrupting the Ras interactions with the membrane and effector proteins. These include the cluster 2 site between helix 3 and helix 4 and the R128 and R135 sites between helix 4 and helix 5 identified by FTMap, in addition to the allosteric site near loop 7 discovered previously.12 The conformational exchange in the effector lobe extends deep into the structure of the G domain reaching cluster 2 and the allosteric site, indicating that these remote hot spots might be involved in allosteric regulation of the effector lobe or might be used for inhibition of its functions. The observation of global conformation exchange in K-Ras-GppNHp that is highly similar to exchange dynamics in H-Ras14 also enables a hypothesis that all isoforms of Ras and perhaps other small monomeric G proteins may possess this feature. The future NMR and X-ray diffraction analyses of conformational diversity in Ras and related GTPases under variable conditions will reveal further details of conformational switching mechanism in the GTP-bound G domains and its functional significance.
Materials and Methods
Multiple solvent crystal structures
H-Ras residues 1–166 were expressed in Escherichia coli BL21 cells and purified in the GDP-bound form as previously described.28 The GDP was exchanged to GppNHp, and the protein was crystallized in 200 mM CaCl2 and 20% polyethylene glycol 3350 as previously published.27
Cross-linking and soaking in organic solvents
H-Ras-GppNHp crystals were transferred to 9-well sitting-drop glass plates containing 50 μL of stabilization buffer [20 mM Hepes (pH 7.5), 50 mM NaCl, 5 mM MgCl2, 1 mM DTT, 200 mM CaCl2 and 25% polyethylene glycol 3350]. The buffer was exchanged at least three times and then exchanged again with stabilization buffer containing 1% glutaraldehyde. The cross-linking reaction was allowed to proceed for 30 min to 1 h before a new exchange into stabilization buffer to remove the unreacted glutaraldehyde. Crystals were then soaked in the organic solvents at concentrations specified in Results.
Data collection and structure refinement
Synchrotron data were collected on the Southeast Regional Collaborative Access Team 22-ID beamline at the Advanced Photon Source (Argonne National Laboratories) with an X-ray wavelength of 1.0 Å and a MAR CCD detector. The protein coordinates taken from PDB ID: 2RGE were used for phasing in each case. The program CNS was used for reciprocal space refinement with 10% of the unique reflections set aside for calculation of Rfree.43 Coot44 was used for visualization of electron density maps. Organic solvent molecules were added to the models after refinement of crystallographic water molecules and were validated as previously described.24,28 Coordinates, topology and parameter files for the solvent molecules were taken from previous publications.24,25 The last round of refinement for each of the models was performed with PHENIX45 using the same Rwork and Rfree reflection sets as used in CNS. The organic solvent molecules were numbered in the PDB according to their binding site, so that all molecules in a given cluster in the superimposed MSCS structures have the same residue number.
FTMap calculations
The general strategy used in the FTMap calculations is summarized in Results. The details of the method were as previously published.8
NMR experiments
The gene for full-length K-Ras was provided by Professor Carol Williams at the Medical College of Wisconsin. The truncated version was obtained by inserting a stop codon after residue 171 (QuikChange II Kit; Stratagene). The gene was then inserted into a pET vector (Novagen) and sequenced. The untagged truncated K-Ras protein was expressed, refolded and purified as described previously for H-Ras.14 The protein yield was ~5 mg per 1 L of synthetic MJ medium. The protein was concentrated to 0.8–1.2 mM and exchanged to a buffer containing 20 mM Tris (pH 7.2), 10 mM NaCl, 5 mM MgCl2, 5 mM DTT and 0.01% NaN3 for NMR data collection.
NMR signal assignment of K-Ras-GppNHp was performed at pH 5.9 with resulting assignments transferred to pH 7.2 for relaxation studies. 13C–15N protein samples at a concentration of 0.6 mM were prepared in a buffer containing 200 mM sodium phosphate at pH 5.9, 10 mM MgCl2, 1 mM DTT, 1 mM GppNHp, 0.01% NaN3 and 10% D2O in a thin-walled Shigemi tube with the final volume of 330 μL. Triple-resonance experiments were performed at 298.15 K on a Bruker Avance II spectrometer equipped with a cryoprobe at the field strength of 14.1 T. Backbone experiments included HNCO, HN(CA)CO, HN(CO)CA, HN(CO)CACB, HNCA and HNCACB with small modifications.46 Spectra were processed with NMRPipe and viewed with Sparky†.47 The PINE (Probabilistic Interaction Network of Evidence) algorithm was used with PINE-SPARKY to automate and import assignments into Sparky.48,49 Backbone 1H, 13C, 15N chemical shifts have been deposited in the Biological Magnetic Resonance Data Bank.50 Transfer of assignments to pH 7.2 was accomplished by pH titrations and further confirmed using three-dimensional 1H–15N–1H nuclear Overhauser enhancement spectroscopy and total correlated spectroscopy heteronuclear single quantum coherence experiments.
Relaxation dispersion measurements
The CPMG spin-relaxation measurements of transverse relaxation constants of the amide 15N nucleus were performed for K-Ras-GppNHp using a procedure similar to that described for H-Ras-GppNHp.14 The relaxation-compensated CPMG (rcCPMG) experiments39 modified for off-resonance artifact suppression as previously described51 were carried out at two static magnetic field strengths of 11.7 T and 14.1 T using Bruker Avance II and Avance III instruments equipped with cryoprobes. Relaxation dispersions of 15N–1H K-Ras, residues 1–171, were recorded at 20 °C, pH 7.2 (the sample buffer contained 10 mM NaCl, 10 mM Tris, 5 mM MgCl2, 1 mM DTT and 0.01% NaN3). The K-Ras (1–171) was complexed with GTP mimic GppNHp. The samples also contained 1 mM GppNHp for sample stability.
To assure accuracy of the CPMG measurements, we performed a series of control experiments to test the effect of fast pulse rates on the cryoprobe performance. The CPMG periods in the pulse sequence used lower power than the rest of the pulse program with 15N 90° refocusing pulses calibrated to 100 μs or 50 μs (while the high-power 90° pulse length was 34.5 μs).
First, we measured the variation of the sample temperature at the practical durations of the CPMG pulse train. The NEOPTIX fiber optics sensor was inserted in the sample tube containing the working buffer, and rcCPMG experiment was initiated. During the pulse program operation, the steady-state temperature of the sample was observed and did not increase beyond 0.1 °C (accuracy of the sensor) from the idle state at both power levels.
Second, we determined whether the radio frequency power applied to the probe coils changed its steady-state performance. The duty cycle compensation was implemented as previously described,52 and the effect of rcCPMG experiment on a steady-state temperature of the cryogenic probe coils was recorded. The strategy of this measurement was based on understanding that the steady-state temperature of the cryogenic coils is maintained at 25 K by continuously cooling the coils in a probe head by a helium gas chilled below 25 K in a cryoplatform. Helium gas temperature is adjusted to a set point of 25 K by a regulated feedback-driven heater. When coil temperature rises, the temperature controller reduces current in the heater, thus reducing temperature of the helium gas coming to the probe head to balance the heating effect of the experiment. The Bruker “NMR Heat” parameter reflects this current, and in all cases, we did not see a drop bigger than 10% of initial value with most of the effect attributed not to the application of CPMG pulse trains but to a routine 15N decoupling during a detection period.
Third, we assessed the degree of transient, short-term detuning of the cryogenic probe coils during the CPMG pulse train at highest (555 and 1110 Hz) CPMG frequencies. Even in the presence of a stream of cold helium, one might expect a fast rise of coil temperature during the pulse train followed by a fast drop afterwards, which would not be compensated by a relatively slow-feedback-driven temperature control. To assess an impact of this effect on the coil tuning, we carried out calibration of the 90° pulse length before and immediately after the CPMG pulse train (by using switchable 270° pulses prior and after the CPMG period). We did not observe any measurable detuning of the Bruker Cryoprobe due to the application of the CPMG pulse trains of up to 30 ms long. These results indicated that cryoprobe head is capable of maintaining its temperature very efficiently during rcCPMG experiments.
Relaxation dispersion analysis
Fitting of the relaxation dispersion profiles was performed using software programs CPMG_FITD8 (Dr. Dmitry Korzhnev) and BiophysicsLab (created in-house). Data were analyzed with a two-state model utilizing the all-timescale analytical solution,53 all-timescale solution for skewed populations (pA > 0.87)54 and the equation for a fast-exchange approximation.55 Fitting of experimental R2 data yielded values of the exchange rate constants (kex), chemical shift difference between the exchanging conformations (Dω) and populations of states (pA and pB) for every resolved peak of sufficient dispersion of R2 values. Interdependence of fitting parameters (correlation) was assessed via a covariation matrix computed by CPMG_FITD8 and directly by Monte Carlo simulations in BiophysicsLab. Analysis of Rex dependence on static magnetic field56 combined with comparative analysis of fitting with multiple equations established fast-intermediate exchange regime for most residues. Monte Carlo analysis with BiophysicsLab resulted in families of fitting results enabling calculations of asymmetric confidence intervals. All data sets were analyzed individually and then globally to determine the consensus exchange parameters.
Accession numbers
Coordinates and structure factors for the 10 MSCS models of Ras-GppNHp were deposited in the PDB with the following PDB IDs (also given in Table 2): cross-linked in aqueous solution, 3RRY; soaked in 50% ETF, 3RS2; soaked in 50% IPA, 3RS7; soaked in 70% GOL, 3RRZ; soaked in 90% RSF, 3RSL; soaked in 20% RSG, 3RSO; soaked in 60% HEZ, 3RS4; soaked in neat YEG, 3RS0; soaked in neat HEX, 3RS3; soaked in 55% DMF, 3RS5.
Backbone 1H, 13C, 15N chemical shifts have been deposited in the Biological Magnetic Resonance Data Bank under accession number 17785.
Acknowledgments
The initial stages of MSCS structure refinement were performed by the following students in the class of C.M. entitled Macromolecular Modeling as part of a project funded by the National Science Foundation: Jason Baucom, Byung Chung, Michelle Dechene, Ralph House, Revonda Pokrzywa, Helen Rho, Austin Rowshan and Cheng Wan. G.B. and Paul Swartz helped teach the laboratory sections of this class. The use of the Advanced Photon Source at the Argonne National Laboratory was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38. This research was funded in part by National Institutes of Health (R01-CA096867) and National Science Foundation (MCB-0818678) grants to C.M.
Abbreviations used
- ETF
2,2,2-trifluoroethanol
- IPA
isopropanol
- GOL
glycerol
- RSF
R,S,R-bisfuranol
- RSG
S, R,S-bisfuranol
- HEZ
1,6-hexanediol
- YEG
cyclopentanol
- HEX
hexane
- DMF
dimethylformamide
- MSCS
multiple solvent crystal structures
- GAP
GTPase-activating protein
- PDB
Protein Data Bank
- CPMG
Carr–Purcell–Meiboom–Gill
- rcCPMG
relaxation-compensated CPMG
Footnotes
Author Contributions. Joint first authors for their respective contributions to MSCS (Greg Buhrman), NMR (Casey O’Connor) and FTMap calculations (Brandon Zerbe).
References
- 1.Bourne HR, Sanders DA, McCormick F. The GTPase superfamily: conserved switch for diverse cell functions. Nature. 1990;348:125–132. doi: 10.1038/348125a0. [DOI] [PubMed] [Google Scholar]
- 2.Boriack-Sjodin PA, Margarit SM, Bar-Sagi D, Kuriyan J. The structural basis of the activation of Ras by Sos. Nature. 1998;394:337–343. doi: 10.1038/28548. [DOI] [PubMed] [Google Scholar]
- 3.Scheffzek K, Ahmadian MR, Kabsch W, Wiesmüller L, Lautwein A, Schmitz F, Wittinghofer A. The Ras–RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science. 1997;277:333–338. doi: 10.1126/science.277.5324.333. [DOI] [PubMed] [Google Scholar]
- 4.Herrmann C. Ras–effector interactions: after one decade. Curr Opin Struct Biol. 2003;13:122–129. doi: 10.1016/s0959-440x(02)00007-6. [DOI] [PubMed] [Google Scholar]
- 5.Cox AD, Der CJ. The dark side of Ras: regulation of apoptosis. Oncogene. 2003;22:8999–9006. doi: 10.1038/sj.onc.1207111. [DOI] [PubMed] [Google Scholar]
- 6.McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007;1773:1263–1284. doi: 10.1016/j.bbamcr.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev, Mol Cell Biol. 2008;9:517–531. doi: 10.1038/nrm2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brenke R, Kozakov D, Chuang GY, Beglov D, Hall D, Landon MR, et al. Fragment-based identification of druggable “hot spots” of proteins using Fourier domain correlation techniques. Bioinformatics. 2009;25:621–627. doi: 10.1093/bioinformatics/btp036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clackson T, Wells JA. A hot spot of binding energy in a hormone–receptor interface. Science. 1995;267:383–386. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- 10.Milburn M, Tong L, deVos AM, Brünger A, Yamaizumi Z, Nishimura S, Kim SH. Molecular switch for signal transduction: structural differences between active and inactive forms of protooncogenic Ras proteins. Science. 1990;247:939–945. doi: 10.1126/science.2406906. [DOI] [PubMed] [Google Scholar]
- 11.Bourne HR, Sanders DA, McCormick F. The GTPase superfamily: conserved structure and molecular mechanism. Nature. 1991;349:117–127. doi: 10.1038/349117a0. [DOI] [PubMed] [Google Scholar]
- 12.Buhrman G, Holzapfel G, Fetics S, Mattos C. Allosteric modulation of Ras positions Q61 for a direct role in catalysis. Proc Natl Acad Sci USA. 2010;107:4931–4936. doi: 10.1073/pnas.0912226107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buhrman G, Kumar VS, Cirit M, Haugh JM, Mattos C. Allosteric modulation of Ras-GTP is linked to signal transduction through RAF kinase. J Biol Chem. 2011;286:3323–3331. doi: 10.1074/jbc.M110.193854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’Connor C, Kovrigin EL. Global conformational dynamics in Ras. Biochemistry. 2008;47:10244–10246. doi: 10.1021/bi801076c. [DOI] [PubMed] [Google Scholar]
- 15.Gorfe AA. Mechanisms of allostery and membrane attachment in Ras GTPases: implications for anti-cancer drug discovery. Curr Med Chem. 2010;17:1–9. doi: 10.2174/092986710789957832. [DOI] [PubMed] [Google Scholar]
- 16.Hancock JF. Ras proteins: different signals from different locations. Nat Rev, Mol Cell Biol. 2003;4:373–384. doi: 10.1038/nrm1105. [DOI] [PubMed] [Google Scholar]
- 17.Abankwa D, Hanzal-Bayer M, Ariotti N, Plowman SJ, Gorfe AA, Parton RG, et al. A novel switch region regulates H-ras membrane orientation and signal output. EMBO J. 2008;27:727–735. doi: 10.1038/emboj.2008.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gorfe AA, Hanzal-Bayer M, Abankwa D, Hancock JF, McCammon JA. Structure and dynamics of the full-length lipid-modified H-Ras protein in a 1,2-dimyristoylglycero-3-phosphocholine bilayer. J Med Chem. 2007;50:674–684. doi: 10.1021/jm061053f. [DOI] [PubMed] [Google Scholar]
- 19.Gorfe AA, Grant BJ, McCammon JA. Mapping the nucleotide and isoform-dependent structural and dynamical features of Ras proteins. Structure. 2008;16:885–896. doi: 10.1016/j.str.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abankwa D, Gorfe AA, Inder K, Hancock JF. Ras membrane orientation and nanodomain localization generate isoform diversity. Proc Natl Acad Sci USA. 2010;107:1130–1135. doi: 10.1073/pnas.0903907107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heesen H, Schlitter AM, Schlitter J. Empirical rules facilitate the search for binding sites on protein surfaces. J Mol Graphics Modell. 2007;25:671–679. doi: 10.1016/j.jmgm.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 22.Jayakanthan M, Wadhwa G, Mohan TM, Arul L, Balasubramanian P, Sundar D. Computer-aided drug design for cancer-causing H-Ras p21 mutant protein. Lett Drug Des Discovery. 2009;6:14–20. [Google Scholar]
- 23.Palmioli A, Sacco E, Abraham S, Thomas CJ, Di Domizio A, De Gioia L, et al. First experimental identification of Ras-inhibitor binding interface using a water-soluble Ras ligand. Bioorg Med Chem Lett. 2009;19:4217–4222. doi: 10.1016/j.bmcl.2009.05.107. [DOI] [PubMed] [Google Scholar]
- 24.Dechene M, Wink G, Smith M, Swartz P, Mattos C. Multiple solvent crystal structures of ribonuclease A: an assessment of the method. Proteins. 2009;76:861–881. doi: 10.1002/prot.22393. [DOI] [PubMed] [Google Scholar]
- 25.Mattos C, Bellamacina CR, Peisach E, Pereira A, Vitkup D, Petsko GA, Ringe D. Multiple solvent crystal structures: probing binding sites, plasticity and hydration. J Mol Biol. 2006;357:1471–1482. doi: 10.1016/j.jmb.2006.01.039. [DOI] [PubMed] [Google Scholar]
- 26.Chuang GY, Mehra-Chaudhary R, Ngan CH, Zerbe BS, Kozakov D, Vajda S, Beamer LJ. Domain motion and interdomain hot spots in a multidomain enzyme. Protein Sci. 2010;19:1662–1672. doi: 10.1002/pro.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buhrman G, Wink G, Mattos C. Transformation efficiency of RasQ61 mutants linked to structural features of the switch regions in the presence of Raf. Structure. 2007;15:1618–1629. doi: 10.1016/j.str.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buhrman G, de Serrano V, Mattos C. Organic solvents order the dynamic switch II in Ras crystals. Structure. 2003;11:747–751. doi: 10.1016/s0969-2126(03)00128-x. [DOI] [PubMed] [Google Scholar]
- 29.Vetter IR, Arndt A, Kutay U, Gorlich D, Wittinghofer A. Structural view of the Ran–importin β interaction at 2.3 Å resolution. Cell. 1999;97:635–646. doi: 10.1016/s0092-8674(00)80774-6. [DOI] [PubMed] [Google Scholar]
- 30.Sasaki AT, Carracedo A, Locasale JW, Anastasiou D, Takeuchi K, Kahoud ER, et al. Ubiquitination of K-Ras enhances activation and facilitates binding to select downstream effectors. Sci Signal. 2011;4:ra13. doi: 10.1126/scisignal.2001518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nassar N, Horn G, Herrmann C, Block C, Janknecht R, Wittinghofer A. Ras/Rap effector specificity determined by charge reversal. Nat Struct Biol. 1996;3:723–729. doi: 10.1038/nsb0896-723. [DOI] [PubMed] [Google Scholar]
- 32.Hu CD, Kariya K, Tamada M, Akasaka K, Shirouzu M, Yokoyama S, Kataoka T. Cysteine-rich region of Raf-1 interacts with activator domain of post-translationally modified Ha-Ras. J Biol Chem. 1995;270:30274–30277. doi: 10.1074/jbc.270.51.30274. [DOI] [PubMed] [Google Scholar]
- 33.Landon MR, Lancia DR, Jr, Yu J, Thiel SC, Vajda S. Identification of hot spots within druggable binding regions by computational solvent mapping of proteins. J Med Chem. 2007;50:1231–1240. doi: 10.1021/jm061134b. [DOI] [PubMed] [Google Scholar]
- 34.Kozakov D, Hall DR, Chuang GY, Cencic R, Brenke R, Grove LE, et al. Structural conservation of druggable hot spots in protein–protein interfaces. Proc Natl Acad Sci USA. 2011 doi: 10.1073/pnas.1101835108. 1101835108, [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spoerner M, Herrmann C, Vetter IR, Kalbitzer HR, Wittinghofer A. Dynamic properties of the Ras switch I region and its importance for binding to effectors. Proc Natl Acad Sci USA. 2001;98:4944–4949. doi: 10.1073/pnas.081441398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spoerner M, Hozsa C, Poetzl JA, Reiss K, Ganser P, Geyer M, Kalbitzer HR. Conformational states of human rat sarcoma (Ras) protein complexed with its natural ligand GTP and their role for effector interaction and GTP hydrolysis. J Biol Chem. 2010;285:39768–39778. doi: 10.1074/jbc.M110.145235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mattos C. Protein–water interactions in a dynamic world. Trends Biochem Sci. 2002;27:203–208. doi: 10.1016/s0968-0004(02)02067-4. [DOI] [PubMed] [Google Scholar]
- 38.Saxena N, Lahiri SS, Hambarde S, Tripathi RP. RAS: target for cancer therapy. Cancer Invest. 2008;26:948–955. doi: 10.1080/07357900802087275. [DOI] [PubMed] [Google Scholar]
- 39.Loria JP, Rance M, Palmer AG. A relaxation-compensated Carr–Purcell–Meiboom–Gill sequence for characterizing chemical exchange by NMR spectroscopy. J Am Chem Soc. 1999;121:2331–2332. [Google Scholar]
- 40.Palmer AG., III NMR characterization of the dynamics of biomacromolecules. Chem Rev. 2004;104:3623–3640. doi: 10.1021/cr030413t. [DOI] [PubMed] [Google Scholar]
- 41.Pacold ME, Suire S, Perisic O, Lara-Gonzalez S, Davis CT, Walker EH, et al. Crystal structure and functional analysis of Ras binding to its effector phosphoinositide 3-kinase γ. Cell. 2000;103:931–943. doi: 10.1016/s0092-8674(00)00196-3. [DOI] [PubMed] [Google Scholar]
- 42.Huang L, Hofer F, Martin GS, Kim SH. Structural basis for the interaction of Ras with RalGDS. Nat Struct Biol. 1998;5:422–426. doi: 10.1038/nsb0698-422. [DOI] [PubMed] [Google Scholar]
- 43.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr, Sect D: Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 44.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 45.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, et al. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr, Sect D: Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 46.Markley JD. Macromolecular structure determination by NMR spectroscopy. Struct Bioinformatics. 2009;2:93–142. [Google Scholar]
- 47.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 48.Bahrami A, Assadi AH, Markley JL, Eghbalnia HR. Probabilistic interaction network of evidence algorithm and its application to complete labeling of peak lists from protein NMR spectroscopy. PLoS Comput Biol. 2009;5:e1000307. doi: 10.1371/journal.pcbi.1000307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee W, Westler WM, Bahrami A, Eghbalnia HR, Markley JL. PINE-SPARKY: graphical interface for evaluating automated probabilistic peak assignments in protein NMR spectroscopy. Bioinformatics. 2009;25:2085–2087. doi: 10.1093/bioinformatics/btp345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ulrich EL, Akutsu H, Doreleijers JF, Harano Y, Ioannidis YE, Lin J, et al. BioMagResBank. Nucleic Acids Res. 2008;36:D402–D408. doi: 10.1093/nar/gkm957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yip GN, Zuiderweg ER. A phase cycle scheme that significantly suppresses offset-dependent artifacts in the R2-CPMG 15N relaxation experiment. J Magn Reson. 2004;171:25–36. doi: 10.1016/j.jmr.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 52.Yip GN, Zuiderweg ER. Improvement of duty-cycle heating compensation in NMR spin relaxation experiments. J Magn Reson. 2005;176:171–178. doi: 10.1016/j.jmr.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 53.Carver JP, Richards RE. A general two-site solution for the chemical exchange produced dependence of T2 upon the Carr–Purcell pulse separation. J Magn Reson. 1972;6:89–105. [Google Scholar]
- 54.Ishima R, Torchia DA. Estimating the time scale of chemical exchange of proteins from measurements of transverse relaxation rates in solution. J Biomol NMR. 1999;14:369–372. doi: 10.1023/a:1008324025406. [DOI] [PubMed] [Google Scholar]
- 55.Luz Z, Meiboom S. Nuclear magnetic resonance study of the protolysis of trimethylammonium ion in aqueous solution—order of the reaction with respect to solvent. J Chem Phys. 1963;39:366–370. [Google Scholar]
- 56.Millet O, Loria JP, Kroenke CD, Pons M, Palmer AG. The static magnetic field dependence of chemical exchange linebroadening defines the NMR chemical shift time scale. J Am Chem Soc. 2000;122:2867–2877. [Google Scholar]